
Abstract
Invariant natural killer T cell (iNKT) subsets are differentially distributed in various immune organs. However, it remains unclear whether iNKT cells exhibit phenotypical and functional differences in different peripheral organs and how thymic iNKT cells emigrate to peripheral organs. Here, we used single-cell RNA-seq to map iNKT cells from peripheral organs. iNKT1 cells from liver, spleen, and lymph node appear to have distinct phenotypic profiles and functional capabilities. However, iNKT17 transcriptomes were comparable across peripheral organs. In addition, by integrating data with a thymic iNKT cell study, we uncovered a transient population of recent thymic emigrants, a cluster of peripheral iNKT cells with high expression of transcription factor Kruppel-like factor 2 (Klf2). Deletion of Klf2 led to a severe impairment of iNKT differentiation and migration. Our study revealed that iNKT subsets are uniquely distributed in peripheral organs with some inter-local tissue variation, especially for iNKT1 cell, and identified Klf2 as a rheostat for iNKT cell migration and differentiation.
Similar content being viewed by others


Introduction
Invariant natural killer T (iNKT) cells are innate-like T cells that are initially selected by CD1d and preferentially use an invariant T cell receptor (TCR) consisting predominantly of the Vα14-Jα18/Vβ8 pair in mice1,2,3. iNKT cells were known to develop in the thymus through a four-stage process in mice: stage 0 (CD24+), stage 1 (CD24−CD44−NK1.1−), stage 2 (CD44+NK1.1−), and stage 3 (CD44+NK1.1+). During development, thymic iNKT cells display substantial functional heterogeneity, with three major subsets, including iNKT1, iNKT2, and iNKT17. These three major functional subsets exhibit distinct transcription factors and cytokine production. For instance, iNKT1 cells are T-bet+ and mainly produce IFN-γ; iNKT2 cells are PLZFhi and mainly produce IL-4; and iNKT17 cells are RORγt+ and produce IL-17. iNKT1 cells develop through all stages and finally mature in stage 3, which also highly express cytolytic effectors (perforin, granzyme B, granzyme A, and FAS ligand) and specific chemokines and their receptors. However, iNKT2 and iNKT17 cells terminate at stage 2. Stage 2 iNKT cells have a high proliferation capability, and most iNKT cells at this stage emigrate to peripheral organs4.
After emigrating from the thymus, iNKT cells are differentially distributed in various peripheral organs. For instance, only a small subset of iNKT cells traffic through the lymph nodes (0.2%–1%), with the largest iNKT cell populations localizing to the liver (12%–30%), lung (5%–10%), and spleen (1%–3%)5,6,7. Most iNKT cells are tissue-resident and noncirculating4,8, with iNKT1 cells being dominant in the liver, while lymph node shows enrichment for iNKT17 cells, and spleen and lungs show preference to iNKT2 cells. This is thought to be mediated by the differences in chemokine receptor expression profiles in iNKT cells and tissue microenvironment9,10,11. Many studies have demonstrated that iNKT cells play a critical role in various pathological conditions, including cancer, autoimmune disease, and infection, and iNKT cells from different organs appear to have distinct functional capabilities2,12,13. More specifically, recent studies suggest that iNKT cells from spleen, thymus, and liver exhibit different anti-tumor activity in varying tumor models9,14, including MCA-1 sarcoma and B16F10 melanoma metastasis models. However, the underlying mechanism of iNKT functional differences in different organs is still unclear.
iNKT cell emigration from the thymus to peripheral organs mainly occurs at stage 2, in a Ccr7-dependent fashion. However, Ccr7 is also a lineage-defining marker for iNKT multipotent precursors15, which makes studying recent thymic emigrants (RTE) difficult. In addition, RTEs are a rare population relative to total peripheral iNKT cells and thus the mechanisms responsible for iNKT thymic emigration and iNKT cell distribution in peripheral organs remain unknown.
Several pioneering studies, including our own, have indicated that thymic iNKT cells are more plastic than their defined iNKT1/2/17 sublineages16. However, whether the thymic iNKT clusters are conserved in peripheral organs is still not quite clear. In this study, we extended our analysis of subsets of iNKT cells in peripheral organs. We performed single-cell RNA-sequencing (scRNA-seq) of iNKT cells from liver, spleen, and lymph node and revisited our scRNA-seq of thymic iNKT cells. We found that there are substantial differences between iNKT cells in the thymus and in peripheral organs. More importantly, in peripheral organs, iNKT cells from liver and spleen showed great commonality, but iNKT cells from lymph node showed great phenotypical differences with iNKT from either liver or spleen, especially for iNKT1 cells. Via integration with thymic iNKT cells, we identified an RTE cluster among peripheral iNKT cells, which highly express Klf2. Studies from Klf2 deletion mouse models indicated that Klf2 is a key regulator for iNKT cell differentiation and migration. Taken together, our data constitute a comprehensive analysis of the similarities and differences among functionally distinct peripheral iNKT cells and provide a valuable resource for future disease model studies.
Results
Overview of the cell types in peripheral iNKT cells identified by scRNA-seq
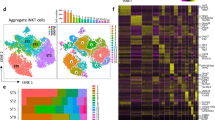
We previously studied cellular heterogeneity of mouse thymic iNKT cells using scRNA-seq. To capture the extent of cellular heterogeneity within mouse peripheral iNKT cells, we applied scRNA-seq (10X genomics chromium) on iNKT cells from six samples representing three peripheral tissues, including spleen, liver, and lymph node (Fig. 1a, b). After quality control (Supplementary Fig. S1a, b), a total of 5570 individual iNKT cells (sample #1: 2760; sample #2: 2810) from liver; 4850 individual iNKT cells (sample #1: 2562; sample #2: 2288) from spleen; and 4897 individual iNKT cells (sample #1: 2408; sample #2: 2489) from lymph node were assessed for single-cell RNA expression. iNKT cells from three organs that we profiled in replicates were well correlated (Spearman’s coefficient: 0.92 between liver iNKT cells; 0.89 between splenic iNKT cells, and 0.91 between lymph node iNKT cells) (Supplementary Fig. S1c).

a iNKT cells collected from peripheral organs including liver, spleen, and lymph node for scRNA-Seq analysis. b Sorting strategy of iNKT cells from liver, spleen, and lymph node post CD8+B220+ deletion enrichment. c UMAP plots from 10X genomics scRNA-Seq dataset from sorted peripheral iNKT cells along with thymic different stages of iNKT cells collected. Displaying relationships between iNKT cell organs origins. d UMAP plots of data identical to those in c, but color coded on iNKT cell clusters. e Heatmap of the top ten differentially regulated genes from each cluster derived from d. Each column represents gene expression for an individual cell with color coded on gene expression profiles. Yellow is upregulated and purple is downregulated. f Bar graph showing the cell number in each of clusters. g The fractions of ten clusters defined in peripheral iNKT cells. h Pearson correlation matrix of the average expression profiles based on all differentially expressed genes from all clusters analyzed.
After processing the sequencing data using the Cell Ranger pipeline (10X Genomics), we performed unbiased clustering analysis of peripheral iNKT cells with thymic iNKT cells (over 17,000 cells) using Seurat. iNKT cells from peripheral organs cluster uniquely from thymic iNKT cells (Fig. 1c), reflecting substantially different transcription programs between thymic iNKT cells and peripheral iNKT cells. Among integrated peripheral iNKT cells, a total of 10 clusters (C1–C10) were identified (Fig. 1c–e), with as few as 123 cells to as many as 4474 cells per cluster (Fig. 1f, g), with some clusters (C4–C10) shared between iNKT cells from different organs.
We used differential gene expression analysis to determine cell type-specific marker genes with highly different transcriptional levels between clusters. The top 10 genes from individual clusters were shown in heatmap (Fig. 1e). Correlation analysis of marker gene signatures revealed that similar cell types clustered together (Supplementary Fig. S1d). For initial analysis, we found that most iNKT cells from liver, spleen, and lymph node were clearly separated, indicating that tissue location is associated with specific transcriptomic program in these iNKT cells (Fig. 1c), e.g., cluster C1 from liver, C2 from spleen, and C3 from lymph node (Fig. 1d), and the similarity of different clusters from different organs were shown in Fig. 1h. These clusters were recognized as main iNKT clusters with natural killer properties; however, we found that the C4–C10 clusters were mixed across all samples (Fig. 1c, d), independent of their location nature. These data suggest that an intrinsic iNKT program is the main determinant of the transcriptional profile of C4–C10 iNKT cells. iNKT cells from lymph node made a major contribution to C4, which highly expressed iNKT17 signature genes, including Rorc, Pxdc1, and Aqp3. Cluster C5 shows high levels of Ccr7 and S1pr1 that are associated with cell homing. C6 shows high levels of Ifit1 and Ifit3. Given that Ifit1/3 function as inhibiting viral replication and translational initiation, we assumed that C6 iNKT cells may be associated with anti-viral function. C7 shows high expression level of Gzma, which is associated with cellular cytotoxic properties. C8–C9 show a great overlap with thymic stage 1 and 2 iNKT cells with high expression of genes regulating the cell cycle (discussed below). Clusters identified in mRNA levels were also verified with available antibodies at protein levels. As shown in Supplementary Fig. S2a–c, RORγt+ iNKT17 cells (C4) in different peripheral organs showed a dramatical increased Aqp3 expression, while IFIT1 (C6) had an increased expression pattern as compared with conventional T cells in different peripheral organs. In addition, Gzma (C7) was identified in iNKT cells from lymph nodes, spleen, and liver, with varying expression enrichment, especially in NK1.1+ mature iNKT cells.
Defining the distinct subsets of iNKT cells in peripheral organs
To explore iNKT cell diversity in more detail, we annotated 10 clusters from peripheral organs with assumed cell-type identities based on known marker genes derived from published expert annotation16. Among these 10 peripheral iNKT cell clusters, we noticed that clusters C8–C9 were mainly proliferating cells showing high expression levels of S and G2M cell-cycle marker genes (Fig. 2a, b; Supplementary Fig. S3a). To avoid the possibility that proliferating genes were affecting cellular cluster analysis, we isolated these clusters and corrected the gene expression levels for cell cycle phase. Subsequent unsupervised clustering analysis revealed that cell clusters were similar to those in primary data (data not shown), indicating that cell-cycle genes did not disturb cell cluster analysis in this study.

a UMAP plots depicting single-cell genes expression trajectory of G1, G2/M, and S phages in peripheral iNKT cell development. b Bar graph represents fraction of G1, G2/M, and S cells in iNKT cells clusters. c Bubble plots showing gene expression in individual clusters (C1–C10) from aggregated iNKT cells. Gene names labeled in blue are iNKT17 signature genes, in red are iNKT1 signature genes and in green are iNKT2 signature genes. X axis shows different clusters identified in Fig. 1d. d Bar graph showing IPA analysis of iNKT1 cells in different peripheral organs.
iNKT1/2/17 subsets have been identified based on their transcription factor and cytokine production profiles. Although these differentiated iNKT subsets initially emerge in the thymus, it is likely that tissue-specific factors and local environmental influences shape the phenotype and function after recruitment to specific tissue sites. Therefore, we dissected peripheral iNKT cells into transcriptionally distinct subpopulations, which could allow us to access the transcriptional landscape in a more detailed iNKT functional model.
Based on published lineage markers of iNKT cells lineages16, we found that clusters C1–C3, clusters C6 (Ifit1/Ifit3), and C7 (Gzma) were categorized into iNKT1 subsets. These cells expressed Ifng and various NK cell lectin-like receptors, such as Klra1, Klrb1, and Klrc2, which may regulate cytotoxic properties. In this study, most iNKT1 cells from different organs were not clustered together, e.g., C1 from liver, C2 from spleen, and C3 from lymph node, indicating that iNKT1 cells exhibit tissue-specific variations in phenotype; clusters C8–C10 from three peripheral organs show highly expressing Gata3, Icos, and Izumol1r (Supplementary Fig. S3b), were assigned as iNKT2 cells; cluster C4, mainly from lymph node, and a small fraction from spleen and liver, were categorized into iNKT17 subset, expressing high levels of Rorc, Il17rb, Il23r, Tmem176b, Ccr6, and Ccr8 (Fig. 2c). Given that iNKT17 cells from all different peripheral organs cluster together as single C4, this suggests that unlike iNKT1 cells, iNKT17 transcriptional programs might not be dependent on specific tissue environment. Cells in C5 had a high expression of the homing marker, Ccr7 (Fig. 1e). Interestingly, this cluster did not stand out in any iNKT subsets, showing an undifferentiated status that could represent RTE iNKT cells. In addition, we found that iNKT17 cells were identical across different peripheral organs and iNKT2 cells showed proliferative activity. However, iNKT1 cells differ from organ to organ. Ingenuity pathway analysis (IPA) data indicated that iNKT1 from lymph nodes enriched in T cell receptor signaling, Tec Kinase signaling, and T helper cell differentiation pathways. iNKT1 from liver showed a higher cellular proliferation status (Fig. 2d).
Comparison of transcriptomic profiles among iNKT cells from different peripheral organs
iNKT cells in different lymphoid organs display distinct tissue tropisms, which might be mediated by local environment. Previous studies have indicated that iNKT cells from different organs mediate a different functional capability2,9,14. Therefore, we compared iNKT cells from different peripheral organs. As shown in Fig. 3a–c, we detected five distinct clusters in peripheral iNKT cells from liver, spleen, and lymph node. Violin plots with the indicated UMAP plot show the top two signatures of each cluster in indicated peripheral organs; the top 10 signatures for each cluster were provided in Supplementary Fig. S4a–c. iNKT1 clusters found in all three organs (Lv_C1 in liver, Spl_C1 in spleen, and Ln_C1 in lymph node) were all assigned to iNKT1, but with different transcriptomic profiles. Ln_C5 in lymph node, Spl_C3 in spleen, and Lv_C4 in liver were identified as an Ifit1/3 cluster, which also showed iNKT1 characteristics; Ln_C4 in lymph node, Spl_C4 in spleen, and Lv_C3 in liver were cell cycle clusters; Ln_C2 in lymph node, Spl_C5 in spleen, and Lv_C5 in liver were identified as iNKT17 cells. In addition, we observed that Lv_C2 with Gadd45b, Icam1, Nfkbia, Irf8, Relb, and Ifng expression was found only in liver (Supplementary Fig. S4d). Further signaling pathway analysis indicated that besides NK cell-mediated cytotoxicity pathway, this cluster was much more enriched in NF-κB signaling pathway and T cell receptor signaling pathway (Supplementary Fig. S4e). This result was initially curious to us, because T cell-NF-κB is important for IFN-γ production and plays an important role in anti-tumor immunity17.

a–c UMAP plots from 10X genomics scRNA-Seq dataset from sorted liver iNKT cell(a), spleen iNKT cell (b), and Lymph node iNKT cells (c) (left). Violin plots showing the top two cluster-specific signatures in iNKT cells from liver (a), spleen (b), and lymph node (c). d Pearson correlation matrix of the average expression profiles based upon all differentially expressed genes from all indicated subpopulations analyzed. e Gene list showing significant change of iNKT1 signatures among iNKT cells from thymus, liver, spleen, and lymph node, extracted from (d).
To test whether iNKT cells in the thymus and in different peripheral organs display similar genomic profiles, we re-visited our previous single-cell study of thymic iNKT cells (data not published). With these data, we carried out a scRNA-seq-based comparison between assigned clusters among iNKT cells from indicated organs (Fig. 3d). These data reflected the extensive transcriptomic differences between iNKT cells from thymus, liver, spleen, and lymph node. There was less overlap of transcriptomic expression between iNKT1 from thymus and peripheral organs (Lv_C1, Spl_C1, and Ln_C1), where the Silhouette coefficient was −0.15 for liver iNKT1 cells, 0 for spleen iNKT1 cells, and −0.05 for lymph node iNKT1 cells relative to thymic iNKT1 cells (Fig. 3d). iNKT1 cells were also shown to be tissue-specific in peripheral organs, where the Spearman’s coefficient was 0.13 between liver and spleen, 0.32 between spleen and lymph node, and 0.09 between liver and lymph node (Fig. 3d). Interestingly, we observed that anti-apoptotic gene Bcl2 and suppressor of cytokine signaling 2 (Socs2) were highly expressed in iNKT cells from liver, but not in iNKT cells from spleen and lymph node (Supplementary Fig. S5a). The differences in Bcl2 and Socs2 expression by iNKT cells in different organs could explain the enrichment of iNKT cell population in liver. Given that the Ifit1/3 cluster was identified in all organs, we explored their similarity among organs, and we observed a strong correspondence of Ifit1/3 clusters among peripheral organs (Spearman’s coefficient was 0.82 between liver and spleen, 0.78 between spleen and lymph node, and 0.81 between liver and lymph node). However, the Spearman’s coefficient between iNKT cells from the thymus and peripheral organs varied from −0.01 to 0.02 (Fig. 3d). A similar phenomenon was also observed in iNKT17 clusters, namely, a high correlation in iNKT17 among peripheral organs (Spearman’s coefficient is 0.69 between liver and spleen; 0.74 between spleen and lymph node; and 0.67 between liver and lymph node); however, the coefficient between thymic iNKT17 cells and peripheral iNKT17 varied from −0.06 to 0.013 (Fig. 3d). These data indicate that iNKT17 cells exhibit a nearly identical transcriptome in peripheral organs.
Comparison of populations in heatmaps is provided in Supplementary Fig. S5b–d. These analyses again highlighted many similarities between iNKT cells from liver, spleen, and lymph node, and showed how transcriptomes change when iNKT cells emigrate from the thymus to different peripheral organs. This includes genes that were differentially expressed between iNKT1 cells from the liver vs the thymus (Fig. 3e; Supplementary Fig. S5b). iNKT17 cells show a lower expression ratio of Furin, Emb, Rorc, Il17a, Avpl1, and Ly6a in peripheral organs, as compared with those from the thymus. However, there was no marked difference in transcriptomic profiles of iNKT17 cells in different peripheral organs (Supplementary Fig. S5c). In addition, the Ifit1/3 cluster from peripheral organs exhibits a great similarity between different organs, but they were more pronounced in iNKT cells from peripheral organs (Fig. 3d; Supplementary Fig. S5d). Overall, these data indicate that iNKT cells from the thymus exhibit a great transcriptomic difference from those in peripheral organs, and transcriptomic profiles of iNKT1 cells correlated poorly within different peripheral organs; however, the transcriptional patterns were highly correlated for iNKT17 cells and the Ifit1/3 cluster among peripheral organs.
Homing signature profiles of iNKT cells from peripheral organs
iNKT cells exhibit altered patterns of tissue localization, suggesting differences in the signals regulating homing and homeostasis. To directly investigate the iNKT cell migration and redistribution mechanism, we integrated different developmental stages of thymic iNKT cells with peripheral iNKT cell (Fig. 4a). We found that cluster C5 iNKT cells from the peripheral organs included candidates for newly arrived iNKT cells that share similarities with stage 1/2 thymic iNKT cell precursors and express high levels of Ccr715. Therefore, this cluster was annotated as an RTE cluster. Consistently, we found multiple homing markers showing similar pseudotime trajectories with Ccr7, including S1pr1, Sell, and Klf2 (Fig. 4b). In addition, pseudotime analysis carried out using Monocle 318,19,20 showed three trajectories branching from this intermediate RTE cluster toward terminally differentiated peripheral iNKT cells (Fig. 4c). These RTEs are the youngest iNKT cells, showing less differentiation/maturity (Fig. 2c). Consistently, we observed that T-bet+ iNKT1, PLZFhi iNKT2, and RORγt+ iNKT17 cells were substantially underrepresented amongst RTE iNKT cells (CCR7+ S1PR1+) compared to non-RTE iNKT cells (CCR7−S1PR1−) (Fig. 4d).

a UMAP plots showing relationships between iNKT cells from different peripheral organs. b UMAP plots showing multiple homing markers expression in integrated thymic and peripheral iNKT cells. c The ordering of iNKT cells along pseudotime in a state-space defined by Monocle 3. Each color represents an iNKT cluster. d Flow cytometry showing RTEs (CCR7+S1PR1+) and non-RTEs (CCR7–S1PR1−) from indicated organs. iNKT1(PLZFloT-bethi), iNKT2 (PLZFhiRORγt−), and iNKT17(PLZFintRORγt+) cells in RTEs and non-RTEs from indicated organs.
To further describe the iNKT cell migration and relocation properties, we characterized the basis of tissue residency of different subsets in spleen, liver, and lymph node by evaluating the expression of the core circulatory and tissue-resident signatures that were reported in a recent study21, on the basis of effector CD8 T cells. A gradient was observed in RTEs with an increased circulatory signature (including Klf2, Sell, S1pr1, and S1pr4) and a decrease of tissue residency signatures (including Atf3, Cd244, Cd69, Fos, and Jun) (Fig. 5a). As RTEs developed and matured in peripheral organs, they lost circulatory signatures, but obtained residency signatures, except cluster C7, which expressed a relative increase in expression of Klf2, S1pr1, Klrb1c, Bin2, and Fam65b (Fig. 5a). We also found that clusters C3 and C4 of iNKT cells from lymph node expressed high levels of most of the tissue residency signature genes, including markers of direct TCR activation (Jun, Fos, Junb, Jund, Dnala1, Dnala4, Dusp1, Icos, Ppp1r15a, Prdx6, Ptp4a1, Opct, Tnfaip3, and Zfp36l1) (Fig. 5a; Supplementary Fig. S6a). iNKT cells from liver and spleen exhibited a higher circulatory property than iNKT cells in lymph node, and this pattern is more pronounced in liver (Fig. 5a).

a Dot plots showing circulatory signatures and residency signature expression in the 10 clusters identified in peripheral iNKT cells. b UMAP plots showing re-cluster analysis of C5_RTEs from Fig. 1d (top). Bar graph showing the fraction of individual clusters occupied in iNKT cells from peripheral and thymus (Stage 0, 1, 2). c Heatmap of the top ten differentially regulated genes from each cluster derived from b. Each column represents gene expression for an individual cell with color coded on gene expression profiles. Yellow is upregulated and purple is downregulated.
To understand the preferential localization of iNKT cells in peripheral organs, we re-clustered RTEs, and three sub-clusters were further identified: C5_1, C5_2, and C5_3 (Fig. 5b, c). Among these sub-clusters, we found the following: spleen RTEs were mainly located in C5_1, expressing high levels of Lgas1 and S100a6, liver RTEs were mainly assigned in C5_2 with high expression of Irf8 and Xcl1, and lymph node RTEs were mainly located in C5_3, expressing high levels of Dapl1, Sell, and Tsc22d3. These data indicated that RTEs, those newly migrated out from thymus, exhibit a greatly different transcriptomic profile. Interestingly, some immature iNKT cells from the thymus were clustered together with lymph node cells in C5_3. The great proximity in a UMAP plot highlighted the similar transcriptional expression program between thymic stage 1/2 iNKT cells and lymph node RTEs. More precisely, we found that RTEs in lymph node show higher levels of Ccr7 and Sell, but lower Zbtb16 and Tbx21 expression levels relative to stage 1/2 iNKT cells. In contrast, RTEs from spleen and liver showed a clear differentiation potential, as judged by higher levels of iNKT subsets signatures (Supplementary Fig. S6b).
Klf2 regulates iNKT cell migration and differentiation
The migration patterns of iNKT cells are associated with the expression of distinct chemokine receptors, but the underlying molecular mechanism for this regulation is unknown. Transcription factor Klf2 has been reported to regulate the migration of conventional αβT cells and γδT cells by restricting chemokine receptor expression patterns22,23,24. In our study, Klf2 was found to be the top gene that was most positively associated with Ccr7 in RTEs (C5). To test the potential role of Klf2 in iNKT cell development and migration, we examined iNKT cell subsets in Lck-Cre Klf2 deletion mice. We first examined the frequency of iNKT cells in the thymus from 6- to 8-week-old Klf2 KO and WT mice by flow cytometry. As shown in Fig. 6a, Klf2 KO mice showed a significantly increased frequency of iNKT cells in the thymus compared to WT, even though the absolute number are comparable. Further analysis on developmental stages showed higher frequencies and absolute numbers of stage 0, 1, and 2 iNKT cells in Klf2 KO mice (Fig. 6b). In contrast, stage 3 iNKT cell frequency and absolute number were significantly reduced in Klf2 KO mice. These data suggest that deletion of Klf2 blocked iNKT cell development prior to terminal stage 3 maturation.

a Representative flow plots of thymic iNKT cells from Klf2 KO and WT mice (left); bar graphs represent means ± SD of frequency and cell number (right), n = 3 for Klf2 KO and WT controls. Data represent two independent experiments. b Representative flow plots of different stages (stage 0: CD24+; stage 1: CD44lo NK1.1−; stage 2: CD44hiNK1.1−; stage 3: CD44hiNK1.1+) of iNKT from Klf2 KO and WT mice (left); bar graphs represent means ± SD of frequencies and cell numbers of different developmental stages of iNKT cells in Klf2 KO and WT controls (right). c, d Representative flow plots of peripheral iNKT cells from Klf2 KO and WT mice. Bar graphs represent means ± SD of frequencies and cells numbers of iNKT cells in indicated organs from Klf2 KO and WT controls. Data represent two independent experiments. e Histogram showing CCR7 expression in thymic iNKT cell from WT and Klf2 KO mice. f Bar graph showing mean fluorescence intensity (MFI) of CCR7 in iNKT cells from WT and Klf2 KO mice. g Representative flow plots showing Ki-67 expression in different developmental stages of thymic iNKT cell from WT and Klf2 KO mice. h Histogram showing PLZF, T-bet, and RORγt expression in different developmental stages of thymic iNKT cell from WT and Klf2 KO mice.
Given that immature iNKT cells with migration potential were blocked and accumulated in the thymus of mice with Klf2 deletion (Fig. 6b), we measured iNKT population in peripheral organs. Consistent with a defect in iNKT cell migration from the thymus, the frequency and absolute numbers of iNKT cells in peripheral organs, including spleen, liver, and lung, were dramatically diminished in Klf2 KO mice as compared with WT controls, with the notable exception of the lymph nodes (Fig. 6c, d). These data indicated that Klf2 mediates iNKT cell migration into peripheral organs, especially for spleen, liver, and lung. Evidence to date supports Klf2 controlling T cell migration by directly regulating the cell surface receptors S1P1 and CCR723,25. To test whether Klf2 controls iNKT cells emigration using a similar mechanism, we measured CCR7 and S1PR1 expression patterns in different developmental stages of iNKT cells from Klf2 KO and WT. As shown in Fig. 6e, f, CCR7 was consistently reduced in immature stages of iNKT cell from Klf2 KO mice. Interestingly, S1PR1 expression in Klf2 KO mice was consistently reduced at stage 1/2 of iNKT cell development but not at terminally differentiated stage 3 (Supplementary Fig. S7a). Taken together, these data suggest that Klf2 promotes thymic egress of stage 1/2 iNKT cells by regulating homing receptor patterns.
Normally, iNKT cells proliferate and expand briskly in stages 1/2. To test whether Klf2 influenced the proliferating capability of iNKT cells, we examined the rate of Ki-67 expression in both Klf2 KO and WT cells. Compared to WT, Klf2 KO iNKT cells exhibited a lower proliferation status at different developmental stages in the thymus (Fig. 6g). Interestingly, we observed that during development from stage 1 to stage 3, Klf2 KO iNKT cells expressed lower levels of PLZF, T-bet, and RORγt, the master regulators for iNKT1/2/17 differentiation (Fig. 6h), and they showed poor differentiation potential (Supplementary Fig. S7b), indicating that Klf2 is required for iNKT cell differentiation. Therefore, not only contributing to tissue localization, Klf2 may also play important roles in regulating iNKT cell effector differentiation.
Discussion
In this study, we systematically analyzed iNKT cell transcriptomic features in various peripheral organs including liver, spleen, and lymph node. A total of ten distinct clusters were identified in integrated peripheral iNKT cells, and iNKT cells from different peripheral organs showed great phenotypic and functional differences, especially iNKT1 cells. In addition, we identified a merged undifferentiated RTE iNKT cluster with high Klf2 expression. More importantly, Klf2 was further recognized as an essential regulator in iNKT migration and differentiation.
Previous studies highlighted a great level of complexity in thymic iNKT cells and presented a model of iNKT cell development. By integrating scRNA-seq of thymic iNKT cells and peripheral iNKT cells, we found that thymic iNKT cells and peripheral iNKT cells are clearly separated in UMAP plot, with a small RTE cluster connecting them.
Peripheral iNKT cells exhibited substantial heterogeneity in their degree of maturation. In contrast to thymic iNKT cells, which require CD1d for their initial selection and maturation, iNKT cells do not require continual CD1d interactions in the periphery to support homeostatic proliferation, long-term survival, or to maintain tissue distribution26. However, iNKT cells require several chemokine receptors and integrins for their maintenance in the specific peripheral organs and these interactions between iNKT cells and the microenvironment help generate organ-specific iNKT populations. For example, iNKT cells located in the spleen partially require Cxcl13; however, Cxcl13 is not required for iNKT cells in liver. Meanwhile, iNKT cells that home to the liver require Cxcr3 and Itgβ2, yet these receptors do not contribute to splenic migration4,27,28. Even though iNKT cells have a restricted TCR profile, there were functional differences from organ to organ. Previous reports have shown that iNKT cells from the liver and spleen have different anti-tumor activities14. More precisely, iNKT cells from liver, including CD4+ iNKT cells and CD4– iNKT cells were better able to reject tumor cells than their counterparts from the spleen or thymus. However, the organ-specific mechanisms for iNKT cells functional differences are still unclear9. In our study, we revealed that transcriptome profiles of iNKT cells were organ-specific, especially for iNKT1 cells. There was a marked phenotypical difference between thymic iNKT1 cells and peripheral iNKT1 cells, even among peripheral organs, iNKT1 cells also exhibited a significant difference. iNKT cells from liver and spleen exhibited a higher circulatory property than iNKT cells in lymph node, and this pattern is more pronounced in liver. In addition, iNKT1 cells in lymph node showed great TCR activation properties. These differences should reflect: (1) anatomical tissue structure, e.g., iNKT cells locate at the parenchyma in spleen, but locate at vasculature in liver29; (2) conventional microbiota in local environment and specific antigenic stimulation in the indicated organs30; and (3) organ-specific antigen-presenting cells (APCs) could also contribute to iNKT cell function and phenotype in local environments. Previous studies suggested that different levels of costimulatory molecules on organ-specific APCs cause different iNKT cell responses. APCs in different organs may also express different tissue-specific glycolipid ligands for iNKT cells31. For example, APCs in the liver might capture and present exogenous glycolipids from the alimentary tract, which could promote IFN-γ-dependent functions32,33. However, unlike iNKT1 cells, which are organ-specific, iNKT17 cells are very similar transcription-wise in different peripheral organs, indicating that Rorc is the major determinant for the iNKT17 cell phenotype.
iNKT cells first exit the thymus in a phenotypically and functionally immature state and require a period of post-thymic maturation before transitioning into the mature/effector iNKT cell compartment. Here, we found that the transcriptional program of RTEs was very different from those of mature iNKT cells in spleen, liver, and lymph node. For example, (1) RTEs in peripheral organs are not well differentiated; (2) RTEs in peripheral organs exhibit a higher proliferating activity; (3) RTEs in peripheral organs exhibit pronounced circulatory properties; (4) RTEs in peripheral organs express high level of homing signatures, including transcription factor Klf2. Previous reports have shown that Klf2 transactivates S1p1r, Ccr7, and Sell promoters, and that deletion of Klf2 leads to accumulation of CD4+ and CD8+ αβT cells in the thymus and preferential homing to peripheral tissues24,25. In this study, we found that Klf2 was mainly expressed by stage 1 and 2 iNKT cells in the thymus and RTEs in peripheral tissues. Loss of Klf2 led to a defect in iNKT cell emigration; a result that is supported by previous studies15. Our scRNA-seq study also offered an exciting opportunity for mapping peripheral RTEs. Pseudotime analysis showed that Klf2 and Ccr7 have a similar expression trajectory and they had a fine correlation during iNKT cell development. Deletion of Klf2 showed severely impaired iNKT cell differentiation and migration. Consistent with this finding, Ccr7 expression was significantly reduced in Klf2-deficient iNKT cells. A previous report indicated that deletion of Klf2 mediated by CD4Cre caused increased expansion or survival of thymic PLZF+ T cells34, and therefore promoted memory-like phenotype (CD44hi, CD122hi) CD8 T cells. However, expressing PLZF+ T cells in mouse thymus may include iNKT cell, γδNKT cells, and MAIT cells, Klf2 function in iNKT cell development were not fully explored. In our current study, we used LckCre Klf2 KO mice, in which Klf2 was deleted in the early stage of T cells development. We found that the frequency of iNKT cells was increased in thymus from Klf2-deficient mice, and most of these iNKT cells were blocked at immature stages (stage 1 and stage 2). Interestingly, peripheral iNKT cells from Lck Cre Klf2 KO mice were significantly reduced. Integrating analysis with scRNA-seq data suggested that Klf2 regulate iNKT cells migration, and deletion of Klf2 blocked thymic iNKT cells outward migration. Therefore, our study further expands previous study and explained the mechanism of Klf2 in regulation thymic iNKT cells.
In addition, it is important to mention that extreme cytotoxic cluster C7, with high levels of Gzma and Ccl5 expression, also exhibit a high expression level of Klf2 and other circulatory signatures but fewer residency signatures. Cluster C7 and C5_RTE clusters showed similar expression programs, judged by their proximity in UMAP plots. We therefore assumed that cluster C7 could be relatively new iNKT cells that directly migrate from thymus, rather than long-term iNKT cells that are from super mature iNKT1 cells. However, their precise developmental trajectory is still unclear. In addition, it is not clear whether specific organ targeting is determined in the thymus by distinct patterns of integrin and chemokine receptor expression or if migration to a given tissue is stochastic, only dependent on subset-specific niches. More important, even though our study revealed the phenotypic differences of iNKT cells in different peripheral organs, we are still unclear about how these phenotypic differences contribute to varying function in different disease models.
In summary, we demonstrate here that iNKT cells from liver, spleen, and lymph node appear to have distinct phenotypic profiles and functional capabilities, especially for iNKT1 cells. Meanwhile, we identified Klf2 in peripheral RTEs as playing a critical role in iNKT cell differentiation and migration.
Materials and methods
Mice
C57BL/6 were purchased from Jackson Laboratory (Bar Harbor, ME). Lck-Cre Klf2 KO mice were provided by Prof. Eric Sebzda (Wayne University, Detroit, MI). Briefly, mice carrying a floxed allele of Klf2 (Klf2fl/fl)35 were mated to transgenic C57BL/6 mice expressing Cre recombinase under the guidance of a proximal Lck promoter (obtained from the Jackson Laboratory), to generate Lck-cre;Klf2fl/fl conditional knockout mice (Klf2 KO). iNKT cells from peripheral organs from 5-week-old C57BL/6 mice were utilized for scRNA-seq study; 6–8-week-old, sex-matched mice were utilized for Klf2 function studies. All studies, protocols, and mouse handling were approved by the Institutional Animal Care and Use Committee.
Flow cytometry gating strategy and antibodies
Single-cell suspensions were washed twice with FACS staining buffer (1× PBS, 2% FBS) and incubated with Fc block (clone 2.4G2). Cells were stained with anti-mouse PBS57-loaded CD1d-tetramer (provided by the NIH Tetramer Core Facility). The following fluorescence conjugated antibodies were used: anti-TCRβ (H57-597), anti-CD24 (M1/69), anti-CD44 (IM7), anti-NK1.1 (PK136), anti-RORγt (B2D), anti-PLZF (Mags.21F7), anti-T-bet (eBio4B10 (4B10)), anti-Ki-67 (520914), anti-CCR7 (4B12), anti-S1PR1 (JM10-66), anti-IFIT1 (OTI3G8), anti-Aqp3 and anti-Gzma (GzA-3G8.5). Cell surface staining was performed with staining buffer; intranuclear staining was performed with eBioscience Fixation/permeabilization buffer. The flow cytometry assay was performed through BD FACSCelesta and data were analyzed using FlowJo V10.2 software. Gating strategy: after gating on lymphocyte, doublets were excluded by using forward scatter (FSC) and side scatter (SSC), mouse iNKT cells were further identified as TCRβ+ CD1d-tetramer+.
Mouse iNKT cell enrichment and sorting
Mouse iNKT cell enrichment and sorting strategy was described previously36. Briefly, peripheral organs from mouse, including spleen, liver, and lymph nodes were harvested from 5-week-old C57BL/6 mice. For the enrichment of iNKT cells, total cells were stained with biotin-conjugated anti-mouse CD8 Ab, anti-mouse B220 Ab, and anti-biotin magnetic beads (eBioscience). Negatively selected CD8− and B220− cells were then stained with anti-mouse TCRβ, CD1d-tetramer Abs. iNKT cells of whole population were further sorted from C57BL/6 mouse spleen, lymph node, and liver using FACSAria II Usage, cells collected with purity > 97%.
scRNA-seq library generation
scRNA-seq library generation was described in our previous published study37. Two biological repeats for each samples (including iNKT cells from spleen, liver, and lymph node) of scRNA-seq libraries were generated using the 10X Genomics Chromium Single Cell 3′ Reagent Kit (v2 Chemistry) and Chromium Single Cell Controller as previously described38.
scRNA-seq data analysis
Sequence reads from scRNA-seq libraries were demultiplexed and aligned to the mm10 mouse reference, barcode processed, and UMI counted using the 10X Genomics Cell Ranger (V3.1.0) pipeline38. Estimated number of cells captured per sample was between 2646 and 2867 with 60,622–70,044 mean reads per cell, 1788–1388 median genes per cell, and 1468–3597 median UMI counts per cell. A total of 16,587 cells with 2179 UMI counts/cell in average were selected via Cell Ranger for further analysis for all of six samples. Datasets were subsequently analyzed using the R Seurat package39,40. Principle Component Analysis (PCA) was employed to analyze combined samples. Quality control metrics employed are as follows. We employed two strategies to identify potential doublets. First, cells expressing both Xist and Y chromosome genes (Kdm5d, Eif2s3y, Gm29650, Uty, and Ddx3y) were excluded from the dataset. Second, cells expressing uncharacteristically high numbers of genes (> 4000) were excluded. Low-quality cells were excluded based on a low number of genes detected (<300) and/or having high mitochondrial genetic content (> 15%). A total of 14,986 genes in 15,317 cells passed these quality control measures. Genes removed include ribosomal structural proteins (as identified by gene ontology term GO: 0003735 and the Ribosomal Protein Gene (RPG) database 4), non-coding rRNAs, Hbb, and genes not expressed in ≥ 3 cells. A total of 14,986 genes in 15,317 cells passed these quality control measures.
A global-scaling normalization method “LogNormalize” in Seurat was employed to normalize gene expression measurements of each cell by the total expression, multiplying this by a factor of 10,000, followed by log-transformation. Highly variable genes in each data analysis were identified, and the intersecting top 3000 genes in each dataset were used for clustering and downstream analyses. Datasets underwent scaling and regressing on the number of detected molecules per cell (nUMI) and the percentage of mitochondrial gene content (pct.mito). The number of principal components (PCs) used to cluster cells was determined by manual inspection of the scree plot. After identifying the number of PCs to be included for downstream analyses (20 PCs), a graph-based clustering approach implemented in Seurat was used to iteratively cluster cells into groups, based on similarities of those components among cells. The UMAP method was utilized to visualize resulting clusters. To assess the effects of cell cycle heterogeneity, cell cycle phase scores (G2/M and S phases) were calculated based on canonical markers and used to regress out the data41. The FindAllMarkers function in Seurat was then implemented to identify differentially expressed genes between clusters with a fold-change of >2 and a Bonferroni adjustment of P value < 0.05 as a statistical significance threshold. To determine if differentially expressed genes belong to identifiable groups, pathway analysis was carried out using the Ingenuity Pathway Analysis (IPA, Qiagen Bioinformatics, Redwood City, CA).
Statistical analysis
For comparison between groups, statistical analysis was performed by unpaired t test with GraphPad Prism 8.0.
Data availability
ScRNA-seq that support the findings of this study have been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE130184 and GSE161495, respectively. All relevant data are available from the authors upon reasonable request.
References
Das, R., Sant’Angelo, D. B. & Nichols, K. E. Transcriptional control of invariant NKT cell development. Immunol. Rev. 238, 195–215 (2010).
Kumar, V. & Delovitch, T. L. Different subsets of natural killer T cells may vary in their roles in health and disease. Immunology 142, 321–336 (2014).
Bendelac, A., Savage, P. B. & Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 25, 297–336 (2007).
Thomas, S. Y. et al. PLZF induces an intravascular surveillance program mediated by long-lived LFA-1-ICAM-1 interactions. J. Exp. Med. 208, 1179–1188 (2011).
Benlagha, K., Weiss, A., Beavis, A., Teyton, L. & Bendelac, A. In vivo identification of glycolipid antigen-specific T cells using fluorescent CD1d tetramers. J. Exp. Med. 191, 1895–1903 (2000).
Hammond, K. J. et al. CD1d-restricted NKT cells: an interstrain comparison. J. Immunol. 167, 1164–1173 (2001).
Matsuda, J. L. et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192, 741–754 (2000).
Crosby, C. M. & Kronenberg, M. Tissue-specific functions of invariant natural killer T cells. Nat. Rev. Immunol. 18, 559–574 (2018).
Seino, K. & Taniguchi, M. Functionally distinct NKT cell subsets and subtypes. J. Exp. Med. 202, 1623–1626 (2005).
Slauenwhite, D. & Johnston, B. Regulation of NKT cell localization in homeostasis and infection. Front Immunol. 6, 255 (2015).
Lee, Y. J. et al. Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity 43, 566–578 (2015).
Torina, A., Guggino, G., La Manna, M. P. & Sireci, G. The Janus face of NKT cell function in autoimmunity and infectious diseases. Int. J. Mol. Sci. 19, 440 (2018).
Berzins, S. P., Smyth, M. J. & Baxter, A. G. Presumed guilty: natural killer T cell defects and human disease. Nat. Rev. Immunol. 11, 131–142 (2011).
Crowe, N. Y. et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J. Exp. Med. 202, 1279–1288 (2005).
Wang, H. & Hogquist, K. A. CCR7 defines a precursor for murine iNKT cells in thymus and periphery. Elife 7, 34793 (2018).
Engel, I. et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat. Immunol. 17, 728–739 (2016).
Barnes, S. E. et al. T cell-NF-kappaB activation is required for tumor control in vivo. J. Immunother. Cancer 3, 1 (2015).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Milner, J. J. et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017).
Odumade, O. A., Weinreich, M. A., Jameson, S. C. & Hogquist, K. A. Kruppel-like factor 2 regulates trafficking and homeostasis of gammadelta T cells. J. Immunol. 184, 6060–6066 (2010).
Carlson, C. M. et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442, 299–302 (2006).
Sebzda, E., Zou, Z., Lee, J. S., Wang, T. & Kahn, M. L. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat. Immunol. 9, 292–300 (2008).
Bai, A., Hu, H., Yeung, M. & Chen, J. Kruppel-like factor 2 controls T cell trafficking by activating L-selectin (CD62L) and sphingosine-1-phosphate receptor 1 transcription. J. Immunol. 178, 7632–7639 (2007).
McNab, F. W. et al. The influence of CD1d in postselection NKT cell maturation and homeostasis. J. Immunol. 175, 3762–3768 (2005).
Johnston, B., Kim, C. H., Soler, D., Emoto, M. & Butcher, E. C. Differential chemokine responses and homing patterns of murine TCR alpha beta NKT cell subsets. J. Immunol. 171, 2960–2969 (2003).
Kim, E. Y., Lynch, L., Brennan, P. J., Cohen, N. R. & Brenner, M. B. The transcriptional programs of iNKT cells. Semin. Immunol. 27, 26–32 (2015).
Liew, P. X. & Kubes, P. Intravital imaging—dynamic insights into natural killer T cell biology. Front. Immunol. 6, 240 (2015).
Mallevaey, T. & Selvanantham, T. Strategy of lipid recognition by invariant natural killer T cells: ‘one for all and all for one’. Immunology 136, 273–282 (2012).
Yang, Y. et al. Control of NKT cell differentiation by tissue-specific microenvironments. J. Immunol. 171, 5913–5920 (2003).
Mattner, J. et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 434, 525–529 (2005).
Kinjo, Y. et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature 434, 520–525 (2005).
Weinreich, M. A., Odumade, O. A., Jameson, S. C. & Hogquist, K. A. T cells expressing the transcription factor PLZF regulate the development of memory-like CD8+ T cells. Nat. Immunol. 11, 709–716 (2010).
Lee, J. S. et al. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev. Cell 11, 845–857 (2006).
Wang, J. et al. miR-183-96-182 cluster is involved in invariant NKT cell development, maturation, and effector function. J. Immunol. 203, 3256–3267 (2019).
Zhou, L. et al. Single-cell RNA-seq analysis uncovers distinct functional human NKT cell sub-populations in peripheral blood. Front. Cell Dev. Biol. 8, 384 (2020).
Zheng, G. X. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Acknowledgements
We thank the National Institutes of Health Tetramer Core Facility for supplying CD1d tetramers for mouse iNKT cell flow cytometry analysis and for cell sorting. This work was partially supported by Henry Ford Immunology Program Grants T71017 (to L.Z.) and T71016 (to Q.-S.M.) and National Institutes of Health Grants 1R01 AI119041-01A1 (to Q.-S.M.), 1R61AR076803-01 (Q.-S.M. and I.A.), 1R56AI119041-01 (to Q.-S.M.).
Author information
Authors and Affiliations
Contributions
Q.-S.M. and L.Z. designed the study; J.W., T.L., K.S., and M.M.H. performed the experiments; X.W. prepared single-cell RNA-seq libraries; I.L., J.W., and I.A. performed single-cell sequence processing with 10X cell Ranger, Seurat, and Signac; E.S. generated LckCre-Klf2 KO mice; J.W., I.L., L.Z., and Q.-S.M. wrote the manuscript, which was reviewed and approved by all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Loveless, I., Adrianto, I. et al. Single-cell analysis reveals differences among iNKT cells colonizing peripheral organs and identifies Klf2 as a key gene for iNKT emigration. Cell Discov 8, 75 (2022). https://doi.org/10.1038/s41421-022-00432-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-022-00432-z
This article is cited by
-
Unique adipose tissue invariant natural killer T cell subpopulations control adipocyte turnover in mice
Nature Communications (2023)
