
Abstract
Secondary resistance to imatinib (IM) represents a major challenge for therapy of gastrointestinal stromal tumors (GISTs). Aberrations in oncogenic pathways, including autophagy, correlate with IM resistance. Regulation of autophagy-related protein 5 (ATG5) by the ubiquitin-proteasome system is critical for autophagic activity, although the molecular mechanisms that underpin reversible deubiquitination of ATG5 have not been deciphered fully. Here, we identified USP13 as an essential deubiquitinase that stabilizes ATG5 in a process that depends on the PAK1 serine/threonine-protein kinase and which enhances autophagy and promotes IM resistance in GIST cells. USP13 preferentially is induced in GIST cells by IM and interacts with ATG5, which leads to stabilization of ATG5 through deubiquitination. Activation of PAK1 promoted phosphorylation of ATG5 thereby enhancing the interaction of ATG5 with USP13. Furthermore, N6-methyladenosine methyltransferase-like 3 (METTL3) mediated stabilization of USP13 mRNA that required the m6A reader IGF2BP2. Moreover, an inhibitor of USP13 caused ATG5 decay and co-administration of this inhibitor with 3-methyladenine boosted treatment efficacy of IM in murine xenograft models derived from GIST cells. Our findings highlight USP13 as an essential regulator of autophagy and IM resistance in GIST cells and reveal USP13 as a novel potential therapeutic target for GIST treatment.
Similar content being viewed by others

Introduction
Gastrointestinal stromal tumors (GISTs) are the most commonly soft tissue sarcoma of the gastrointestinal tract. GISTs originate from the interstitial cells of Cajal (ICCs) or other precursor cells and are driven mainly by overexpressed KIT tyrosine kinase that is implicated in lineage commitment and survival [1]. Despite targeted drugs have revolutionized treatment of GIST, many patients experience tumor recurrence within 2 years due to drug resistance [2,3,4] Thus, drug resistance mechanisms must be characterized to provide successful long-term treatments.
The pivotal role that autophagy plays in GIST progression has emerged recently [5]. Autophagy is a highly conserved catabolic process [6], which is modulated by a range of autophagy-related genes (ATGs), ensuring the ordered circulation and degradation of cytoplasmic proteins and impaired intracellular organelles [7]. ATG5 plays a critical role in regulating autophagosome formation [8]. Although accumulating evidence indicates that IM causes protective autophagy in GIST [9], the mechanisms that underlie this protection and the dysregulation of ATG5 in GIST have not been deciphered.
Deubiquitinating enzymes (DUBs) are a diverse set of proteases that catalyze the removal of ubiquitin moieties from target proteins to offset the action of substrate ubiquitination [10,11,12], which commonly are misregulated in many tumors and play critical roles in tumorigenesis and progression [13, 14]. USP13 is an important member of the USP subfamily that controls the ubiquitination status of diverse substrates that are involved in multiple processes, including cell cycle regulation, autophagy, metabolism, DNA repair response, and innate antiviral immunity [15,16,17,18,19]. However, whether USP13 plays a role in GIST biology remains unclear.
Here, we identify USP13 as a key factor that mediates drug resistance through regulating the polyubiquitylation of ATG5. As PAK1-mediated phosphorylation at residue T101 protects ATG5 from ubiquitination-dependent degradation [20], we further validate that this phosphorylation is critical for the binding and deubiquitination of ATG5 with USP13. Methyltransferase-like 3 (METTL3) is an N6-methyladenosine (m6A) that acts widely on RNA. We demonstrate that aberrant expression of USP13 in GIST is related to METTL3-induced m6A modification which maintains the stability of USP13 mRNA.
Materials and methods
Cell culture
The human cell lines GIST-T1 and GIST-882 were maintained in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37 °C in 5% CO2. The KIT exon 13 deletion GIST-882 cell line and KIT exon 11 deletion GIST-T1 were supplied by Biowit Technologies (Shenzhen, China).
Construction of drug-resistant cell lines
GIST-882R and GIST-T1R were stable IM-resistant cell lines that originated from the respective sensitive parental lines after screening for 3 years. Next-generation sequencing validated that no secondary mutations occurred in the GIST cell lines.
Antibodies and reagents
The following antibodies were used: rabbit monoclonal anti-ATG5 (ab221604; Abcam, Cambridge, UK); mouse monoclonal anti-USP13 (ab109264; Abcam); rabbit monoclonal anti-LC3B (ab192890; Abcam); rabbit polyclonal anti-β actin (66009-1-Ig; Proteintech, Wuhan, China); rabbit monoclonal anti-IGF2BP2 (ab124930, UK); rabbit polyclonal anti-P62 (18420-1-AP; Proteintech); rabbit polyclonal anti-USP3 (12490-1-AP; Proteintech); rabbit polyclonal anti-OTUD4 (25070-1-AP; Proteintech); mouse monoclonal anti-USP32 (sc-374465; Santa Cruz Biotechnology, Dallas, USA); rabbit monoclonal anti-Myc (ab32072; Abcam); rabbit monoclonal anti-GST (ab138491; Abcam); rabbit monoclonal anti-PAK1 (ab223849; Abcam); rabbit monoclonal anti-cleaved Parp (5625; Cell Signaling Technology, Massachusetts, USA); rabbit monoclonal anti-cleaved Caspase3 (9664; Cell Signaling Technology); rabbit monoclonal anti-Flag (14793; Cell Signaling Technology); rabbit monoclonal anti-HA (ab236632; Abcam); and, mouse monoclonal anti-Ubiquitin (646302; Biolegend, San Diego, USA). 3-Methyladenine (3-MA), Chloroquine (CQ), MG-132 proteasome inhibitor and cycloheximide (CHX) protein synthesis inhibitor were purchased from Selleckchem (Houston, USA) and Cell Signaling Technology, respectively. When indicated, the medium contained 20 μM MG-132 or 40 μg/mL CHX.
Plasmids, lentiviral vectors, shRNA and transfection
Lentiviral expression vectors with Flag-labeled USP13, Flag-labeled USP13 C345A, and His-labeled ATG5 were generated by inserting the relevant genes with the Flag or His tag into the pCDH-CMV-MCS-EF1α-Puro vector. HA-labeled ubiquitin-Lys48(K48) and HA-labeled ubiquitin-Lys63(K63) were obtained from Addgene (Watertown, USA), in which one Lys (K48 or K63) was retained and the rest were replaced with Arg residues. All constructs were verified by sequencing. Lentiviral shRNA plasmids that target ATG5 and USP13 together with the nonspecific control shRNA were obtained from Dharmacon (Shanghai, China). The shRNA targeted sequences were as follows: 5′-TGATCCTTCAATCTGTTGG-3′ (shATG5-1), 5′-GCCTGTCAAATCATAGTAT -3′ (shATG5-2), 5′-GAGCCAACATCAAAACCTG-3′ (shUSP13-1), 5′-TTCCTACATACGCTCTCTG-3′ (shUSP13-2), 5′-AGGAGCCAGCCAAGAAAUCAATT-3′(shMETTL3-1), 5′-CUGCAAGUAUGUUCACUAUGATT-3′(shMETTL3-2), 5′-GGATTCTGTGCACAGATAA-3′(shPAK1), 5′-CAUGCCGCAUGAUUCUUGATT-3′(shIGF2BP2). Transfection of plasmid and shRNA was performed with Lipo3000 (Invitrogen, Carlsbad, USA) following the manufacturer’s instructions. LV-Flag-labeled USP13, LV-Flag-labeled USP13 C345A and LV-His-labeled ATG5 were seeded in GIST-882 and GIST-T1 cells with a multiplicity of infection. Transfection with an empty vector was used as a control. After puromycin screening for 5–7 days until a final concentration of 2 μg/mL was reached, the cells that overexpressed Flag-labeled USP13, Flag-labeled USP13 C345A, His-labeled ATG5, and control cells were harvested. ATG5 knockout GIST-882 and GIST-T1 cell lines were screened with puromycin 2 days after lentivirus infection.
RNA isolation and real-time RT-PCR
Total RNA from cell lines and tissues was isolated utilizing the Total RNA Purification Kit (Norgen, Thorold, Canada). Quantitative real-time RT-PCR (qRT-PCR) was executed on a StepOnePlus™ Real-Time PCR System (ThermoFisher Scientific, Waltham, USA). Targeted mRNA levels were standardized against GAPDH level. Primers utilized for qPCR were as follows: ATG5, 5′-AAAGATGTGCTTCGAGATGTGT-3′ (forward) and 5′-CACTTTGTCAGTTACCAACGTCA-3′ (reverse); and GAPDH, 5′-GAGTCAACGGATTTGGTCGT-3′ (forward) and 5′-GACAAGCTTCCCGTTCTCAG-3′ (reverse); and METTL3, 5′-TTGTCTCCAACCTTCCGTAGT-3′ (forward) and 5′-CCAGATCAGAGAGGTGGTGTAG-3′ (reverse); and USP13 5′-GGTCTACAAGAACGAGTG-3′ (forward) and 5′-TTCTCTCGCGCATGTCTT-3′ (reverse) and IGF2BP2, 5′-GCCCCTCATTAAGCCCAAG-3′, (forward) and 5′-TTGTGGTGGTCTGACAGTTCG-3′) (reverse).
IC50 and resistance index calculation
IC50 and resistance index of GIST cell lines were determined by CCK-8 assay (CK04, Dojindo, Tokyo, Japan). In brief, GIST cells were seeded in 96-well plates. Each well was supplemented with DMEM medium (100 μL) together with CCK-8 reagent (10 μL) and co-cultured for 2 h. OD450 values of each well were determined with a microplate reader (ThermoFisher Scientific, USA). Assays were performed at least three times in duplicate.
Patients and tissues
Drug-sensitive GIST tissues were derived from 21 patients who received curative surgery between February 2015 and February 2020 at the Department of General Surgery of the First Affiliated Hospital of Nanjing Medical University. Patients were treated with IM following surgery and none developed tumor recurrence for at least 1 year after surgery based on CT scans. Drug-resistant GIST tissues were obtained by surgery or CT-guided needle biopsy from 21 patients who demonstrated disease progression while treated with IM. All tissues were stored in liquid nitrogen immediately after resection. Consent for tissue experiments was obtained from the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University. The clinicopathological features of GIST patients are summarized in Table S1.
Cell cycle and apoptosis
Cell cycle analysis was performed with the Cell Cycle Staining Kit (MultiSciences Biotech, Hangzhou, China) according to the manufacturer’s protocol. GIST cells subsequently were assessed by fluorescence activated cell sorter (FACS). For cell apoptosis analysis, GIST cells were stained with Annexin V-FITC and PI (Keygentec, Nanjing, China) according to the supplier’s protocol and were assessed by FACS.
Colony formation assay
Cells were seeded in a six-well plate (1000 cells/well) combined with 2 mL complete DMEM medium. Visible colonies were fixed with 4% paraformaldehyde. The fix solution was removed, samples were flushed slowly in tap water, and the colonies were stained with crystal violet and photographed 14 days later after desiccation at room temperature.
Western blot
GIST cells and tissues were lysed to acquire proteins using RIPA Lysis Buffer (Beyotime, Nanjing, China) following the manufacturer’s instructions. The lysates were subjected to SDS-PAGE and then transferred to PVDF membranes (Millipore, Massachusetts, USA). The membranes were incubated at 4 °C with primary antibodies. After incubation with secondary antibody against mouse or rabbit immunoglobulin at room temperature, the membranes were processed with the Super ECL Plus Kit (US EVERBRIGHT INC, Suzhou, China). Normalization was carried out by blotting identical samples with antibody against β-actin.
Co-immunoprecipitation
Cells were lysed on ice for 30 min with NP-40 Lysis Buffer (Beyotime). Lysates were centrifuged at 12000 g for 15 min and the supernatant was incubated with the targeted antibody with rotation at 4 °C overnight. Protein A/G Plus-Agarose (ThermoFisher Scientific) was added followed by continued rotation at 4 °C overnight. The Protein A/G Plus-Agarose was washed throughly three times with wash buffer (1 mL) at 4 °C for 20 min. The wash buffer was removed and 2X SDS-PAGE Sample Buffer (Beyotime) was added to the sample with incubation at 100 °C for 10 min. Co-immunoprecipitated proteins were subjected to standard western blot analysis.
Denature-IP assay
All the ubiquitin assays in our study were performed in denaturing conditions. Ubiquitination was detected using d-IP. Cells were lysed in SDS-denaturing buffer (62.5 mM Tris-HCl pH6.8, 2% SDS, 10% glycerol, 1.5% β-mercaptoethanol) and boiled for 10 min. Cell lysates were then diluted tenfold to fortyfold in native lysis buffer (50 mM Tris-HCl pH7.4, 0.5% Triton X-100, 200 mM NaCl, 10% glycerol). After centrifugation at 13,000 r.p.m. for 5 min, the supernatants were immunoprecipitated. The lysates and immune complexes were subjected to western blot analysis.
GST-tagged protein purification and GST pull-down assays
For GST pull-down assays, bacterially-expressed GST, GST-ATG5, wild-type GST-USP13, or GST-USP13-C345A bound to glutathione-Sepharose 4B beads (GE Healthcare, Chicago, USA) were incubated with wild-type Flag-USP13, Flag-USP13-C345A, or His-ATG5 expressed in HEK293T cells for 2 h at 4 °C. Subsequently, complexes were washed with GST-binding buffer at least four times and eluted repeatedly with TNGT elution buffer (0.5 mL) at least five times. Elution fractions were collected and subjected to western blot analysis.
Protein half-life assay
For the ATG5 half-life assay, the indicated GIST cells were treated with CHX (80 μg/ml) for the indicated durations before gathering cell lysates.
Transmission electron microscopy (TEM)
GIST cells were digested with trypsin, centrifuged at 300 g for 5 min, and resuspended with serum in a 1.5 mL microcentrifuge tube. The cells were fixed in EM fixation buffer overnight at 4 °C. After staining with OsO4 (1%), the samples were sliced into ultrafine sections and subjected to electron microscopy (JEM-F200, Japan).
EdU incorporation
EdU assay was performed with the Click-iTCell-Light EdU Apollo567 In Vitro Kit (Ribobio, Guangzhou, China) according to the manufacturer’s instruction. In brief, digested GIST cells were seeded at 5000 cells per well in 96-well plate. EdU was added to the medium for 3 h at a final concentration of 50 μmol/L. 4% paraformaldehyde in PBS was utilized to fix cells and the reaction was quenched with 50 μL of glycine solution (2 mg/mL). Cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) and one field of view per well was examined for EdU-positive cells.
Immunohistochemistry
Human GIST tissues and GIST subcutaneous murine tumor models were fixed and embedded in paraffin for immunohistochemistry (IHC). The sections (4 μm) derived from paraffin-embedded tissues were incubated with the indicated antibody overnight at 4 °C and then with a secondary antibody HRP conjugate at room temperature for 1 h. IHC used KI67 and TUNEL antibodies. Immunohistochemical staining intensity scores were indicated as: negative (0), weak staining (1), moderate staining (2) and strong staining (3), and the extent of stained cells were indicated as: 0% = 0, 1–24% = 1, 25–49% = 2, 50–74% = 3, 75–100% = 4. All cases were independently scored by two experienced pathologists blinded to the clinical data in Jiangsu Province Hospital & First Affiliated Hospital of Nanjing Medical University. The final scores were defined by multiplying the intensity scores by the scores of the extent of stained cells (0–12). Cutoff values to define the high and low expression of targets were chosen on the basis of a measurement of heterogeneity with the log-rank test statistic with respect to progression-free survival. Because the optimal cutoff thresholds were identified from the current study as 4, tumors with IHC scores of 4 or more were considered as high expression, and tumors with IHC scores of <4 were considered as low expression.
Subcutaneous tumor xenograft models
6 × 106 logarithmically growing GIST cells resuspended in 100 μL PBS were injected subcutaneously into the flank of 6-week-old female nude mice for tumor growth assays. The following treatments were initiated when the tumor volume reached approximately 300 mm3. (1) IM-sensitive GIST cell lines and IM-resistant GIST cell lines were used to establish a subcutaneous xenograft model of GIST (n = 6 mice/group). When the tumors grew to the required size, mice were treated with IM via drinking water; (2) GIST cells were pretreated with USP13 lentivirus or USP13 control lentivirus. In addition, IM-resistant GIST cells were pretreated with USP13 inhibitors or control. These lentivirus-transfected cells were implanted into subcutaneous mice tumors to construct the USP13 positive, USP13-negative, and control groups (n = 6 mice/group). When the transplanted tumor grew to the required volume, mice in each group were treated with IM via drinking water. (3) IM-resistant GIST cells-xenografted mice and control mice were treated with USP13 inhibitors or with USP13 inhibitors and autophagy inhibitors in combination therapy to assess reversal of IM resistance (n = 6 mice/group). Mice were treated with IM (45 mg/kg/day) and with or without 3-MA (15 mg/kg/day) via drinking water when the tumors grew to the required size. The USP13 inhibitor Spautin-1 was used at 20 mg/kg/day when required.
Isobaric tags for relative and absolute quantitation (iTRAQ)
iTRAQ data analysis was performed by Gene Create Biolabs Inc (Wuhan, China). Total protein extracts were obtained from IM-sensitive and IM-resistant GISTs of patients using RIPA lysis buffer (Sigma, Schwieberdingen, Germany). After reduction and alkylation, protein samples were mixed with trypsin at a final trypsin: protein ratio of 1:10 and digested overnight at 37 °C. Tryptic peptides were dried by vacuum centrifugation, reconstituted in labeling buffer (70% ethanol/25 mM TEAB), and labeled with iTRAQ reagents according to the manufacturer’s protocol (AB-Sciex, Framingham, USA). Protein identification and quantification were performed by LC-MS/MS analysis using a nanoLC Ultra 1D plus/Triple TOF 5600 analyzer (AB-Sciex).
m6A sequencing and m6A-RNA immunoprecipitation (RIP)-qPCR
A MeRIP-qPCR assay was performed to detect m6A modifications of individual gene transcripts. Briefly, Protein A/G magnetic beads were prewashed and incubated with 5 μg of rabbit IgG or anti-m6A antibody (Synaptic Systems, Goettingen, Germany) for 2 h at 4 °C with continuous rotation. The antibody-bead complex was mixed with purified RNA and immunoprecipitation (IP) buffer containing RNase inhibitors (Beyotime). After IP, the proteins were digested with proteinase K and RNA enrichment was examined by qPCR analysis.
Luciferase reporter assay
Cells were seeded into 24-well plates and transfected with luciferase reporter vectors as described above. After 48 h, the cells were lysed and tested using a dual-luciferase reporter assay kit (Promega, Madison, USA) according to the manufacturer’s instructions. Independent experiments were conducted in triplicate
Statistical analysis
Data are expressed as the mean ± SD or as mean ± SEM of at least three independent experiments. All statistical analyses were accomplished by GraphPad Prism 7.0 and SPSS 20.0 software. All statistical tests were two-tailed Student’s t test or one-way ANOVA with P < 0.05 representing statistical significance.
Results
ATG5 promotes IM resistance and the malignant proliferation of GIST
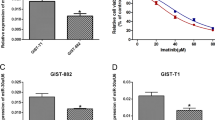
iTRAQ technique was used to screen differentially expressed proteins in patient-derived IM-sensitive and -resistant tissues, which revealed that ATG5 was among the top 10 highly expressive proteins in resistant tissues. (Fig. 1A, B) and proteomic data-analysis was shown in Table S3. Next, IHC staining demonstrated that the expression of ATG5 and CD117 were associated with increased IM resistance in GIST (Fig. 1C). Assessments of protein and mRNA levels revealed a marked increase in ATG5 protein in IM-resistant GIST cells and tissues, but not in mRNA (Figs. 1D–G, S1A). To interrogate the relationship between ATG5 expression and IM resistance in GIST, GIST-T1R and GIST-882R cells were transfected with a short hairpin RNA (shRNA) for ATG5 or ATG5 overexpression plasmid and the efficiency was exhibited in Fig. S1B, C. ATG5 knockdown significantly decreased half-maximal inhibitory concentration (IC50) values for IM. In addition, overexpressed ATG5 increased IC50 values in GIST-882 and GIST-T1 cells (Figs. 1H, I, S1D). As the preceding experiments suggest an important role for ATG5 in GIST, we utilized receiver operating characteristic curve analysis to assess the role of ATG5 in the prognosis of patients. The area under the curve was 0.696 (95% CI, 0.535–0.857), which indicates that ATG5 may be a sensitive biomarker for IM resistance diagnostics in GIST (Fig. 1J, K). Collectively, the above findings indicate that high ATG5 expression is associated with IM resistance in GIST.

A Heatmap representation of differentially expressed proteins in patients with IM-resistant and IM-sensitive GIST. B Volcano plot for differentially expressed proteins in patients with IM-resistant and IM-sensitive GIST. C IHC for ATG5 and CD117. D Protein level of ATG5 in human GIST tissue from IM-resistant and IM-sensitive patients. E Quantification of ATG5 protein level in GIST cells. F RT-qPCR analyses of ATG5 mRNA levels in sensitive and resistant GIST tissues. G Quantitative RT-PCR analysis of ATG5 target genes from IM-sensitive and IM-resistant cells. H, I CCK8 proliferation assay verified the effect of ATG5 expression levels on the sensitivity of GIST cells to IM. J Kaplan–Meier plot of progression-free survival by ATG5 expression. K Receiver operating characteristic analysis of the risk of patients with IM-sensitive and IM-resistant GIST. L, M Cell cycle distribution and apoptosis rate of GIST cells transfected with vector, ATG5, shCtrl, or shATG5. Error bars represent the mean (n = 3) ±S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
To further elaborate the effect of ATG5 on proliferation, flow cytometry assays showed that ATG5 overexpression weakened cell cycle arrest on G0/1 phase and apoptosis induced by IM, whereas sh-ATG5 strengthened the effect of IM (Fig. 1L, M). Moreover, results of colony formation and EdU assays were consistent with above, indicating the key role of ATG5 in proliferation in GIST (Fig. S1E, F). Xenograft models were employed to explore the oncogenic role of ATG5 in GIST. The shATG5 significantly attenuated xenograft tumor growth in nude mice (Fig. S1G, H). Assessment of cellular proliferation with IHC assays also substantiated the role of ATG5 in promoting xenograft tumor proliferation (Fig. S1I). In summary, the preceding data demonstrates that ATG5 enhances the malignant proliferation and facilitates IM resistance of GIST cells.
Protective autophagy induces IM resistance in GIST
Autophagy plays a critical role in IM resistance in GIST [9]. IHC revealed that the LC3 level was significantly higher in IM-resistant xenografts (Fig. S2A). This result was similar to that in patient-derived tumors (Fig. S2B). In addition, common markers of autophagy, including ATG5 and LC3-II/I, which were elevated in IM-resistant cell lines with or without chloroquine (CQ) treatment, demonstrated that autophagy was involved in IM resistance (Fig. S2C–E). 3-methyladenine (3-MA) inhibits phosphoinositide 3-kinase which is a key factor in autophagy. Treatment with 3-MA led to a decrease in the IC50 (Fig. S2F). Furthermore, more LC3 fluorescent spots were found in IM-resistant cells (Fig. S2G, H). Evidence of IM-induced autophagy was also obtained by direct observation of autophagosome formation by TEM (Fig. S2I, J). In summary, the preceding results suggest that protective autophagy is critical for IM resistance in GIST.
USP13 maintains ATG5 stability
In contrast to ubiquitin modification, the role of deubiquitination in protein modification is less well understood. As noted in the preceding experiments, a significant difference existed in ATG5 protein levels, but not mRNA, between IM-resistant and -sensitive GIST samples. We blocked de novo protein synthesis using CHX and chased the subsequent ATG5 protein levels in HEK293T and ICCs cells. ATG5 was degraded gradually and treatment with the proteasome inhibitor MG-132 caused a significant increase in ATG5 protein levels (Fig. S3A). In view of these observations, we conjectured that ATG5 is regulated by the ubiquitin-proteasome system in post-translational level. We screened an siRNA library consisting of 98 human DUBs in HEK293T cells to identify potential deubiquitinating enzymes for ATG5. USP3, USP13, USP32, and OTUD4 each impacted ATG5 expression in this screening (Fig. S3B). Notwithstanding the co-expression of these DUBS and ATG5 in HEK293T cells, we observed a direct interaction only between ATG5 and USP13 in IP assays (Fig. 2A). The interaction between ATG5 and USP13 was examined further by transfecting HEK293T cells with Flag-labeled wild-type or a catalytically inactive C345A point mutant USP13. Expression of wild-type USP13, but not the C345A mutant, enhanced ATG5 protein level in a dose-dependent manner (Fig. 2B), indicating that USP13 modulates ATG5 via its deubiquitinating activity. Moreover, USP13 and ATG5 are expressed highly in IM-resistant GIST cell lines (Figs. 2C, S3C). Subsequently, the loss of USP13 induced a concomitant reduction in ATG5 expression which also was attenuated by MG-132 or by overexpression of wild-type USP13, but not USP13-C345A mutant (Figs. 2D, E, S3D, E). In addition, CHX pulse-chase experiments revealed that overexpression of USP13, but not the USP13-C345A mutant, dramatically increased the stability of ATG5 in HEK293T cells (Fig. 2F, G), whereas USP13 knockdown in resistant cell lines destabilized the ATG5 protein (Figs. 2H, I, S3F). Taken together, these results demonstrate that USP13 regulates ATG5 stability.

A Flag-labeled DUBs (USP3, USP13, USP32, and OTUD4) were expressed in HEK293T cells, and cell lysates were analyzed by IP with Flag beads followed by western blotting with antibodies against ATG5 and Flag. B Increasing amounts of Flag-labeled wild-type USP13 or USP13-C345A were transfected into HEK293T cells, and cell lysates were analyzed by western blotting with antibody against ATG5. C Western blot analysis of ATG5 from IM-sensitive and IM-resistant cell lines. D GIST-T1R cells transfected with two independent USP13 shRNA were treated with or without the proteasome inhibitor MG-132 (20 μM for 8 h) and then USP13 and ATG5 were analyzed. E Western blotting analysis of ATG5 levels in GIST-T1R cells transfected with USP13 shRNA together with either shRNA-resistant (sh-res) Flag-labeled wild-type USP13 or USP13-C345A. F, G HEK293T cells were co-transfected with His-labeled ATG5 and Flag-labeled wild-type USP13 or USP13-C345A, treated with CHX (40 μg/ml), collected at the indicated times, and then subjected to western blotting with antibodies against His and Flag. Quantification of ATG5 levels relative to β-actin are shown. H, I GIST-T1R cells stably expressing control shRNA or shRNA-USP13 were treated with CHX (40 μg/ml), harvested at the indicated times, and then subjected to western blotting with antibodies against ATG5 and USP13. Quantification of ATG5 levels relative to β-actin are shown. J HEK293T cells were transfected with His-ATG5 alone or in combination with Flag-labeled wild-type USP13 or USP13-C345A. Cell lysates were analyzed by IP with Flag beads followed by western blotting with antibodies against His and Flag. a, vector; b, Flag-USP13; c, Flag-USP13-C345A. K Confocal images showing colocalization of USP13 (red) and ATG5 (green) in GIST cells. Nuclei were counterstained with DAPI (blue). L, M Cell lysates from GIST-T1R cellls were analyzed by IP using antibodies against USP13 and ATG5, then subjected to western blotting analysis. IgG was used as the isotype control. N Purified Flag-labeled wild-type USP13 or USP13-C345A were incubated with GST or GST-ATG5 coupled to glutathione-sepharose beads. Proteins retained on sepharose were then subjected to western blotting with the indicated antibodies. Recombinant GST-ATG5 was purified from bacteria and analyzed by SDS-PAGE and Coomassie blue staining. O Schematic representation of full-length (FL) Flag-labeled USP13, His-labeled ATG5, and various deletion mutants. P HEK293T cells were co-transfected with His-ATG5 and FL Flag-labeled USP13 or deletion mutants. Cell lysates were analyzed by IP with Flag beads followed by western blotting with antibodies against His and Flag. Q HEK293T cells were co-transfected with Flag-USP13 and His-labeled FL ATG5 or deletion mutants. Cell lysates were analyzed by IP with His beads followed by western blotting with antibodies against Flag and His. Error bars represent the mean (n = 3) ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
USP13 interacts with ATG5
In view of the direct interaction that was observed between ATG5 and USP13 in IP assays (Fig. 2A), we investigated whether the catalytic activity of USP13 was required for this binding. IP assays showed that His-labeled ATG5 interacts with both Flag-labelled wild-type USP13 or USP13-C345A in HEK293T cells which demonstrated that the deubiquitinating activity of USP13 does not grossly alter the binding efficiency (Fig. 2J). Subsequently, we examined the subcellular colocalization of USP13 and ATG5. Confocal images showed colocalization of USP13 (red) and ATG5 (green) in cytoplasm of GIST cells (Figs. 2K, S3G). Analogously, we validated that endogenous USP13 associated with ATG5 protein physically in GIST cells (Figs. 2L, M, S3H, I). Moreover, we performed pull-down assays in which purified GST-ATG5 was incubated with purified recombinant Flag-labelled wild-type or mutant USP13. GST-ATG5, but not GST alone, interacted with wild-type USP13 and the USP13-C345A mutant (Fig. 2N) which confirmed that USP13 interacted with ATG5 directly and that the deubiquitinating activity of USP13 was not required for this interaction. To map the interacting regions on USP13 and ATG5, various Flag-labelled USP13 and His-ATG5 deletions were tested (Fig. 2O). IP assays demonstrated that the M1 region (amino acids 1–184) of ATG5 and the amino terminus of USP13 (amino acids 300–863) are the principal regions that mediate the interaction (Fig. 2P, Q).
USP13 regulates the deubiquitination of ATG5
The interaction between USP13 and ATG5 was probed further. The ubiquitination level of ATG5 in resistant cells decreased compared with the level in sensitive cells. The addition of IM alone led to a concomitant decrease in the amount of ATG5 ubiquitination. The concentration of USP13 was elevated in the corresponding drug-resistant cells, and the addition of the drug alone also led to an increase in USP13 levels (Figs. 3A, S4A). We co-transfected wild-type or the C345A mutant of USP13 in HEK293T cells to test whether USP13 targets ATG5 for deubiquitination. ATG5 from cells treated with MG132 was heavily ubiquitinated in IP analysis. In contrast, the ubiquitination of ATG5 was almost eliminated by co-transfection with wild-type USP13, but not USP13-C345A (Fig. 3B, C). In support of these observations, ubiquitination of ATG5 was promoted by shUSP13 in resistant cells (Figs. 3D, S4B). To determine whether ATG5 is a direct substrate of USP13, purified USP13 and ubiquitinated ATG5 were incubated in a cell-free system. Purified wild-type USP13, but not the catalytically inactive C345A mutant, decreased ATG5 polyubiquitination in vitro (Fig. 3E). Therefore, USP13 directly deubiquitinates ATG5. Two main forms of polyubiquitin chains constituted by Lys48(K48) or Lys63(K63) linkages have been described. To investigated which type of polyubiquitin modifications on ATG5 are influenced by USP13. we used a panel of ubiquitin mutants in which one Lys residue was retained and the rest were replaced with Arg residues, and we observed that expression of USP13 inhibited the Lys48(K48)-linked but not Lys63(K63)-linked ubiquitination of ATG5. (Figs. 3F, S4C). In addition, the expression of ATG5 was attenuated by the enforced expression of a Lys48-resistant (K48R) version of ubiquitin (Figs. 3G, S4D). We generated three lysine-to-arginine substitutions mutants of ATG5 according to GPS-Uber (http://gpsuber.biocuckoo.cn/wsresult.php) which predicts general and E3-specific lysine ubiquitination sites. These mutants (ATG5-K5R, -K171R and -K201R) were used in denaturing-IP assays (Fig. S4E). Without USP13 overexpression, all three mutants were ubiquitinated, albeit less strongly than wild-type ATG5. Comparing the amount of ATG5-K5R-ubiquitin from cells in which USP13 was expressed with the equivalent signal without USP13 expression demonstrated clearly that USP13 targeted this mutant for deubiquitylation. As ATG5 ubiquitinated on other lysines did not appear resistant to USP13 deubiquitylation, USP13 may have the highest activity for K5 ubiquitination (Fig. 3H). In addition, the expression of His-ATG5, which was downregulated by USP13 depletion, was attenuated by the K5R mutation (Fig. 3I). Taken together, our results reveal that USP13 stabilizes ATG5 by removing K48-linked polyubiquitin chains at residue K5.

A GIST-T1S and GIST-T1R were co-transfected with HA-ubiquitin (HA-Ub), and cell lysates were subjected to denature-IP with ATG5 antibody, followed by western blotting with antibodies against HA and ATG5. Cells were treated with 20 μM MG-132 and with or without low-dose IM for 8 h before harvesting. B, C HEK293T cells or ICCs were co-transfected with His-ATG5, HA-ubiquitin (HA-Ub), and Flag-labeled wild-type USP13 or USP13-C345A, and cell lysates were subjected to denature-IP with His beads followed by western blotting with antibodies against HA and His. Cells were treated with 20 μM MG-132 for 8 h before harvesting. D GIST-T1R was co-transfected with the indicated shRNA and HA-Ub, and cell lysates were subjected to denature-IP with ATG5 antibody, followed by western blotting with antibodies against HA and ATG5. Cells were treated with 20 μM MG-132 for 8 h before harvesting. E Unubiquitylated or ubiquitylated His-ATG5 was incubated with wild-type GST-USP13 or GST-USP13-C345A coupled to glutathione-sepharose beads. His-ATG5 was subjected to denature-IP with His beads followed by western blotting with antibodies against HA and His. Recombinant GST-USP13 or GST-USP13-C345A were analyzed by SDS-PAGE. F GIST-T1S cells were co-transfected with His-ATG5, Flag-USP13, and HA-Ub Lys0, Lys48-only, or Lys63-only plasmids, and then the ATG5 ubiquitylation linkage was analyzed. G GIST-T1S cells transfected with wild-type Ub or Ub-Lys48R were cultured for 72 h in the presence of control shRNA or USP13 shRNA. Cell lysates were analyzed by western blotting using antibodies against ATG5 and USP13. H HEK293T cells were transfected with the vector plasmid or Flag-USP13, HA-Ub and wild-type His-ATG5 or K-to-R mutants and treated with a low-dose of IM. Samples were subjected to denature-IP with anti-Flag beads and then analyzed by immunoblot with an anti-HA or anti-Flag antibody. I HEK293T transfected with wild-type Ub, wild-type His-ATG5 or His-ATG5-K5R were cultured for 72 h in the presence of control shRNA or USP13 shRNA. Cell lysates were analyzed by western blotting using antibodies against ATG5 and USP13.
USP13 enhances IM-resistance and malignant cell proliferation by the induction of protective autophagy in vitro and in vivo
Compared with IM alone, combined administration of 3-MA and IM decreased cell viability (Figs. 4A, B, S5A, B) which suggests that USP13 promotes IM resistance by inducing protective autophagy. Moreover, elevated USP13 induced the overexpression of ATG5, enhanced transformation of LC3-I to LC3-II, and down-regulated P62. Conversely, down-regulation of autophagy levels was observed in IM-resistant cells transfected with shUSP13 (Figs. 4C, S5C). In addition, USP13 overexpression in IM sensitive cells inhibited apoptosis, whereas the knockdown of USP13 in IM resistant cells induced an opposite effect (Figs. 4D, S5D).

A, B GIST-T1S cells were transfected with a USP13 overexpression vector and empty control (NC), and GIST-T1R cells were transfected with USP13 shRNA and control (NC). Inhibition curves of GIST cells after treatment are indicated. C Western blot detected autophagy element expression levels in GIST-T1S and GIST-T1R cells after transfection as indicated. D Apoptosis-related protein levels were detected by western blot. E–G The autophagy flow and autophagosome in GIST cells were detected by confocal microscopy and transmission electron microscopy. H GIST-T1S was co-transfected with His-ATG5, HA-Ub, and Flag-USP13 in the absence or presence of Spautin-1 (1 μM) or a low-dose of IM and cell lysates were subjected to denature-IP with His beads followed by western blotting with antibodies against HA and His. Cells were treated with 20 μM MG-132 for 8 h before harvesting. I Western blot analysis of ATG5 in GIST-T1S treated with Spautin-1 (1 μM) or vehicle with or without MG-132. J GIST-T1S cells were treated with Spautin-1 (1 μM) or vehicle for 24 h, followed by CHX (40 μg/ml), harvested at the indicated times, and then subjected to western blotting with antibodies against ATG5. SE, short exposure; LE, long exposure. K–M Representative images from tumors in nude mice bearing GIST-T1S or GIST-T1R cells in different groups and the expression of autophagy markers monitored by western blot (n = 6 mice/ group). Scale bars: 1 cm. N–P Representative images of tumors in nude mice formed by the GIST-T1R cells in the different subgroups and the expression of autophagy and apoptosis markers monitored by western blot. Error bars represent the mean (n = 3) ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001, respectively.
We utilized a tandem GFP-mCherry-LC3 construct to examine the association between autophagic flux and USP13 expression. More autophagosomes and autolysosomes were observed in cells with elevated USP13 expression, whereas knock-down of USP13 decreased the level of autophagy (Figs. 4E, S5E). Subsequently, TEM analysis was exploited to study autophagic activity which demonstrated that USP13 overexpression contributed to enhanced autophagosome levels (Figs. 4F, G, S5F). In addition, EdU assays demonstrated that USP13 overexpression enhanced IM resistance, while USP13 knockdown increased the killing effect of IM (Fig. S5G, H). In summary, these data demonstrate that enhanced IM resistance and malignant proliferation in GIST cells involve protective autophagy induced by USP13.
To examine the preceding observations in a more clinical-like setting, we determined the impact of pharmacologic inhibition of USP13 on cell-derived GIST models. A small molecule inhibitor of USP13, Spautin-1, first was investigated for attenuation of the deubiquitinating activity of USP13 on ATG5. This molecule almost abolished the deubiquitination ability of USP13 at 10 μmol/L (Figs. 4H, S5I). Spautin-1 also reduced ATG5 protein levels in IM resistant cell lines and this reduction was counteracted by MG-132. Thus, Spautin-1, similar to the effect of USP13 knockdown, facilitates the ubiquitin-proteasome degradation of ATG5 (Figs. 4I, S5J). Moreover, a marked decrease in the half-life of the ATG5 protein was induced by co-administration of Spautin-1 and CHX (Figs. 4J, S5K).
In view of the preceding in vitro results, we established murine GIST xenografts for in vivo studies. Overexpression of USP13 in these xenografts diminished sensitivity to IM and increased tumor volume. Accordingly, an increased sensitivity to IM was observed in xenografts treated with Spautin-1 (Figs. 4M, S5N, O). Co-administration of USP13 lentivirus with IM upregulated the level of autophagy and ATG5 (Fig. 4K), which were attenuated by the USP13 inhibitor (Fig. 4L). Furthermore, USP13 lentivirus inhibited apoptosis and promoted IM resistance in the xenografts (Fig. S5L). Conversely, with administration of the Spautin-1, apoptosis was enhanced and IM resistance was impaired (Fig. S5M). In summary, USP13 overexpression triggered autophagy through the USP13/ATG5 pathway in xenografts of GIST tumors.
Following the preceding xenograft studies, co-administration of Spautin-1 and 3-MA blocked autophagy effectively. Interestingly, the antitumor effect of IM was not enhanced by co-administration of 3-MA, whereas addition of Spautin-1 and 3-MA attenuated protective autophagy and slowed down tumor growth by enhancing apoptosis (Figs. 4N, O, P, S5P). Overexpressed USP13 promoted autophagy, IM resistance, and tumor propagating potential which were largely reversed by knockdown of ATG5 (Fig. S6A–H).
PAK1 promotes ATG5 binding to and deubiquitination by USP13
PAK1-mediated phosphorylation at residue T101 decreased ubiquitination of the ATG5 [20]. Hence, we hypothesized that PAK1 may function in cooperation with DUBs to deubiquitinate ATG5. As outlined above, analysis of truncated mutants demonstrated that the M1 region (amino acids 1–184) of ATG5 is critical for direct interaction with USP13 (Fig. 2Q). As the T101 phosphorylation site of ATG5 is located in the M1 domain, we reasoned that PAK1-mediated phosphorylation at this residue may affect binding and deubiquitination of ATG5 with USP13. Knockdown of PAK1 or the PAK1 inhibitor FRAX597 (1 μmol) in GIST-T1R cells attenuated the interaction between endogenous USP13 and ATG5 (Fig. 5A–D). In addition, PAK1-K420R is a dominant-negative mutant, which could inhibit PAK1 activation. The levels of PAK1 (K420) acetylation significantly correlate with ATG5 (T101) phosphorylation. PAK1-K420R could suppress PAK1 activation by repressing its phosphorylation and inhibiting its dimerization [20]. The interaction between the two proteins was reduced after transfection of Myc-PAK1-K420R (Fig. 5E, F). Furthermore, the T101A mutation in ATG5 significantly decreased the interaction between ATG5 and USP13 in the presence of PAK1 (Fig. 5G, H). Accordingly, deubiquitination of ATG5 by USP13 was attenuated after PAK1 knockdown (Fig. 5I). In addition, the deubiquitination of ATG5 by USP13 was reduced after transfection with PAK1-K420R (Fig. 5J). The T101A mutation also attenuated deubiquitination of ATG5 by USP13 in the presence of PAK1 (Fig. 5K). Finally, deubiquitination of ATG5 by USP13 was reduced by FRAX597 (Fig. 5L). In summary, the results above validate that PAK1-dependent phosphorylation of ATG5 is critical for binding and deubiquitination by USP13.

A, B GIST-T1R cells were transfected with control shRNA or PAK1 shRNA, and cell lysates were analyzed by IP using antibodies against USP13 or ATG5, then subjected to western blot analysis. IgG was used as the isotype control. C, D GIST-T1R cells were treated with or without PAK1 inhibitor, and cell lysates were analyzed by IP using antibodies against USP13 and ATG5, then subjected to western blotting analysis. IgG was used as the isotype control. E, F HEK293T cells were transfected with Flag-USP13, His-ATG5, and wild-type PAK1 (WT) or PAK1 (K420R). Cell lysates were analyzed by IP and western blots were performed as indicated. G, H HEK293T cells were transfected with Flag-USP13, wild-type His-ATG5, His-ATG5-101A, His-ATG5-101D, or Myc-PAK1. Cell lysates were analyzed by IP and western blots were performed as indicated. I HEK293T cells were transfected with HA-Ub and Flag-USP13 plus control shRNA or PAK1 shRNA for 36 h. Then the cells were treated with MG-132 for 8 h. Cell lysates were denature-IP using an anti-His antibody and then analyzed by western blotting using an anti-HA antibody. J HEK293T cells were transfected with HA-Ub, and wild-type Myc-PAK1 or Myc-PAK1(K420R), and then treated with MG-132 for 8 h. Cell lysates were denature-IP using an anti-His antibody then analyzed by western blotting using an anti-HA antibody. K HEK293T cells were transfected with HA-Ub, Myc-PAK1, wild-type His-ATG5 His-ATG5-101A, or His-ATG5-101D, and then treated with MG-132 for 8 h. Cell lysates were denature-IP using an anti-His antibody then analyzed by western blotting using an anti-HA antibody. L HEK293T cells were transfected with HA-Ub, His-ATG5, Flag-USP13 with or without PAK1 inhibitor for 36 h. Then the cells were treated with MG-132 for 8 h. Cell lysates were denature-IP using an anti-HA antibody and then analyzed by immunoblotting.
Knockout of PAK1 reverses the effect of USP13 on ATG5 in vitro and in vivo
Our preceding experiments indicate that USP13 stabilizes ATG5 in a PAK1-dependent manner to enhance autophagy and promote IM resistance in GIST. In support of this hypothesis, we demonstrated that the effect of overexpressed USP13 on autophagy and IM resistance was largely rescued by shPAK1 (Fig. 6A–G). For in vivo experiments, we used GIST cells that stably overexpressed USP13 to establish xenografts in mice. Bioluminescence imaging demonstrated that USP13 overexpression promoted tumor growth and that the effect was rescued by silencing of PAK1 (Fig. 6H, I). Taken together, these observations indicate the significance of the USP13-PAK1-ATG5 axis in IM resistance and malignant proliferation of GIST cells.

A, B The expression of autophagy and apoptosis marker proteins monitored by western blot. C Inhibition curves of GIST cells after treatment as indicated. D The effect of USP13 expression levels on the proliferation of GIST cells was examined by clone formation assay. E–G Cell cycle distribution, apoptosis rate, proliferation and the autophagy flow were assessed in GIST-T1S cells transduced with empty vector or USP13, and reconstituted with shCtrl or shPAK1. Error bars represent the mean (n = 3) ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001, respectively. H–I Bioluminescence imaging of xenografted tumors and and quantitative analysis of tumor volume. Error bars represent the mean (n = 3) ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001, respectively.
METTL3-mediated m6A modification of USP13 mRNA maintains IGF2BP2-dependent stability
We previously reported that methylation of mRNA m6A by METTL3 stimulates IM resistance in GIST [21]. Here, USP13 mRNA levels were differential in IM-sensitive and IM-resistant GIST cells and tissues (Fig. S7A, B). Our previous m6A-seq analysis showed significant enrichment of m6A in the 3′-UTR region of USP13 mRNA (Fig. 7A). To interrogate any role for this region of USP13 mRNA in GIST, we examined the expression levels of METTL3 and USP13 in GIST patients and observed a significant positive correlation (R = 0.5819) (Fig. S7C). Consistent with above, cells that overexpressed METTL3 showed significantly increased USP13 mRNA and protein levels (Fig. 7B, C). Conversely, METTL3 deficiency exerted the opposite effects (Figs. 7D, E, S7D). Furthermore, m6A-RIP-qPCR analysis demonstrated that METTL3 overexpression significantly enhanced m6A methylation of USP13 mRNA (Figs. 7F, S7E), whereas knockout of METTL3 reduced this methylation (Figs. 7G, S7F). To probe whether METTL3 leads to attenuation of the USP13 mRNA levels, USP13 mRNA stability was examined in GIST cells. Treated with the transcriptional inhibitor actinomycin D, METTL3 deficiency attenuated the stability of USP13 mRNA, but overexpressed METTL3 enhanced stability (Figs. 7H, S7G). We further observed that overexpression of METTL3 enhanced ATG5 protein level which was reversed by knockdown of USP13 (Figs. 7I, S7H). Moreover, based on m6A-seq data and RMBase database, we identified two m6A peaks in the 3′-UTR of USP13 mRNA (Fig. S7I).

A m6A peaks are enriched in the 3′-UTR region of USP13 mRNA as determined by m6A-seq. B–E qRT-PCR and western blot analysis of METTL3 and USP13 after METTL3 knockout or overexpression. F–G RNA immunoprecipitation-qPCR showing the enrichment of USP13 m6A after METTL3 knockout or overexpression. H The decay rate of USP13 mRNA after treatment with actinomycin D (2.5 mmol/L) for the indicated times with METTL3 knockout or overexpression. I Western blot analysis of ATG5 after METTL3 overexpression with or without shUSP13. J The mRNA levels in sh-Ctrl or sh-METTL3 GIST cells transfected with pGL3-USP13-3′-UTR wild-type or pGL3-USP13-3′-UTR-Mut1/2 reporters for 24 h. K–M USP13 mRNA levels in sh-Ctrl or sh-METTL3 GIST cells transfected with pcDNA-USP13-3′-UTR-WT/Mut1/2 for 24 h and then treated with actinomycin D for the indicated times determined by RT-qPCR. N The mRNA levels of USP13 in IGF2BP1/2/3 knockdown GIST cells were detected by qRT-PCR. O RNA immunoprecipitation-qPCR showing the enrichment of USP13 m6A after IGF2BP2 knockout. P The decay rate of USP13 mRNA after treatment with actinomycin D (2.5 mmol/L) for the indicated times with IGF2BP2 knockout. Q IGF2BP2 expression correlates positively with USP13 expression in GIST patients (linear regression). Error bars represent the mean (n = 3) ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001.
We next constructed reporters in which either the wild-type USP13 3′-UTR or mutant 1/2 was inserted downstream of the firefly luciferase reporter gene in pGL3. Each mutant contains a single base-pair change compared to the wild-type sequence (Fig. S7J). Luciferase assays revealed that mutation of the m6A motif (3′-UTR-Mut1) but not that of the site (3′-UTR-Mut2) partially abolished the difference in reporter gene expression between sh-METTL3 and control cells (Figs. 7J, S7K). In addition, the stability of the USP13 3′-UTR was reduced significantly in the sh-METTL3 background (Figs. 7K, S7L). The 3′-UTR-Mut1 mutation, but not 3′-Mut2 alteration, partially abolished the difference in stability of the USP13 mRNA 3′-UTR in sh-METTL3 and control (Figs. 7L, M, S7M, N). Taken together, the above observations suggest that m6A in the 3′-UTR is involved in the stability of USP13 mRNA via a role for an m6A-modified 5′-TAGAACTG′-3 sequence in the secondary structure of the 3′-UTR.
The regulatory effects of “m6A readers” on m6A-modifed transcripts have been studied recently. IGF2BP proteins, including IGF2BP1/2/3, are a distinct family of m6A readers that can recognize and bind to thousands of mRNA targets through the m6A motif in mRNA stabilization [22]. Hence, specific shRNAs against IGF2BP1/2/3 were designed and results showed that silencing of IGF2BP2 markedly suppressed USP13 mRNA expression (Figs. 7N, S7O). An RNA IP analysis further confirmed the interaction between IGF2BP2 and USP13 mRNA in GIST cells (Figs. 7O, S7P). The mRNA stability analysis indicated the key role of IGF2BP2 in USP13 mRNA stability (Figs. 7P, S7Q). The correlation between IGF2BP2 and USP13 was also confirmed in GIST tissues (Figs. 7Q, S7R). Together, our findings indicate that METTL3-mediated m6A modification maintains USP13 expression via IGF2BP2-dependent USP13 mRNA stability.
Correlation between METTL3, USP13, PAK1 and ATG5 expression and association with GIST patient progression-free survival
We assessed the expression of METTL3, USP13, PAK1, and ATG5 in a cohort of 42 GIST specimens by IHC to explore the potential clinical relevance of our findings. METTL3, USP13, PAK1 and ATG5 were upregulated significantly in resistant tumors (Fig. 8A). Moreover, the results revealed a positive correlation for the expression of each of these proteins (Fig. 8B–E), and that increased expression of each protein correlated with reduced progression-free survival of GIST patients (Fig. 8F–H).

A Representative images of IHC staining of METTL3, USP13, PAK1, and ATG5 in clinical GIST samples. Scale bars: 50 µm. B–E Correlation analyses of IHC data of GIST in (A). Statistical significance was determined by the chi-square test. A total of 42 GIST specimens were analyzed. F–H Kaplan–Meier analyses for GIST in (A). A total of 42 GIST specimens were analyzed. I The mechanistic scheme of this study.
Discussion
Resistance to IM is a major clinical problem that leads to poor prognosis in patients with GIST [23]. Autophagy plays a crucial role in drug resistance in tumors [24, 25], but the underlying molecular mechanisms that mediate autophagy-induced IM resistance in GISTs are not well-characterized. In the present study, we verified that the key autophagy protein ATG5 is essential for IM resistance in GIST. Screening a siRNA library that comprised 98 human DUBs revealed USP13 as a new bona fide deubiquitinase that interacts with and reverses ubiquitination of ATG5, and stabilizes the protein in a PAK1-dependent manner. Moreover, this is the first report that the METTL3 methyltransferase regulates m6A modification of USP13 mRNA thereby enhancing USP13 protein production.
Recycling of damaged molecules and organelles is required for tumor progression. Autophagy plays a critical role in the efficient turnover of intracellular components, which suppresses tumor initiation and reduces genomic instability, but promotes the survival of established tumors [26,27,28,29,30]. IM-induced activation of autophagy also is a key survival pathway in quiescent GIST cells [31]. Autophagy biogenesis is characterized by the formation of an isolation membrane phagophore whose production is regulated by ATG genes through ubiquitination-like conjugation steps [32]. ATG5 is involved in the extension of the phagophore membrane in autophagic vesicles, which is relatively scarce [33,34,35]. Several mechanisms potentially are involved in the regulation of ATG5 expression. For example, the Gadd45ß-MEKK4-p38MAPK kinase phosphorylates ATG5 which inactivates the protein and inhibits autophagy [36], whereas deacetylation of ATG5 by the Sirt1 deacetylase induces autophagy [37]. Moreover, methylation of the ATG5 promoter downregulates the expression of ATG5 and also blocks autophagy [38]. Furthermore, overexpression of the immunoproteasome catalytic β5i subunit causes the degradation of ubiquitinated ATG5 which inhibits the induction of autophagy [39]. Here, we identify USP13 as a crucial deubiquitinase that stabilizes and maintains the activity of ATG5.
Previous studies have implicated USP13 in cancer development via the regulation of oncogene or tumor suppressor protein stability [15,16,17,18,19]. However, the pathological function of USP13 in the tumorigenesis of different cancers is controversial. For example, USP13 deubiquitinates and stabilizes microphthalmia-associated transcription factor (MITF) which is a lineage-specific master regulator of human melanoma [40]. In addition, USP13 plays an oncogenic role by stabilization of the c-MYC transcription factor in glioblastoma, and by upregulation of ATP citrate lyase and oxoglutarate dehydrogenase in ovarian cancer [15, 41]. In contrast, USP13 was discovered as a deubiquitinase of the PTEN phosphatase which is a tumor-suppressing protein in breast and other cancers [42]. Here, we demonstrated that USP13 is a novel ATG5 regulator that stabilizes ATG5 through deubiquitination in autophagy-mediated IM resistance. Interestingly, Beclin1, which is a subunit of the PI3K-III complex of intracellular signal transducer enzymes, also controls the stabilities of the USP10 and USP13 proteins by regulating their deubiquitinating activities [16]. Beclin1 and ATG5 are key regulatory proteins in the nucleation stage and extension stages of autophagy, respectively. Hence, USP13 potentially may serve as the molecular bridge between these stages.
RNA m6A modification is involved in diverse cellular processes, and dysregulation of this modification is a common feature of many tumors [43, 44]. The studies here revealed that USP13 is regulated by METTL3-induced m6A modification, thereby mediating ATG5 stabilization which promotes autophagy and IM resistance in GIST cells. Protein interactions frequently are facilitated by post-translational modifications such as phosphorylation [45]. Accordingly, PAK1-mediated phosphorylation of ATG5 at T101 decreased its ubiquitination [20]. Here we identified USP13 as a crucial deubiquitinase that stabilizes ATG5 in a PAK1-dependent manner to enhance autophagy and promote IM resistance in GIST cells. These findings hold considerable promise for filling the void in understanding the impact of posttranscriptional modifications on ATG5 function and stability.
In conclusion, the study revealed that USP13 induces autophagy to promote proliferation and drug resistance via deubiquitylation and stabilization of ATG5 in GISTs. Activation of PAK1 phosphorylates ATG5 at residue T101 which mediates the interaction between USP13 and ATG5, and which reduces ATG5 ubiquitination. Furthermore, METTL3 upregulates the USP13 expression through m6A modification. Therefore, specifically targeting the USP13-ATG5 axis may be a potential therapeutic strategy for future treatment of GIST (Fig. 8I).
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References
Dekker TJA. Ripretinib for advanced gastrointestinal stromal tumours. Lancet Oncol. 2020;21:e414.
Glod J, Arnaldez FI, Wiener L, Spencer M, Killian JK, Meltzer P, et al. A phase II trial of vandetanib in children and adults with succinate dehydrogenase-deficient gastrointestinal stromal tumor. Clin Cancer Res. 2019;25:6302–8.
Joensuu H, Eriksson M, Hall KS, Reichardt A, Hermes B, Schütte J, et al. Survival outcomes associated with 3 years vs 1 year of adjuvant IM for patients with high-risk gastrointestinal stromal tumors: an analysis of a randomized clinical trial after 10-year follow-up. JAMA Oncol. 2020;6:1241–6.
Raut CP, Espat NJ, Maki RG, Araujo DM, Trent J, Williams TF, et al. Efficacy and tolerability of 5-year adjuvant IM treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: the PERSIST-5 clinical trial. JAMA Oncol. 2018;4:e184060.
Ravegnini G, Sammarini G, Nannini M, Pantaleo MA, Biasco G, Hrelia P, et al. Gastrointestinal stromal tumors (GIST): facing cell death between autophagy and apoptosis. Autophagy 2017;13:452–63.
Li Z, Nakatogawa H. Degradation of nuclear components via different autophagy pathways. Trends Cell Biol. 2022;32:574–84.
Grunwald DS, Otto NM, Park JM, Song D, Kim DH. GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy 2020;16:600–14.
Cui T, Wang Y, Song P, Yi X, Chen J, Yang Y, et al. HSF1-dependent autophagy activation contributes to the survival of melanocytes under oxidative stress in vitiligo. J Investig Dermatol. 2022;142:1659–69.
Ni B, Li Q, Zhuang C, Huang P, Xia X, Yang L, et al. The nerve-tumour regulatory axis GDNF-GFRA1 promotes tumour dormancy, imatinib resistance and local recurrence of gastrointestinal stromal tumours by achieving autophagic flux. Cancer Lett. 2022;535:215639.
Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbé S. Deubiquitylases from genes to organism. Physiol Rev. 2013;93:1289–315.
Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat Chem Biol. 2017;13:1207–15.
Rehman SAA, Kristariyanto YA, Choi SY, Nkosi PJ, Weidlich S, Labib K, et al. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Mol Cell. 2016;63:146–55.
Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014;74:495549–66.
Suresh B, Lee J, Kim H, Ramakrishna S. Regulation of pluripotency and differentiation by deubiquitinating enzymes. Cell Death Differ. 2016;23:1257–64.
Han C, Yang L, Choi HH, Baddour J, Achreja A, Liu Y, et al. Amplification of USP13 drives ovarian cancer metabolism. Nat Commun. 2016;7:13525.
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34.
Sun H, Zhang Q, Jing YY, Zhang M, Wang HY, Cai Z, et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat Commun. 2017;8:15534.
Li Y, Luo K, Yin Y, Wu C, Deng M, Li L, et al. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nat Commun. 2017;8:15752.
Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat Commun. 2018;9:215.
Feng X, Zhang H, Meng L, Song H, Zhou Q, Qu C, et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy 2021;17:723–42.
Xu K, Zhang Q, Chen M, Li B, Wang N, Li C, et al. N6-methyladenosine modification regulates IM resistance of gastrointestinal stromal tumor by enhancing the expression of multidrug transporter MRP1. Cancer Lett. 2022;53:85–99.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Prim. 2021;7:22.
Li H, Roy M, Liang L, Cao W, Hu B, Li Y, et al. Deubiquitylase USP12 induces pro-survival autophagy and bortezomib resistance in multiple myeloma by stabilizing HMGB1. Oncogene 2022;41:1298–308.
Ianniciello A, Helgason GV. Targeting ULK1 in cancer stem cells: insight from chronic myeloid leukemia. Autophagy. 2022;18:1734–6.
Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb Perspect Biol. 2012;4:a008821.
Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226:255–73.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64.
Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99:287–92.
Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–39.
Gupta A, Roy S, Lazar AJF, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci USA. 2010;107:14333–8.
Zhang F, Kumano M, Beraldi E, Fazli L, Du C, Moore S, et al. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun. 2014;5:5775.
Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Investig. 2013;123:5284–97.
Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA. 2011;108:4123–8.
Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2017;13:619–24.
Keil E, Höcker R, Schuster M, Essmann F, Ueffing N, Hoffman B, et al. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 2013;20:321–32.
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374–79.
Liu H, He Z, Simon HU. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 2014;10:372–3.
Xie X, Bi HL, Lai S, Zhang YL, Li N, Cao HJ, et al. The immunoproteasome catalytic β5i subunit regulates cardiac hypertrophy by targeting the autophagy protein ATG5 for degradation. Sci Adv. 2019;5:eaau0495.
Zhao X, Fiske B, Kawakami A, Li J, Fisher DE. Regulation of MITF stability by the USP13 deubiquitinase. Nat Commun. 2011;2:414.
Fang X, Zhou W, Wu Q, Huang Z, Shi Y, Yang K, et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J Exp Med. 2017;214:245–67.
Zhang J, Zhang P, Wei Y, Piao HL, Wang W, Maddika S, et al. Deubiquitylation and stabilization of PTEN by USP13. Nat Cell Biol. 2013;15:1486–94.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014;505:117–20.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015;161:1388–99.
Zhang A, Huang Z, Tao W, Zhai K, Wu Q, Rich JN, et al. USP33 deubiquitinates and stabilizes HIF-2alpha to promote hypoxia response in glioma stem cells. EMBO J. 2022;41:e109187.
Acknowledgements
We would like to give our special thanks to Jin Xu for his support in Statistical analysis and Shuxian Dai for her stimulating discussions. This work was supported by grants from National Natural Science Foundation of China (grant number: 82072708); Youth Program of National Natural Science Foundation of China (grant number: 81902461); Natural Science Foundation of Province (grant number: BK20191495, BK20191073); The Priority Academic Program Development of Jiangsu Higher Education Institutions (grant number: PAPD, JX10231801); Jiangsu Key Medical Discipline (General Surgery) (grant number: ZDXKA2016005).
Author information
Authors and Affiliations
Contributions
Conception and design: ZSG; Data acquisition, analysis, and interpretation: CL, HX, ZKX; Investigation: YBB, ZWC, NFW, ZJW, YY, ZHL, ZYH, and BWL; Acquisition of patient specimens: FYL, ZL, LJW, ZKX and LY; Paper drafting and revising: ZSG, HX, and CL; and paper writing: ZSG. All authors approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The collection of specimens and animal handling for the study have been reviewed and approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by M. Piacentini
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gao, Z., Li, C., Sun, H. et al. N6-methyladenosine-modified USP13 induces pro-survival autophagy and imatinib resistance via regulating the stabilization of autophagy-related protein 5 in gastrointestinal stromal tumors. Cell Death Differ 30, 544–559 (2023). https://doi.org/10.1038/s41418-022-01107-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-01107-8
This article is cited by
-
Crosstalk between m6A modification and autophagy in cancer
Cell & Bioscience (2024)
-
Drug resistance mechanisms and treatment strategies mediated by Ubiquitin-Specific Proteases (USPs) in cancers: new directions and therapeutic options
Molecular Cancer (2024)
-
N6-methyladenosine-modified MIB1 promotes stemness properties and peritoneal metastasis of gastric cancer cells by ubiquitinating DDX3X
Gastric Cancer (2024)
-
New insights into the regulation of METTL3 and its role in tumors
Cell Communication and Signaling (2023)
-
USP13 promotes proliferation and angiogenesis of diffuse large B-cell lymphoma cells by promoting MCL-1 expression
Molecular & Cellular Toxicology (2023)
