
Abstract
Mutations in genes encoding general transcription factors cause neurological disorders. Despite clinical prominence, the consequences of defects in the basal transcription machinery during brain development are unclear. We found that loss of the TATA-box binding protein-associated factor TAF8, a component of the general transcription factor TFIID, in the developing central nervous system affected the expression of many, but notably not all genes. Taf8 deletion caused apoptosis, unexpectedly restricted to forebrain regions. Nuclear levels of the transcription factor p53 were elevated in the absence of TAF8, as were the mRNAs of the pro-apoptotic p53 target genes Noxa, Puma and Bax. The cell death in Taf8 forebrain regions was completely rescued by additional loss of p53, but Taf8 and p53 brains failed to initiate a neuronal expression program. Taf8 deletion caused aberrant transcription of promoter regions and splicing anomalies. We propose that TAF8 supports the directionality of transcription and co-transcriptional splicing, and that failure of these processes causes p53-induced apoptosis of neuronal cells in the developing mouse embryo.

Similar content being viewed by others


Introduction
For transcription of RNA polymerase II dependent genes, binding of the general transcription factor IID complex (TFIID) to the promoter is the first step in the assembly of the pre-initiation complex, followed by the binding of the other general transcription factor complexes and then RNA polymerase II. TFIID consists of the TATA-box binding protein (TBP) and up to 13 different TBP-associated factors (TAFs) [1]. TAF8 has been shown to bind to TAF10 via its histone fold domain in yeast [2] and mammalian cells [3, 4]. The additions of a TAF8-TAF10 heterodimer or both, the TAF8-TAF10 and TAF3-TAF10 heterodimers together have been proposed to be important for the breaking of the symmetry of the TFIID complex [1, 5] and for the recruitment of TBP and other TAFs to the core complex [5, 6]. However, other studies proposed that TAF8 was an important part of TFIID but questioned the existence of a symmetric core TFIID in cells [7]. TAF8 has been proposed to be essential for nuclear import of TAF2 and TAF10 [6], yet others suggest that TAF8 might be dispensable for TFIID function in certain cell types [3]. Since no consensus had been reached from cell biology and structural biology studies, the role of TAF8 in the structure and function of TFIID remained unclear.
TAF8 (TBN) is essential for embryonic development beyond E4.5 [8]. Blastocysts lacking TAF8 displayed extensive apoptosis of the inner cell mass (ICM) characterised by cleaved (active) caspase-3 and DNA fragmentation. Interestingly, trophectoderm cells of Taf8–/– blastocysts remained intact in vivo and could be cultured in vitro for at least 10 days [8], suggesting a cell type-specific requirement for TAF8.
Mutations in genes encoding members of the TFIID protein complex cause neurological disorders. Poly-glutamine expansions in the TBP protein cause spinocerebellar ataxia [9]. TBP mutations were observed in patients diagnosed with Huntington’s disease [10,11,12] and are associated with Parkinson’s disease [13] and cognitive dysfunction [14]. TAF1 mutations cause X-linked dystonia-parkinsonism [15] and are associated with intellectual disability [16]. TAF2 mutations are associated with cognitive disorders [17, 18]. Mutations in TAF6 are associated with two neurological disorders, Alazami-Yuan [19] and Cornelia de Lange syndrome [20]. Variants of the TAF13 gene are associated with cognitive dysfunction [21]. TAF8 mutation is associated with intellectual disability [22].
An overall picture emerges suggesting that general transcriptional disturbance, “transcriptomopathies” [20, 23], could cause intellectual disability and other neurological disorders. Despite considerable interest in this concept, the cellular and molecular consequences of broad transcription anomalies remain unclear.
We investigated the cellular and molecular effects of impaired TFIID function on brain development. We selectively deleted Taf8 in the developing mouse central nervous system. We assessed the role of TAF8 in TFIID integrity and function in vivo and determined the consequences of the loss of TAF8 on general and gene-specific aspects of transcription, as well as on cellular functions in the developing brain.
Results
Deletion of Taf8 results in extensive cell death in the developing cerebral cortex
Taf8 is uniformly expressed in the developing nervous system [8]. We deleted Taf8 in the developing central nervous system (CNS) using our Taf8 conditional allele (Supplementary Fig. 1) and a nestin-Cre (NesCre) transgene [24]. Genomic deletion of Taf8 was complete from embryonic day 12.5 onward (E12.5; Supplementary Fig. 1); mRNA levels fell below 10% at E14.5 in Taf8lox/–NesCreT/+ brains vs. control brains (Fig. 1A). TAF8 protein levels were reduced to 30% in the cortex and 56% in the “brain minus cortex” (BMC) at E14.5, and to 14% in the cortex and 36% in the BMC at E16.5 in Taf8lox/–NesCreT/+ brains compared to control brains (Fig. 1B, C). Non-neural cell types in the brain are not expected to activate the NesCre-transgene to delete Taf8, including blood vessel endothelium and blood.

The brains were separated into two areas: the cerebral cortex (including the hippocampus) and the remainder of the brain excluding the cortex (“brain minus cortex” = BMC). A Assessment of Taf8 mRNA levels in Taf8lox/–NesCreT/+ and Taf8lox/+NesCre+/+ control cortex and BMC by RT-qPCR. B Assessment of the TAF8 protein levels in the cortex and BMC by Western blotting. C Assessment of TAF8 protein levels in (B) by densitometry. D Representative examples of H&E-stained serial sections of Taf8lox/–NesCreT/+ and Taf8lox/+NesCreT/+ control brain regions as indicated. E Enumeration of pyknotic nuclei indicating cell death in Taf8lox/–NesCreT/+ and Taf8lox/+NesCreT/+ control brain regions as indicated. F TUNEL and bisbenzimide DNA counterstain of Taf8lox/–NesCreT/+ and Taf8lox/+NesCreT/+ control brain regions as indicated. G Cleaved (active) caspase-3 (brown) immunohistochemistry on paraffin embedded sections of E14.5 Taf8lox/–NesCreT/+ and Taf8lox/+NesCreT/+ control brain regions as indicated, counterstained with haematoxylin. Related data are displayed in Supplementary Fig. 1. N = 3 animals per data point, presented as mean ± SEM. Data were analysed using two-tailed Student’s T-tests. 3V, 3rd Ventricle; 4V, 4th ventricle; CP, cortical plate; dTne, dorsal telencephalon neuroepithelium; Htne, hypothalamic neuroepithelium; IZ, intermediate zone; LV, Lateral ventricle; Mene, medullary neuroepithelium; Pone, pontine neuroepithelium; Tene, tegmental neuroepithelium; Thne, thalamic neuroepithelium; VZ; ventricular zone; white arrowheads, TUNEL positive cells and condensed nuclei; white arrows, autofluorescent nucleated embryonic and anuclear, definitive erythrocytes; black arrowheads indicate active-caspase-3 positive cells.
Taf8lox/–NesCreT/+ mice developed to term but died at birth, failing to establish normal breathing. Taf8lox/–NesCreT/+ brains were indistinguishable from controls on histological sections up to E12.5 (Supplementary Fig. 1). At E13.5 (Supplementary Fig. 1), the Taf8lox/–NesCreT/+ cerebral cortex had a few pyknotic nuclei and at E14.5 (Fig. 1D, E) many pyknotic nuclei, which were TUNEL positive (Fig. 1F), and cells that were positive for cleaved (active) caspase-3 (Fig. 1G). The dying cells made up more than 20% of cells in the E14.5 Taf8lox/–NesCreT/+ cortex (Fig. 1E) and spanned all layers of the cortex from the ventricular zone to the pial surface. In contrast, control animals did not display measurable cell death (Fig. 1D–G). By E18.5, pyknotic cells were no longer detected in the Taf8lox/–NesCreT/+ cortex, but the Taf8lox/–NesCreT/+ cortex had a 29% reduction in the number of cells compared to controls (Supplementary Fig. 1). Intriguingly, brain areas other than the cerebral cortex were relatively unaffected by loss of Taf8, for example, the diencephalon (Fig. 1D–F), hindbrain regions (Fig. 1D, F) and neural retina (Supplementary Fig. 1).
Loss of TAF8 causes nuclear p53 accumulation and upregulation of p53 target genes
The finding of extensive cell death with DNA fragmentation and caspase-3 activation indicated apoptosis in the Taf8lox/–NesCreT/+ cortex. BCL-2 family regulated apoptosis is important during embryonic development, including to ensure tissue cell homeostasis [25]. Pro-apoptotic BCL-2-family proteins (BH3-only proteins, including BIM, PUMA, NOXA) inhibit the pro-survival BCL-2 family proteins (including BCL-2, MCL-1, BCL-XL), which in healthy cells suppress the effectors of apoptosis, the multi-BH domain proteins BAX and BAK (Fig. 2A). Pro-apoptotic BH3-only proteins have diverse potency to inhibit pro-survival proteins (Fig. 2B). Some BH3 only proteins have also been shown to directly activate BAX and BAK. Once unleashed, BAX and BAK form pores in the mitochondrial outer membrane, resulting in the release of cytochrome C, followed by its association with the adaptor protein APAF-1, caspase-9 activation, effector caspases activation and demolition of the cell.

A Simplified schematic drawing of the BCL-2 protein family regulated apoptotic pathway (reviewed in [65]). B Simplified schematic drawing of the specificity of antagonism between pro-survival (green) and pro-apoptotic BH3-only BCL2-family members [based on [66, 67]]. C Schematic drawing of p53 and examples of its target genes inducing either cell cycle arrest or apoptosis. D Western blot and densitometry of nuclear p53 protein from cortex and brain minus cortex (BMC) of the indicated genotypes and lamin B1 loading control. N = 3 pools per genotype of 2 animals, i.e. 6 animals per genotype. Control whole cell lysate from WT MEFs and WT MEFs treated with Doxorubicin to induce p53 protein and from Mdm2;p53 double knockout MEFs. E RT-qPCR of p53 target genes Noxa, Puma, Bax and p21 in E14.5 and E16.5 cortex and BMC of the indicated genotypes. Normalised to the mean of 3 housekeeping genes (Pgk1, Hsp90ab1, Gapdh). N = 4 animals per genotype. F Western blot and densitometry of PUMA protein in E14.5 cortex and BMC of the indicated genotypes. N = 3 individual brains per genotype. G Western blot and densitometry of MCL-1 protein in E14.5 cortex and BMC. N = 3 animals per genotype. Related data are displayed in Supplementary Fig. 2. Data are presented as mean ± SEM. Data were analysed using two-tailed Student’s T-tests.
The transcription factor p53 can activate genes that induce cell cycle arrest (e.g. Cdkn1a encoding p21) or apoptosis (e.g. Puma, Noxa, Bax; Fig. 2C; [26]). At E13.5, one day before the substantial increase in apoptotic cells was observed in the Taf8lox/–NesCreT/+ cortex (Supplementary Fig. 1F vs. Fig. 1D), nuclear p53 was elevated 27-fold in the Taf8lox/–NesCreT/+ cortex compared to controls, while nuclear p53 levels in the BMC (Fig. 2D), cytoplasmic p53 levels and p53 mRNA levels were much less affected or unchanged (Supplementary Fig. 2).
At E14.5 the p53 target genes were significantly affected by the loss of TAF8: Noxa, Puma, Bax and Cdkn1a mRNA levels were 17-fold, 3-fold, 2-fold and 11-fold upregulated, respectively, in the Taf8lox/–NesCreT/+ cortex compared to control cortex (Fig. 2E); PUMA protein was also elevated (Fig. 2F). MCL-1 protein, which is targeted by both NOXA and PUMA (Fig. 2B), was reduced by ~30% in Taf8lox/–NesCreT/+ brain compared to controls (Fig. 2G). Other BCL-2 family genes were affected to a lesser extent by the absence of TAF8 (Supplementary Fig. 2).
Loss of p53 prevents apoptosis in the Taf8 lox/– NesCre T/+ cortex at E14.5
The cell death observed in the E14.5 Taf8lox/–NesCreT/+ cortex was completely prevented in Taf8lox/–NesCreT/+p53–/– cortex, and Taf8lox/–NesCreT/+Puma–/– brain regions had ~76–97% fewer pyknotic nuclei compared to control brains (Fig. 3A, B). Despite the substantial elevation in Noxa mRNA in Taf8lox/–NesCreT/+cortex (Fig. 2E), the absence of Noxa did not significantly affect cell death caused by Taf8 deletion (Fig. 3A, B).

A Representative images of H&E-stained serial sections of E14.5 brain regions and genotypes as indicated. Arrowheads indicate pyknotic nuclei. B Quantification of pyknotic nuclei in E14.5 brain region and genotypes as indicated. Cells were counted blinded to genotype. C p53 chromatin immunoprecipitation followed by qPCR of p53 response elements in the Noxa and Puma locus in E14.5 cortex and BMC (on the Puma-deficient background for reduced cell death, but retaining p53 response elements). p53-deficient brains were used as negative controls. D Enumeration of BrdU positive cells in brain regions and genotypes as indicated. E Enumeration of cells in differentiation zones of brain regions and genotypes as indicated. Related data are displayed in Supplementary Fig. 3. N = 3 animals per genotype presented as mean ± SEM. Data were analysed using two-tailed Student’s T-tests.
To assess p53 occupancy of the p53-binding element at the Noxa and Puma loci, we performed anti-p53 chromatin immunoprecipitation (ChIP) followed by qPCR using E14.5 Taf8 NesCre brains on the Puma-deficient background, where the two p53 binding elements in the Puma locus [27] remain intact [28], to avoid the confounding effects of simultaneous apoptosis. p53 occupancy was ~2-fold and ~4.5-fold elevated at the Noxa and Puma locus, respectively, in the in the absence of TAF8 compared to controls brains (Fig. 3C).
Loss of TAF8 results in a failure of cell differentiation in the cortex
On histological sections, the E14.5 Taf8lox/–NesCreT/+p53–/– brains looked significantly healthier than Taf8lox/–NesCreT/+p53+/+ brains. However, they appeared developmentally delayed with a marked reduction in size of the differentiation zones of the neocortex and the hippocampus (Supplementary Fig. 3). This suggested an additional function of TAF8 in the progression of brain development. Furthermore, like Taf8lox/–NesCreT/+ mice, Taf8lox/–NesCreT/+p53–/– mice developed to term but died at birth. We examined cell proliferation and differentiation in E14.5 Taf8lox/–NesCreT/+ and control brains in the presence or absence of p53. BrdU incorporation marking DNA synthesis was not significantly affected in Taf8lox/–NesCreT/+ compared to control brains in the neocortex, hippocampus (including subiculum) and was increased in the diencephalon irrespective of p53 status (Fig. 3D). Cells in the ventricular proliferation zone can be clearly distinguished from cells in the peripheral differentiation zones, allowing counting of total cell number and the number of differentiating cells in different brain regions. We observed a reduction in the percentage of differentiating cells in Taf8lox/–NesCreT/+ compared to control brains in neocortex, hippocampus (including subiculum), but the percentage of differentiating cell in the diencephalon was not affected, irrespective of p53 status (Fig. 3E). This indicates that a primary effect of loss of TAF8 was a failure of differentiation that, unlike the defects in cell survival, was not rescued by loss of p53.
To examine the extent of the cortical cell differentiation defect in the absence of TAF8, we took advantage of the rescued survival in the absence of p53 and assessed neuronal and cortical layer markers, as well as glia markers (Fig. 4A–K). Although present in the correct position, Taf8lox/–NesCreT/+p53–/– brains had fewer TBR2+ subventricular zone cells (Fig. 4D–F). The cortical layers were severely disorganised in the Taf8lox/–NesCreT/+p53–/– brains (Fig. 4G, H) and contained very few SATB2 single positive cells. Instead, the marker proteins were commonly co-expressed in individual cells, indicating a disrupted cell differentiation process (Fig. 4G, H). The total number of neurons was slightly reduced in the cortical plate of Taf8lox/–NesCreT/+p53–/– brains compared to Taf8lox/+NesCreT/+p53–/– control brains (Fig. 4I). GFAP+ astrocytes and GFAP/S100ß+ maturing astrocytes were reduced in the Taf8lox/–NesCreT/+p53–/– brains compared to Taf8lox/+NesCreT/+p53–/– control brains (Fig. 4J, K).

A–K Immunofluoresence detection and enumeration of neuronal and glia markers of the ventricular and subventricular zone and the superficial, intermediate and deep layers of the developing cortex on frozen sections of E18.5 Taf8lox/–NesCreT/+p53–/– and Taf8lox/+NesCreT/+p53–/– control cortices. A–C Ventricular zone marker PAX6 (red), counterstained with DAPI (blue). D–F Subventricular zone marker TBR2 (red), counterstained with DAPI (blue). G, H Markers of the superficial (SATB2, green), intermediate (CTIP2, red) and deep layers (TBR1, blue) of the developing cortex. I Enumeration of the total number of neuronal cells per pia-to-ventricle bin. J Marker of astrocytes (GFAP, green) and maturing astrocytes (S100ß, red), counterstained with DAPI (blue). K Enumeration of the proportions of GFAP and GFAP/S100ß double positive cells. Schematic diagrams illustrate locations of marker proteins in the wild-type cortex. Proportion of PAX6 (B) and TBR2-positive cells (E) were assessed in 20 µm deep and 120 µm wide bins beginning at the ventricular surface. The average proportion per 50 µm by 120 µm was also determined (C, F). White boxes in the lower magnification images (G) indicate the position of the higher magnification images (also in G). White arrow heads (G) indicate cells double positive for SATB2/CTIP2, CTIP2/TBR1 and SATB2/TBR1. Marker protein positive cells were enumerated in 10 equal depth bins assigned numbering from the pial to the ventricular surface (H). N = 3 animals per genotype presented as mean ± SEM. Data were analysed using two-way ANOVA with genotype and bin as the independent factors (I) and with Sídák correction for multiple testing (B, E) or two-tailed Student’s T-tests (C, F, K).
Elevated levels of nuclear p53 in the developing cerebral cortex
The developing cerebral cortex has been reported to display higher levels of apoptosis during normal development than other areas of the developing brain [29]. To investigate factors that might predispose the cortex to apoptosis, we examined p53 and the pro-survival BCL-2 family protein BCL-XL, which has been reported to be important for cell survival during brain development [30,31,32] in E14.5 and E16.5 wild-type cortex and compared these to the other brain areas (BMC). p53 mRNA was moderately elevated, and Bclx mRNA was modestly reduced in the cortex compared to BMC at both developmental stages (Fig. 5A). Nuclear p53 protein levels were 3 to 8-fold higher in the cortex than in BMC, although this did not reach significance due to high variation (p = 0.05 Mann–Whitney test; Fig. 5B). In contrast, cytoplasmic p53 levels and BCL-XL protein levels were not strikingly different in the cortex compared to BMC (Fig. 5B, C).

A RT-qPCR of p53 and Bclx mRNA levels in wild-type E14.5 and E16.5 cortex and BMC normalised to 3 housekeeping genes. B Western blot analysis and densitometry of p53 protein levels in E14.5 nuclear and cytoplasmic fractions of wild-type cortex and BMC. Probing for lamin B1 and tubulin was used as loading controls, doxorubicin treated wild-type MEFs and p53–/– MEFs were used as a positive and negative control, respectively. C Western blot of wild-type BCL-XL protein levels in wild type E14.5 whole cell lysate from cortex and BMC, with probing for tubulin used as a loading control. N = 3 animals per genotype presented as mean ± SEM. Data were analysed using two-tailed Student’s T-test.
TFIID proteins can associate in the absence of TAF8
To determine if the TFIID complex was formed/remained intact in the absence of TAF8, we assessed TBP, TAF8, TAF5 and TAF1, as well as representative TAFs (bold) for pairs of TAFs joined by their histone-folds (TAF6-TAF9, TAF4-TAF12, TAF3-TAF10). In total E16.5 cell lysates TAF8 protein was readily detectable in control brains, but undetectable in the Taf8lox/–NesCreT/+p53–/– brains (Fig. 6A). Protein levels of other TFIID subunits were either unchanged or up to 10-fold elevated in Taf8lox/–NesCreT/+p53–/– brains compared to controls (Fig. 6A). Similarly, mRNA level of genes encoding TFIID subunits were unchanged or up to 2.4-fold elevated (Supplementary Fig. 4). TFIID complex proteins remained associated with TBP, as assessed by immunoprecipitation with anti-TBP antibodies followed by Western blotting for TAF1, TAF5, TAF6, TAF10 and TAF12 mostly at similar abundance levels in Taf8lox/–NesCreT/+p53–/– compared to controls, all relative to input (Fig. 6B).

A Western blots and densitometry of nuclear lysates of E16.5 Taf8lox/–NesCreT/+p53–/– and Taf8lox/+NesCre+/+p53–/– control cortex and BMC probed for TAF1, TAF5, TAF6, TAF8, TAF10, TAF12 and TBP. * represents residual TAF5 signal from previous probing. These lysates represent 1/5 of the input for the immunoprecipitation in (B). The densitometric readings were normalised to control protein levels to allow display within one graph. B Western blots and densitometry of anti-TBP antibody immunoprecipitated proteins from E16.5 Taf8lox/–NesCreT/+p53–/– and Taf8lox/+NesCre+/+p53–/– control nuclear lysates of cortex and BMC probed for TAF1, TAF5, TAF6, TAF8, TAF10, TAF12 and TBP. There is some non-specific binding as indicated by the signal in the IgG negative control, but this is never above the signal from the anti-TBP-immunoprecipitation. The densitometric readings were normalised to input protein levels and control levels. Related data are displayed in Supplementary Fig. 4. N = 3 animals per genotype presented as mean ± SEM. Data were analysed using two-tailed Student’s T-tests. * represents residual TAF5 signal from previous probing.
Loss of TAF8 does not affect global mRNA abundance
To test if loss of TAF8 had a broad effect on TFIID function and the expression of most RNA polymerase II transcribed genes, we first spiked Taf8lox/–NesCreT/+p53–/– and Taf8lox/+NesCre+/+p53–/– control brain tissue samples with 10 µg of E. coli RNA prior to RNA isolation. E. coli 16S and 23S ribosomal RNA were used to compare the amount of rRNA isolated between the samples (Supplementary Fig. 5). We found no difference in RNA polymerase I transcribed rRNA between Taf8lox/–NesCreT/+p53–/– and control brains in the cortex or the BMC (Supplementary Fig. 5). Next, we subjected total RNA, which contains ~5% mRNA, from cortex and BMC of E14.5 Taf8 NesCre foetuses on the p53-deficient background to Northern blotting probing with poly-dT probes to detect mRNA transcripts (and poly-A-tracts in other RNA species). No difference in poly-A + RNA between Taf8lox/–NesCreT/+p53–/– brains compared to controls in the cortex or BMC was observed (Supplementary Fig. 5).
Loss of TAF8 does not affect transcription at all gene loci
In the absence of global effects of loss of TAF8 on RNA levels, we determined effects on individual gene loci by RNA-sequencing of E14.5 Taf8lox/–NesCreT/+p53–/– and Taf8+/+NesCreT/+p53–/– control cortices. The p53-deficient background was used to avoid complications due to apoptosis, but prevented us from examining changes in p53-induced gene transcription. Taf8 deleted and control samples clustered within genotype and segregated between genotypes in the first dimension (Fig. 7A). The mRNA levels of 4815 and 4280 genes were downregulated and upregulated, respectively, in the Taf8lox/–NesCreT/+p53–/– vs. Taf8+/+NesCreT/+p53–/– control cortices (FDR < 0.05; Fig. 7B; Supplementary Table 2). RNA-seq data were confirmed for a collection of 16 genes by RT-qPCR (Supplementary Fig. 5; Supplementary Table 2). The effects of loss of TAF8 on the four biological replicates per genotype were consistent (Fig. 7C). While downregulated genes were predominantly associated with nervous system development, upregulated genes were not nervous system specific (Fig. 7D, Supplementary Fig. 5). Gene ontology term analysis of genes downregulated in the absence of TAF8 indicated that neuronal function would be affected, including synapses and neuron projections (Fig. 7D; Supplementary Table 3). Pathways significantly enriched among downregulated genes included neuroactive ligand-receptor interactions, ribosomes and axon guidance (Fig. 7E, F). When compared with gene expression datasets following neuronal differentiation from embryonic stem (ES) cells to neuronal rosettes [33], the Taf8 deleted cortex displayed upregulation of genes marking primitive ectoderm and downregulation of genes marking all steps of neuronal differentiation (Fig. 7G). Similarly, genes that are expressed at higher levels in cells leaving the ventricular proliferation zone and moving into the subventricular zone and then the cortical plate to differentiate [34] were significantly enriched among the genes downregulated in the absence of TAF8 (Fig. 7H). In contrast, genes upregulated in the absence of TAF8, were much less specific to neuronal functions and involved general cell processes and the transcription factor TFIID complex (Supplementary Fig. 5; Supplementary Table 3).

Transcriptome analysis comparing E14.5 Taf8lox/–NesCreT/+p53–/– and Taf8+/+NesCreT/+p53–/– control cerebral cortices. RNA-seq data were analysed as described in the methods section. The p53–/– background was used to avoid the confounding effects of extensive cell death in the absence of TAF8 and presence of p53 (see Fig. 3). N = 4 animals per genotype. A Multidimensional scaling plot of the leading gene expression differences between samples in pair-wise comparisons showing Taf8lox/–NesCreT/+p53–/– and Taf8+/+NesCreT/+p53–/– control samples segregate in dimension one. B Plot of log2-fold change in gene expression between Taf8lox/–NesCreT/+p53–/– and Taf8+/+NesCreT/+p53–/– control samples over average expression, showing a large number of upregulated and downregulated genes. C Heatmap of the top 50 differentially expressed genes (FDR 3 × 10−10 to 2 × 10−8) showing consistency between animals within genotype and differences between different genotypes. D GO annotations significantly enriched among genes >20% downregulated in the absence of TAF8. E KEGG pathways significantly enriched among genes >20% downregulated in the absence of TAF8 (FDR < 0.05); the numbers of downregulated genes in each of the pathways are shown. F Effects of loss of TAF8 on genes expressing the ephrin/EPH, ROBO/SLIT and semaphorin axon guidance proteins (FDR < 0.05; >20% DE). G Proportion and direction of change in expression of cardinal gene data sets [33] expressed at stage of neuronal differentiation from embryonic stem cell (ESC) via primitive ectoderm (PE), transient neuroepithelial progenitors (TNEP), committed neurogenic progenitors (NP) to maturing neuronal rosettes (NR) affected by loss of TAF8. H Barcode plot displaying the positive correlation between genes that are expressed at higher levels in the subventricular zone (SVZ) and cortical plate (CP) than in the ventricular proliferation zone [VZ; [34]] with genes downregulated in the absence of TAF8. Roast p value is shown. Related data are displayed in Supplementary Fig. 6 and Supplementary Tables 2, 3.
Loss of TAF8 results in transcription defects and aberrant mRNA splicing
TFIID binds to three major promoter motifs, the TATA box, directly bound by TBP, the initiator (INR), associated with the dimeric complex of TAF1 and TAF2, and the downstream promoter element (DPE), previously thought to be bound by TAF6 and TAF9 (reviewed by [35]), although this was recently revised [36]. Only 20–30% of mammalian genes contain a TATA box [37]. The other binding motifs are thought to mediate binding in TATA-less promoters. We examined if the presence or absence of these binding motifs influenced transcription outcome in the absence of TAF8. We found that genes upregulated in the absence of TAF8 were twice as likely to contain a TATA box than genes that were unaffected or downregulated (Fig. 8A). The presence or absence of an INR or DPE motif appeared to have comparably modest effects (Fig. 8A). Genes differentially expressed in the absence of TAF8 correlated positively with differential gene expression in the absence of TAF4 [38], TAF9B [39] and TAF10 [40] (Fig. 8B). Single exon genes were approximately twice as likely to be upregulated than downregulated in the absence of TAF8, while multi-exon genes were not significantly favoured in either direction (Fig. 8C).

The RNA-seq data E14.5 Taf8lox/–NesCreT/+p53–/– and Taf8+/+NesCreT/+p53–/– control cerebral cortices described in Fig. 7 were used to assess the general parameters of transcription. The analyses are described in the methods section. N = 4 animals per genotype. A Schematic drawings of core promoter elements and their association with expression changes in the absence of TAF8. TATA box, initiator (INR) and downstream promoter element (DPE). B Barcode plots comparing the genes that are differentially expressed in the absence of TAF8 with genes differentially expressed in the absence of TAF4 [38], TAF9B [39] and TAF10 [40]. C Directional effects of the loss of TAF8 on the expression of genes with only one exon (no intron splicing required) vs. multi-exon genes. D Number of introns with increased and decreased numbers of sequencing reads. In the absence of TAF8, the 8528 introns displayed an increase in reads and 1392 introns showed a decrease in reads (p < 0.0001; binomial test). E Number of promoter regions (−5 kb to TSS) with increased and decreased numbers of sequencing reads and direction of reads. In the absence of TAF8, the 4136 promoter regions displayed an increase in reads and 944 showed a decrease in reads (p < 0.0001; binomial test). The reads observed were in the opposite direction to the main transcript of the locus at 5049 promoter regions, and in the same direction at 31 promoter regions (p < 0.0001; binomial test). F Representative genome browser image of increased sequence reads mapping to promoter sequences in the absence of TAF8 in the Mcl1 locus, typically in opposite direction of the normal transcript of the affected locus. The blue reads covering the gene body represent the Mcl1 mRNA; reads depicted in red are in the opposite direction. G Schematic drawing of the roles of MDM2 and MDM4 in opposing p53 function. H Effects of loss of TAF8 on splicing out of exon 6 in the Mdm4 gene as assessed by exon 5-to-exon 7 fusion RNA-seq read counts. I Working model of the effects caused by loss of TAF8 in the presence or absence of p53. Related data are displayed in Supplementary Fig. 6 and Supplementary Tables 2 and 4.
We noted that loss of TAF8 caused an increase in RNA-seq reads outside of exons. Taf8lox/–NesCreT/+p53–/– and Taf8+/+NesCreT/+p53–/– control cerebral cortex samples clustering within genotype when reads were mapped to either exons or promoter regions and introns (Supplementary Fig. 6). Introns were 6-fold more likely to display an increase than a decrease in RNA-sequencing reads in the absence of TAF8 (Fig. 8D). The promoter regions of genes (−5 kb to TSS) were more likely to display an increase in reads than a decrease (Fig. 8E). The aberrant promoter region expression proceeded overwhelmingly in the opposite direction of the normal transcript (Fig. 8E) and was strong in some gene loci (examples displayed in Fig. 7F, Supplementary Fig. 6).
We identified 1763 exons in 1267 genes (Sime’s method) and 2265 genes (F-tests) that were differentially spliced as a result of Taf8 deletion (FDR < 0.05; Supplementary Table 4). Aberrant splicing is a candidate mechanism underlying p53 stabilisation and activation. MDM4 (also called MDMX), together with MDM2, is an essential regulator of p53 protein activity and abundance (Fig. 8G), including in the embryonic central nervous system [41]. Induction of Mdm4-short mRNA with exon 6 spliced out has been associated with a reduction in MDM4-FL (MDM4-full length) protein and is thought to serve as a mechanism to de-repress p53 [42, 43]. We examined Mdm4 exon usage in our RNA-seq data. RNA-sequencing reads spanning the alternative exon 5 to exon 7 boundary (omitting exon 6) were ~5-fold elevated in Taf8lox/–NesCreT/+p53–/– compared to Taf8+/+NesCreT/+p53–/– control cerebral cortices (Fig. 8H), which was confirmed by conventional PCR (Supplementary Fig. 6) and RT-qPCR (Supplementary Fig. 6). Reduction in MDM4-FL has been shown to result in stabilisation of its binding partner MDM2 [44], and indeed we observed a ~3-fold increase in cytoplasmic MDM2 protein in the Taf8lox/–NesCreT/+ cortex compared to Taf8lox/+NesCre+/+ controls (Supplementary Fig. 6), but no changes in Mdm2 mRNA (Supplementary Fig. 6). These findings suggest an overall decrease in MDM4-mediated repression of the p53 pathway in the cortex.
Discussion
Loss of TAF8 resulted in extensive apoptotic cell death in the cortex, but not in other brain regions, and in upregulation of nuclear p53 protein and p53 target genes Noxa, Puma and Bax (Fig. 8I). The cell death caused by Taf8 gene deletion was completely prevented by loss of p53 and to a large extent rescued by loss of PUMA. The rescue shows that loss of TAF8 caused cell death specifically via the activation of p53, which then causes an increase in PUMA to initiate apoptosis (Fig. 8I). The Taf8 and p53 double deleted neocortex and hippocampus displayed a neuronal differentiation defect that was evident through abnormal morphology, cortex marker protein expression and reduction in the expression of neuronal differentiation genes (Fig. 8I). Our findings show that TFIID without TAF8 can assemble and mediates normal or even elevated mRNA expression from approximately three quarters of expressed gene loci, congruent with earlier work in yeast showing that depletion of individual TFIID components did not result in a global reduction in mRNA abundance [45]. Nevertheless, loss of TAF8 did cause functional disruption at many gene loci, which included reduced levels of mRNA expression, aberrant transcription of promoters and intergenic regions, as well as pre-mRNA splicing anomalies.
The genes most prominently affected by the absence of TAF8 were neuronal differentiation genes that are subject to de novo gene activation during brain development. In contrast, many of the genes that were not affected by loss of TAF8 were genes that were already active at the time when TAF8 was deleted. Based on this we hypothesise that loss of TAF8 might affect de novo gene activation (Fig. 8I), whereas the TAF8-less TFIID is capable of maintaining pre-established gene activity.
TFIID consists of three lobes, A, B and C [46]. While TFIID is not bound to DNA, (reviewed in [36]), lobe A has been reported to form a nucleosome-like octamer of four pairs of TAFs, each interacting with their histone-fold domains (TAF6-TAF9, TAF4-TAF12, TAF3-TAF10, TAF11-TAF13) as well as TBP, TAF5 and part of TAF1; lobe B contains TAF5 and three pairs of histone-fold TAFs (TAF6-TAF9, TAF4-TAF12, TAF8-TAF10), lobe C, joining lobes A and B, contains TAF1, TAF2, TAF7 and part of TAF6 and TAF8. In this model, an extension of the TAF8 histone-fold domain interacts with TAF6 and TAF2 in lobe C. Both TAF8 and TAF6 bridge lobes B and C [47]. Therefore, the model allows for the possibility that TFIID remains intact in the absence of TAF8. In contrast, when TAF10 was depleted from mouse hepatocytes, no TFIID subunits were immunoprecipitated using antibodies against TAF4 [40], suggesting that the entire complex fails to form in the absence of TAF10 in vivo. TAF8 has been proposed to be important for TAF10 and TAF2 nuclear localisation and incorporation into TFIID [6, 48]. We found that loss of TAF8 did not affect the association of TAF1, TAF5, TAF6, TAF10 or TAF12 with TBP. Our findings suggest that TAF8 is dispensable for the addition of TAF10. Nevertheless, we cannot assume that the TFIID attains its normal structure in the absence of TAF8. TAF8 is thought to stabilise the connection between lobes B and C [47] and so its absence could result in more flexibility, similar to the junction between lobes A and C.
In the current study, the activation of p53 and p53-mediated transcriptional induction of Puma, Noxa and Bax and apoptosis was curiously restricted to the cortex. Underpinning this tissue specificity in response to loss of TAF8, we found that nuclear p53 protein levels were already elevated in the developing wild-type cortex compared to other regions, which would predispose the cortex for more immediate activation of the p53-dependent apoptotic pathway. A predilection of the cortex to undergo developmental apoptosis had been reported previously [29].
The upregulation of nuclear p53 protein with only a modest increase in p53 mRNA suggested substantial post-transcriptional regulation, which nevertheless must presumably indirectly be controlled by TFIID and thus sensitive to loss of TAF8. A candidate mechanism was an impairment of the p53 regulation system orchestrated by MDM4 and MDM2. MDM2:MDM4 heterodimers are more effective at inhibiting p53 compared to MDM2:MDM2 homodimers [44]. The Mdm4 RNA is subject to alternative mRNA splicing [49]. The splice product Mdm4-short was shown to serve as a mechanism to reduce MDM4-FL (encoding full-length protein), thereby de-repressing p53 [42, 43]. We found that in the absence of TAF8 the Mdm4 locus produced Mdm4-short mRNA. The induction of MDM4 exon 6 skipping has been proposed as a new therapeutic approach for killing malignant cells through activation of wild-type p53 [50], and aberrant skipping of Mdm2 and Mdm4 exons has also been reported to result in motor neuron death in a mouse model of spinal muscular atrophy [51]. We hypothesise that loss of TAF8 may cause neuronal cell death through a failure of p53 repression that is at least in part caused by alternative splicing at the Mdm4 locus. The aberrant splicing of Mdm4 pre-mRNA is not an isolated case, but part of a more widespread defect in splicing, overall affecting introns in 23% of all genes expressed in Taf8 mutant cortices. Although aberrant splicing could indicate a general problem in co-transcriptional splicing, the fact that 77% of genes are spliced correctly in the absence of TAF8, suggests to some degree specific, rather than general effects of loss of TAF8 on splicing.
We observed anomalous transcripts in promoter regions and 3’ of the normal transcription termination regions. While the aberrant transcripts could be due to a failure of the normal RNA degradation process, their nature suggests a more specific defect: the aberrant transcripts were only present at ~15% of all gene loci. Abnormal 5’ transcripts were generated in the opposite direction of the normal transcript, suggesting a relaxation of the directionality of the transcriptional machinery when TAF8 is absent. The potential loss of the stabilising effect of TAF8 on the connection between lobes B and C of TFIID (discussed above) may contribute to such relaxation of directionality. However, despite the ambivalence displayed with respect to direction of transcription, the absence of TAF8 did not typically affect the levels of the normal transcript of the same locus.
In summary, we have shown that (1) TFIID requires the presence of TAF8 for accurate transcription at only a subset of gene loci; (2) the absence of TAF8 causes multiple defects in transcription, and (3) the absence of TAF8 leads to p53-mediated induction of Puma expression and apoptosis largely restricted to the cortex. By deleting Taf8 in the developing central nervous system, we have provided a model of transcription dysfunction during brain development in the mouse and have shown the cellular and molecular consequences that are relevant to a number of cognitive congenital [14, 16,17,18,19,20,21,22] and neurodegenerative disorders [9,10,11,12,13, 15, 52, 53], caused by mutations in TFIID components and in other transcription factors.
Materials and methods
Generation of the Taf8 conditional knockout allele and CNS specific deletion
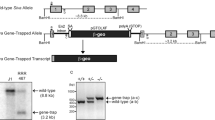
A conditional Taf8 allele was generated by homologous recombination in E. coli. LoxP sites flanking exons 1 to 7 of Taf8 were introduced, and a neomycin resistance cassette, flanked on each side by Frt recombinase sites, was introduced between exons 7 and 8 (Supplementary Fig. 1). Southern blot analysis performed on tail DNA revealed that the LoxP site and the neo cassette were correctly inserted (Supplementary Fig. 1). Taf8+/lox mice were crossed with Cre-deleter mice [54], which deletes loxP flanked sequences in the germline. This resulted in Taf8+/– mice, which were used to generate Taf8lox/– mice. Primers used to distinguish Taf8+, Taf8lox and Taf8– alleles are listed in Supplementary Table 1 and Supplementary Fig. 1 includes PCRs of tails DNA showing the different Taf8 alleles. To achieve deletion of Taf8 in the CNS, from embryonic day 9 (E9.0) onward, Taf8lox/– mice were crossed with Nestin-Cre (NesCre) transgenic mice [24]. Primers used to detect the presence of the NesCre transgene are listed in Supplementary Table 1. Animals were used in order of their birth and assigned to treatment groups according to their genotypes. No animal was excluded. Where possible animals were examined before their genotypes were known.
Histology and TUNEL
Tissues were fixed in 4% PFA or Bouin’s fixative for 24 h, embedded in paraffin, sectioned and stained with H & E. Pyknotic nuclei were counted blinded to genotype, in selected anatomical regions including the hippocampus with the future subiculum and the neocortex in the forebrain and the superior collicullus in the midbrain. TUNEL staining was carried out on paraffin embedded sections using an Apotag Kit (Roche).
Immunohistochemistry and immunofluorescence
For immunohistochemistry, paraffin sections of E14.5 heads were dewaxed, treated with antigen unmask solution (for BrdU only) and processed with intervening wash steps through 0.25% (w/v) gelatine, primary and secondary antibody and colour reaction was observed. Sections were counterstained with 0.3% methyl green or haematoxylin and mounted.
For immunofluorescence, frozen sections of E18.5 heads were processed with intervening wash steps through 1–4% (w/vol) PFA, 0.25 to 1% (w/vol) Triton X-100, 10% (w/vol) pre-immune serum, primary and secondary antibody, mounted and imaged using a fluorescent microscope (Axioplan2, Zeiss) and a digital camera (AxioCam HRC, Zeiss).
Western blot, co-immunoprecipitation (Co-IP), and chromatin-immunoprecipitation (ChIP)
Protein whole cell lysate was extracted with KALB (1% Triton X, 1 mM EDTA, 150 mM NaCl, 50 mM Tris HCl pH 7.4, 0.02% NaN3) supplemented with protease inhibitors (Roche). Protein nuclear and cytoplasmic fractions were isolated using Thermo Scientific kit (78833). Protein was run on 4–12% NuPAGE Bis-Tris gel (Life Technologies). All antibodies are listed in Supplementary Table 5. For co-immunoprecipitation (Co-IP), protein (100 µg nuclear extract) was incubated with antibody (5 µg) overnight then with Protein-G magnetic beads (Millipore) for 1 h. Beads were washed 5 times for 20 min each with PBST (Tween 0.1%) supplemented with protease inhibitors at 4 oC. Immunoprecipitated proteins were eluted from beads by incubating at 95 oC for 15 min in 2x protein reducing buffer. Uncropped Western blots are provided in the Supplementary Material.
Chromatin-immunoprecipitation (ChIP) was performed as previously described [55], however, a different sonicator (Covaris) was used for shearing chromatin, and magnetic beads instead of agarose beads were used for the precipitation step. The antibody for ChIP is described in Supplementary Table 5 and qPCR primers are described in Supplementary Table 6.
RT-qPCR and Northern blot analyses
For gene expression analysis RNA was isolated with the RNeasy Kit (Qiagen) and cDNA was generated using Superscript III (Roche) according to manufacturer’s instructions. RT-qPCR was performed using Sensimix (HiRox, Bioline, QT605-05) and a LightCycler 480 (Roche). RT-qPCR primers are listed in Supplementary Table 7. MDM4 primers were the same as in [42, 43]. TFIID primers were the same as in [40], except for TAF1 variant 1 and 2 common primers. For analysis of total mRNA by Northern blotting, brains were first weighed then spiked with 10 µg E. coli RNA to track the efficiency of RNA isolation by Bioanalyzer (Agilent Technologies), then RNA was isolated with Trizol (Life Technologies), subjected to Northern blotting and probed with a poly d-T probe synthesised using poly-A primer and TdT enzyme.
RNA-sequencing library preparation
RNA was isolated with RNeasy Kit (Qiagen). Unstranded RNA-seq libraries were prepared using a TruSeq RNA Sample Preparation kit version 2 and sequencing was performed on an HiSeq 2000 to produce paired-end 100-bp reads. We observed aberrant RNA-seq reads outside of the gene bodies at many gene loci. To examine this phenomenon further, we prepared stranded libraries from the same RNA samples with a TruSeq Stranded mRNA Library Preparation kit (Illumina). Sequencing was performed on an Illumina NextSeq 500 to produce paired-end 81-bp reads to a depth of 40 × 106 reads per sample. This allowed detailed analysis of aberrant reads and distinction of reads aligning to the plus and minus strand, generated in the same or opposite direction of the transcription of adjacent genes.
RNA-sequencing data analysis
Reads were aligned to the mouse genome mm10 using subjunc [56]. Read counts were summarised at both the gene level and the exon level using the featureCount function of the Rsubread package together with Rsubread’s built-in mm10 RefSeq annotation [57]. For the stranded RNA-seq, strand-specific counting was used. Gene annotation was downloaded from the NCBI (ftp://ftp.ncbi.nih.gov/gene/DATA/GENE_INFO/). Downstream analysis was conducted using the limma and edgeR packages [58, 59]. Genes that failed to achieve ≥0.1 counts-per-million (cpm) in at least 4 samples were filtered from the analysis. Library sizes were normalised using the TMM method [60]. Differential expression between Taf8 deleted and Taf8 intact samples was assessed using edgeR’s quasi-likelihood pipeline with an FDR of 5% [61]. edgeR’s glmTreat function was used to narrow down the list of DE genes by focusing on genes that showed fold-changes significantly greater than 1.2. Gene set tests were conducted using roast [62].
Aberrant transcripts were assessed (1) between the TSS and 5000 bp upstream of the TSS using SeqMonk (https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/), as well as (2) between the TES and 1000 bp downstream of the TES and (3) introns using featureCounts to summarise the read counts. KEGG pathway and tissue signature analyses were performed using DAVID [63, 64].
Statistical analysis
Experiments were conducted blinded where possible, e.g. before genotyping or by automated analysis, with N = 3 to 6 animals per genotype. Results are presented as mean ± SEM and were compared by Student’s T test, two-way ANOVA with Sídák correction for multiple testing.
Data availability
Complete results from our RNA-sequencing studies, including complete lists of differentially expressed genes, differentially enriched GO terms and KEGG pathways, are included in the in the Supplementary Tables. Raw data: GEO series GSE198505.
References
Papai G, Weil PA, Schultz P. New insights into the function of transcription factor TFIID from recent structural studies. Curr Opin Genet Dev. 2011;21:219–24.
Gangloff YG, Sanders SL, Romier C, Kirschner D, Weil PA, Tora L, et al. Histone folds mediate selective heterodimerization of yeast TAF(II)25 with TFIID components yTAF(II)47 and yTAF(II)65 and with SAGA component ySPT7. Mol Cell Biol. 2001;21:1841–53.
Guermah M, Ge K, Chiang CM, Roeder RG. The TBN protein, which is essential for early embryonic mouse development, is an inducible TAFII implicated in adipogenesis. Mol Cell. 2003;12:991–1001.
Soutoglou E, Demeny MA, Scheer E, Fienga G, Sassone-Corsi P, Tora L. The nuclear import of TAF10 is regulated by one of its three histone fold domain-containing interaction partners. Mol Cell Biol. 2005;25:4092–104.
Bieniossek C, Papai G, Schaffitzel C, Garzoni F, Chaillet M, Scheer E, et al. The architecture of human general transcription factor TFIID core complex. Nature. 2013;493:699–702.
Trowitzsch S, Viola C, Scheer E, Conic S, Chavant V, Fournier M, et al. Cytoplasmic TAF2-TAF8-TAF10 complex provides evidence for nuclear holo-TFIID assembly from preformed submodules. Nat Commun. 2015;6:6011.
Louder RK, He Y, López-Blanco JR, Fang J, Chacón P, Nogales E. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature. 2016;531:604–9.
Voss AK, Thomas T, Petrou P, Anastassiadis K, Scholer H, Gruss P. Taube nuss is a novel gene essential for the survival of pluripotent cells of early mouse embryos. Development. 2000;127:5449–61.
Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, et al. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet. 1999;8:2047–53.
Bauer P, Laccone F, Rolfs A, Wullner U, Bosch S, Peters H, et al. Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington’s disease-like phenotype. J Med Genet. 2004;41:230–2.
Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, Dode C, et al. Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003;126:1599–603.
Toyoshima Y, Yamada M, Onodera O, Shimohata M, Inenaga C, Fujita N, et al. SCA17 homozygote showing Huntington’s disease-like phenotype. Ann Neurol. 2004;55:281–6.
Wu YR, Lin HY, Chen CM, Gwinn-Hardy K, Ro LS, Wang YC, et al. Genetic testing in spinocerebellar ataxia in Taiwan: expansions of trinucleotide repeats in SCA8 and SCA17 are associated with typical Parkinson’s disease. Clin Genet. 2004;65:209–14.
Rooms L, Reyniers E, Scheers S, van Luijk R, Wauters J, Van Aerschot L, et al. TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. Eur J Hum Genet. 2006;14:1090–6.
Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80:393–406.
O’Rawe JA, Wu Y, Dörfel MJ, Rope AF, Au PY, Parboosingh JS, et al. TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am J Hum Genet. 2015;97:922–32.
Hellman-Aharony S, Smirin-Yosef P, Halevy A, Pasmanik-Chor M, Yeheskel A, Har-Zahav A, et al. Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatr Neurol. 2013;49:411–6.
Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63.
Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–61.
Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015;125:636–51.
Tawamie H, Martianov I, Wohlfahrt N, Buchert R, Mengus G, Uebe S, et al. Hypomorphic pathogenic variants in TAF13 are associated with autosomal-recessive intellectual disability and microcephaly. Am J Hum Genet. 2017;100:555–61.
El-Saafin F, Curry C, Ye T, Garnier JM, Kolb-Cheynel I, Stierle M, et al. Homozygous TAF8 mutation in a patient with intellectual disability results in undetectable TAF8 protein, but preserved RNA polymerase II transcription. Hum Mol Genet. 2018;27:2171–86.
Olley G, Ansari M, Bengani H, Grimes GR, Rhodes J, von Kriegsheim A, et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome. Nat Genet. 2018;50:329–32.
Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103.
Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, et al. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell. 2018;173:1217–30.
Massagué J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306.
Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci USA. 2007;104:4054–9.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8.
Haydar TF, Kuan CY, Flavell RA, Rakic P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex. 1999;9:621–6.
Fogarty LC, Flemmer RT, Geizer BA, Licursi M, Karunanithy A, Opferman JT, et al. Mcl-1 and Bcl-xL are essential for survival of the developing nervous system. Cell Death Differ. 2019;26:1501–15.
Grabow S, Kueh AJ, Ke F, Vanyai HK, Sheikh BN, Dengler MA, et al. Subtle changes in the levels of BCL-2 proteins cause severe craniofacial abnormalities. Cell Rep. 2018;24:3285–95.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–10.
Abranches E, Silva M, Pradier L, Schulz H, Hummel O, Henrique D, et al. Neural differentiation of embryonic stem cells in vitro: a road map to neurogenesis in the embryo. PLoS ONE. 2009;4:e6286.
Fietz SA, Lachmann R, Brandl H, Kircher M, Samusik N, Schroder R, et al. Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc Natl Acad Sci USA. 2012;109:11836–41.
Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–78.
Patel AB, Greber BJ, Nogales E. Recent insights into the structure of TFIID, its assembly, and its binding to core promoter. Curr Opin Struct Biol. 2020;61:17–24.
Jin VX, Singer GA, Agosto-Pérez FJ, Liyanarachchi S, Davuluri RV. Genome-wide analysis of core promoter elements from conserved human and mouse orthologous pairs. BMC Bioinforma. 2006;7:114.
Alpern D, Langer D, Ballester B, Le Gras S, Romier C, Mengus G, et al. TAF4, a subunit of transcription factor II D, directs promoter occupancy of nuclear receptor HNF4A during post-natal hepatocyte differentiation. eLife. 2014;3:e03613.
Herrera FJ, Yamaguchi T, Roelink H, Tjian R. Core promoter factor TAF9B regulates neuronal gene expression. eLife. 2014;3:e02559.
Tatarakis A, Margaritis T, Martinez-Jimenez CP, Kouskouti A, Mohan WS 2nd, Haroniti A, et al. Dominant and redundant functions of TFIID involved in the regulation of hepatic genes. Mol Cell. 2008;31:531–43.
Xiong S, Van Pelt CS, Elizondo-Fraire AC, Liu G, Lozano G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci USA. 2006;103:3226–31.
Bardot B, Bouarich-Bourimi R, Leemput J, Lejour V, Hamon A, Plancke L, et al. Mice engineered for an obligatory Mdm4 exon skipping express higher levels of the Mdm4-S isoform but exhibit increased p53 activity. Oncogene. 2015;34:2943–8.
Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, et al. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013;27:1903–16.
Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA. 2003;100:12009–14.
Moqtaderi Z, Bai Y, Poon D, Weil PA, Struhl K. TBP-associated factors are not generally required for transcriptional activation in yeast. Nature. 1996;383:188–91.
Andel F 3rd, Ladurner AG, Inouye C, Tjian R, Nogales E. Three-dimensional structure of the human TFIID-IIA-IIB complex. Science. 1999;286:2153–6.
Patel AB, Louder RK, Greber BJ, Grünberg S, Luo J, Fang J, et al. Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science. 2018;362:eaau8872.
Demeny MA, Soutoglou E, Nagy Z, Scheer E, Janoshazi A, Richardot M, et al. Identification of a small TAF complex and its role in the assembly of TAF-containing complexes. PLoS ONE. 2007;2:e316.
Mancini F, Di Conza G, Moretti F. MDM4 (MDMX) and its transcript variants. Curr Genomics. 2009;10:42–50.
Marine JC, Jochemsen AG MDMX (MDM4), a promising target for p53 Reactivation Therapy and Beyond. Cold Spring Harb Perspect Med. 2016;6:a026237.
Van Alstyne M, Simon CM, Sardi SP, Shihabuddin LS, Mentis GZ, Pellizzoni L. Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. 2018;32:1045–59.
Shatunov A, Fridman EA, Pagan FI, Leib J, Singleton A, Hallett M, et al. Small de novo duplication in the repeat region of the TATA-box-binding protein gene manifest with a phenotype similar to variant Creutzfeldt-Jakob disease. Clin Genet. 2004;66:496-501.
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441-8.
Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–1.
Voss AK, Dixon MP, McLennan T, Kueh AJ, Thomas T. Chromatin immunoprecipitation of mouse embryos. Methods Mol Biol. 2012;809:335–52.
Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108.
Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25.
Chen Y, Lun AT, Smyth GK. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016;5:1438.
Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, Smyth GK. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26:2176–82.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13.
Voss AK, Strasser A. The essentials of developmental apoptosis. F1000Res. 2020;9:F1000 Faculty Rev-148.
Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17:1270–81.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403.
Acknowledgements
We thank Carmen Gatt and Faye Dabrowski for expert animal care, Melissa Becroft and Natalie Downer for technical assistance, Günther Schütz for the nestin-Cre transgenic mice, Philippe Bouillet for helpful advice and discussion, David Segal and David Huang for provision of BCL-2, and BCL-XL antibodies, Lorraine O’Reilly for BCL-W antibodies and a positive control, and Cassandra Vandenberg for MDM2 antibodies. We thank Ygal Haupt and Sue Haupt for Mdm2;p53 double knockout MEFs.
Funding
This work was supported by the National Health and Medical Research Council (NHMRC) Project grants 215301 and 1084504 (A.K.V. and T.T.), NHMRC Program grant (1016701 [A.S.]), NHMRC Research Fellowships (1003435 [T.T.], 575512 and 1081421 [A.K.V.], 1020363 [A.S.], 1154970 [G.K.S.]), NHMRC Investigator Grants (1176789 [A.K.V.], 1176199 [Y.C.]), Independent Research Institutes Infrastructure Support Scheme from the Australian Government’s NHMRC, and a Victorian State Government Operational Infrastructure Support Grant.
Author information
Authors and Affiliations
Contributions
FE, MIB, REM, PE, MD and SW performed experiments and analysed data. YC, SH, MJD, GKS analysed data. AS provided critical discussion, advice and reagents. TT and AV conceived and supervised the project. FE, MIB, TT and AV wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All animal experiments were approved by the Walter and Eliza Hall Institute Animal Ethics Committee and conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G Melino
Rights and permissions
About this article

Cite this article
El-Saafin, F., Bergamasco, M.I., Chen, Y. et al. Loss of TAF8 causes TFIID dysfunction and p53-mediated apoptotic neuronal cell death. Cell Death Differ 29, 1013–1027 (2022). https://doi.org/10.1038/s41418-022-00982-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-022-00982-5
