
Abstract
Developing methods to improve the regenerative capacity of somatic stem cells (SSCs) is a major challenge in regenerative medicine. Here, we propose the forced expression of LIN28A as a method to modulate cellular metabolism, which in turn enhances self-renewal, differentiation capacities, and engraftment after transplantation of various human SSCs. Mechanistically, in undifferentiated/proliferating SSCs, LIN28A induced metabolic reprogramming from oxidative phosphorylation (OxPhos) to glycolysis by activating PDK1-mediated glycolysis-TCA/OxPhos uncoupling. Mitochondria were also reprogrammed into healthy/fused mitochondria with improved functional capacity. The reprogramming allows SSCs to undergo cell proliferation more extensively with low levels of oxidative and mitochondrial stress. When the PDK1-mediated uncoupling was untethered upon differentiation, LIN28A-SSCs differentiated more efficiently with an increase of OxPhos by utilizing the reprogrammed mitochondria. This study provides mechanistic and practical approaches of utilizing LIN28A and metabolic reprogramming in order to improve SSCs utility in regenerative medicine.
Similar content being viewed by others


Introduction
Undifferentiated stem/precursor cells found in adult somatic tissues, also called somatic stem cells (SSCs), are responsible for regenerating damaged tissues [1]. However, the intrinsic regenerative capacity of SSCs declines during culturing; thus, the tissue regeneration attained by transplanting cultured SSCs is not satisfactory in most cases. In vitro expansion of SSCs is a critical step to establish scalable SSCs cultures for therapeutic purposes. However, akin to biological aging, cellular senescence inevitably accumulates during cell proliferation, and that is understood to be the main cause of the observed decline in the intrinsic repair capacity of SSCs. The leading theory about the mechanism of cellular aging is called the free radical theory of aging, and it proposes that cellular activities such as aerobic respiration inevitably produce reactive oxygen species (ROS) and oxidative stress that cause mitochondrial dysfunction, DNA damage, telomere shortening, and protein oxidation [2, 3]. At the same time, the accumulation of damaged mitochondria causes increased production of mitochondrial ROS, toxic protein aggregate formation, and inflammation, which further aggravate cell senescence (reviewed in [4]). In addition, accumulating evidence has indicated the importance of mitochondrial function in stem cell activation, fate decisions, and defense against senescence (reviewed in [5]). Therefore, metabolic and mitochondria reprogramming in SSCs to enable efficient bioenergy production with low ROS and improved mitochondrial function could help to improve the therapeutic capacity of SSCs.
Lin28a and its paralog Lin28b are heterochronic RNA-binding proteins that inhibit the processing of let7 miRNA [6] and regulate mRNA translation [7]. Lin28 expression is mostly detected during embryonic development to regulate embryogenesis, developmental timing, as well as organogenesis and its expression rapidly diminishes after birth [8,9,10]. Surprisingly, forced expression of Lin28a in postnatal tissues promotes tissue repair in mice, including from ear and digit injuries [11]. These findings suggest that LIN28A has a great potential to be utilized in regenerative medicine. Although, directly inducing the expression of LIN28 in damaged tissues is not practically applicable in clinics due to the oncogenic property of LIN28 (reviewed in [12]). Instead, mechanistic insight of LIN28A-mediated repair in SSCs will help to develop effective regenerative strategies that circumvent LIN28A oncogenic potential.
Despite being extensively studied, the mechanism for LIN28A-mediated tissue repair is still ambiguous. Regulation of glucose metabolism and increased ATP production has been suggested as the main mode of action of LIN28A [11]. However, the existing studies reported conflicting results, some studies claimed that LIN28A increased glycolysis and oxidative phosphorylation (OxPhos) [11, 13], enhanced glycolysis at the expense of OxPhos [14], no effect [15], and even decreased glucose metabolism [16]. Furthermore, how LIN28A-mediated metabolic regulation is linked to tissue regeneration remains to be unraveled. We have previously shown LIN28A prevented culture-dependent loss of stem cell capacity in the embryonic rat neural stem cells through regulation of developmental timing genes [8, 15, 17]. Moreover, significant part of LIN28A studies were done on iPSC, cancer cell line, or primary cancer tissue which are metabolically different from SSCs. Therefore, the biological context of LIN28A in the clinically relevant cells such as SSCs needs to be explored further.
In this study, we showed that LIN28A expression in cultured SSCs greatly enhanced their self-renewal and proliferative capacity. In contrast to the common idea that cellular stress accumulates after cellular activities, the LIN28A-expressing SSCs maintained low oxidative/mitochondrial stress and cell senescence, even after undergoing extensive cell division. Furthermore, SSCs expanded with LIN28A expression exhibited improved differentiation into their tissue-specific lineage and cell engraftment after transplantation. We concluded that metabolic reprogramming is the main driver of the effects of LIN28A. The LIN28A-expressing SSCs predominantly used glycolysis for bioenergy production instead of mitochondrial OxPhos. Ultimately, LIN28A reprogrammed mitochondria have greater metabolic plasticity which can efficiently execute OxPhos which is a metabolic requirement for stem cell differentiation. These observations collectively indicate the potential future usefulness of LIN28A-engineered SSCs and LIN28A-mediated regenerative mechanisms in regenerative medicine.
Results
LIN28A enhances SSC self-renewal while maintaining a low level of cell senescence and improving SSC-specific marker expression
Improving the therapeutic capacity of SSCs is a prime challenge in regenerative medicine. In this study, we asked whether this issue could be addressed by engineering human SSCs, particularly SSCs already being used clinically, to express LIN28A. The SSCs we tested in this study were mesenchymal stem cells derived from bone marrow (BM-MSCs), those from umbilical cord (UC-MSCs), and muscle stem cells from skeletal muscle (SkMuSCs). To assess the effect of forced LIN28A expression, those human SSC cultures were transduced with lentiviruses carrying a doxycycline-inducible LIN28A expression vector (Dox-on-system, Fig. 1A) or an empty (or GFP) vector as a control. The transduced cultures were expanded in vitro in the presence of doxycycline (LIN28A expression) through serial cell passages, and cell proliferation and marker expression were assessed at each passage (Fig. 1B). Given that the function of Lin28 is associated with blocking the terminal differentiation of SSCs [18, 19], the continuation of exogenous Lin28a expression could interfere with SSC differentiation. Therefore, to induce SSC differentiation without exogenous LIN28A expression, doxycycline was removed from the differentiation media (Fig. 1C and Fig. S1A).

A–C Vector design for LIN28A expression and experimental scheme. Various SSC cultures were transduced with lentivirus carrying an inducible LIN28A expression vector (Dox-on system) (A) or mock vector (control). The transduced SSCs were expanded with cell passages in the presence of doxycycline (1 µg/ml, Dox+). Differentiation of the LIN28A-transduced SSCs was induced in the absence of exogenous LIN28A expression by withdrawing the doxycycline supplementation (Dox−) (B). Exogenous LIN28A induction and disappearance were confirmed in Dox(+) proliferating and Dox(−) differentiating cultures of LIN28A-transduced UC-MSCs and SkMuSCs (C).a (D–I) Analyses of undifferentiated/proliferating UC-MSC and SkMuSC cultures. Experiments were performed using SSC cultures at passage 5–6 (P5–6), except that P9 cells were used in (H–I). D–F Cell proliferation, as estimated by the cell growth curve and population doubling level (PDL, D), marker expressions specific for cell proliferation (E), and %Ki67+ cells (F) in undifferentiated cultures of UC MSCs and SkMuSCs. The inset in (F) contains DAPI+ cell images from identical microscopic fields. n = 4 (D) and 3 (E, F) independent cultures.a,c (G) Superoxide anions measured by CellROXTM in undifferentiated UC-MSC and SkMuSC cultures. n = 15.b,c (H) Cell senescence assayed using the SA-β-galactosidase assay. n = 3.a,c (I) Immunoblotting of p16 and p21 protein expression in UC-MSCs and SkMuSCs. J–N Differentiation of cultured UC-MSCs and SkMuSCs in vitro (J–L) and in vivo after transplantation (M, N). J Osteogenesis assessed by alizarin red staining in UC MSC cultures (P10) 14 days after the induction of in vitro osteogenic differentiation. n = 3.a (K) Cartilage-specific gene expression in differentiated LIN28A (mock)-transduced UC-MSC cultures (P11) after 21 days of chondrogenic differentiation.a (L) Skeletal muscle differentiation of LIN28A- and control-SkMuSC cultures (P6) in vitro. Fusion index of MF20-stained myotubes was estimated at differentiation day 4. n = 3.a,c (M) In vivo bone formation capacities of transplanted LIN28- and control-UC-MSCs. Undifferentiated UC-MSCs (P6) were grafted into rats with critical calvarial defects. Newly formed bone (highlighted in green) was detected 2 months post-transplantation in µCT-scans. n = 4 (sham), 4 (mock-transduced UC-MSC transplant), 9 (LIN28A-UC-MSC transplant).a (N) In vivo integration and muscle differentiation of LIN28A-SkMuSC grafts. Undifferentiated LIN28A (or control)-SkMuSCs at P6 were transplanted into the tibialis anterior muscles of mice. Five weeks after transplantation, grafted cell survival and muscle differentiation were assessed using the areas co-stained with Stem121 (human-specific) and Collagen VI (muscle specific). n = 6 for each group.b,c aData shown are the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-tailed Student’s t testing or one-way ANOVA followed by Tukey’s post-hoc testing. bData shown are the median. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by the Mann–Whitney U testing. cScale bar, 100 µm.
In UC-MSC and SkMuSC cultures, LIN28A overexpression drastically elevated cell expansion as estimated by total viable cell counts and the population doubling levels (PDLs, Fig. 1D), and the onset of replicative senescence was delayed by 1–2 passages. Consequently, compared with the control cultures, LIN28A-expressing UC-MSC and SkMuSC cultures yielded 11.7 ± 2.3 and 23.3 ± 1.6 fold more cells, respectively, before reaching replicative senescence. LIN28A-mediated cell expansion was also manifested in the cultures of BM-derived MSCs (Fig. S1B). Along with enhanced expression of the proliferative markers E2F1, MCM2, and MCM7 in the UC-MSC and SkMuSC cultures (Fig. 1E), the % Ki67+ proliferating cells was greater in the LIN28A-expressing cultures than the control cultures (Fig. 1F). There was no significant difference in apoptotic, necrotic, and viable cells between LIN28A and control cultures on annexin V/ propidium iodide (PI) flow cytometry assay (Fig. S2), indicating that LIN28-induced cell expansion was mainly attributed to cell proliferation.
It has been postulated that a main source of cellular damage contributing to cellular aging is the ROS generated from the cell metabolism. As the consequence, the more metabolically active cells inevitably produce more ROS. Interestingly, despite greater cell proliferative activity, superoxide levels (estimated by CellROXTM) were lower in LIN28A-expressing UC-MSC and SkMuSC cultures than in the control cultures (Fig. 1G). Accordingly, we also observed that LIN28A group has significantly lower senescence as demonstrated by several senescence assays, such as %β-galactosidase staining (Fig. 1H), p16 and p21 immunoblot (Fig. 1I), and quantification of senescence-associated secretory phenotype (SASP) molecules (IL1A, IL6, IL8. MMP3) (Fig. S3). These findings collectively suggest that LIN28A expression in cultured SSCs enhances cell proliferation while maintaining low levels of oxidative stress and cellular senescence.
Improved differentiation and repair capacity of SSCs cultured with LIN28A expression
The differentiation potential of SSCs declines with age [20]. Consistently, bone and cartilage differentiation were poorly induced in BM-MSC cultures derived from old donors [21]. The forced expression of LIN28A in MSCs derived from the BM of old donors greatly enhanced the differentiation toward bone and chondrocytes (Fig. S1D–H). Similar to the age-dependent changes in SSC properties, SSC differentiation capacity is also lost during in vitro cell expansion [2, 22, 23]. Differentiation potential gradually declined during cell expansion and was substantially lost in the late passages of UC-MSC cultures (P10) (Fig. 1J), but bone and cartilage differentiation were much more efficient in the late passages UC-MSC cultures expanded with LIN28A expression (Fig. 1J, K), collectively indicating that the age- and culture-dependent losses of MSC differentiation potential are rescued by LIN28A expression. We also used the myoblast fusion index to assess in vitro muscle differentiation in long-term passaged SkMuSCs (P6). Cells were induced to differentiate and stained with MF20, which reacts with the sarcomeric myosin heavy chain. The forced expression of LIN28A promoted muscle cell fusion and generated larger myotubes with more nuclei than the control cells (% of MF20 + myotube containing >4 nuclei: 58.7% ± 6.3% (LIN28A) vs 28.0% ± 3.8% (control)) (Fig. 1L).
Tissue regeneration in cell replacement therapy largely depends on the capacity of the donor SSC to efficiently differentiate and survive long-term in the host tissues. The effects of LIN28A on cell senescence and differentiation in vitro indicate that donor SSCs cultured with LIN28A could show improved tissue regeneration upon transplantation. To test that possibility, we transplanted cultured UC-MSCs into rats with critical calvarial defects. Eight weeks post-transplantation, bone regeneration, estimated using micro-CT to examine the density of newly generated bone, was dramatically greater in the rats grafted with LIN28A-expressing UC-MSCs than in those in the control conditions (p < 00001, n = 9 (LIN28A), 4 (control), 4 (sham-operated)) (Fig. 1M). Similarly, upon transplantation into mouse tibialis anterior (TA) muscles, SkMuSCs expanded with LIN28A (P6) were efficiently integrated and survived for at least for five weeks after grafting, and myocyte-specific collagen VI immunoreactivity was co-localized in portions of the human-specific STEM121+ cells grafted (Fig. 1N). By contrast, hardly any viable STEM121+ cells were detected in any of the 6 mice grafted with the control SkMuSCs. These findings together indicate that LIN28A expression could be used to increase the therapeutic utility of SSC cultures.
RNA replicon-mediated gene delivery as a clinical system for LIN28A expression
The clinical application of LIN28A expression will require safe gene delivery methods with an equivalent efficacy. RNA-based gene delivery, which is virus- and integration-free, appears to be an inherently safe method for future clinical applications. Due to rapid degradation, RNA delivery methods are inefficient and require repetitive daily transfection, but this problem has recently been solved by the development of a synthetic, self-replicative RNA system called the RNA replicon [24]. Therefore, with clinical application in mind, we tested RNA replicon-mediated gene delivery for LIN28A expression (Fig. S4A–C). Transfection is inevitably toxic to cells, and thus it is frequently a critical reason for reductions in the naïve repair capacity of SSCs. After testing various transfection methods, RNA replicon-mediated gene delivery with the RiboJuiceTM transfection reagent turned out to be efficient and caused no cell toxicity in UC-MSC cultures (data not shown). The RNA replicon-mediated transgene expression was maintained for at least 14 days and completely subsided during the next several days. During the days (with 3 passages) of LIN28A expression induced by the RNA replicon system, 1.4 ± 0.04 and 1.9 ± 0.1 fold more cells were yielded compared with the mock-transfected and untransfected control cultures, respectively (Fig. S4D). In addition, the effects of LIN28A on the cell senescence and bone differentiation of the UC-MSCs were replicated with RNA replicon-mediated LIN28A expression (Fig. S4E, F).
LIN28A induces reprogramming of glucose metabolism in SSCs
An understanding of the mechanisms underlying the effects of LIN28A could also be useful in regenerative medicine. Therefore, we conducted experiments to uncover how LIN28A enhances the regenerative capacity of SSCs. Due to a limited supply of BM tissues the following mechanism studies were carried out using only UC-MSC and/or SkMuSC cultures. It has been shown that tissue repair is attained with metabolic reprogramming [11, 25,26,27,28]. Glucose metabolism is a key player to control the fates and behaviors of stem cells [29]. Lin28a, Lin28b, and let7 miRNA have been reported to regulate glucose metabolism, and transcripts encoding mitochondrial OxPhos and glycolysis enzymes are among the top mRNAs bound by Lin28a [30, 31]. These findings together suggest that metabolic reprogramming could underlie the LIN28A effects we observed.
Because the cell proliferation promoted by LIN28A is a biological process that demands a large amount of energy, we expected the bioenergy production in the LIN28A-expressing SSC cultures to be greater than in the controls. As expected, intracellular ATP levels during cell proliferation were much greater in the LIN28A-transduced UC-MSCs and SkMuSCs than in their respective controls (Fig. 2A). Surprisingly, LIN28A expression in the proliferating SSCs dramatically lowered oxygen consumption (Fig. 2B). Specifically, decreased basal OxPhos and oxygen-linked ATP production indicated that the LIN28A-expressing SSCs depended less than the controls on OxPhos to fulfill their energy demand. By contrast, glycolysis was enhanced by LIN28A expression, with the maximum glycolytic capacity being significantly greater in the LIN28A-SSCs than in the controls (Fig. 2C). To confirm that finding, we performed a metabolomics study using LC/MS-MS. The metabolomics data show that LIN28A increased the relative abundance of glycolysis intermediates such as fructose bi-phosphate, 3-phosphoglyceric acid, and lactate (Fig. 2D–F). Combined, those findings indicate that LIN28A causes a metabolic switch from OxPhos to glycolysis in proliferating SSCs, which is the characteristic metabolic feature of highly proliferating cancer/stem cells [32], known as Warburg’s effect. Considering that mitochondrial OxPhos produces a lot of ROS, the switch in the bioenergy production pathway from OxPhos to glycolysis enables the LIN28A-SSCs to undergo cell proliferation with low ROS production and cell senescence. The enhanced glycolytic capacity probably also contributes to the observed efficiency in the engraftment of LIN28A-UC-MSCs and LIN28A-SkMuSCs after transplantation (Fig. 1M, N) because it has been reported that grafted cell survival and engraftment are positively associated with the glycolytic capacity of donor cells [33].

A Total intracellular ATP levels in proliferating/undifferentiated UC-MSCs and SkMuSCs transduced with LIN28A (or mock). n = 3 independent cultures. B,C Seahorse analyses to measure OxPhos (oxygen consumption rate, OCR, B) and glycolytic capacity (extracellular acidification rate, ECAR, C) in undifferentiated UCMSCs and SkMuSCs. n = 6. D Heatmap of metabolic profiles from LIN28A-overexpressing UC-MSCs and control UCMSCs. n = 4. GLC glucose, FBP fructose bi-phosphate, PGA 3-phosphoglyceric acid, PYR pyruvate, LAC lactate, CIT citrate, AKG α-ketoglutarate, SUC succinate, FUM fumarate, MAL malate. E Comparison of metabolic profiles between LIN28A-overexpressed UC-MSCs and control UC-MSCs. n = 4. Numbers on the top row represent the mean ± SEM fold change of metabolite peak intensity in LIN28A-overexpressing cells compared with control cells. Numbers on the bottom row are the p value as determined by two-tailed Student’s t testing. F Extracellular lactate concentrations in undifferentiated UC-MSC and SkMuSC cultures. n = 3. G–H OCR and ECAR in UC-MSC and SkMuSC cultures during differentiation, which was induced without exogenous LIN28A expression by withdrawing the doxycycline. The Seahorse analyses were conducted 4 days after differentiation. n = 3. All experiments were done in SSC cultures at P5–6. Data shown are the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-tailed Student’s t testing.
It has also been reported that OxPhos increases during SSC differentiation and that OxPhos is therefore a metabolic signature of cell differentiation (reviewed in [29, 32]). Consistently, when differentiation of UC-MSCs and SkMuSCs was induced by the withdrawal of mitogens and doxycycline (no LIN28A expression, Fig. 1B, C), an increase of OxPhos occurred and the glycolytic capacity in the differentiating SSC cultures decreased (Fig. S5). Of note, OxPhos increases in the cultures differentiated from LIN28A-SSCs were much greater than those in the control cultures (Fig. 2G). In addition, although basal respiration did not differ significantly between the LIN28A and control cultures, spare respiratory capacity was much greater in the differentiated LIN28A cultures (Fig. 2G). Spare respiratory capacity is a reserve capacity that allows mitochondria to respond to sudden energy demands and has been shown to be positively linked with enhanced cell survival [34, 35] and inversely related to cellular aging [36]. The glycolytic capacities of the differentiating cultures for the control and LIN28A-SSCs were indistinguishable (Fig. 2H). These findings together suggest that the greater increase in OxPhos during the differentiation of LIN28A-SSCs was associated with their differentiation capacity, which was greater than that of the control SSCs (Fig. 1J–N).
Mechanism underlying LIN28A-induced metabolic reprogramming of undifferentiated SSCs
Insulin is the major extracellular molecule that activates glycolysis. Levels of activated (phosphorylated) AKT (p-AKT), the signal transducer downstream of insulin/insulin receptor binding, were significantly greater in LIN28A-UC-MSC and SkMuSC cultures than in their respective controls (Fig. 3A). Across the insulin concentrations tested, the levels of Akt activation were greater in LIN28A-SSCs (Fig. 3B), suggesting a LIN28A-mediated increase in insulin sensitivity [31, 37]. It is well acknowledged that AKT signal activation promotes glucose transport by causing trans-localization of glucose transporters to the plasma membrane [38, 39]. Consistently, glucose uptake in LIN28A-expressing UC-MSC cultures was greater than in the control cultures (Fig. 3C), and it was followed by an increase in the intracellular glucose levels in LIN28A-expressing cultures (Fig. 2D). Given that glucose is an allosteric activator of glucokinase/hexokinase, the key enzyme of the glycolytic pathway, we suggest that the increase in intracellular glucose is a major mechanism underlying LIN28A-induced glycolysis in undifferentiated SSC cultures. In addition, transcripts of several glycolytic enzymes were significantly upregulated by LIN28A (Fig. 3D), which is consistent with the observed increase in glycolysis (Fig. 2C). The upregulated glycolytic enzyme expression was likely mediated by the Lin28/Let7 axis [11, 31] and/or activated AKT signaling [40, 41].

A, B Immunoblotting for intracellular AKT signaling activated in LIN28A-overexpressed UCMSC and SkMuSC cultures. The activated (phosphorylated) AKT (p-AKT) and total AKT protein levels in (A) were determined in undifferentiated SSCs cultured in serum-containing medium in which the insulin concentration is unidentified. To determine insulin sensitivity, the undifferentiated UC-MSC cultures were serum-starved for 24 h, and then the levels of AKT activation were determined as the ratio of p-AKT/total AKT in cultures treated with the indicated doses of insulin for 5 min (B). n = 3. (C) Glucose uptake activity assessed in undifferentiated UC-MSCs. n = 3. (D) mRNA expression of the key glycolysis genes (HK1, HK2, PFKM, PFKP, LDHA) (E) Venn diagram showing common gene identified from published LIN28A in stem cell transcriptome data. Numbers are representing the number of genes that significantly downregulated in Lin28KO cells compared to WT control. F qRT-PCR of PDHK1 genes in UC-MSCs and SkMuSCs. n = 3. G Immunoblotting and quantification of PDK1 protein expression in LIN28A-expressing SSCs. n = 3. H Extracellular lactate concentration in control and LIN28A-overexpressed UC-MSCs after PDK1 inhibitor treatment (DCA) for 72 h. n = 3. I Total cell number of control and LIN28A-overexpressing UC-MSCs after PDK1 inhibitor treatment for 1 passage. n = 3. J qRT-PCR of proliferation markers (MKI67, MCM2, MCM7) in UC-MSCs after PDK1 inhibitor treatment. n = 3. K SA-β-galactosidase assay in UC-MSCs after PDK1 inhibitor treatment. n = 3. Data shown are the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-tailed Student’s t testing. Scale bar, 100 µm.
Next, to gain a mechanistic understanding of the LIN28A-mediated decrease of OxPhos, we analyzed transcriptome data obtained from Lin28aKO and Lin28a,bDKO mouse induced pluripotent stem cells (miPSCs) (GSE67568) and polysome profiles from Lin28a,bDKO mouse neural precursor cells (mNPCs) (GSE131536). Notably, one of ten genes commonly and significantly upregulated in all the datasets was pyruvate dehydrogenase kinase 1 (PDK1) (Fig. 3E). Consistently, we found that the protein and mRNA levels of PDK1 were upregulated in LIN28A-transduced UC-MSC and SkMuSC cultures (Fig. 3F, G). PDK1 is the enzyme catalyzing the uncoupling of glycolysis and the tricarboxylic acid (TCA) cycle by inhibiting the activity of the pyruvate dehydrogenase (PDH), thereby shutting down pyruvate entry into the TCA-OxPhos pathway. Thus, we strongly suggest that the upregulated PDK1 expression is responsible for the observed LIN28A activity to inhibit OxPhos. When the PDK1 inhibitor dichloroacetic acid (DCA) was administered to LIN28A-transduced UC-MSC cultures, the level of lactate, an indicator of cellular glycolytic activity, was decreased significantly (Fig. 3H). Along with the decreased lactate level, the LIN28A-induced UC-MSC proliferation was abolished in the assays we used to determine PDLs and cell-cycle gene expression (Fig. 3I, J). Furthermore, the LIN28A-mediated anti-senescence effect was almost completely removed by PDK1 inhibitor treatment (Fig. 3K). Together, these findings strongly suggest that LIN28A-mediated upregulation of PDK1 is the critical mediator in the OxPhos-to-glycolysis metabolic switch and that PDK1-mediated metabolic reprogramming is responsible for enhanced SSC self-renewal with low levels of oxidative stress, which is directly associated with the therapeutic utility of LIN28A-SSC cultures.
Mitochondrial reprograming underlies enhanced OxPhos during differentiation of LIN28A-SSCs
Interestingly, PDK1 signaling, estimated using PDK1 protein levels, was drastically downregulated upon the induction of differentiation in UC-MSCs (Fig. S6). Given the role of PDK1 in uncoupling glycolysis from the TCA cycle, the downregulation of this signaling molecule could contribute to the transition from glycolysis-dependent bioenergy production in undifferentiated/proliferating SSCs to OxPhos-dependent production during differentiation (Fig. S4).
One remaining question is how OxPhos was greatly increased during the differentiation of LIN28A-transduced SSC cultures compared with the control SSC differentiation (Fig. 2G). Because exogenous LIN28A expression was turned off during differentiation by withdrawing doxycycline, the observed increase of OxPhos in the differentiating LIN28A-SSC cultures is unlikely to be mediated by a direct action of LIN28A. Because mitochondria are responsible for the TCA-OxPhos process, we postulated that LIN28A action on mitochondria during the undifferentiated/proliferating stage of the SSC cultures could contribute to increased OxPhos during differentiation.
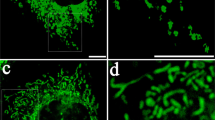
First, we observed that mitochondrial content, estimated using the mitochondrial DNA copy number and flow cytometry-based quantification of MitoTrackerTM-labeled mitochondria (Fig. S7A, B), was indistinguishable between undifferentiated controls and LIN28A-UC-MSCs. Interestingly, MitoTrackerTM + mitochondria in the LIN28A-cells were preferentially localized in the peri-nuclear area, whereas those in the control cells were distributed throughout the cell body (Fig. 4A). The pattern of the mitochondria distribution was further confirmed by a super-resolution stochastic optical reconstruction microscopy (STORM)-based image analysis of mitochondria labeled with TOM20, a mitochondrial outer membrane protein. In addition, TOM20 + mitochondria in LIN28A-expressing cells were highly fused and interconnected, while those in the control cells were fragmented with a radiated strand-like arrangement (Fig. 4B, C and S7C, D). Consistently, the protein levels of Mitofusin-2 (MFN2) and Optic Atrophy 1 (OPA1), positively associated with mitochondrial fusion, were greater in the LIN28A-UC-MSCs than the controls, while decreased level of Dynamin-1-like protein (DRP1), which regulates mitochondrial fission, was manifested in the LIN28A cells (Fig. 4D). Further interestingly, we observed that TOM20+ mitochondria in LIN28A cultures were arranged into multiple “ring”-like structures that had been previously observed in uncoupled mitochondria (mitochondria unengaged in mitochondrial ATP generation) [42]. Upon differentiation induction by withdrawal mitogens and doxycycline (without LIN28A expression), the interconnected mitochondrial structures were abolished (Fig. S7E, F). This morphologic characteristic further supports that mitochondria in undifferentiated LIN28A cultures are not mainly recruited for OxPhos-ATP generation.

A MitoTrackerTM red staining of control and LIN28A-overexpressing UC-MSCs (left). Ratio of mitochondrial peri-nuclear and network localization calculated from MitotrackerTM staining using ImageJ (right). n = 13.a (B) Super-resolution STORM imaging of mitochondria as labeled by outer membrane protein TOM20. (C) Mitochondria morphometry analysis of reconstructed STORM images. Scale bar, 5 µm. n = 8.b (D) Immunoblotting of fusion proteins (MFN2 and OPA1) and fission proteins (p-DRP1 and DRP1) in mitochondrial extract of proliferating UC-MSCs and SkMuSCs. n = 3. E Immunoblotting of cytosolic cytochrome C release in UC-MSCs and SkMuSCs. n = 3. F Mitochondrial ROS measurement one week after transient H2O2 treatment using MitoSOXTM in UC-MSCs and SkMuSCs. n = 3.a(G) Mitochondrial oxidative stress shown by red/green signal intensity ratio in Mitotimer. Scale bar, 100 µm. n = 15.b (H) The NADPH level and NADPH/total NADPH ratio in undifferentiated UC-MSC and SkMuSC cultures. n = 4. a(I) GSH/GSSG ratio in cultured UC-MSCs and SkMuSCs. n = 3. a(J) Antioxidant capacity, as determined by superoxide radical scavenging capacity in cultured UC-MSCs. n = 4. a(K) ATP synthase activity determined in mitochondria isolated from undifferentiated LIN28A- and control-UC-MSCs. The activities were calculated from the time course curves for ATP production normalized with citrate synthase activity(right). aData are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-tailed Student’s t testing. bData are shown as the median. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by Mann–Whitney U testing.
Peri-nuclear localization of mitochondria detected in the LIN28A-UC-MSCs has been suggested as a characteristic of mitochondria that are newly synthesized, immature, or mostly undamaged [43,44,45]. By contrast, mitochondrial fission and fragmentation, more prominent in the control cells, was detected in damaged mitochondria associated with cell apoptosis (reviewed in [46, 47]). Consistently, cytosolic levels of cytochrome C, which is released from damaged mitochondria [48], were significantly lower in undifferentiated LIN28A-UC-MSC cultures than in the controls (Fig. 4E). In addition, the LIN28A-SSC cultures had lower mitochondrial ROS levels (MitoSOXTM, Fig. 4F) than the controls. Next, we used Mitotimer, which encodes the COX8 mitochondrial protein tagged with a DsRed mutant (DsRed1-E5) that emanates green fluorescence when newly synthesized and shifts irreversibly to red when oxidized [49]. We found that undifferentiated LIN28A-SSCs showed a higher intensity of green fluorescent puncta than the controls (Fig. 4G). These findings together suggest that mitochondria in the undifferentiated LIN28A-SSCs are healthier and more intact than those in the control SSCs.
In healthy mitochondria, the ROS produced from the OxPhos reaction were rapidly neutralized through a combination of the mitochondrial glutathione peroxidase, peroxiredoxin, and thioredoxin systems [50]. These ROS scavenging systems require reduced glutathione (GSH) and NADPH, a crucial co-factor for catalyzing the reduction of glutathione disulfide (GSSG) to its sulfhydryl form (GSH) [51]. NADPH is principally produced in the pentose-phosphate pathway, which branches from glucose-6-phosphate, an intermediate glycolytic pathway, indicating that NADPH production is increased in LIN28A-SSCs via increased glycolysis (Fig. 2C). As expected, undifferentiated UC-MSC and SkMuSC cultures transduced with LIN28A had increased NADPH and NADPH/NADP ratios (Fig. 4H) and a significantly greater GSH/GSSG ratio than the controls (Fig. 4I). Consequently, the capacity to scavenge superoxide anions was significantly greater in LIN28A-expressing UC-MSC and SkMuSC cultures than in the controls (Fig. 4J). These findings collectively suggest a close link between LIN28A-mediated metabolic and mitochondrial reprogramming, in which the metabolic OxPhos-to-glycolysis switch in LIN28A-SSCs contributes to remodeling of healthy and intact mitochondria, not only through low mitochondrial occupancy in the ROS-producing OxPhos process, but also by increasing the mitochondrial antioxidant capacity.
Mitochondrial protein folding stress is induced during stem cell transition from quiescence to proliferation [52]. Therefore, LIN28A might reduce mitochondrial stress by controlling mitochondria protein folding stress. However, no LIN28A-mediated changes were detected in the expression of HSP60 protein and the genes involved in the mitochondrial unfolded protein response (UPRmt) (Fig. S8), suggesting that UPRmt is not involved in the LIN28A effect relieving mitochondrial stress.
Mitochondria with fused morphology has been suggested to execute OxPhos more efficiently than the fragmented mitochondria [53, 54], indicating the mitochondria in LIN28A-cells acquire the greater functional capacity to execute ATP production via OxPhos. Therefore, our next question was whether the mitochondria reprogrammed in LIN28A cells are more functional. To address it, mitochondria were isolated from undifferentiated LIN28A- and control-UC-MSC, and subjected to mitochondrial functional analyses in the cell-free condition. Interestingly, the rate cell-free ATP synthesis (final step of OxPhos) from LIN28A mitochondria was much greater than those from the mitochondria isolated from the control cells (Fig. 4K). Possibly indicating a more robust mitochondrial integrity. Collectively, these findings strongly suggest that mitochondria in undifferentiated LIN28A-SSCs were reprogrammed into fused/healthy mitochondrial with greater OxPhos functionality, but that the mitochondria in undifferentiated LIN28A-SSCs are not engaged in OxPhos-ATP production due to PDK1-mediated glycolysis-OxPhos uncoupling.
The observed LIN28A effects are partially recapitulated in late passaged SSCs transplanted with the reprogrammed mitochondria
As the final experiment in this study, we assessed the contribution of the mitochondrial reprogramming to the observed LIN28A-mediated increase of the SSC therapeutic capacities. To this end, we isolated mitochondria from the LIN28A (or mock)-transduced UC-MSC cultures, and long-term passaged UC-MSCs (or SkMuSCs) were transplanted with the isolated mitochondria (Fig. 5A) [55]. MitotrackerTM-labeled mitochondria from the donor cells were efficiently internalized and detected in the recipient late-passage UC-MSCs (Fig. 5B). Of note, UC-MSCs transplanted with the mitochondria isolated from LIN28A-cells showed higher proliferation rates as indicated by increased PDL and % Ki67+ proliferating cells (Fig. 5C, D). Moreover, the LIN28A-mitochondrial transplantation significantly reduced cellular senescence in the late-passage UC-MSC (Fig. 5E). However, the Lin28-mitochondria transplantation did not have a significant effect on UC-MSC or SkMuSC differentiation (Fig. 5F, G). This suggests that mitochondrial reprogramming alone is insufficient to recapitulate LIN28A-mediated increase of SSC differentiation. Alternatively, considering short-term maintenance of mitochondrial integrity and functionality after isolation and transplantation [56], the mitochondrial transplantation we applied may not provide the recipient cells with mitochondria with sufficient levels of functionality and integrity which could be sustained for the time period required for the cell differentiation effect. Collectively, the mitochondrial transplantation experiment confirms that the mitochondrial reprogramming is at least in part responsible for the LIN28A-mediated improvement of the therapeutic capacity of cultured SSCs.

A Schematic drawing of mitochondria isolation and transplantation experiments. Mitochondria from the LIN28A (mock)-transduced UC-MSC cultures, and long-term passaged UC-MSCs (or SkMuSCs) were transplanted with the isolated mitochondria. B MitoTrackerTM-labeled donor mitochondria in UC-MSC 24 h after transplantation. C Accumulated population doubling of UC-MSC for two passages after transplantation. n = 3. D KI67 staining of UC-MSC and its quantification. n = 3. E Senescence-associated β-galactosidase staining of UC-MSC and its quantification. n = 3. F qRT-PCR of osteocytes specific gene in UC-MSC 14 days after the induction of osteogenic differentiation. n = 3. G MF20 staining after skeletal muscle differentiation and its quantification. n = 3. Data are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by two-tailed Student’s t testing. Scale bar, 100 µm.
Discussion
In this study, we have shown that LIN28A expression greatly enhances the self-renewal and proliferation capacities of SSCs cultured from human somatic tissues, which enabled the production of larger numbers of SSCs by culturing. Notably, even though the LIN28A-expressing SSCs underwent more extensive cellular activities by performing more numerous rounds of cell cycles, their levels of oxidative and mitochondrial stress and senescence were substantially lower than those of control SSCs. Thus, SSCs expanded in the presence of LIN28A exhibited improved integration and differentiation in vitro and in vivo after transplantation into two animal models. The effects of LIN28A reported here were commonly manifested in cultures of BM-MSCs, UC-MSCs, and SkMuSCs, all of which are currently used in clinics to treat intractable degenerative disorders, though limited cell supplies and repair capacities remain critical challenges to be addressed [57]. With further clinical applications in mind, we have here proposed RNA replicon-mediated transfection as a safe and efficient LIN28A delivery method.
LIN28 is an RNA-binding protein whose functions were identified in early embryogenesis and pluripotent stem cells [58]. LIN28A expression ends after development in most of tissues, and its re-expression in adult transgenic mice has been reported to enhance the regeneration of various somatic tissues by acting on SSCs harbored within those tissues [11]. Therefore, in addition to ex vivo LIN28 expression in SSC cultures, directly inducing the expression of LIN28 in damaged tissues is another therapeutic candidate for treating degenerative disorders, though it is not practically applicable in clinics due to the oncogenic property of LIN28. Instead, mechanistic information about the downstream signals involved in LIN28A-mediated repair functions will help to develop regenerative strategies that circumvent the oncogenic hazard. In this study, we have attempted to understand how LIN28A expression can cause paradoxical phenomena, such as enhanced SSC proliferation with low ROS levels and senescence, and how LIN28A-mediated SSC proliferation is linked to the efficient differentiation of SSCs. This study is of value to provide a comprehensive understanding for the LIN28A actions in cultured SSCs, with the mechanisms involving metabolic and mitochondrial reprogramming, which are closely interconnected.
We identified reprogramming of the bioenergy-producing metabolism from OxPhos to glycolysis as the central mechanism underlying LIN28A-induced SSC proliferation with low oxidative stress and senescence. Glycolytic reprogramming was attained because LIN28A enhanced glucose uptake and the expression of several glycolytic enzymes, probably through Akt signal activation and LET7 mRNA regulation. Furthermore, the activation of PDK1, an enzyme that uncouples glycolysis-TCA/OxPhos, was critical for the LIN28A-mediated glycolysis increase at the expense of OxPhos. Glycolysis and its side pathways produce metabolites that are used as building blocks for cell growth and expansion. On the other hand, OxPhos, albeit more efficient at producing energy per glucose molecule, completely oxidizes glucose into CO2 and water, which cannot be used for cell growth. In addition, inefficiency and defects in the electron transport chain system inevitably result in the production of ROS. Therefore, glycolysis is a desirable energy generating pathway for maintaining cell proliferation with low cellular stress. This raises question of how the high energy demand required for enhanced cell proliferation could be attained with glycolysis, which is a less efficient pathway to yield bioenergy than OxPhos. The contradiction could be explained by the catalytic speed of glycolysis, which is tens of fold faster than that of OxPhos [59]. In addition to the naïve, low ROS generation in glycolysis, we found that the antioxidation capacity of LIN28A-SSCs was enhanced by greater NADPH production via the PPP, a pathway branched from glycolysis. Given the beneficial aspects of glycolysis-dependent bioenergy production, high metabolic flux through glycolysis is regarded as a metabolic feature of pluripotent stem cells and SSCs in young adult tissues exhibiting high repair capacity [60,61,62,63,64,65,66,67,68].
Given mitochondria-independent glycolysis is the main energy metabolism in stem cells, mitochondria had been regarded until recently as an organelle less important in stem cell activities. However, recent studies have shown the importance of mitochondria in stem cell fate decisions and defense against senescence, with a decline in stem-cell mitochondrial activity contributing to the age-related deterioration of stem cell capacity in multiple different tissues [69,70,71,72,73,74]. However, the activity of mitochondria in stem cell functions is ill-defined. In this study, we have shown that mitochondria in LIN28A-SSCs were reprogrammed to have a fused morphology and healthier phenotype than we found in the control cells. Without being needed for TCA-OxPhos, the mitochondria in the undifferentiated LIN28A-SSCs probably carried out other mitochondria-specific functions, such as anti-oxidation and Ca2+ buffering, more efficiently, which might be associated with the maintenance of stemness and self-renewal capacity. Isolated mitochondria from LIN28A-SSCs could partially reproduced LIN28A effects when transplanted to late-passage recipient cells, further suggesting mitochondrial reprogramming as one of the mechanisms for the LIN28A-mediated improvement of therapeutic capacity in cultured SSCs. More importantly, the healthy mitochondria were spared and preserved for efficient OxPhos during differentiation. Indeed, upon the induction of differentiation in LIN28A-SSCs, PDK1-mediated uncoupling of glycolysis-TCA/OxPhos was relieved and followed by an increase of OxPhos to a level greater than in the control SSCs, indicating that LIN28A enhances metabolic plasticity in SSCs. Whereas glycolysis is the major energy metabolism in undifferentiated stem cells, OxPhos is the metabolic signature for differentiating stem cells and is closely associated with stem cell differentiation potential [34, 35].
In conclusion, our data demonstrate that LIN28A can reprogram cellular metabolism by uncoupling glucose oxidation from TCA cycle, which caused SSCs to be more glycolytic and acquire less oxidative metabolism. This, in turn, improves self-renewal and differentiation potentials of SSCs over long-term passaging due to lesser oxidative damage to the mitochondria. The practical and mechanistic information attained in this study will help build up future strategies to improve SSC-based cell therapy, especially through SSCs metabolic engineering.
Experimental procedures
Ethical statement for SSC isolation from human tissues
BM-MSCs
Human bone marrow was obtained from three patients (age range: 54–72 years) undergoing total hip replacement due to osteoarthritis, with prior consent obtained from all donors. All experiments were carried out in accordance with the guidelines and regulations of the Dongguk University Ilsan Hospital Institutional Review Board (approved protocol DUIH 2012-34). The BM MSCs were isolated as previously described [75].
UC-MSCs
Human umbilical cords were provided by Samsung Medical Center in South Korea with prior consent from all donors. All experiments were carried out in accordance with the guidelines and regulations under the auspices of the Samsung Medical Center Institutional Review Board approved protocol 2016-07-102. Isolation of UC-MSCs was performed as previously described [76].
Cell culture
BM-MSCs were cultured in low glucose DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. UC-MSCs were cultured in alpha-MEM (Gibco) containing 10% FBS (Gibco), glutaMAXTM-I (Gibco), and 1 µg/mL doxycycline (Sigma). SkMuSCs were cultured in F-10 medium (Gibco) containing 20% FBS (Gibco), 20 ng/mL bFGF (Peprotech), and 1 µg/mL doxycycline (Sigma). Osteogenic differentiation of MSCs was induced by alpha-MEM (Gibco) containing 10% FBS (Gibco) supplemented with 100 µM L-ascorbate-2-phosphate (Sigma), 10 mM β-glycerophosphate (Sigma), and 100 nM dexamethasone (Sigma) for 14 days. Chondrogenic differentiation was performed as previously described [75] using chondrogenic medium (100 µM L-ascorbate-2-phosphate (Sigma) and 100 nM dexamethasone (Sigma) were added to DMEM/F12 (Gibco) containing penicillin/streptomycin, 1.25 mg/mL BSA (Sigma), ITS-G (Gibco), 1 mM sodium pyruvate (Sigma), and 100 µM L-proline (Sigma)) for 21 days. SkMuSC differentiation was induced through growth factor withdrawal by switching to DMEM/F12 containing N2 supplements (Gibco) for 4 days.
Virus production and transduction
Lentiviruses expressing human LIN28A under the Dox-on system were generated and used to transduce in vitro cultures, as previously described [15]. Titers of the lentiviruses were determined using a QuickTiterTM HIV Lentivirus Quantitation Kit (Cell Biolabs), and 10 µL of 106 transducing units/mL/well were used for each transduction reaction.
Cell proliferation analysis
Cell expansion of each MSC or SkMuSC passage was estimated by PDL, which was determined as log(N/No)/log2 where N is the number of cells at the end of each passage, and No is the number of cells plated initially.
Quantitative RT-PCR analysis
Total RNA was prepared using TRIzolTM reagent (Invitrogen) and the RNA isolation protocol. For general mRNA and pre-LET7 miRNA, cDNA synthesis was carried out using a SuperScriptTM kit (Invitrogen). Quantitative RT-PCR was performed on a CFX96TM Real-Time System using iQTM SYBR® Green Supermix (Bio-Rad), and gene expression levels were determined relative to beta-actin levels. Primer information is shown in Table S1.
Immunostaining
Cultured cells and cryo-sectioned muscle slices were fixed with 4% paraformaldehyde (PFA) in PBS and blocked in 0.3% Triton X-100 with 1% BSA for 1 h, then incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: KI67 (1:200, Abcam), MF20 (1:10, Developmental Studies Hybridoma Bank), STEM121 (1:200, Cellartis), and COLVI (1:2000, Abcam). The secondary antibodies used for visualization were Cy3 (1:500, Jackson Immunoresearch Laboratories) or Alexa Fluor 488 (1:500, Life Technologies). The stained cells were mounted using VECTASHIELD with DAPI mounting solution (Vector Laboratories), and photographs were obtained using an epifluorescence microscope (Leica).
Mitochondrial, cytosol, and nuclear fractionation
Cells were lysed by mechanical homogenization using a 26 G syringe in mitochondrial extraction buffer (20 mM HEPES-KOH pH 7.5, 220 mM sorbitol, 10 mM potassium acetate, 70 mM sucrose) containing proteinase inhibitor cocktail and phosphatase inhibitor. Lysates were centrifuged at 600 g and 4 °C for 15 m. The supernatant was retained, and the pellet was processed further using an EpiQuikTM nuclear extraction kit I (Epigentek) to isolate the nuclear fraction. The supernatant was centrifuged at 7000 g and 4 °C for 15 m. The supernatant was retained as the cytosol fraction, and the pellet was resuspended in mitochondrial extraction buffer as the mitochondrial fraction.
Mitochondria functional analysis in cell-free condition
Mitochondria were isolated from cultured UC-MSCs by differential centrifugation as described previously [55]. In brief, cells were harvested, homogenized in SHE buffer (0.25 M sucrose, 20 mM K-HEPES (pH 7.4), 2 mM K-EGTA) containing 0.1% defatted bovine serum albumin (BSA), and centrifuged at 1100 x g for 3 min at 4 °C. The supernatant was collected and then centrifuged at 12,000 x g for 10 min at 4 °C to pellet mitochondria. Next, mitochondria pellet was resuspended in 1 mL SHE buffer containing 0.1% defatted BSA and centrifuged at 20,000 x g for 5 min at 4 °C. Then pellet was resuspended in 1 mL SHE buffer and recentrifuged at 20,000 x g for 5 min at 4 °C. Mitochondria pellet was resuspended in 50 µL SHE buffer or PBS and quantified by determining the protein concentration using a bicinchoninic acid assay. Citrate synthase activity assay was performed as described previously [77]. Briefly, 2 µg of isolated Mitochondria sample was added into 96-wells plate and mixed with 100 µL of Tris buffer containing Triton-X 100, 6 µL of 10 mM Acetyl-CoA and 20 µL of 1 mM DTNB. The volume was adjusted with distilled water up to 192 µL. To initiate the reaction, 10 µL of 10 mM oxaloacetic acid was added for each sample. The absorbance (OD450nm) was measured for 20 min. The citrate synthase activity of the isolated mitochondria can be calculated based on GSH standard curve. A total of 50 mM GSH solution and its serially diluted solutions were used as standard. ATP synthase activity assay was conducted using CellTiter-Glo Luminescence cell viability kit (Promega) based on the manufacturer’s protocol with modification. Briefly, the ATP standard solutions were prepared with serial dilution of 2 mM ATP stock solution. For the sample measurement, 5 µg of isolated mitochondria was added into the well contains 10 µL of 500 µM ADP, and the volume was adjusted up to 100 µL with distilled water. Finally, 100 µL of CellTiter-Glo Reagent was added to both standard curve and sample wells. The luminescence was detected immediately for 18 min in kinetic mode.
Mitochondria transplantation
Mitochondria transplantation was conducted by adding 10 µg of isolated mitochondria to the 1 × 105 UC-MSCs. For MitoTrackerTM-labeled donor mitochondria, Mock or LIN28A-overexpressed cells were treated with MitoTrackerTM before mitochondria isolation. Cell proliferation analysis, KI67 staining, and SA-β-galactosidase assay were performed two passages after transplantation.
Cell death assay
Cell death assay was performed using the Annexin-V-FLUOS staining kit (Roche) according to the manufacturer’s protocol and counted on a FACSCanto II Flow Cytometer (BD Bioscience) according to the manufacturer’s instructions. Obtained data were processed using FlowJo.
Western blotting
Cell lysates were prepared in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and 1 mM EDTA) containing a proteinase inhibitor cocktail and phosphatase inhibitor unless otherwise indicated, followed by sonication. Lysates were cleared of insoluble material by centrifugation at 15,000 g and 4 °C for 5 m. Protein concentration was determined using the Pierce BCA assay (Thermo Fisher) or protein assay dye (Bio-Rad). SDS-PAGE was conducted followed by electrophoretic transfer to nitrocellulose or PVDF membranes. Immunoblots were blocked with 5% BSA dissolved in TBST for 1 h, then incubated with primary antibodies overnight at 4 °C. The primary antibody information is shown in Table S1. For immunodetection of the primary antibodies, a goat-anti-rabbit-HRP conjugate or goat-anti-mouse-HRP conjugate (Cell Signaling Technology) was used at 1:1,000 in 5% BSA dissolved in TBST. HRP was detected using West-Pico chemiluminescence substrate (Thermo Fisher). For the p-AKT and AKT immunoblots, cultured cells were pretreated with 4 µg/mL insulin (Gibco) for 5 m unless otherwise indicated.
SA-β-galactosidase assay
The senescence-associated-β-galactosidase assay was performed using a Senescence β-galactosidase staining kit (Cell Signaling Technology) on late-passage cultured cells according to the manufacturer’s protocol and photographed using a camera attached to a light microscope (Olympus).
Scavenging capacity assay
The scavenging capacity assay was performed using a CheKineTM superoxide anion scavenging capacity assay kit (Abbkine). Samples and reagents were prepared according to the manufacturer’s protocol. Absorbance measurement was conducted twice, at time points 0 and 16 h after reagent addition.
Superoxide anion analysis
Cultured cells were treated with 100 µM menadione for 1 h at 37 °C, then incubated with 5 µM CellROXTM green reagent (Invitrogen) for 30 m. The stained cells were mounted using VECTASHIELD with DAPI mounting solution (Vector Laboratories), and photographs were obtained using an epifluorescence microscope (Leica).
Alizarin red and safranin O staining
vTerminally differentiated MSC-derived osteoblasts were fixed with 4% PFA in PBS and stained with alizarin red, pH 4.1–4.3 (Sigma), for 30 m at room temperature. MSC-derived chondroblasts were fixed with 4% PFA in PBS and embedded in Tissue-Tek OCT compound (Sakura Finetek, Inc., Torrance, CA). The embedded chondroblasts were cryo-sectioned into 10 µm-thick slices and stained with safranin-o (Sigma) for 10 m at room temperature.
Alkaline phosphatase staining
BM-MSCs were cultured in osteogenic medium for 7 days. The differentiated cells were fixed with 4% PFA in PBS and permeabilized with 0.1% Triton-X for 30 m, then stained with nitro blue tetrazolium and 5-bromo-4-chroro-3-indolyl phosphate (Sigma).
Transplantation and histological procedures
All procedures for animal experiments were approved by the Hanyang College of Medicine Institutional Animal Care and Use Committee (approval number, 2017-0182A and 2018-0217A). For the calvarial defect model, the scalps of adult female Sprague-Dawley rats (220–250 g) were exposed, and a single 8-mm diameter critical-sized calvarial defect was created in each one using a dental trephine in the left posterior quadrant of the calvaria. Then, one million MSCs were suspended in a fibrin glue scaffold and placed in the calvarial hole. The animals were euthanized 8-weeks post-surgery, and new bone formation was measured using μ-CT (Quantum FX micro CT, Perkin Elmer). Muscle regeneration models were made using 12-week-old female ICR mice. Muscle injury was induced using 10 μL of 1.2% BaCl2 injected intramuscularly into the TA muscle. Twenty-four hours after the injury, 1 × 104 human skeletal muscle stem cells were injected in the injured muscle. The mice were euthanized 4-weeks post transplantation, and the TA muscles were subjected to immunostaining.
In vitro transfection using synthetic self-replicating RNA
Human LIN28A or enhanced GFP was inserted into multiple cloning sites of pVEE-IRES-Puro (a kind gift from Dr. Steven F. Dowdy [24]). Plasmids were linearized by MluI, followed by RNA synthesis using a MEGAscript T7 kit (Invitrogen). 5′ capping and poly(A) tailing modification were done with a ScriptCapTM m7G capping system (Cellscript), ScriptCapTM 2‘-O-methyltrasferase kit (Cellscript), and A-PlusTM poly(A) polymerase tailing kit (Cellscript). Cells were transfected with synthetic self-replicating RNA using a RiboJuiceTM mRNA transfection kit (Merck).
Extracellular lactate measurement
Cells were cultured for 8 h in serum-free DMEM (without phenol red or sodium pyruvate) containing 20 ng/mL bFGF (Peprotech), 20 ng/mL EGF (Peprotech), 20 ng/mL IGF-1 (Peprotech), 20 ng/mL PDGF-BB (Peprotech), 20 nM insulin, and 1 µg/mL doxycycline. Supernatant was collected and deproteinized using an Amicon Ultra-0.5 mL centrifugal filter (Merck). Extracellular lactate was measured using a Lactate assay kit II (Sigma) according to the manufacturer’s protocol.
Metabolites profiling using UPLC-Orbitrap-MS
For sample preparation, 100 μL ice-cold 70% methanol containing an internal standard (2 μg/mL reserpine) was added to the cell pellets (1.2 × 106) and vortexed for 30 s. Cells were lysed with liquid nitrogen in 3 consecutive freeze/thaw cycles. The lysate was centrifuged for 10 m at 14,000 rpm and the supernatant was used for analysis. Quality control (QC) samples for instrument conditioning and evaluating the repeatability prepared by pooling equal volumes of each sample. An Ultimate 3000 UHPLC system coupled to an LTQ Orbitrap Velos Pro™ system mass spectrometer (Thermo Fisher Scientific) equipped with a heated electrospray ionization source (HESI) was used for instrumental analysis. Chromatographic separation were performed with an Acquity® UPLC HSS T3 column (1.8 μm particle size, 2.1 mm × 100 mm, Waters), and the autosampler and column oven temperature were maintained 5 °C at 40 °C during analysis, respectively. The gradient elution was performed using mobile phase A (0.1% formic acid in distilled water) and mobile phase B (0.1% formic acid in methanol) at a flow rate of 0.4 mL/min as follows: 99% A and 1% B (v/v), were maintained from 0 to 1 min, and a linear gradient increase to 20% B occurred over 2 min. An increase to 70% B for 4 min was then performed, and mobile phase B was increased to 100% over 6 min. The status was maintained for 2.5 min and re-equilibrated to the initial conditions within for 3.5 min. The samples were injected 10 μL and analyzed in positive and negative ionization mode. The detailed mass spectrometry parameters were as follows: heater temperature, 200 °C; sheath gas flowrate, 35 arb (arbitrary units); auxiliary gas flow rate, 5 arb; sweep gas flowrate, 10 arb; capillary temperature, 320 °C; and S-lens RF level, 67.5%. Xcalibur 2.2 and Compound Discoverer 2.1 (Thermo Fisher Scientific) were used for data acquisition and peak alignments in the centroid mode at a resolution of 60,000 using a mass range of m/z 50–1000. The metabolites related to the glycolysis and TCA cycle were identified by using database, such as HMDB (http://www.hmdb.ac), Metlin (http://metlin.scripps.edu) and Lipidblast (Fiehn lab, UC Davis) based on the m/z of the adducts, MS/MS fragmentation and retention time matching. The identified metabolites were normalized by the areas of the internal standard for statistical analysis.
Extracellular metabolic flux analysis
Cultured cells were seeded onto an XF96 Cell Culture Microplate (Seahorse Bioscience) at a density of 3.5 × 104 cells/well and incubated overnight. The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience) according to the manufacturer’s protocol with a Seahorse XF Cell Mito Stress Test kit and Glycolysis Stress Test kit, respectively. The OCR and ECAR were normalized to the protein concentration using a Pierce BCA assay (Thermo Fisher). For the OCR, cells were sequentially treated with 2 µM oligomycin, 2 µM FCCP, and 1.5 µM rotenone + 0.5 µM antimycin. For the ECAR, cells were sequentially treated with 10 mM glucose, 2 µM oligomycin, and 50 mM 2-deoxy-D-glucose (2-DG).
ATP measurement
Cells were lysed by mechanical homogenization using a syringe in 10 mM Tris-Cl pH 7.5, followed by centrifugation at 15,000 g and 4 °C for 5 m. The supernatant was retained and diluted 10–100 times for ATP measurement, which was carried out using an ATP determination kit (Invitrogen) according to the manufacturer’s protocol.
Insulin sensitivity assay
Cells were cultured in DMEM/F12 (Gibco) for 24 h to induce starvation. Then, MSCs or SkMuSCs were treated with 0, 10, and 100 nM insulin or 0, 5, and 10 nM insulin (Gibco), respectively, for 5 m, followed by western blotting.
Transcriptome analysis
Normalized microarray data for Lin28aKO and Lin28a,bDKO mouse embryonic fibroblast (MEF)-derived mIPSCs were obtained from GSE67568 and RNA-seq count data of polysome-associated RNA of Lin28a,bDKO mouse NPC were obtained from GSE131536. Microarray data was analyzed using R package “limma” with significance criteria determined as FDR < 0.01 and at least 1.5-fold lower expression than WT control [78]. Polysome RNA-seq data was analyzed by using R package “edgeR” and significance criteria was FDR < 0.01 [79].
Glucose uptake assay
Cultured cells were seeded onto a black 96-well plate at a density of 1 × 104 cells/well and incubated overnight. The glucose uptake assay was performed using a glucose uptake-Glo assay kit (Promega) according to the manufacturer’s protocol.
NADPH and total NADP assay
The NADPH and total NADP assay were conducted using an NADP/NADPH assay kit (Abcam) according to the manufacturer’s protocol.
Glutathione measurement
Reduced GSH was measured using a CheKineTM reduced GSH colorimetric assay kit (Abbkine) according to the manufacturer’s protocol. Total glutathione was determined using a CheKineTM GSSG colorimetric assay kit (Abbkine) based on the manufacturer’s protocol but discarding the inhibitor treatment. Oxidized glutathione was calculated as (total glutathione − reduced glutathione) / 2.
MitoSOXTM-based flow cytometry analysis
Cultured cells were treated with 0, 100, 1,000, and 10,000 nM H2O2 for 1 h, followed by culturing for 1 week. Then the cells were treated with 5 µM MitoSOXTM at 37 °C for 10 m, followed by 3 washes with PBS. The treated cells were counted on a FACSCanto II Flow cytometer (BD Bioscience) according to the manufacturer’s instructions. Obtained data were processed using FlowJo.
mtDNA copy number
The mtDNA copy number was measured using quantitative RT-PCR. Cells were lysed in 100 µL of lysis buffer (25 mM NaOH and 0.2 mM EDTA) at 95 °C for 30 m, followed by the addition of 100 µL of neutralization buffer (40 mM Tris-Cl pH 5.5). Primers for NADH dehydrogenase subunit 1 (ND1) and Beclin-1 (BECN1) were used to determine the mtDNA copy number (Table S1). PCR conditions were 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 15 s at 60 °C. Quantitative RT-PCR was conducted in triplicate and performed on a CFX96TM real-time system using iQTM SYBR green Supermix (Bio-Rad). The CT values of ND1 were normalized to nuclear genomic DNA (BECN1).
MitoTrackerTM-based flow cytometry analysis
Cultured cells were treated with 250 nM MitoTrackerTM at room temperature for 30 m, followed by 3 washes with PBS. The treated cells were counted on a FACSCanto II Flow cytometer (BD Bioscience) according to the manufacturer’s instructions. Obtained data were processed using FlowJo.
MitoTrackerTM analysis
Cultured cells were seeded onto a 24-well plate containing a cover slip at a density of 1 × 104 cells/well. Then, the cells were treated with MitoTrackerTM according to the manufacturer’s protocol. The cells were fixed with 4% PFA in PBS and mounted using VECTASHIELD with DAPI mounting solution (Vector Laboratories). Photographs were obtained using an epifluorescence microscope (Leica), and the captured images were processed using ImageJ.
Mitotimer analysis
Lentiviruses expressing human COX8-Mitotimer were generated and used to transduce in vitro cultures as previously described [49]. Cultured cells were seeded onto a 24-well plate containing a cover slip at a density of 5 × 103 cells/well. The cells were transduced with lentiviruses expressing human COX8-Mitotimer and cultured for 1 week before being fixed with 4% PFA. The fixed cells were mounted using VECTASHIELD with DAPI mounting solution (Vector Laboratories), and photographs were obtained on a confocal microscope (Leica). Captured images were processed using ImageJ.
STORM imaging and morphometry analysis
Undifferentiated LIN28A-transduced UC-MSC cultures were stained with anti-TOM20 (F-10) (Santa Cruz Biotechnology) antibody for mitochondria and anti-LIN28A (Proteintech) antibody for LIN28A protein. STORM (Stochastic Optical Reconstruction Microscopy) imaging was performed with an inverted microscope (Nikon) equipped with a 100× and 1.49 NA oil immersion objective lens (Nikon). Lasers with wavelengths at 488 nm (OBIS; Coherent, 110 mW) and at 647 nm (OBIS; Coherent, 120 mW) were used as excitation lasers. The excitation lasers were delivered to the sample by the multi-band dichroic mirror (Semrock). The illumination angle was adjusted for total internal reflection fluorescence (TIRF). Fluorescence of the sample was filtered by the multi-band emission filter (Semrock) and detected using an Electron Multiplying CCD (EM-CCD) camera (Andor) at a frame rate of 60 Hz. During image acquisition, the CRISP Autofocus system (ASI) maintained the focal plane by the feedback of the IR LED reflected signal at the interface between the cover glass and the imaging buffer. In STORM image analysis, the pixelated point spread function of single fluorophores in each frame was fitted to a Gaussian function to determine the spatial position of individual fluorophores, and the reconstructed STORM image from the collection of fluorophore positions was rendered after the drift-correction. As reconstructed STORM images are collection of “dots” instead of continuous lines, high-resolution STORM images were traced manually to specifically select for mitochondria and exclude noise. For morphometry analysis, ImageJ macro tool “MiNA” was used [80]. For calculating the number of TOM20 puncta and fragments, native ImageJ function “Analyze Particle” was used after applying background correction and CLAHE.
Statistical analysis
Statistical analyses were performed with Prism software version 6 (GraphPad Software) or IBM SPSS Statistics 26 (IBM) using two-tailed unpaired t testing or one-way analysis of variance (ANOVA) for verifying differences between normally distributed samples, or Mann–Whitney U testing for verifying differences between samples without Gaussian distribution. In all cases, results were considered statistically significant when p < 0.05. Error bars show the mean ± standard error of the mean (SEM) or the median, and p values are represented as: p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Data availability
All data are present in the paper and Supplementary Materials. Additional data may be requested upon reasonable request.
References
Ermolaeva M, Neri F, Ori A, Rudolph KL. Cellular and epigenetic drivers of stem cell ageing. Nat Rev Mol Cell Biol. 2018;19:594–610.
Bonab MM, Alimoghaddam K, Talebian F, Ghaffari SH, Ghavamzadeh A, Nikbin B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006;7:14.
Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–81.
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95.
Zhang H, Menzies KJ, Auwerx J. The role of mitochondria in stem cell fate and aging. Development. 2018;145:dev143420.
Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA.2008;14:1539–49.
Balzer E, Moss EG. Localization of the developmental timing regulator Lin28 to mRNP complexes, P-bodies and stress granules. RNA Biol. 2007;4:16–25.
Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell.1997;88:637–46.
Richards M, Tan SP, Tan JH, Chan WK, Bongso A. The transcriptome profile of human embryonic stem cells as defined by SAGE. Stem Cells. 2004;22:51–64.
Yokoyama S, Hashimoto M, Shimizu H, Ueno-Kudoh H, Uchibe K, Kimura I, et al. Dynamic gene expression of Lin-28 during embryonic development in mouse and chicken. Gene Expr patterns: GEP. 2008;8:155–60.
Shyh-Chang N, Zhu H, Yvanka de Soysa T, Shinoda G, Seligson MT, Tsanov KM, et al. Lin28 enhances tissue repair by reprogramming cellular metabolism. Cell.2013;155:778–92.
Wang X, Weng M, Jin Y, Yang W, Wang X, Wu D, et al. Beyond an oncogene, Lin28 is a master regulator of cancer progression. Histol Histopathol. 2018;33:327–34.
Shinoda G, Shyh-Chang N, Soysa TY, Zhu H, Seligson MT, Shah SP, et al. Fetal deficiency of lin28 programs life-long aberrations in growth and glucose metabolism. Stem Cells. 2013;31:1563–73.
Docherty CK, Salt IP, Mercer JR. Lin28A induces energetic switching to glycolytic metabolism in human embryonic kidney cells. Stem Cell Res Ther. 2016;7:78.
Rhee YH, Kim TH, Jo AY, Chang MY, Park CH, Kim SM, et al. LIN28A enhances the therapeutic potential of cultured neural stem cells in a Parkinson’s disease model. Brain.2016;139:2722–39.
Zhang J, Ratanasirintrawoot S, Chandrasekaran S, Wu Z, Ficarro SB, Yu C, et al. LIN28 regulates stem cell metabolism and conversion to primed pluripotency. Cell Stem Cell. 2016;19:66–80.
Ambros V, Horvitz HR. Heterochronic mutants of the nematode Caenorhabditis elegans. Science.1984;226:409–16.
Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes & development. 2014;28:971–82.
Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet. 2012;44:1199–206.
Carlson ME, Conboy IM. Loss of stem cell regenerative capacity within aged niches. Aging Cell. 2007;6:371–82.
Mendes SC, Tibbe JM, Veenhof M, Bakker K, Both S, Platenburg PP, et al. Bone tissue-engineered implants using human bone marrow stromal cells: effect of culture conditions and donor age. Tissue Eng. 2002;8:911–20.
Jang YC, Sinha M, Cerletti M, Dall’Osso C, Wagers AJ. Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function. Cold Spring Harb Symp Quant Biol. 2011;76:101–11.
Kretlow JD, Jin YQ, Liu W, Zhang WJ, Hong TH, Zhou G, et al. Donor age and cell passage affects differentiation potential of murine bone marrow-derived stem cells. BMC Cell Biol. 2008;9:60.
Yoshioka N, Gros E, Li HR, Kumar S, Deacon DC, Maron C, et al. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell. 2013;13:246–54.
Ash SR, Cuppage FE. Shift toward anaerobic glycolysis in the regenerating rat kidney. Am J Pathol. 1970;60:385–402.
Fu X, Zhu MJ, Dodson MV, Du M. AMP-activated protein kinase stimulates Warburg-like glycolysis and activation of satellite cells during muscle regeneration. J Biol Chem. 2015;290:26445–56.
Chen F, Zhou J, Li Y, Zhao Y, Yuan J, Cao Y, et al. YY1 regulates skeletal muscle regeneration through controlling metabolic reprogramming of satellite cells. EMBO J. 2019;38:e99727.
Honkoop H, de Bakker DE, Aharonov A, Kruse F, Shakked A, Nguyen PD, et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. 2019;8:e50163.
Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–56.
Peng S, Chen LL, Lei XX, Yang L, Lin H, Carmichael GG, et al. Genome-wide studies reveal that Lin28 enhances the translation of genes important for growth and survival of human embryonic stem cells. Stem Cells. 2011;29:496–504.
Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell.2011;147:81–94.
Intlekofer AM, Finley LWS. Metabolic signatures of cancer cells and stem cells. Nat Metab. 2019;1:177–88.
Moya A, Paquet J, Deschepper M, Larochette N, Oudina K, Denoeud C, et al. Human mesenchymal stem cell failure to adapt to glucose shortage and rapidly use intracellular energy reserves through glycolysis explains poor cell survival after implantation. Stem Cells. 2018;36:363–76.
Nickens KP, Wikstrom JD, Shirihai OS, Patierno SR, Ceryak S. A bioenergetic profile of non-transformed fibroblasts uncovers a link between death-resistance and enhanced spare respiratory capacity. Mitochondrion.2013;13:662–7.
Pfleger J, He M, Abdellatif M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015;6:e1835.
Porter C, Hurren NM, Cotter MV, Bhattarai N, Reidy PT, Dillon EL, et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am J Physiol Endocrinol Metab. 2015;309:E224–32.
Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl Acad Sci USA. 2011;108:21075–80.
Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20:436–50.
Hajduch E, Litherland GJ, Hundal HS. Protein kinase B (PKB/Akt)-a key regulator of glucose transport? FEBS Lett. 2001;492:199–203.
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9.
Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–10.
Miyazono Y, Hirashima S, Ishihara N, Kusukawa J, Nakamura KI, Ohta K. Uncoupled mitochondria quickly shorten along their long axis to form indented spheroids, instead of rings, in a fission-independent manner. Sci Rep. 2018;8:350.
Ahlijanian MK, Striessnig J, Catterall WA. Phosphorylation of an alpha 1-like subunit of an omega-conotoxin-sensitive brain calcium channel by cAMP-dependent protein kinase and protein kinase. J Biol Chem. 1991;266:20192–7.
Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol. 2010;48:725–34.
Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010;28:721–33.
Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–80.
Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–6.
Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–33.
Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 2014;289:12005–15.
Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–54.
Mailloux RJ, McBride SL, Harper ME. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem Sci. 2013;38:592–602.
Mohrin M, Widjaja A, Liu Y, Luo H, Chen D. The mitochondrial unfolded protein response is activated upon hematopoietic stem cell exit from quiescence. Aging Cell. 2018;17:e12756.
Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain.2008;131:352–67.
Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife. 2019;8:e41351.
Kim MJ, Hwang JW, Yun CK, Lee Y, Choi YS. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci Rep. 2018;8:3330.
Lightowlers RN, Chrzanowska-Lightowlers ZM, Russell OM. Mitochondrial transplantation-a possible therapeutic for mitochondrial dysfunction?: Mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep. 2020;21:e50964.
Abdallah BM, Kassem M. Human mesenchymal stem cells: from basic biology to clinical applications. Gene Ther. 2008;15:109–16.
Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell.2010;140:445–9.
Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science.2001;292:504–7.
Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606.
Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, et al. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011;14:264–71.
Varum S, Rodrigues AS, Moura MB, Momcilovic O. Easley CAt, Ramalho-Santos J, et al. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE. 2011;6:e20914.
Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–90.
Turner WS, Seagle C, Galanko JA, Favorov O, Prestwich GD, Macdonald JM, et al. Nuclear magnetic resonance metabolomic footprinting of human hepatic stem cells and hepatoblasts cultured in hyaluronan-matrix hydrogels. Stem Cells. 2008;26:1547–55.
Chen CT, Shih YR, Kuo TK, Lee OK, Wei YH. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2008;26:960–8.
Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pr Cardiovasc Med. 2007;4:S60–7.
Kondoh H, Lleonart ME, Nakashima Y, Yokode M, Tanaka M, Bernard D, et al. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal. 2007;9:293–9.
Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park DJ, et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–8.
Ahlqvist KJ, Hamalainen RH, Yatsuga S, Uutela M, Terzioglu M, Gotz A, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15:100–9.
Anso E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol. 2017;19:614–25.
Khacho M, Clark A, Svoboda DS, MacLaurin JG, Lagace DC, Park DS, et al. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum Mol Genet. 2017;26:3327–41.
Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011;14:623–34.
Stoll EA, Cheung W, Mikheev AM, Sweet IR, Bielas JH, Zhang J, et al. Aging neural progenitor cells have decreased mitochondrial content and lower oxidative metabolism. J Biol Chem. 2011;286:38592–601.
Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science.2016;352:1436–43.
Kang ML, Ko JY, Kim JE, Im GI. Intra-articular delivery of kartogenin-conjugated chitosan nano/microparticles for cartilage regeneration. Biomaterials.2014;35:9984–94.
Kwon S, Ki SM, Park SE, Kim MJ, Hyung B, Lee NK, et al. Anti-apoptotic effects of human Wharton’s Jelly-derived mesenchymal stem cells on skeletal muscle cells mediated via secretion of XCL1. Mol Ther. 2016;24:1550–60.
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–46.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics.2010;26:139–40.
Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017;119:315–26.
Acknowledgements
We are grateful to Prof. Koh Hyun-Chul (Hanyang University) and Park Aeri (Hanyang University) for research information and experimental materials.
Funding
This work was supported by the grants 2017R1A5A2015395, 2020M3A9D8039925, 2017M3A9B4062415, and 2018R1C1B6003436 funded by the National Research Foundation of Korea (NRF) of the Ministry of Science and ICT, Republic of Korea.
Author information
Authors and Affiliations
Contributions
SHL supervised the project. SHL, YAS, KP conceived and designed experiments, and drafted and revised the manuscript. YAS, KP performed experiments, analyzed and interpreted data, and performed statistical analyses. NW, WHWD, JYK, JWC, MJK, MRL, SAL, HL, GL, BHJ, HP, GK, DK, GC, CHK, DDL, and KSP performed research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Isolation of BM-MSCs were carried out with informed consent in accordance with the guidelines and regulations of the Dongguk University Ilsan Hospital Institutional Review Board. Isolation of UC-MSCs were carried out with informed consent in accordance with the guidelines and regulations under the auspices of the Samsung Medical Center Institutional Review Board. All procedures for animal experiments were approved by the Hanyang College of Medicine Institutional Animal Care and Use Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by L. Scorrano
Supplementary information
Rights and permissions
About this article

Cite this article
Pieknell, K., Sulistio, Y.A., Wulansari, N. et al. LIN28A enhances regenerative capacity of human somatic tissue stem cells via metabolic and mitochondrial reprogramming. Cell Death Differ 29, 540–555 (2022). https://doi.org/10.1038/s41418-021-00873-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00873-1
