
Abstract
The F-box and WD-repeat-containing protein 2 (FBXW2) plays a crucial role as an E3 ligase in regulating tumorigenesis. However, the functions of FBXW2 in breast cancer are still unknown. Here, we find that nuclear factor-kB (NF-κB) p65 is a new substrate of FBXW2. FBXW2 directly binds to p65, leading to its ubiquitination and degradation. Interestingly, p300 acetylation of p65 blocks FBXW2 induced p65 ubiquitination. FBXW2-p65 axis is a crucial regulator of SOX2-induced stemness in breast cancer. Moreover, FBXW2 inhibits breast tumor growth by regulating p65 degradation in vitro and in vivo. FBXW2 overexpression abrogates the effects of p65 on paclitaxel resistance in vitro and in vivo. Furthermore, FBXW2 induced p65 degradation is also confirmed in FBXW2-knockout mice. Our results identify FBXW2 as an important E3 ligase for p65 degradation, which provide insights into the tumor suppressor functions of FBXW2 in breast cancer.
Similar content being viewed by others


Introduction
Breast cancer is a leading cause of cancer death in women worldwide, with a steady increase in incidence [1]. In developed countries, breast cancer treatment is continually being improved to further reduce mortality [2]. Breast cancer affects approximately one in eight women in developed countries, remaining the primary burden of cancer-related diseases in women [3]. Prevention provides the most cost-effective strategy to reduce the public health cost of breast cancer. However, no clear approach to prevent breast cancer has been defined.
The SCF (SKP1-Cullin1-F-box protein) ubiquitin complex, also known as CRL1 (Cullin-RING ligase 1), consists of S-phase kinase-associated protein 1 (SKP1), Cullin-1, Ring box protein-1 (RBX1)/ROC1, and an F-box protein, which recognizes the specificity of the substrates [4]. Part of a huge family, SCF ubiquitin ligase plays an important role in regulating cancer progression [5]. One 67KD F-box protein, F-box and WD-repeat-containing protein 2 (FBXW2), has been identified as a new tumor suppressor in multiple cancers [6]. As a housekeeping gene, FBXW2 induces the degradation of S-phase kinase-associated protein 2 (SKP2) and decreases lung tumor growth [7]. Furthermore, FBXW2 inhibits migration and invasion of lung cancer cells through the ubiquitination and degradation of β-catenin [8]. The receptor for activated C kinase 1 (RACK1) binds to FBXW2, leading to increased stability of glial cells missing transcription factor 1 (GCM1) and enhancing placental cell migration and invasion [9]. In addition, FBXW2 interacts with a known transcription repressor MSX2, promoting its ubiquitination and degradation, consequently leading to an increased expression of sex-determining region Y-box 2 (SOX2) [10]. Furthermore, FBXW2–MSX2–SOX2 axis regulates stem cell property and drug resistance [10]. However, the role of FBXW2 in breast tumorigenesis remains largely unknown.
The transcription factor nuclear factor-kB (NF-κB) plays an important role in multiple cancers [11]. It composes of five members, p65 (RELA), p50 (RELB), c-REL, NF-κB1, and NF-κB2. NF-κB mainly acts as an oncogene, playing a very complicated role in tumors [12]. The most abundant form of NF-κB is a heterodimer of p50 and p65 subunits. The p65 subunit has a transcriptional activation domain and is involved in cell survival, invasion, proliferation, metastasis, angiogenesis, and chemoresistance [13]. Ubiquitination of p65 plays an important role in regulating its function. PDLIM2 acts as a nuclear ubiquitin E3 ligase and targets the p65 subunit of NF-κB, negatively regulating NF-κB activity [14]. Furthermore, PPARγ, also acting as an E3 ubiquitin ligase, interacts with p65 and inhibits NF-κB activation by inducing its proteasome-dependent degradation [15]. However, the upstream E3 ligases that regulate p65 ubiquitination are still poorly understood.
In our study, we demonstrate that FBXW2 E3 ligase ubiquitinates p65 at K122, leading to a decrease in SOX2 expression. Degradation of p65 by FBXW2 decreases stemness, breast tumor growth, and paclitaxel resistance in vitro and in vivo. Furthermore, the level of FBXW2 expression correlates negatively with p65 level in breast cancer tissues. Collectively, our work discovers a new signaling cascade of the FBXW2-p65-SOX2 axis that regulates breast tumorigenesis.
Materials and methods
Cell culture
HEK293T, MCF-7, and MDA-MB231 cell lines were cultured in DMEM medium containing 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin at 37 °C and 5% CO2. MCF10A cell line was cultured in DMEM/Ham’s F12 supplemented with 5% horse serum (Gibco), hydrocortisone, insulin, epidermal growth factor (EGF) (Sigma), and 1% penicillin-streptomycin at 37 °C and 5% CO2. All cell lines were passaged no more than 15 times for use in experiments and were authenticated by short tandem repeat analysis. All cell lines were found to be negative for Mycoplasma by PCR assays every 2 months.
RT-PCR
Total RNA was extracted from indicated cells as previously described [16], and RNA was reversely transcribed using PrimeScript™ RT reagent Kit (Takara) according to the manufacturer’s instructions. RT-PCR was performed by TB Green® Fast qPCR Mix (Takara). β-actin was used as an endogenous control. Sequence information for primers was listed in Supplementary materials.
Immunoprecipitation (IP), GST pull-down assays and western blotting
The indicated cells were lysed in NP40 buffer with inhibitors (Sigma-Aldrich) [17]. Cell lysates were incubated with indicated beads and antibodies overnight at 4 °C. Then the beads were washed 5 times with lysis buffer, and used for indicated experiments. The indicated proteins were purified from E. coli BL21 (DE3), and GST-pull down assays were performed as described previously [17,18,19]. For western blotting, cells were lysed in cell lysis buffer and then were centrifuged at 13,000 rpm at 4 °C for 10 min. The supernatant was quantified using BCA protein assay, and boiled with 5× loading buffer. The samples were loading on SDS-PAGE gels and transferred onto PVDF membrane. The membranes were incubated with indicated primary antibody at 4 °C overnight after blocking with 5% non-fat milk in room temperature [20]. The proteins were visualized by odyssey instrument. The information of antibodies was provided in Supplementary materials.
In vitro ubiquitylation assay
HA-FBXW2 proteins were purified from transfected 293T cells by IP assays and then eluted with 3× HA peptide. Flag-tagged p65 proteins were pulled down by Flag beads from transfected 293T cells. The reaction was performed at 37 °C for 1 h in 50 μl reaction buffer (40 mM Tris-HCl (pH = 7.5), 5 mM MgCl2, 2 mM DTT, 2 mM ATP) in the presence of E1, E2 and HA-FBXW2. Poly-ubiquitinated p65 in the Flag beads was washed 3 times and resolved by SDS-PAGE loading buffer, and then used for WB.
Generation of stable cells using lentivirus
The 293T cells were transfected with the expression and the packaging plasmids (pMD2.G and psPAX2). The viruses in the supernatants were used to infect indicated cells. Stable knockdown transfection cell lines were selected with puromycin (1 μg/mL) or hygromycin (200 μg/mL) for two weeks. Stable overexpression transfection cell lines were selected with G418 (1 mg/mL) for two weeks. The stable cells were cultured in a 96-well plate to separate individual clones. The indicated protein expressions were verified by western blotting.
Xenograft tumor studies
The animal experiments were performed according to the Institutional Animal Care and Use Committee of Weifang Medical University. In total, 5 × 106 MCF-7 shFBXW2 cells with p65 knockdown were subcutaneously injected into the flanks of athymic nude mice (4 week old females). Tumor volume was recorded by formula: tumor volume = (length × width2)/2. Tumors were dissected and weighed after 3 weeks.
For paclitaxel resistant experiments, 1 × 107 MCF-7/TaxR cells with HA-FBXW2 overexpression were subcutaneously injected into the flanks of athymic nude mice (4 week old females). After 7 days, the mice were randomly divided into four groups: PBS (control), paclitaxel (10 mg/kg) alone, HA-FBXW2 overexpression alone and HA-FBXW2 overexpression combined with paclitaxel, and given intraperitoneally three times each week for treatment. After 14 days of treatment, the mice were sacrificed, and tumor volume, body, and tumor weights were recorded.
Statistical analysis
Statistical analyses were performed with Graphpad Prism 7.0 software or SPSS software version 17.0 and presented as mean ± sem. Differences between variables were done by using the 2-tailed Student’s t-test, and were considered to be statistically significant at P < 0.05. *P < 0.05 and ns was not significant.
Results
p65 is a new substrate of FBXW2
Recent studies have shown that FBXW2 played an important role in regulating cancer progression [7, 8, 10]. To study the clinical significances of FBXW2, we first evaluated FBXW2 expression in different types of human cancer using the TIMER database. The mRNA levels of FBXW2 were significantly higher in normal breast tissues than in breast cancer tissues (Supplementary Fig. S1A). Results from the UALCAN database also demonstrated that FBXW2 mRNA levels were significantly lower in breast cancer tissues than in normal breast tissues (Supplementary Fig. S1B). In addition, analyses of FBXW2 expression in 12 paired breast cancer samples and adjacent normal samples indicated a marked decrease in FBXW2 expression in breast cancer (Supplementary Fig. S1C). Consistently, the expression of FBXW2 was higher in normal breast cell line than in breast cancer cell line (Supplementary Fig. S1D).
To study the functions of FBXW2 in breast cancer, we used FBXW2 as the bait to identify interacting proteins through mass spectrometry analyses in MCF-7 cells. Surprisingly, we found p65 associated with FBXW2 (Supplementary Fig. S1E and Table S1). Ectopically expressed FBXW2 and p65 showed its interaction in 293T cells (Fig. 1A, B). We also found endogenous FBXW2 binding to p65 in MCF-7 breast cells (Fig. 1C, D). GST-pull down assay showed that FBXW2 is directly bound to p65 in vitro (Fig. 1E). Moreover, FBXW2 co-localized with p65 both in the cytoplasm and nucleus of MCF-7 cells (Fig. 1F and Supplementary Fig. S1F). Next, we mapped the binding regions between FBXW2 and p65. The full-length FBXW2 protein was cut into three truncated fragments, amino acids 1–100 (F1), 101–350 (F2), and 351–483 (F3). Based on its structure, p65 protein was also cut into three fragments, amino acids 1–291 (P1), 292–428 (P2), and 429–551 (P3). The data showed that F1 of FBXW2 and P1 of p65 were required for their interaction (Fig. 1G, H). We next tested whether p65 bound to other F-box proteins. We constructed Flag-tagged seven F-box proteins (FBXW1, FBXW4, FBXW5, FBXW7, FBXW8, FBXL3, and FBXO4) and determined their binding to HA-tagged p65. We found HA-tagged p65 bound only to Flag-tagged FBXW2 (Fig. 1I). Analysis of p65 protein sequence revealed one conserved amino-acid sequence (SDRELS), which resembled the recently defined canonical FBXW2 degradation motif TSXXXS [8] (Fig. 1J). Before being targeted for ubiquitination and degradation by the SCF ubiquitin ligase, a substrate needed to be phosphorylated in order to interact with an F-box protein [7]. To determine whether p65 binding to FBXW2 is phosphorylation dependent, we constructed several variants of p65 with different mutations in its FBXW2 binding motifs. As expected, p65-WT and the constitutively active forms of p65, p65-S276D and p65-S3D (S276D, D277D, and S281D) [21,22,23,24,25], interacted with FBXW2, whereas the non-phosphorylatable mutant forms of p65, p65-S276A, and p65-S3A (S276A, D277A, and S281A) showed no interaction (Fig. 1K). A previous study showed that PKA phosphorylated p65 at S276, regulating NF-κB transcriptional activity by modulating its interaction with CBP/p300 [26]. To study whether PKA is required for FBXW2-mediated degradation of p65, we reduced the activity of protein kinase A (PKA) by knockdown or with a PKA inhibitor (H-89). Indeed, PKA knockdown or PKA inhibitor (H-89) treatment decreased FBXW2-p65 binding (Supplementary Fig. S1G), suggesting that p65 was a new substrate of FBXW2. Taken together, these results indicate that p65 is a new substrate of FBXW2.

Immunoprecipitation and immunoblotting analyses were performed with the indicated antibodies. A, B Immunoprecipitation (IP) and western blot analysis of the exogenous FBXW2/p65 proteins interaction in the 293T cells co-transfected with Flag-tagged FBXW2 and HA-tagged p65. C, D IP and western blot analysis of the endogenous FBXW2/p65 proteins interaction in the MCF-7 cells. E GST-pull down assay analysis of FBXW2/p65 proteins interaction using purified GST-tagged FBXW2 and His-p65. F Confocal immunofluorescence microscopy analysis of the p65/FBXW2 proteins interaction in the MCF-7 cells. G IP and western blot analysis of the HA-tagged p65/GFP-tagged FBXW2 fragments proteins interaction in the 293T cells. H IP and western blot analysis of the HA-tagged FBXW2/GFP-tagged p65 fragments proteins interaction in the 293T cells. I IP and western blot analysis of the HA-tagged p65/Flag-tagged FBXs proteins interaction in the 293T cells. J Evolutionary conservation of FBXW2 degron motif on p65. K IP and western blot analysis of the HA-tagged p65 (WT, 3D (S276D, D277D and S281D), 3 A (S276A, D277A and S281A), S276A or S276D)/Flag-tagged FBXW2 proteins interaction in the 293T cells.
FBXW2 enhances p65 degradation via the ubiquitin-proteasome pathway
As an E3 ligase, FBXW2 regulated biological process through ubiquitination of a series of substrates [10]. To assess the role of FBXW2 in regulating p65 protein stability, we manipulated FBXW2 expression through knockdown or overexpression. Overexpression of FBXW2 decreased p65 protein stability (Fig. 2A and Supplementary Fig. S2A, B). Furthermore, wild-type FBXW2 promoted p65 protein degradation, whereas FBXW2-ΔF, which lacked the ability to recruit other components of SCF ubiquitin ligase, had little effect [8] (Fig. 2B). Similarly, FBXW2 did not alter the stability of p65-S3A mutant, which lacked the ability to bind to FBXW2 (Supplementary Fig. S2C). Moreover, knockdown or overexpression of FBXW2 affected p65 protein stability in breast cancer cells (Fig. 2C–G), but did not affect its mRNA levels (Supplementary Fig. S2D). The knockdown efficiency was verified by western blotting (Supplementary Fig. S2E). Furthermore, we constructed a FBXW2-knockout mouse model and got mouse embryonic fibroblasts (MEFs) cells from mouse embryos. Consistent with the results from our previous experiments, FBXW2-knockout MEFs expressed increased p65 protein level (Fig. 2H). To test whether the proteasome pathway is involved in FBXW2-induced degradation of p65, we treated the transfected cells with the proteosomal inhibitor MG132. Interestingly, MG132 blocked the FBXW2-mediated degradation of p65 protein, suggesting that FBXW2 regulated p65 protein degradation via the ubiquitin proteasome pathway (Fig. 2I). Furthermore, we treated indicated cells with cycloheximide (CHX) to examine the half-life of p65 protein. Consistent with our previous observed effect of FBXW2 on p65 protein, FBXW2 clearly decreased the half-life of p65 compared with the control (Fig. 2J–L). However, inhibition of PKA abrogated this effect (Supplementary Fig. S2F). Collectively, these results suggest that FBXW2 promotes p65 degradation via the ubiquitin proteasome pathway.

Immunoblotting analyses were performed with the indicated antibodies. A The 293T cells were overexpressed with the indicated HA-tagged p65 and Flag-tagged FBXW2 (0, 0.5, or 1 μg) proteins. Immunoblotting analyse was performed. B The 293T cells were overexpressed the indicated HA-tagged p65 and Flag-tagged FBXW2 (WT or ΔF) proteins. Immunoblotting analyse was performed. C, E MCF-7 or MB231 cells were overexpressed the indicated Flag-tagged FBXW2 proteins. Immunoblotting analyse was performed. D, F MCF-7 or MB231 cells were knocked down FBXW2 with shRNA. Immunoblotting analyse was performed. G MCF-7 or MB231 cells were knocked down FBXW2 with shRNA. Immunofluorescence was performed. H FBXW2 depletion caused increase of p65 protein level in MEFs cells. Immunoblotting analyse was performed. I The 293T cells with overexpression of the indicated both Flag-tagged FBXW2 and HA-tagged p65 proteins was treated with MG132 for 8 h. Immunoblotting analyses were performed. J MCF-7 cells with overexpression of Flag-tagged FBXW2 were treated with CHX for the indicated time. Immunoblotting analyse was performed. K MCF-7 cells with knockdown of FBXW2 were treated with CHX for indicated time. Immunoblotting analyse was performed. L MEFs cells with knockout of FBXW2 were treated with CHX for indicated time. Immunoblotting analyse was performed.
FBXW2 ubiquitination p65 at K122 site depends on p300 induced acetylation
As our data showed that FBXW2 decreased p65 protein stability via the ubiquitin proteasome pathway, we next determined whether FBXW2 could directly regulate the ubiquitination level of p65. We found that knockdown or overexpression of FBXW2 in MCF-7 cells altered the ubiquitination level of p65 (Fig. 3A, B). Moreover, FBXW2-knockout MEFs showed reduced ubiquitination level of p65 (Fig. 3C). Furthermore, FBXW2 could directly ubiquitinate p65 in vitro (Fig. 3D). Interestingly, FBXW2 ubiquitination of p65 required PKA (Supplementary Fig. S3A). FBXW2 induced ubiquitination of p65-S3A mutant was markedly less than its ubiquitination of the wild-type p65 (Supplementary Fig. S3B). Furthermore, we isolated nucleus and cytoplasm proteins from FBXW2 knockdown MCF-7 cells. The data showed that FBXW2 promoted ubiquitination of p65 both in the cytoplasm and nucleus, which was consistent with the PLA results. To identify the lysine residues on p65 protein ubiquitinated by FBXW2, we mutated all lysine residues on p65 protein. Interestingly, FBXW2 did not affect the protein stability of p65-K122R mutant (Fig. 3E, F), also failing to ubiquitinate it (Fig. 3G). That FBXW2 could not ubiquitinate p65-K122R mutant suggested that FBXW2 ubiquitinated p65 at K122, thereby altering p65 stability. To determine which lysine (K)-linked polyubiquitin chain conjugated with p65, we overexpressed several ubiquitin mutants. We found that p65 was conjugated with lysine (K) 48- or 63-linked polyubiquitin chains (Fig. 3H).

Immunoblotting analyses were performed with the indicated antibodies. A MCF-7 cells were stably expressed shRNA-FBXW2. Cells were treated with MG132 for 8 h followed by IP and western blot. B MCF-7 cells were transfected with Flag-tagged FBXW2. After 48 h transfection, cells were treated with MG132 for 8 h followed by IP and western blot. C The indicated MEFs cells were treated with MG132 for 8 h followed by IP and western blot. D FBXW2 proteins were purified from 293T cells transfected with HA-tagged FBXW2 by IP with HA beads and elution with 3× HA peptide. p65 proteins were purified from 293T cells with Flag-tagged p65 transfected by IP with Flag beads. FBXW2 and p65 on Flag beads were added into a reaction mixture containing ATP, His-ubiquitin, E1 and E2, followed by constant mixing at 37 °C for 60 min. Immunoblotting analyse was performed. E, F The 293T cells were co-overexpressed both Flag-tagged FBXW2 and different mutants of HA-tagged p65. Immunoblotting analyse was performed. G The 293T cells were co-overexpressed both HA-tagged FBXW2 and Flag-tagged p65 (WT or K122R). After 48 h transfection, cells were treated with MG132 for 8 h followed by IP and western blot. H 293T cells transfected with different ubiquitin mutants were co-overexpressed both HA-tagged FBXW2 and Flag-tagged p65. After 48 h transfection, cells were treated with MG132 for 8 h followed by IP and western blot.
K122 site of p65 was demonstrated to be ubiquitinated [27] or acetylated [28, 29] by mass spectrum. Because p65 was acetylated at K122 by p300 [30], we clarified the relationship between ubiquitination and acetylation of p65 with the following experiments. We manipulated p300 activity by overexpression, knockdown or with a p300 inhibitor (C646). Our results showed that p300 blocked the effect of FBXW2 on p65 protein stability in MCF-7 cells (Supplementary Fig. S4A, B). Similarly, p300 inhibited FBXW2 ubiquitination of p65 (Supplementary Fig. S4C). To further study the mechanisms underlying p300 role on FBXW2 ubiquitination of p65, we performed IP assay. Interestingly, p300 reduced FBXW2 interaction with p65 in studies using overexpression, knockdown, or inhibitor of p300 (Supplementary Fig. S4D, E). Consistent with its effect on FBXW2 binding to p65, p300 evidently counteracted the effect of FBXW2 on the protein half-life of p65 (Supplementary Fig. S4F). Together, these results suggest that FBXW2 ubiquitination of p65 at K122 depends on p300 regulated p65 acetylation.
FBXW2 decreases transcriptional activity of p65
With FBXW2 significantly altering p65 protein stability, we speculated that inactivation of FBXW2 would regulate p65 global transcriptional program. To this end, we performed RNA-sequencing analysis of FBXW2-depleted MCF-7 cells (Supplementary Fig. S4G, H), using Volcano-plot identified the significantly regulated genes with a middle-high expression basement (Fig. 4A). From the list of 30 top differentially expressed genes, we found that FBXW2-depleted cells showed increased expression of previously reported p65 transcriptional target genes such as LIN28B [31], QPCT [32], SCN5A [33], and WT1 [34] (Fig. 4B). Furthermore, FBXW2 depletion significantly upregulated NF-κB-induced gene expression programs (Fig. 4C). Moreover, RT-PCR data confirmed that depletion of FBXW2 increased RNA expression of LIN28B, QPCT, SCN5A, and WT1 in breast cancer cells (Fig. 4D, E). In addition, luciferase reporter assays showed that FBXW2 significantly reduced p65 transcriptional activity in these cells (Fig. 4F, G). To determine whether FBXW2 reduces p65 binding to its target gene promoters, we performed chromatin-immunoprecipitation (CHIP) assay. Our results showed that FBXW2 decreased p65 binding to the promoter of QPCT, previously reported to be a p65 target gene (Fig. 4H, I). Thus, FBXW2 impairs the transcriptional activity of p65.

A The volcano map results of knocking down FBXW2 in MCF-7 cells. B Heat map representing the most significantly regulated genes detected in RNA-seq analysis with knockdown of FBXW2 in MCF-7 cells. C Gene set enrichment analysis (GSEA) of regulated genes in FBXW2 knockdown MCF-7 cells. D, E Examination of the mRNA levels of LIN28B, QPCT, SCN5A, and WT1 by RT-PCR analyses in breast cancer cells. F, G Luciferase reporter assays were performed in FBXW2 knockdown breast cancer cells. H, I CHIP assays were performed using FBXW2 knockdown breast cancer cells. The results were normalized against the values of IgG controls. (All data represent mean ± SEM n = 3), *p < 0.05.
The FBXW2–p65 axis regulates breast cancer stemness via upregulation of SOX2
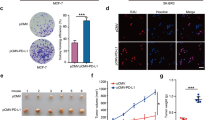
Our RNA-sequencing results showed that FBXW2 knockdown upregulated expression of SOX2, a key transcription factor in maintaining stemness. To examine whether FBXW2 regulates SOX2 expression, we knocked down and restored FBXW2 in breast cancer cells. We found that FBXW2 inhibited SOX2 expression (Fig. 5A–D). It also inhibited breast cancer cell sphere formations (Fig. 5E). As p65 was a transcriptional factor which governed breast cancer stemness [35,36,37], we asked whether p65 could directly regulate SOX2 expression. Indeed, p65 knockdown dramatically reduced both RNA and protein levels of SOX2 in breast cancer cells (Supplementary Fig. S5A–C). In addition, luciferase reporter assay and CHIP assay showed that p65 directly bound to the promoter of SOX2, which suggested that SOX2 was a direct target gene of p65 (Supplementary Fig. S5D–F). Furthermore, we determined whether FBXW2 regulation of SOX2 expression required p65. We found that knockdown of p65 blocked FBXW2-regulated expression of SOX2 (Fig. 5F, G). Moreover, knockdown of FBXW2 could promote p65 binding to SOX2 promoter (Supplementary Fig. S5G). Our data suggest that the FBXW2-p65 axis regulates breast cancer stemness via upregulation of SOX2.

A, B SOX2 mRNA levels were quantitated by RT-PCR in FBXW2-depleted breast cancer cells rescued with rFBXW2. C, D Immunofluorescence analysis of FBXW2-depleted breast cancer cells rescued with rFBXW2. E Tumor sphere formation assays were performed in FBXW2-depleted breast cancer cells rescued with rFBXW2. F, G Breast cancer cells were overexpressed Flag-tagged FBXW2 with shRNA-control or shRNA-p65 lentiviral. (All data represent mean ± SEM n = 3), *p < 0.05.
p65 is involved in FBXW2 inhibiting breast tumor growth in vitro and in vivo
Previous studies showed that FBXW2 was a tumor suppressor in multiple cancers [8]. To further clarify the role of the FBXW2/p65 signaling pathway in tumor growth, FBXW2-knockdown breast cancer cells were stably knocked down p65 (Fig. 6A). Knocking down FBXW2 significantly increased cell proliferation but knocking down both FBXW2 and p65 no longer produced this effect (Fig. 6B). Colony formation assay also showed that FBXW2 affected cell proliferation through its regulation of p65 (Fig. 6C). Furthermore, the cell-scratch test and transwell migration assay demonstrated that FBXW2 required p65 in its regulation of cell migration (Fig. 6D, E). To test whether FBXW2 regulates breast tumor growth via p65 in vivo, we employed an in vivo xenograft tumor model. Comparing tumors derived from cells with stable knockdown of FBXW2 alone and cells with knockdown of both FBXW2 and p65, we found that FBXW2 mediated tumor growth in vivo through its regulation of p65 (Fig. 6F–H). Moreover, immunohistochemistry (IHC) staining of these tumors for the expression of the proliferation biomarker Ki67 showed that knockdown of p65 significantly reduced the effect of FBXW2 on cancer cell proliferation in vivo (Fig. 6I). As p300 acetylation of p65 inhibited FBXW2 ubiquitination of p65, we proceeded to determine whether p65 K122 acetylation affected tumor growth. We knocked down p65 and reconstituted expression of p65 (WT, K122R or K122Q) in MCF-7 cells (Supplementary Fig. S6A). Compared with cells expressing p65-WT or p65-K122Q, cells with p65-K122R mutant showed reduced proliferation (Supplementary Fig. S6B, C). Moreover, to study the role of p65 acetylation on K122 in tumor growth in vivo, we injected the manipulated MCF-7 cells subcutaneously into athymic nude mice and evaluated growth of the xenografted-tumors. Tumors from p65-K122R mutant cells were smaller than tumors from p65-WT or p65-K122Q expressing cells (Supplementary Fig. S6D–F). Taken together, our results clearly demonstrate that p65 is involved in FBXW2 inhibiting tumor growth in vitro and in vivo in breast cancer.

A MCF-7 or MB231 cells with FBXW2 depletion reconstituted stable knockdown of p65. Total cell lysates were prepared, and immunoblotting analyse was performed. B MCF-7 or MB231 cells with FBXW2 depletion reconstituted stable knockdown of p65. CCK-8 assays were performed. C MCF-7 or MB231 cells with FBXW2 depletion reconstituted stable knockdown of p65. Clone formation assays were performed. D MCF-7 or MB231 cells with FBXW2 depletion reconstituted stable knockdown of p65. Wound healing assays were performed. E MCF-7 or MB231 cells with FBXW2 depletion reconstituted stable knockdown of p65. Cell invasion assays were performed. F–H MCF-7 cells with FBXW2 depletion reconstituted stable knockdown of p65 were subcutaneously injected into nude mice. After 3 weeks, the mice were sacrificed and dissected at the endpoint. Tumor growth and weight were examined. I Representative images of H/E staining and Ki67 staining of tumor samples (Scale bar, 20 μm). (All data represent mean ± SEM n ≥ 3), *p < 0.05.
FBXW2-p65 axis is involved in regulating breast cancer cell paclitaxel resistance
Paclitaxel was a chemotherapy drug commonly used to treat breast cancers [38]. However, paclitaxel resistance was still the biggest obstacle to successful clinical treatment [39,40,41]. With p65 reported to play an important role in paclitaxel resistance [42], we determined whether FBXW2-p65 axis was involved in regulating paclitaxel resistance in breast cancer cell. To this end, we determined the expression levels of FBXW2 and p65 in MCF-7/TaxR or MCF-7 cells. As expected, FBXW2 expression was reduced in MCF-7/TaxR cells, whereas p65 level was high in MCF-7/TaxR cells (Fig. 7A, B). To study the effects of FBXW2 and p65 on paclitaxel resistance, we knocked down p65 or overexpressed FBXW2 in MCF-7/TaxR cells. As expected, p65 knockdown or FBXW2 overexpression decreased the MCF-7/TaxR cell proliferation rate (Fig. 7C, D). Because FBXW2 led to p65 degradation, we focused on the effect of FBXW2 overexpression on paclitaxel resistance. We overexpressed FBXW2 in MCF-7/TaxR cells following paclitaxel treatment. Compared with the control cells, cells with FBXW2 overexpression showed decreased resistance to paclitaxel, suggesting that FBXW2 played an important role in the paclitaxel resistance of breast cancer cells (Fig. 7E). To better understand the role of FBXW2 in paclitaxel resistance in vivo, we used a xenograft nude mouse model. Tumors derived from MCF-7/TaxR overexpressing FBXW2 showed lower tumor volume and weight than the control tumors, indicating that FBXW2 was effective in suppressing MCF-7/TaxR tumor growth (Fig. 7F–H). Taken together, our data demonstrate that FBXW2-p65 axis contributes to paclitaxel resistance in breast cancer cells.

A Total cell lysates from MCF-7 and MCF-7/TaxR cells were prepared. Immunoblotting analyses were performed. B MCF-7 and MCF-7/TaxR cells were prepared. Immunofluorescence was performed. C p65 was knocked down in MCF-7/TaxR cells. Cell proliferation rates were measured by cell counting. D FBXW2 was stably overexpressed in MCF-7/TaxR cells. Cell proliferation rates were measured by cell counting. E MCF-7/TaxR cells with stable expression of FBXW2 were treated with 10 nM paclitaxel for two days. Cell proliferation rates were measured by cell counting. F-H MCF-7/TaxR cells were injected into nude mice. After 7 days, the mice were randomly assigned to four groups: PBS (control), paclitaxel, HA-FBXW2 overexpression and HA-FBXW2 overexpression combined with paclitaxel, and given intraperitoneally three times each week for treatment. After 14 days of treatment, the mice were sacrificed, and the tumor weight was measured. Tumor volume was measured during the tumor growth for 3 weeks. (All data represent mean ± SEM n ≥ 3), *p < 0.05.
FBXW2 expression is negatively correlated with p65 in breast cancer tissues
To evaluate the expression of FBXW2 and p65 in breast cancer tissues, we performed IHC analyses of breast cancer tissues from 90 cases. Notably, consistent with our previous observations, FBXW2 was lowly expressed in breast cancer tissues, whereas p65 was highly expressed (Fig. 8A). Furthermore, the protein levels of FBXW2 and p65 negatively correlated with each other (Fig. 8B). To determine whether the expression of FBXW2 correlates with patient survival, we performed the Kaplan–Meier survival analysis. Of the 1764 breast cancer samples, we found that high FBXW2 expression (n = 868) predicts better patient survival (Fig. 8C). All of these data support the clinical potential of targeting FBXW2-p65 axis as breast cancer therapeutics.

A Representative images of IHC staining of tumor tissues from two breast cancer patients were shown (scale bar, 20 µm). B Pearson correlation analysis was performed to determine correlation between FBXW2 and p65 expression in breast cancer tissues. C Kaplan–Meier curves of overall survival were performed on breast cancer patients. D Schematic model of mechanism that FBXW2 functioned as a tumor suppressor to regulate breast cancer stemness and paclitaxel resistance via ubiquitylation of p65. (All data represent mean ± SEM n ≥ 3), *p < 0.05.
Discussion
NF-κB signaling transcription factors are composed of Nfkb1, Nfkb2, Rela (p65), c-Rel, and Relb [43]. As a member of NF-κB family, p65 locates mainly in the cytoplasm in a non-induced state [44]. p65 is highly expressed in diverse cancer types and can transcriptionally activate downstream genes to perform various functions including regulating cell proliferation, apoptosis, and immunity [45]. Posttranslational modifications of NF-κB p65 subunit provide essential mechanisms to differentially regulate NF-κB signaling activity in many cancer types [46]. Although these modifications have a critical role in affecting p65 functions, the role of p65 ubiquitination is remains elusive. Here, we find that FBXW2 acts as an E3 ligase, ubiquitinating p65 at K122 and promoting its degradation (Fig. 8D). Furthermore, FBXW2 ubiquitination of p65 modulates breast cancer stemness and paclitaxel resistance.
SOX2 plays a critical role in cancer cell stemness and maintenance [47]. The functions of SOX2 depend mainly on its transcriptional and post-transcriptional modifications [48, 49]. At the transcriptional level, T-box transcription factor 6 (TBX6) binds to a specific enhancer of the SOX2 gene, thereby reducing SOX2 expression [50]. In addition, MSX2 binds to the promoter of SOX2 and represses its expression. Moreover, some cytokines such as IL-4, IL-6 and TGFβ, induce expression of SOX2 [51]. Furthermore, E2f3a and E2f3b transcription factors differentially regulate SOX2 and cell fate in neural stem cells [52]. Posttranslational regulation of SOX2 is reported to be mediated by FBXW2-MSX2 axis. FBXW2 E3 ligase ubiquitinates and destabilizes MSX2. The consequently, low expression of MSX2 leads to high expression of SOX2 in tamoxifen resistance breast cancer cells [10]. Other studies investigate FBXW2 promotes SOX2 expression in tamoxifen resistance breast cancer cells. Our studies, however, focus on FBXW2 suppression of SOX2 expression in breast cancer cells. In addition, SOX2 is highly expressed in breast cancer and reportedly acts as an oncogene [53,54,55]. These conclusions are consistent with our experimental results that FBXW2 inhibits SOX2 expression via degradation of p65, contributing to the stemness of breast cancer cells.
F-box proteins are the key component of SCF ubiquitin ligase complexes. They directly bind to substrates through specific domains, leading to ubiquitin-mediated proteolysis of their substrates [56]. F-box proteins play an important role in cancer development and progression [57]. Chemotherapy is still the main treatment strategy for many types of cancer. However, due to the development of drug resistance in patients, preventing or overcoming drug resistance is still the key to improve chemotherapy effectiveness. F-box proteins have rarely been reported to be involved in drug resistance. FBXL7 phosphorylation by AURKA increases paclitaxel resistance, which is associated with poor prognosis in ovarian cancer patients, thus making FBXL7 a potential biomarker for selecting paclitaxel therapy [58]. SKP2 promotes paclitaxel resistance in lung cancer by reducing the expression of MAD2 and enhancing the phosphorylation of p27 and pRB [59]. In addition, SKP2 is highly expressed in paclitaxel-resistant prostate cancer cells, and knockdown of FBXL1 renders prostate cancer cells sensitive to paclitaxel [60]. Moreover, because inhibition of FBXW7 in cancer cells promotes resistance to paclitaxel, overexpression of FBXW7 could potentially reverse drug resistance [61]. Our results show that FBXW2 regulates paclitaxel resistance via degradation of p65. Our findings expand the understanding of F-box protein functions in drug resistance. Thus, targeting FBXW2-p65 axis could be a promising strategy to overcome drug resistance.
In summary, our results demonstrate that FBXW2 functions as a tumor suppressor, affecting cancer development by altering stemness and paclitaxel resistance through the ubiquitination of p65. Our data further suggest that the FBXW2 plays an important role in the ubiquitination of p65 upon its acetylation by p300. Thus, our findings prove that FBXW2 is a tumor suppressor in breast cancer.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References
Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nat Rev Cancer. 2020;20:417–36.
Hanker AB, Sudhan DR, Arteaga CL. Overcoming endocrine resistance in breast cancer. Cancer Cell. 2020;37:496–513.
Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Prim. 2019;5:66.
Schulman BA, Carrano AC, Jeffrey PD, Bowen Z, Kinnucan ER, Finnin MS, et al. Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature. 2000;408:381–6.
Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014;13:889–903.
Yang F, Sun Y. FBXW2 suppresses proliferation and invasion of lung cancer cells by targeting SKP2 and beta-catenin. Mol Cell Oncol. 2019;6:1607458.
Xu J, Zhou W, Yang F, Chen G, Li H, Zhao Y, et al. The beta-TrCP-FBXW2-SKP2 axis regulates lung cancer cell growth with FBXW2 acting as a tumour suppressor. Nat Commun. 2017;8:14002.
Yang F, Xu J, Li H, Tan M, Xiong X, Sun Y. FBXW2 suppresses migration and invasion of lung cancer cells via promoting beta-catenin ubiquitylation and degradation. Nat Commun. 2019;10:1382.
Wang CC, Lo HF, Lin SY, Chen H. RACK1 (receptor for activated C-kinase 1) interacts with FBW2 (F-box and WD-repeat domain-containing 2) to up-regulate GCM1 (glial cell missing 1) stability and placental cell migration and invasion. Biochem J. 2013;453:201–8.
Yin Y, Xie CM, Li H, Tan M, Chen G, Schiff R, et al. The FBXW2-MSX2-SOX2 axis regulates stem cell property and drug resistance of cancer cells. Proc Natl Acad Sci USA. 2019;116:20528–38.
Aggarwal BB, Sung B. NF-kappaB in cancer: a matter of life and death. Cancer Discov. 2011;1:469–71.
Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-kappaB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209.
Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–24.
Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–91.
Hou Y, Moreau F, Chadee K. PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nat Commun. 2012;3:1300.
Lu C, Ren C, Yang T, Sun Y, Qiao P, Han X, et al. Fructose-1, 6-bisphosphatase 1 interacts with NF-kappaB p65 to regulate breast tumorigenesis via PIM2 induced phosphorylation. Theranostics. 2020;10:8606–18.
Yang T, Ren C, Lu C, Qiao P, Han X, Wang L, et al. Phosphorylation of HSF1 by PIM2 induces PD-L1 expression and promotes tumor growth in breast cancer. Cancer Res. 2019;79:5233–44.
Han X, Ren C, Yang T, Qiao P, Wang L, Jiang A, et al. Negative regulation of AMPKalpha1 by PIM2 promotes aerobic glycolysis and tumorigenesis in endometrial cancer. Oncogene. 2019;38:6537–49.
Lu C, Ren C, Yang T, Sun Y, Qiao P, Wang D, et al. A noncanonical role of fructose-1, 6-bisphosphatase 1 is essential for inhibition of notch1 in breast cancer. Mol Cancer Res. 2020;18:787–96.
Lu C, Qiao P, Sun Y, Ren C, Yu Z. Positive regulation of PFKFB3 by PIM2 promotes glycolysis and paclitaxel resistance in breast cancer. Clin Transl Med. 2021;11:e400.
Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 2003;22:1313–24.
Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. Pim-1 controls NF-kappaB signalling by stabilizing RelA/p65. Cell Death Differ. 2010;17:689–98.
Hochrainer K, Racchumi G, Anrather J. Site-specific phosphorylation of the p65 protein subunit mediates selective gene expression by differential NF-kappaB and RNA polymerase II promoter recruitment. J Biol Chem. 2013;288:285–93.
Seldon MP, Silva G, Pejanovic N, Larsen R, Gregoire IP, Filipe J, et al. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. J Immunol. 2007;179:7840–51.
Hochrainer K, Racchumi G, Anrather J. Hypo-phosphorylation leads to nuclear retention of NF-kappaB p65 due to impaired IkappaBalpha gene synthesis. FEBS Lett. 2007;581:5493–9.
Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–71.
Akimov V, Barrio-Hernandez I, Hansen SVF, Hallenborg P, Pedersen AK, Bekker-Jensen DB, et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat Struct Mol Biol. 2018;25:631–40.
Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013;4:842–51.
Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–7.
Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci USA. 1997;94:2927–32.
Zhou J, Chooi JY, Ching YQ, Quah JY, Toh SH, Ng Y, et al. NF-kappaB promotes the stem-like properties of leukemia cells by activation of LIN28B. World J Stem Cells. 2018;10:34–42.
Zhao T, Bao Y, Gan X, Wang J, Chen Q, Dai Z, et al. DNA methylation-regulated QPCT promotes sunitinib resistance by increasing HRAS stability in renal cell carcinoma. Theranostics. 2019;9:6175–90.
Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, et al. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008;294:C372–379.
Dehbi M, Hiscott J, Pelletier J. Activation of the wt1 Wilms’ tumor suppressor gene by NF-kappaB. Oncogene. 1998;16:2033–9.
Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, et al. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010;70:10464–73.
Kendellen MF, Bradford JW, Lawrence CL, Clark KS, Baldwin AS. Canonical and non-canonical NF-kappaB signaling promotes breast cancer tumor-initiating cells. Oncogene. 2014;33:1297–305.
Shostak K, Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13:214.
Yu KD, Ye FG, He M, Fan L, Ma D, Mo M, et al. Effect of adjuvant paclitaxel and carboplatin on survival in women with triple-negative breast cancer: a phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1390–6.
Khongkow P, Gomes AR, Gong C, Man EP, Tsang JW, Zhao F, et al. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene. 2016;35:990–1002.
Ajabnoor GM, Crook T, Coley HM. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis. 2012;3:e260.
Yang T, Ren C, Qiao P, Han X, Wang L, Lv S, et al. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene. 2018;37:5997–6009.
Dong QG, Sclabas GM, Fujioka S, Schmidt C, Peng B, Wu T, et al. The function of multiple IkappaB: NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene. 2002;21:6510–9.
Eluard B, Thieblemont C, Baud V. NF-kappaB in the new era of cancer therapy. Trends Cancer. 2020;6:677–87.
Verzella D, Pescatore A, Capece D, Vecchiotti D, Ursini MV, Franzoso G, et al. Life, death, and autophagy in cancer: NF-kappaB turns up everywhere. Cell Death Dis. 2020;11:210.
Dimitrakopoulos FD, Kottorou AE, Kalofonou M, Kalofonos HP. The fire within: NF-kappaB involvement in non-small cell lung cancer. Cancer Res. 2020;80:4025–36.
Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res. 2014;2:823–30.
Zhang S, Xiong X, Sun Y. Functional characterization of SOX2 as an anticancer target. Signal Transduct Target Ther. 2020;5:135.
Novak D, Huser L, Elton JJ, Umansky V, Altevogt P, Utikal J. SOX2 in development and cancer biology. Semin Cancer Biol. 2020;67:74–82.
Sharma NS, Gupta VK, Dauer P, Kesh K, Hadad R, Giri B, et al. O-GlcNAc modification of Sox2 regulates self-renewal in pancreatic cancer by promoting its stability. Theranostics. 2019;9:3410–24.
Takemoto T, Uchikawa M, Yoshida M, Bell DM, Lovell-Badge R, Papaioannou VE, et al. Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature. 2011;470:394–8.
Wu Q, Zhang L, Su P, Lei X, Liu X, Wang H, et al. MSX2 mediates entry of human pluripotent stem cells into mesendoderm by simultaneously suppressing SOX2 and activating NODAL signaling. Cell Res. 2015;25:1314–32.
Julian LM, Vandenbosch R, Pakenham CA, Andrusiak MG, Nguyen AP, McClellan KA, et al. Opposing regulation of Sox2 by cell-cycle effectors E2f3a and E2f3b in neural stem cells. Cell Stem Cell. 2013;12:440–52.
Piva M, Domenici G, Iriondo O, Rabano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6:66–79.
Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283:17969–78.
Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–65.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9.
Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–47.
Kamran M, Long ZJ, Xu D, Lv SS, Liu B, Wang CL, et al. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis. 2017;6:e298.
Huang T, Yang L, Wang G, Ding G, Peng B, Wen Y, et al. Inhibition of Skp2 sensitizes lung cancer cells to paclitaxel. Oncol Targets Ther. 2017;10:439–46.
Yang Y, Lu Y, Wang L, Mizokami A, Keller ET, Zhang J, et al. Skp2 is associated with paclitaxel resistance in prostate cancer cells. Oncol Rep. 2016;36:559–66.
Sailo BL, Banik K, Girisa S, Bordoloi D, Fan L, Halim CE, et al. FBXW7 in cancer: what has been unraveled thus far? Cancers. 2019;11:e246.
Funding
The study was supported by research grants from the National Natural Science Foundation of China (Grant no. 81972489 and 82003201), the National Natural Science Foundation of Shandong Province (Grant no. ZR2020YQ58 and ZR2020QH255), Taishan Scholar Program of Shandong Province (Grant no. tsqn202103113), Shandong Province College Science and Technology Plan Project (Grant no. J17KA254), Projects of medical and health technology development program in Shandong province (Grant no. 2018WS057).
Author information
Authors and Affiliations
Contributions
CR, XH, CL, and TY performed experiments and analyzed data. PQ and YS provided access to material and facilities and contributed reagents. ZY designed, supervised the project, and wrote the manuscript.
Corresponding author
Ethics declarations
Ethics statement
The procedures related to human subjects were approved by the Ethics Committee of Weifang Medical University. Animal experiments were performed according to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals, and were approved by the ethics committee of Weifang Medical University (Weifang, China).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by C. Borner
Rights and permissions
About this article

Cite this article
Ren, C., Han, X., Lu, C. et al. Ubiquitination of NF-κB p65 by FBXW2 suppresses breast cancer stemness, tumorigenesis, and paclitaxel resistance. Cell Death Differ 29, 381–392 (2022). https://doi.org/10.1038/s41418-021-00862-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-021-00862-4
This article is cited by
-
O-GlcNAcylation of melanophilin enhances radiation resistance in glioblastoma via suppressing TRIM21 mediated ubiquitination
Oncogene (2024)
-
GSK3β and UCHL3 govern RIPK4 homeostasis via deubiquitination to enhance tumor metastasis in ovarian cancer
Oncogene (2024)
-
Arginine methylation of ALKBH5 by PRMT6 promotes breast tumorigenesis via LDHA-mediated glycolysis
Frontiers of Medicine (2024)
-
Biological effects and mechanisms of fisetin in cancer: a promising anti-cancer agent
European Journal of Medical Research (2023)
-
FBXW2 suppresses breast tumorigenesis by targeting AKT-Moesin-SKP2 axis
Cell Death & Disease (2023)
