
Abstract
Axonal degeneration and neuronal cell death are fundamental processes in development and contribute to the pathology of neurological disease in adults. Both processes are regulated by BCL-2 family proteins which orchestrate the permeabilization of the mitochondrial outer membrane (MOM). MOM permeabilization (MOMP) results in the activation of pro-apoptotic molecules that commit neurons to either die or degenerate. With the success of small-molecule inhibitors targeting anti-apoptotic BCL-2 proteins for the treatment of lymphoma, we can now envision the use of inhibitors of apoptosis with exquisite selectivity for BCL-2 family protein regulation of neuronal apoptosis in the treatment of nervous system disease. Critical to this development is deciphering which subset of proteins is required for neuronal apoptosis and axon degeneration, and how these two different outcomes are separately regulated. Moreover, noncanonical BCL-2 family protein functions unrelated to the regulation of MOMP, including impacting necroptosis and other modes of cell death may reveal additional potential targets and/or confounders. This review highlights our current understanding of BCL-2 family mediated neuronal cell death and axon degeneration, while identifying future research questions to be resolved to enable regulating neuronal survival pharmacologically.
Similar content being viewed by others
Facts
-
Neuronal apoptosis and axon degeneration both occur naturally in development, but also contribute to the pathology of nervous system disease.
-
Anti-apoptotic proteins such as BCL-XL and BCL-W can prevent both axon degeneration and apoptosis.
-
Many stressors induce neuronal cell death in a PUMA- and BAX-dependent manner.
-
Transcriptional upregulation of PUMA is required for axon degeneration.
-
BAX, not BAK, is the primary executor of MOMP in neurons.
Open questions
-
Intracellularly, where does MOMP occur after NGF deprivation from axons?
-
How do neurons prevent apoptosis while undergoing axon degeneration?
-
Why do many stressors trigger neuronal cell death and/or axon degeneration that is PUMA and BAX dependent rather than using other BCL-2 family proteins to?
-
Do CNS and PNS axons degenerate through similar mechanisms?
-
Can pharmaceutical inhibition of BAX prevent neuronal cell death and improve outcome in neurological events like stroke?
-
Does prevention of axon degeneration halt or delay the pathology of neurodegenerative disease?
Introduction
For many cells, the balance between life and death is regulated by the BCL-2 family of proteins. As reviewed previously, a dance occurs within the BCL-2 family on the mitochondrial outer membrane (MOM) dance floor [1]—with the outcome of this dance ultimately deciding whether a cell will live or die. But what if the dance floor were extended unimaginably? Neurons are morphologically unique cells with long cytoplasmic extensions called axons. The vast distance axons span results in a separation of mitochondrial populations within a single cell—one population within the cell body, and another that extends down the length of the axon. The integrity of both mitochondrial populations is vital to neuronal health [2]. The same BCL-2 family dance-of-death occurs within neurons but now the dance floor has been extended; and as a result, the BCL-2 family can regulate axonal degeneration in addition to life and death. Mitochondria are the “power-house” organelle of the cell, but ironically, the MOM is also the platform to initiate BCL-2-protein regulated cell death [1]. MOM permeabilization (MOMP) results in the release of pro-apoptotic factors into the cytoplasm thereby committing a neuron to die through apoptosis or degenerate only the axon. It remains unclear how the BCL-2 family of proteins regulates this dichotomy of programmed cell death and degeneration, yet it is widespread in the nervous system during development [3] and disease.
Neuronal cell death and axon degeneration in development
Knockout studies in rodents have solidified the importance of BCL-2 family proteins in the development of the nervous system. Selective deletion of the pro-apoptotic BCL-2 family protein BAX prevents the normal cell death that occurs in the cerebellum and retina, and these mice are reported to have increases in hippocampal and dorsal root ganglion (DRG) neurons [4,5,6,7]. Selective deletion of a BAX-activating, pro-apoptotic protein called p53 upregulated modulator of apoptosis (PUMA), also prevents apoptosis of DRG neurons in the peripheral nervous system (PNS). This results in an increased number of neurons and innervating branches during development [8, 9], with no change in expected birth frequency for PUMA knockout mice [10, 11]. Anti-apoptotic proteins of the BCL-2 family also play key roles in development. For example, single or double deletion of the genes encoding proteins BCL-XL and/or MCL-1 results in massive neuronal cell death in the developing central nervous system (CNS) [12,13,14]. The loss of one allele for mcl1 plus one allele for bcl-X (BCL-XL gene) is sufficient to cause severe brain and craniofacial abnormalities in mouse development suggesting apoptosis must be tightly regulated during development [15]. Moreover, the selective deletion of the BAX-activating protein BIM results in a significantly reduced birth frequency [16]. However, single allele deletion for the bim gene rescues the brain and craniofacial abnormalities observed in mcl1+/−and bcl-x+/− mice [15], suggesting that BIM may play a role in nervous system development.
BCL-W is an anti-apoptotic protein that contributes to the maintenance of axons [17]. BCL-W knockout mice demonstrate progressive nociceptor sensory neuropathy, and as a result, fail to quickly respond to thermosensation [18]. Intriguing, this neuropathy is due to axon degeneration and occurs without cell body loss, demonstrating a separation between axon degeneration and neuronal cell death [18]. Nevertheless, BCL-W knockout mice are still born at the expected frequency [19], while BCL-XL deficient mice are not viable due to massive apoptosis in the CNS [20]. This suggests BCL-W may play more of a role in pathological settings rather than during development.
Key components of the apoptotic pathway, such as caspases and pro-apoptotic BCL-2 proteins, are downregulated during organismal maturation; rendering most adult tissues, including the brain, resistant to apoptotic stimuli [21]. Additionally, primary cultures of murine hippocampal neurons taken at the embryonic stage develop resistance to apoptotic stimuli as they mature in vitro [21]. Furthermore, we have observed that mature cultures of primary cortical neurons resist death in response to the expression of truncated BIM, a protein with reduced pro-apoptotic activity [22]. However, these cultures of mature neurons remain sensitive to the expression of full-length BIM, demonstrating that mature neurons are resistant, but not entirely refractory to apoptotic stimuli [22]. Resistance to apoptotic stimuli correlating with the maturation of neurons has also been shown in vitro with cultures of sympathetic neurons of the PNS [23,24,25]. Although mature sympathetic neurons resist apoptosis, they remain permissive to axon degeneration [24] demonstrating that that cell death and axon degeneration can be regulated separately in a single cell. This change in the regulation that occurs during maturation in vitro means that it is important to account for the level of maturity and/or differentiation in experimental design and to report this in experiments using neuron cultures. For instance, stroke mainly occurs in older adults [26], thus it is important for stroke research to be conducted in mature cultures of neurons that have the apoptotic machinery profile more representative of an adult. Additionally, due to the anatomical and biochemical differences between rodent and human neurons [27], mature cultures of human neurons would be the most representative albeit impractical model system. Consequently, there is emerging emphasis on understanding cell death in neurons derived from human stem cell cultures, and grown as brain organoids. Indeed, brain organoids are already proving to be valuable tools for the study of pathology of diseases such as cerebral malaria [28] and microcephaly induced by Zika virus [29].
Neuronal cell death and axon degeneration in disease
Neuronal cell death and axon degeneration occur naturally during development, however, in the adult they contribute to the pathology of terrible neurodegenerative diseases. In amyotrophic lateral sclerosis (ALS), motor neuron cell death was thought to be the major contributor to disease pathology [8]. However, neuromuscular denervation via axon degeneration prior to cell death has also been shown to be a primary contributor to disease pathology [30]. The genetic mutations that cause ALS prevent proper shuttling of mRNA within axons, disrupting function and leading to cell death [31]. The selective deletion of pro-apoptotic BCL-2 family proteins, such as BAX, BIM, or PUMA, significantly delays disease onset in animal models of ALS [30, 32, 33].
Parkinson’s disease (PD) develops from the specific loss of dopaminergic neurons in the substantia nigra. In pharmaceutically induced models of PD this cell death is regulated by BCL-2 family proteins [34, 35]. Axonal degeneration is also evident in PD and is not just limited to dopaminergic neurons. The early axonal degeneration of serotonergic neurons may contribute to non-motor-related pathologies of PD including anxiety and depression [36].
Apoptosis and axonal degeneration also occur in more acute pathologies such as stroke. Outside of the necrotic core of the infarct in stroke, neurons within the penumbra die from delayed apoptosis, which can be prevented in mouse models by the genetic deletion of BAX [37]. Axonal degeneration has been reported in murine models of stroke [38] and may occur in distinct phases [39].
Wallerian degeneration is a type of axonal degeneration that is thought to occur independent of BCL-2 family proteins [40]. Curiously, Wallerian degeneration can even occur in axons devoid of mitochondria [41] and appears to be independent of BAX and BAK [40]. In response to nerve crush or axotomy, the axon distal to the site of injury undergoes Wallerian degeneration, which involves the activation of calcium-dependent cysteine proteases called calpains that degrade the axon. The portion of the axon proximal to the injury, and still connected to the cell body, remains intact [42]. Here, we mainly discuss BCL-2 family-regulated axon degeneration that requires MOMP and caspase activation (commonly referred to as “pruning”). The contribution of Wallerian degeneration to neurodegenerative disease and injury is discussed elsewhere [43].
BCL-2 family regulation of MOMP
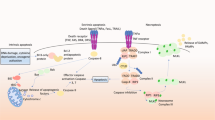
MOMP is regulated by interactions among anti-apoptotic (BCL-2, BCL-XL, BCL-W, and MCL-1) and pro-apoptotic members of the BCL-2 family proteins. The pro-apoptotic proteins are generally subdivided on the basis of function and the presence of BCL-2 homology (BH) motifs [44] into the pore-formers (BAX and BAK), and the BH3-only proteins (BID, BIM, PUMA, NOXA, BAD, BIK, HRK, etc.). Anti-apoptotic proteins and the pro-apoptotic pore formers contain all four BH motifs (BH1–4), while the BH3-only proteins, as implied from their name, possess only the BH3 motif. Abundance, relative affinity and post-translational modifications all dictate how BCL-2 family proteins interact with each other, ultimately leading to the execution or prevention of MOMP [1]. The embedded together model, reviewed elsewhere [1, 45], posits that these interactions are competitive binding interactions that result in either activation or mutual sequestration mediated inactivation [46,47,48,49], the affinities of the requisite interactions are altered by binding to the mitochondrial membrane as the active platform. MOMP occurs upon the activation and oligomerization of the pore-forming proteins BAX and/or BAK on the MOM. BAX and BAK become activated through binding BH3-only “activator” proteins such as BID, BIM, and PUMA. MOMP can be prevented by anti-apoptotic proteins such as BCL-XL and BCL-W by binding to BH3-only activators and/or activated BAX or BAK. As the resulting heteromeric complexes are neither pro nor anti-apoptotic, we refer to this as mutual sequestration [50]. Finally, “sensitizer” BH3-only proteins such as BAD and NOXA promote MOMP by binding to select anti-apoptotic proteins, resulting in the displacement of BH3 activators and active pore formers. Upon BAX activation and subsequent MOMP, cytochrome c is released from the mitochondrial intermembrane space to the cytoplasm where it binds with seven Apaf-1 and caspase-9 molecules in a large complex called an apoptosome. Complex formation activates caspase-9, which, in turn, activates the executioner caspase-3 that degrades many cellular proteins, contributing to apoptosis of the cell (Fig. 1). In addition to cytochrome c, other proteins released into the cytoplasm by MOMP contribute to cell death. Released proteins include apoptosis inducing factor (AIF), endonuclease G (EndoG), SMAC (also called DIABLO), and Omi (also called HtrA2). AIF and EndoG translocate to the nucleus to induce chromatin condensation and DNA fragmentation [51]. SMAC binds to and inhibits x-linked inhibitor of apoptosis protein (XIAP), which normally prevents caspase activity [52]. Omi is a serine protease that cleaves XIAP as well as other target proteins [53].

In neuronal apoptosis it remains uncertain if PUMA activates BAX directly or indirectly. Unknown mechanisms are indicated by a “?”.
PUMA and BAX are the main regulators of neuronal MOMP
The rate limiting step for MOMP is the activation of one or more of the multi-BH motif pore-forming proteins BAX, BAK, or BOK [54]. Surprisingly, in a variety of different neuron cultures the deletion of BAX is sufficient to confer full protection from apoptosis (Table 1), while the deletion of BAK offers no protection [21, 55, 56] and BOK is reported to have no apoptotic role in neocortical neurons [57]. A neuron-specific splice variant of BAK, called N-BAK, has been identified [58], however, the expression of the protein is largely suppressed, and therefore it likely has no apoptotic role [59]. Despite this, in mouse knockout studies the double deletion of both BAX and BAK results in a further accumulation of neurons in the CNS compared to BAX deletion alone [60]. Furthermore, the added deletion of BAK lowers the number of pups that survive into adulthood to <10%. The triple deletion of BAX, BAK, and BOK results in: abnormal brain development and even fewer survivors, but 1% do survive to adulthood [61]. Overall, these knockout studies suggest that BAK and BOK may have a relevant role in the normal apoptosis of the nervous system throughout development. It would be interesting therefore to determine the extent to which BAK and BOK may contribute to axon degeneration rather than playing a major role in survival.
The proteins responsible for BAX activation are the BH3-only “activators” BID, BIM, and PUMA. Despite being a potent direct activator of BAX, there is limited evidence to suggest that BID plays a fundamental role in neuronal cell death [62, 63], and no indication it contributes to axon degeneration [24, 64]. The expression of BIM has been shown to increase in neurons deprived of nerve growth factor (NGF), and in neuronal cultures undergoing ER or oxidative stress [35, 65, 66]. The selective deletion of BIM can delay neuronal apoptosis in some circumstances in vitro, and may play a role in the progression of neurodegenerative disease, as knockout of BIM increases lifespan and delays disease onset in a mouse model of ALS [33]. However, while BIM knockout has been shown to delay neuronal apoptosis upon NGF deprivation, complete protection is afforded by the genetic deletion of BAX [67], suggesting that other factors, in addition to BIM, can result in BAX activation and neuronal apoptosis. Indeed, there is a growing list of publications that demonstrate both neuronal apoptosis and axon degeneration are highly dependent on the BH3-only protein PUMA (Tables 1 and 2). Moreover, numerous publications demonstrate that unlike BIM the single deletion of PUMA prevented both neuronal apoptosis [35, 65, 66, 68,69,70,71] and axon degeneration [64] in vitro.
PUMA was first discovered as a potent apoptosis inducing BCL-2 family member transcriptionally regulated by p53 [72, 73] hence its name; the p53 upregulated modulator of apoptosis (PUMA). However, other transcription factors, including CHOP [65] and Foxo3a [35], also regulate the expression of PUMA, enabling its expression in response to multiple varieties of stress in addition to DNA damage [65, 70, 74]. Deficiency of PUMA and/or BAX is sufficient to prevent neuronal cell death in response to oxidative stress, ER stress, DNA damage, environmental toxins, proteasomal inhibition, stroke-like stress, trophic-factor deprivation, pan-kinase inhibition, and death receptor activation (Table 1). PUMA and BAX are also required for axon degeneration induced by local deprivation of NGF (Table 2). The local application of small-molecule inhibitors of anti-apoptotic proteins (termed BH3 mimetics) on axons is sufficient to induce PUMA- and BAX-dependent axon degeneration (Table 2). PUMA and BAX have been demonstrated to be required for apoptosis and axon degeneration in a variety of neuronal cell types from both the CNS and PNS, and across different species (mouse and rat) by multiple independent groups (Tables 1 and 2), suggesting that they play a significant role in regulating neuronal MOMP.
Regulation of cell death and axon degeneration
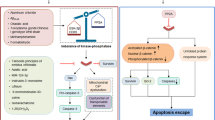
Often axon degeneration occurs without the death of the neuronal cell body. In order to prevent the neuronal cell death and axon degeneration that contributes to neurodegenerative pathology, it is important to understand what factors determine whether axon degeneration or neuronal apoptosis occurs. One component may be the location of stress. Whole cell trophic-factor deprivation causes both PNS and CNS neurons to die in a PUMA- and BAX-dependent manner [7, 71, 75], however, trophic-factor deprivation exclusively from axons results in degeneration [24]. Using microfluidic chambers that enable separate treatment of neuronal cell bodies and axons, trophic-factor deprivation from axons results in transcriptional upregulation of key genes required for axon degeneration such as bbc3, encoding PUMA [9, 64] (Fig. 2). PUMA expression de novo in the cell body is required for axon degeneration as application of the transcriptional inhibitor Actinomycin D prevents trophic-factor withdrawal-induced axon degeneration [9, 64]. PUMA is a BH3-only protein and, thus, is predicted to function as either a direct activator or a sensitizer (indirect activator) of BAX and BAK. Numerous publications have shown that the BH3-domain of PUMA can directly bind to and activate BAX, resulting in MOMP [75,76,77]. In addition, PUMA can also bind to and inhibit all anti-apoptotic proteins [78]. Therefore, PUMA has two apoptotic functions to execute MOMP (activator and sensitizer), but which of these functions occur during neuronal apoptosis or axon degeneration? The reported role for PUMA in apoptosis and axon degeneration may be related to the location of BCL-2 family proteins. Biochemical fractionation has determined that the anti-apoptotic proteins BCL-2 and MCL-1 are primarily localized within the cell body of cultured murine DRG neurons, while BCL-XL and BCL-W can be found in both the cell body and axon [18, 64]. Localization of mitochondria within the axon may also play a relevant role. For example, in PNS neurons, accumulations of mitochondria are consistently observed at nodal junctions between myelin on the axon and at the synapse, both areas that are rich in membrane channel proteins and participate in the generation of action potentials [79]. However, CNS neurons appear to have more axonal mitochondria at intermodal areas [80]. Could MOMP selectively occur to one cluster of mitochondria within the axon, and not perpetuate down the length of the axon? Or does an apoptotic trigger wave occur spreading down the entire length of the axon [81]? It makes intuitive sense that MOMP occurring in the cell body would induce apoptosis, while selective MOMP in the axon could result in axon degeneration (Fig. 2). Indeed, selective application of BH3-mimetic to axons results in caspase activation and subsequent degeneration [17, 64, 82]; suggesting that axonal MOMP is sufficient to induce degeneration. Additionally, because BH3 mimetics work by inhibiting anti-apoptotic proteins, these data suggest that the sensitizer function of PUMA would be sufficient to induce axon degeneration. Immunofluorescence assays have also shown that trophic-factor deprivation from axons results in cytochrome c release [24], but whether MOMP is exclusive to the axon remains to be determined. Western blot and mass spec demonstrate that PUMA (protein) can be detected in the cell body while evidence for synthesis in or localization to the axon is less clear [9, 64]. PUMA is required for CNS neurons to die from trophic-factor deprivation (Table 1), however, a role for PUMA in CNS neuron axon degeneration has not been determined. In PNS neurons, selective application of the translational inhibitor cycloheximide to the cell body prevents axon degeneration, thus de novo PUMA expression in the cell body is required for axon degeneration [64]. However, it is unknown how the expression of PUMA in the cell body of PNS neurons results in axonal MOMP, while sparing the cell body (Fig. 2). Furthermore, neurons somehow restrict the spillover of active caspases from the axon into the cell body. A role has been speculated for the endogenous caspase inhibitor XIAP in preventing active caspases reaching the cell body [24], but how XIAP is regulated in apoptosis versus axon degeneration remains unclear. Additionally, it is unclear how neuronal cell death is prevented in the cell body subsequent to MOMP mediated release of other mitochondrial proteins including many implicated in cell death-including cytochrome c, EndoG, Smac, and Omi (Fig. 2). One key difference between the CNS and PNS is the remarkable ability of PNS neurons to regenerate after xonal degeneration [83]. In the PNS, regeneration of the axon is possible as long as the neuronal cell body remains alive. Thus, future research should investigate whether in CNS neurons the cell bodies die after axon degeneration, potentially explaining one reason why regeneration in the CNS is so poor. If so then understanding of how PNS neuronal cell bodies survive becomes even more important.

However, there are many unknowns in the regulation of selective degradation of axons including: the location of MOMP, how caspase-9 is activated, and how neuronal cell bodies remain alive when PUMA is expressed and caspases activated. Unknown mechanisms are indicated by a “?”. The numbers “1” and “2” indicate the extension of activation and inhibition signals down the axon, respectively.
Another relevant factor may be assembly of the apoptosome at the onset of neuronal apoptosis [24, 64] (Fig. 1), but not axonal degeneration [24] (Fig. 2). Mature neurons restrict the expression of Apaf-1 through chromatin remodeling limiting caspase-9 activation by MOMP [84]. However, during neuronal degeneration, activation of caspase-9 occurs by an as yet unknown mechanism [24, 82]. The activation of executioner caspases and subsequent apoptosis has been reported to occur in other cell types genetically deficient in either Apaf-1 or caspase-9 [85].
Pharmacological inhibition of apoptotic machinery—implications for neurodegenerative disease
Activation of BAX is required in both axon degeneration and neuronal cell death. However, attempts at therapies preventing cell death in response to neurological damage have targeted steps either upstream or downstream of the coordination of BCL-2 family proteins at the mitochondria. For example, Foretinib, a pan-kinase inhibitor, was identified in a screen for kinase inhibitors that could reduce cell death and axon degeneration in rat superior cervical ganglion cultures [86]. Foretinib inhibits both trophic-factor deprivation-induced axon degeneration and Wallerian degeneration (due to axotomy). However, Foretinib delayed but did not prevent axon degeneration in vivo upon sciatic nerve-crush experiments in rats [86].
In contrast, in ischemia and reperfusion injury, the inhibition of JNK signaling has been an attractive target to prevent apoptosis for the protection of tissues [87, 88]. Inhibiting JNK3 has been proposed for treating ALS as JNK inhibition prevents the apoptosis of motor neurons derived from human iPS cells [89]. Consistent with potential utility in ALS, the selective JNK inhibitor SP600125 protects CNS neurons from axon degeneration induced by trophic-factor withdrawal [90]. Although cardioprotective and neuroprotective activities have been observed for synthetic small-molecule inhibitors of JNKs [87], systemic administration of current JNK inhibitors is expected to suffer from on-target side effects because different members of the JNK family exert diverse physiological properties. Therefore, to be useful as neuroprotectants it will be necessary to make inhibitors with exquisite isoform specificity. Recent structure activity relationship studies resulted in an inhibitor that is somewhat more specific for JNK3 suggesting selectivity may be achievable [91].
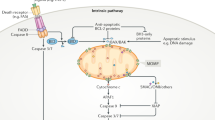
In contrast to these inhibitors that act upstream of MOMP, attempts to inhibit the function of caspases downstream of MOMP met limited success. Treatment of NGF deprived murine neurons with pan-caspase inhibitor Z-VAD-FMK only partially protected neurons from cell death or axonal degradation compared to BAX knockout neurons [92]. This result is consistent with the release of multiple pro-apoptotic molecules from mitochondria by MOMP (Fig. 3) and suggests that the prevention of MOMP would be a more effective strategy than inhibiting apoptosis downstream of MOMP [93]. Thus, pharmacologic targeting of BAX may be an efficient way to limit cell death, as BAX activation is required for MOMP induced apoptosis (Fig. 3).

Upstream signaling pathways converge at the MOM with BAX activation as the commitment step in apoptosis. Preventing caspase activation is not sufficient as MOMP causes the release of pro-apoptotic proteins that can lead to caspase independent cell death.
Until recently only the inhibitory BCL-2 family proteins have been selectively targeted by compounds. The success of the BCL-2 inhibitor Venetoclax, in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma, and as a combinatorial therapy in acute myeloid leukemia [94,95,96], clearly demonstrates that the protein–protein interactions of BCL-2 family proteins are druggable. Venetoclax and other inhibitors of anti-apoptotic proteins are referred to as BH3 mimetics because they were designed to mimic the BH3 motif of BAD, and thereby function as competitive inhibitors for binding of BH3 proteins and active BAX and BAK to anti-apoptotic proteins [97]. As activation of BAX is rate limiting for MOMP and is regulated by multiple BCL-2 family members upstream of oligomerization, Bax is an ideal post-insult target (Tables 1 and 2). Genetic deletion of BAX does not prevent cell injury but has been shown to prevent neuronal cell death in mouse models of stroke and traumatic brain injury [37, 98]. Thus, small peptides based on BH3 motifs that inhibit rather than activate BAX, similar to those identified for BAK [99], or synthetic small molecules that bind interfaces on BAX allosteric to the canonical BH3-groove interface would be very useful tools. The small-molecule inhibitor BAI1 binds at a novel, allosteric pocket between the α5–α6 hairpin and the loop between α3 and α4 [100]. Microscale thermophoresis measurements and chemical shift perturbations in 15N-1H HSQC nuclear magnetic resonance experiments demonstrate that BAI1 binds BAX directly. Although BAI1 appears to inhibit BAX and not BAK, given the similar fold between BCL-2 family members it remains to be formally determined whether BAI1 will have off-target effects due to binding of other BCL-2 family proteins [101]. The in vivo activity of BAI1 to prevent apoptosis is limited. Nonetheless, in zebrafish and mice, BAI1 protects against doxorubicin-induced cardiomyopathy as measured by a reduction in caspase-3 cleavage and retention of mitochondrial polarization [102]. The effects of BAI1 in cerebral injury or ischemia-reperfusion models have yet to be demonstrated but provide an exciting opportunity that may drive further development of this early stage synthetic.
Inhibition of BAX prior to activating conformation changes such as oligomerization and insertion into membranes is attractive because in neurons it would prevent all of the pro-apoptotic BH3 proteins from triggering MOMP and therefore also prevent release of other pro-apoptotic proteins from mitochondria (Fig. 3). In addition, BAX activation can occur independent of BH3-only proteins by heat or a variety of small molecules suggesting there may be as yet undiscovered effectors of BAX in cells [103, 104]. BAX and BAK execution of MOMP has also been demonstrated in cultured cells in which the genes encoding all known BH3-only proteins have been deleted [105], further stressing the importance of targeting the executioner proteins directly as opposed to other upstream signaling proteins.
Currently, there is only one study demonstrating that it is possible to inhibit both BAX and BAK with a single compound. MSN-50 and MSN-125 were identified as BAX oligomerization inhibitors and shown to prevent cell death of cortical neurons from glutamate excitotoxicity [106]. Inhibition of BAX and BAK with these molecules protected cells long enough that they were able to recover from an otherwise lethal exposure to Actinomycin D or Staurosporine.
BAI1 and the oligomerization inhibitors MSN-50 and MSN-125 are early stage tool compounds with micromolar affinities- with off-target effects at effective concentrations. Nonetheless, it may be possible to use BAI1 to probe the role of BAX in axonal degeneration. Most important, BAI1 enabled identification of the binding pocket on BAX which will likely lead to optimization of these inhibitors, or new molecules.
Given the emergence of these inhibitors, the question remains if pharmacological BAX inhibition protects against neurological events such as stroke. After the excitotoxic cell death characteristic of ischemic injury from a stroke, there is a transitional period where a reduction of local cerebral blood flow results in programmed cell death over a number of days, to weeks [107,108,109]. This time period provides a unique opportunity for a BAX inhibitor to limit the amount of cell death following stroke. However, it remains unknown whether pharmaceutical inhibition of BAX can prevent axonal degeneration, and if preventing this axonal degeneration will translate to better outcome for patients. If BAX inhibition is demonstrated to provide therapeutic benefit it would provide the impetus for optimization of specificity, affinity, and pharmacological properties of a BAX inhibitor.
The initial reaction to inhibiting BAX and BAK as a treatment for neurological diseases of all kinds including neurodegenerative diseases such as ALS has been skepticism as BAX and BAK are predicted to be tumor suppressors. However, BAX deficiency alone does not result in an increased risk to spontaneous cancer formation in mice [110]. Additionally, an acute insult such as ischemia or stroke would only require transient BAX inhibition, further limiting risk to cancer development. Given the exquisite dependence of neuronal cell death on PUMA and BAX an attractive approach would be to target this protein pair specifically, which may limit undesirable on-target effects in other tissues. Only with the development of better tool compounds will it be possible to address both the potential benefits and issues related to inhibiting neuronal apoptosis.
Noncanonical roles of BCL-2 family proteins in neurons
BCL-2 family proteins are well known for their role in regulating MOMP, resulting in caspase activation and committing a neuron to either apoptosis or axon degeneration. However, other types of cell death exist which proceed independently of caspase activation [111, 112], but may still be regulated in part by noncanonical functions of BCL-2 family proteins. Indeed, other types of cell death such as necroptosis and ferroptosis contribute to the pathology of neurological disease and trauma, such as hemorrhagic stroke [113]. Necroptosis signaling has been shown to contribute to the progression of Wallerian degeneration of both CNS and PNS neurons [114], a process that when induced through nerve-crush experiments, also results in a transcriptional upregulation of PUMA [9]. In colorectal cancer cell lines PUMA can enhance necroptosis signaling by inducing the release of mitochondrial DNA to the cytoplasm where it is recognized by DNA sensors DAI/Zbp1 and STING, leading to enhanced signaling by RIP3 and phosphorylation of MLKL [115]. Curiously, genetic deletion of BAX and BAK had no effect on inhibiting this necroptosis, suggesting that PUMA acts in a noncanonical fashion to induce necroptosis [115]. This is in stark contrast with another study showing that the genetic deletion of BAX is sufficient to prevent necroptosis in mouse embryonic fibroblasts [116]. Further research is required to determine whether PUMA and/or BAX contribute to necroptosis signaling in neuronal cell death and/or axon degeneration (Wallerian), and whether pharmacological inhibition of either protein can prevent non-apoptotic neuron death. If so, inhibition of BAX may be sufficient to prevent multiple types of neuronal cell death, and multiple types of axon degeneration. It is also vital to understand the mechanism by which BAX is required for necroptosis, as current small-molecule inhibitors such as MSN-125 prevent BAX oligomerization. If BAX oligomerization is not required for necroptosis, an alternative strategy will be needed to inhibit this form of cell death. Ferroptosis is a MOMP independent, iron-dependent form of programmed cell death that occurs upon the accumulation of lipid peroxidation [117]. Conditional deletion of the antioxidant enzyme glutathione peroxidase 4 results in rapid motor neuron cell death through ferroptosis and paralyzes mice, suggesting that the inhibition of ferroptosis may play a role in the response to oxidative stress in adult motor neurons in vivo [118]. Moreover, ferroptosis kills a large percentage of the cells during transdifferentiation of somatic cells, such as fibroblasts, into neurons [119]. Surprisingly, overexpression of either BCL-2 or BCL-XL improved the efficiency of neuron conversion by reducing the levels of reactive oxygen species (ROS), and preventing ferroptosis [119]. How anti-apoptotic proteins reduce ROS is unknown, however, the expression of BCL-2 mutants such as one in which serine 70 was replaced with alanine exhibited reduced BAX binding but increased activity at preventing ferroptosis, suggesting that BCl-2 and BCL-XL prevent ROS accumulation through a noncanonical role [119].
Chemotherapy-induced peripheral neuropathy is a form of pathological axon degeneration that occurs in patients treated with the chemotherapeutic paclitaxel. Paclitaxel treatment of cultured sensory neurons prevents the axonal transport of bclw mRNA, reducing protein levels and resulting in peripheral neuropathy [120]. BAX may also be involved as knockdown significantly reduced axon degeneration induced by paclitaxel. Interestingly, loss of axonal BCL-W was reported to result in aberrant calcium signaling, possibly from the IP3 receptor at the ER, and subsequently calpain activation [120]. Consistent with this, release of cytochrome c from axonal mitochondria was not observed in cultured neurons treated with paclitaxel [120]. Together, these data suggest that BCL-W and BAX act independently of MOMP to regulate peripheral neuropathy. Thus, caspase inhibitors are unlikely to be useful as MOMP and subsequent caspase activation do not contribute to this form of axon degeneration. However, a BAX inhibitor may be of clinical use because the knockdown of BAX protected cultured axons from neuropathy.
Conclusion
Unlike other cells, neurons regulate the degeneration of a large part of the cell, the axon, without the cell dying. Surprisingly, neurons use BCL-2 family proteins to regulate both cell death and axon degeneration. Moreover, despite the redundancy that exits within the BCL-2 family that presumably enables cells to respond differently to many stimuli, several types of neurons require only PUMA and BAX to execute many instances of apoptotic and axon degeneration events (Fig. 4). This unique reliance on only two BCL-2 family proteins affords the possibility of pharmaceutical intervention without serious effects in other tissues where the role of PUMA and BAX is more redundant. To take the advantage of the unique regulation of apoptosis and axon degeneration in neurons, it is important to more fully understand the detailed mechanism(s) of PUMA- and BAX-induced MOMP (Fig. 4). Such information is a key to the development of BAX inhibitors that will ultimately save neurons in a wide variety of neurological conditions with minimal effects on other tissues. In addition to PUMA and BAX, other BCL-2 family proteins have been shown to contribute to the regulation of both apoptosis and other types of neuronal death (necroptosis and ferroptosis) and Wallerian axon degeneration. Thus, a more detailed understanding of these processes will likely reveal other potential therapeutic targets. Importantly the imminent development of specific small-molecule inhibitors of BAX will provide the initial tools needed to parse mechanistic details of the coordinate regulation of apoptosis, necroptosis, and other forms of cell death in the pathology of neurodegenerative disease.

This unique reliance on only two proteins affords the possibility of pharmacological inhibition to prevent both process from occurring with minimal effects in other tissues. However, detailed understanding of the mechanism(s) of PUMA and BAX induced MOMP and necroptosis are first required.
References
Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80. https://doi.org/10.1038/cdd.2017.186.
Misgeld T, Schwarz TL. Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron. 2017;96:651–66.
Yuan J, Yanker BA. Apoptosis in the nervous system. Nature. 2000;407:802–9.
Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011;6:50. https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/1750-1326-6-50.
Bar-Peled O, Knudson M, Korsmeyer SJ, Rothstein JD. Motor neuron degeneration is attenuated in bax-deficient neurons in vitro. J Neurosci Res. 1999;55:542–56.
White FA, Keller-Peck CR, Michael Knudson C, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax- deficient mice. J Neurosci. 1998;18:1428–39.
Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–11.
Sun W, Gould TW, Vinsant S, Prevette D, Oppenheim RW. Neuromuscular development after the prevention of naturally occurring neuronal death by Bax deletion. J Neurosci. 2003;23:7298–310.
Maor-Nof M, Romi E, Shalom HS, Ulisse V, Raanan C, Nof A, et al. Axonal degeneration is regulated by a transcriptional program that coordinates expression of pro- and anti-degenerative factors. Neuron. 2016;92:991–1006. https://doi.org/10.1016/j.neuron.2016.10.061.
Villunger A, Michalak EM, Coultas L, Müllauer F, Ausserlechner MJ, Adams JM, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. vol. 302. American Association for the Advancement of Science; 2003. p. 1036–8. http://www.jstor.org/stable/3835665.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8.
Fogarty LC, Flemmer RT, Geizer BA, Licursi M, Karunanithy A, Opferman JT. et al. Mcl-1 and Bcl-xL are essential for survival of the developing nervous system. Cell Death Differ. 2019;26:1501–15. https://doi.org/10.1038/s41418-018-0225-1.
Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung ECC. et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. 2008;28:6068–78. https://doi.org/10.1523/JNEUROSCI.4940-07.2008.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–10.
Grabow S, Kueh AJ, Ke F, Vanyai HK, Sheikh BN, Dengler MA, et al. Subtle changes in the levels of BCL-2 proteins cause severe craniofacial abnormalities. Cell Rep. 2018;24:3285–95.e4. https://doi.org/10.1016/j.celrep.2018.08.048.
Bouillet P, Metcalf D, Huang DCS. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–8.
Cosker KE, Pazyra-Murphy MF, Fenstermacher SJ, Segal RA. Target-derived neurotrophins coordinate transcription and transport of Bclw to prevent axonal degeneration. J Neurosci. 2013;33:5195–207.
Courchesne SL, Karch C, Pazyra-Murphy MF, Segal RA. Sensory neuropathy attributable to loss of Bcl-w. J Neurosci. 2011;31:1624–34.
Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, et al. Apoptosis regulator Bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA. 1998;95:12424–31.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x- deficient mice. Science. 1995;267:1506–10.
Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell. 2016;31:142–56. http://linkinghub.elsevier.com/retrieve/pii/S1535610816305542.
Chi X, Pemberton J, Nguyen D, Osterlund EJ, Liu Q, Brahmbhatt H, et al. The carboxyl-terminal sequence of Bim enables Bax activation and killing of unprimed cells. Elife. 2020;9. http://biorxiv.org/content/early/2019/02/19/554907.abstract.
Deshmukh M, Johnson EM. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705.
Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun. 2013;4. http://www.ncbi.nlm.nih.gov/pubmed/23695670.
Annis RP, Swahari V, Nakamura A, Xie AX, Hammond SM. Mature neurons dynamically restrict apoptosis via redundant premitochondrial brakes. FEBS J. 2016;283:4569–82.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics—2020 update. Circulation. 2020;141:139–596.
Kalmbach BE, Buchin A, Long B, Close J, Nandi A, Miller JA. et al. h-Channels contribute to divergent intrinsic membrane properties of supragranular pyramidal neurons in human versus mouse cerebral cortex. Neuron. 2018;100:1194–208.e5. https://doi.org/10.1016/j.neuron.2018.10.012.
Harbuzariu A, Pitts S, Cespedes JC, Harp KO, Nti A, Shaw AP, et al. Modelling heme-mediated brain injury associated with cerebral malaria in human brain cortical organoids. Sci Rep. 2019;9:1–14.
Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 2016;165:1238–54. https://doi.org/10.1016/j.cell.2016.04.032.
Gould TW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci.2006;26:8774–86. https://doi.org/10.1523/JNEUROSCI.2315-06.2006.
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW. et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–43. https://doi.org/10.1016/j.neuron.2013.12.018.
Kieran D, Woods I, Villunger A, Strasser A, Prehn JHM. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci USA. 2007;104:20606–11. http://www.pnas.org/content/104/51/20606.full.
Hetz C, Thielen P, Fisher J, Pasinelli P, Brown RH, Korsmeyer S, et al. The proapoptotic BCL-2 family member BIM mediates motoneuron loss in a model of amyotrophic lateral sclerosis. Cell Death Differ. 2007;14:1386–9.
Bernstein AI, Garrison SPS, Zambetti GP, O’Malley KL. 6-OHDA ge nerated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol Neurodegener. https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/1750-1326-6-2.
Steckley D, Karajgikar M, Dale LB, Fuerth B, Swan P, Drummond-Main C, et al. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J Neurosci. 2007;27:12989–99.
Grosch J, Winkler J, Kohl Z. Early degeneration of both dopaminergic and serotonergic axons—a common mechanism in Parkinson’s disease. Front Cell Neurosci. 2016;10:1–8. https://doi.org/10.3389/fncel.2016.00293/full.
D’Orsi B, Kilbride SM, Chen G, Perez Alvarez S, Bonner HP, Pfeiffer S, et al. Bax regulates neuronal Ca2+ homeostasis. J Neurosci. 2015;35:1706–22. https://www.jneurosci.org/content/35/4/1706.
Ito U, Kuroiwa T, Nagasao J, Kawakami E, Oyanagi K. Temporal profiles of axon terminals, synapses and spines in the ischemic penumbra of the cerebral cortex: ultrastructure of neuronal remodeling. Stroke. 2006;37:2134–9.
Hinman JD. The back and forth of axonal injury and repair after stroke. Curr Opin Neurol. 2014;27:615–23.
Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–1.
Kitay BM, McCormack R, Wang Y, Tsoulfas P, Zhai RG. Mislocalization of neuronal mitochondria reveals regulation of wallerian degeneration and NMNAT/WLDS-mediated axon protection independent of axonal mitochondria. Hum Mol Genet. 2013;22:1601–14.
Gerdts J, Summers D, Milbrandt J, Aaron D. Axon self destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron. 2016;89:1–13.
Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15:394–409.
Auoacheria A, Combet C, Tompa P, Hardwick JM. Redefining the BH3 death domain as a “short linear motif” Abdel. Trends Biochem Sci. 2015;40:736–48.
Kale J, Liu Q, Leber B, Andrews DW. Shedding light on apoptosis at subcellular membranes. Cell. 2012;151:1179–84. https://doi.org/10.1016/j.cell.2012.11.013.
Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–7. https://doi.org/10.1073/pnas.0438011100.
Wilfling F, Weber A, Potthoff S, Vögtle F-N, Meisinger C, Paschen SA, et al. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 2012;19:1328–36. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3392640&tool=pmcentrez&rendertype=abstract.
Liu Q, Oesterlund EJ, Chi X, Pogmore J, Leber B, Andrews DW. Bim escapes displacement by BH3-mimetic anti-cancer drugs by double-bolt locking both Bcl-XL and Bcl-2. Elife. 2019;8:1–30.
Singh PK, Roukounakis A, Frank DO, Kirschnek S, Das KK, Neumann S, et al. Dynein light chain 1 induces assembly of large bim complexes on mitochondria that stabilize Mcl-1 and regulate apoptosis. Genes Dev. 2017;31:1754–69.
Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:1–21.
Muñ Oz-Pinedo C, Guío-Carrió A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR, et al. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci USA. 2006;103:11573–8.
Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionary conserved mechanism of IAPs. EMBO J. 2005;24:645–55.
Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–96.
Kale J, Chi X, Leber B, Andrews D. Examining the molecular mechanism of bcl-2 family proteins at membranes by fluorescence spectroscopy. Methods Enzymol. 2014;544:1–23. https://doi.org/10.1016/B978-0-12-417158-9.00001-7.
Harris CA, Johnson EM. BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–60.
Crowther AJ, Gama V, Bevilacqua A, Chang SX, Yuan H, Deshmukh M. et al. Tonic activation of Bax primes neural progenitors for rapid apoptosis through a mechanism preserved in medulloblastoma. J Neurosci. 2013;33:18098–108. https://doi.org/10.1523/JNEUROSCI.2602-13.2013.
D’Orsi B, Engel T, Pfeiffer S, Nandi S, Kaufmann T, Henshall DC, et al. Bok is not pro-apoptotic but suppresses poly ADP-ribose polymerase-dependent cell death pathways and protects against excitotoxic and seizure-induced neuronal injury. J Neurosci. 2016;36:4564–78. http://www.ncbi.nlm.nih.gov/pubmed/27098698.
Sun Y-F, Yu L-Y, Saarma M, Timmusk T, Aruma¨e U. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001;276:16240–7.
Jakobson M, Lintulahti A, Arumäe U. MRNA for N-Bak, a neuron-specific BH3-only splice isoform of Bak, escapes nonsense-mediated decay and is translationally repressed in the neurons. Cell Death Dis. 2012;3:1–7.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99.
Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S. et al. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell. 2018;173:1217–30.e17. https://doi.org/10.1016/j.cell.2018.04.036.
Leonard JR, D’Sa C, Cahn BR, Korsmeyer SJ, Roth KA. Bid regulation of neuronal apoptosis. Dev Brain Res. 2001;128:187–90.
Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, et al. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:15318–23.
Simon DJ, Pitts J, Hertz NT, Te M, Molina H, Tessier-lavigne M. Axon degeneration gated by retrograde activation axon degeneration gated by retrograde activation of somatic pro-apoptotic signaling. Cell. 2016;164:1–15.
Galehedar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30:16938–48.
Ghosh AP, Klocke BJ, Ballestas ME, Roth KA. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS ONE. 2012;7:1–11.
Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, et al. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–28.
Wyttenbach A, Tolkovsky AM. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J Neurochem. 2006;96:1213–26.
Ghosh AP, Walls KC, Klocke BJ, Toms R, Strasser A, Roth KA. The proapoptotic BH3-only, Bcl-2 family member, Puma is critical for acute ethanol-induced neuronal apoptosis. J Neuropathol Exp Neurol. 2009;68:747–56. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2745204&tool=pmcentrez&rendertype=abstract.
Tuffy LP, Concannon CG, D’Orsi B, King MA, Woods I, Huber HJ, et al. Characterization of Puma-dependent and Puma-independent neuronal cell death pathways following prolonged proteasomal inhibition. Mol Cell Biol. 2010;30:5484–501. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2976426&tool=pmcentrez&rendertype=abstract.
Ambacher KK, Pitzul KB, Karajgikar M, Hamilton A, Ferguson SS, Cregan SP. The JNK- and AKT/GSK3B-Signaling pathways converge to regulate puma induction and neuronal apoptosis induced by trophic factor deprivation. PLoS ONE. 2012;7:1–14.
Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–94.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–82. http://linkinghub.elsevier.com/retrieve/pii/S1097276501002131.
Ming L, Sakaida T, Yue W, Jha A, Zhang L, Yu J. Sp1 and p73 activate PUMA following serum starvation. Carcinogenesis. 2008;29:1878–84.
Ren D, Tu H-C, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–3. http://www.sciencemag.org/content/330/6009/1390.long.
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJD, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–58.
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP. et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–99. https://doi.org/10.1016/j.molcel.2009.09.030.
Edwards AL, Gavathiotis E, Labelle JL, Braun CR, Opoku-nsiah KA, Bird GH. et al. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol. 2013;20:888–902. https://doi.org/10.1016/j.chembiol.2013.06.007.
Perkins GA, Ellisman MH. Mitochondrial configurations in peripheral nerve suggest differential ATP production. J Struct Biol. 2011;173:117–27. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf.
Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, et al. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci. 2011;31:7249–58.
Cheng X, Ferrell JE. Apoptosis propagates through the cytoplasm as trigger waves. Science. 2018;361:607–12. http://science.sciencemag.org/content/361/6402/607/tab-pdf.
Simon DJ, Weimer RM, Mclaughlin T, Kallop D, Stanger K, Yang J, et al. A caspase cascade regulating developmental axon degeneration. J Neurosci. 2012;32:17540–53.
Curcio M, Bradke F. Axon regeneration in the central nervous system: facing the challenges from the inside. Annu Rev Cell Dev Biol. 2018;34:495–521.
Wright KM, Smith MI, Farrag L, Deshmukh M. Chromatin modification of Apaf-1 restricts the apoptotic pathway in mature neurons. J Cell Biol. 2007;179:825–32.
Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG, et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419:634–7.
Feinberg K, Kolaj A, Wu C, Grinshtein N, Krieger JR, Moran MF, et al. A neuroprotective agent that inactivates prodegenerative TrkA and preserves mitochondria. J Cell Biol. 2017;216:3655–75.
Shvedova M, Anfinogenova Y, Atochina-Vasserman EN, Schepetkin IA, Atochin DN. c-Jun N-terminal kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front Pharm. 2018;9:1–18.
Shvedova MV, Anfinogenova YD, Shchepetkin IA, Atochin DN. c-Jun N-terminal kinases and their pharmacological modulation in ischemic and reperfusion brain injury. Neurosci Behav Physiol. 2018;48:721–8.
Wu C, Watts ME, Rubin LL. MAP4K4 activation mediates motor neuron degeneration in amyotrophic lateral sclerosis. Cell Rep. 2019;26:1143–56.e5. https://doi.org/10.1016/j.celrep.2019.01.019.
Magnifico S, Saias L, Deleglise B, Duplus E, Kilinc D, Miquel MC, et al. NAD acts on mitochondrial SirT3 to prevent axonal caspase activation and axonal degeneration. FASEB J. 2013;27:4712–22.
Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, MacHleidt T. et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140–54. https://doi.org/10.1016/j.chembiol.2011.11.010.
Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, et al. Axonal degeneration is regulated by the apoptotic machinery or a NAD +-sensitive pathway in insects and mammals. J Neurosci. 2010;30:6375–86.
Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8.
Zhu H, Almasan A. Development of venetoclax for therapy of lymphoid malignancies. Drug Des Devel Ther. 2017;11:685–94.
DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8.
Tehranian R, Rose ME, Vagni V, Pickrell AM, Griffith RP, Liu H. et al. Disruption of Bax protein prevents neuronal cell death but produces cognitive impairment in mice following traumatic brain injury. J Neurotrauma. 2008;25:755–67. https://doi.org/10.1089/neu.2007.0441.
Brouwer JM, Lan P, Cowan AD, Bernardini JP, Birkinshaw RW, van Delft MF. et al. Conversion of Bim-BH3 from activator to inhibitor of Bak through structure-based design. Mol Cell. 2017;68:659–72.e9. https://doi.org/10.1016/j.molcel.2017.11.001.
Garner TP, Amgalan D, Reyna DE, Li S, Kitsis RN, Gavathiotis E. Small-molecule allosteric inhibitors of BAX. Nat Chem Biol. 2019;15:322–30.
Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94.
Amgalan D, Garner TP, Pekson R, Jia XF, Yanamandala M, Paulino V, et al. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat Cancer. 2020;1:315–28.
Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM, et al. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA. 2005;102:17975–80.
Brahmbhatt H, Uehling D, Al-awar R, Leber B, Andrews D. Small molecules reveal an alternative mechanism of Bax activation. Biochem J.2016;473:1073–83. https://doi.org/10.1042/BCJ20160118.
O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30:973–88.
Niu X, Brahmbhatt H, Mergenthaler P, Zhang Z, Sang J, Daude M. et al. A Small-Molecule Inhibitor of Bax and Bak oligomerization prevents genotoxic cell death and promotes neuroprotection. Cell Chem Biol. 2017;24:493–506.e5. https://doi.org/10.1016/j.chembiol.2017.03.011.
Li H, Zhang N, Lin HY, Yu Y, Cai QY, Ma L, et al. Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis-induced ischemia in adult mice. BMC Neurosci. 2014;15:1–13.
Ferrer I. Apoptosis: future targets for neuroprotective strategies. Cerebrovasc Dis. 2006;21:9–20.
Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:331–9.
Knudson CM, Johnson GM, Lin Y, Korsmeyer SJ. Bax accelerates tumorigenesis in p53-deficient mice. Cancer Res. 2001;61:659–65.
Galluzzi L, Vitale I. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. 2018;98:813–80. https://doi.org/10.1152/physrev.00011.2017.
Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48:1033–43.
Arrázola MS, Saquel C, Catalán RJ, Barrientos SA, Hernandez DE, Martínez NW, et al. Axonal degeneration is mediated by necroptosis activation. J Neurosci. 2019;39:3832–44.
Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci. 2018;115:3930–5. https://www.pnas.org/content/115/15/3930.
Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD. Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLoS ONE. 2015;10:1–12.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11. https://doi.org/10.1038/s41419-020-2298-2.
Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–106.
Gascón S, Murenu E, Masserdotti G, Ortega F, Russo GL, Petrik D, et al. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell. 2016;18:396–409.
Pease-Raissi SE, Pazyra-Murphy MF, Li Y, Wachter F, Fukuda Y, Fenstermacher SJ. et al. Paclitaxel reduces axonal bclw to initiate IP3R1-dependent axon degeneration. Neuron. 2017;96:373–86. https://doi.org/10.1016/j.neuron.2017.09.034.
Baxter PS, Bell KFS, Hasel P, Kaindl AM, Fricker M, Thomson D, et al. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat Commun. 2015;6:6761. https://www.nature.com/articles/ncomms7761.
Fricker M, Papadia S, Hardingham GE, Tolkovsky AM. Implication of TAp73 in the p53-independent pathway of Puma induction and Puma-dependent apoptosis in primary cortical neurons. J Neurochem. 2010;114:772–83.
Kim TW, Moon Y, Kim K, Lee JE, Koh HC, Rhyu IJ, et al. Dissociation of progressive dopaminergic neuronal death and behavioral impairments by bax deletion in a mouse model of parkinson’s diseases. PLoS ONE. 2011;6:1–10.
Smith MI, Deshmukh M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 2007;14:1011–9. https://doi.org/10.1038/sj.cdd.4402089.
Concannon CG, Ward MW, Bonner HP, Kuroki K, Tuffy LP, Bonner CT, et al. NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER stress and PUMA independent manner. J Neurochem. 2008;105:891–903.
Uo T, Kinoshita Y, Morrison RS. Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J Neurosci. 2007;27:12198–210.
Deshmukh M, Johnson EM. Staurosporine-induced neuronal death: multiple mechanisms and methodological implications. Cell Death Differ. 2000;7:250–61.
Hertz NT, Adams EL, Weber RA, Shen RJ, O’Rourke MK, Simon DJ. et al. Neuronally enriched RUFY3 is required for caspase-mediated axon degeneration. Neuron. 2019;103:412–22.e4. https://doi.org/10.1016/j.neuron.2019.05.030.
Nikolaev A, McLaughlin T, O’Leary DDM, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–9. https://doi.org/10.1038/nature07767.
Léveillé F, Papadia S, Fricker M, Bell KFS, Soriano FX, Martel M-A, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci. 2010;30:2623–35.
Guadagno J, Xu X, Karajgikar M, Brown A, Cregan SP. Microglia-derived TNFα induces apoptosis in neural precursor cells via transcriptional activation of the Bcl-2 family member Puma. Cell Death Dis. 2013;4:e538. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3613837&tool=pmcentrez&rendertype=abstract.
Bernstein AI, O’Malley KL. MPP+-induces PUMA- and p53-dependent, but ATF3-independent cell death. Toxicology Letters 2013;219:93–8.
Acknowledgements
We would like to thank Dr. Justin Kale and Dr. Philipp Mergenthaler for their critical review and editing of this paper. Research in the Andrews’ laboratory is supported by a Canadian Institutes of Health Research Foundation grant (FDN143312). DWA holds the Tier 1 Canada Research Chair in Membrane Biogenesis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pemberton, J.M., Pogmore, J.P. & Andrews, D.W. Neuronal cell life, death, and axonal degeneration as regulated by the BCL-2 family proteins. Cell Death Differ 28, 108–122 (2021). https://doi.org/10.1038/s41418-020-00654-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00654-2
