
Abstract
Neoepitopes or neoantigens are a spectrum of unique mutations presented in a particular patient’s tumor. Neoepitope-based adoptive therapies have the potential of tumor eradication without undue damaging effect on normal tissues. In this context, methods based on the T cell receptor (TCR) engineering or chimeric antigen receptors (CARs) have shown great promise. This review focuses on the TCR-like CARs and TCR-CARs directed against tumor-derived epitopes, with a concerted view on neoepitopes. We also address the current limitations of the field to know how to harness the full benefits of this approach and thereby design a sustained and specific antitumor therapy.
Similar content being viewed by others


Introduction
Malignancies result from the accumulation of a variety of mutations and epigenetic changes. Mutations can be appeared in cell surface proteins or presented as neoepitopes by the major histocompatibility complex (MHC), making tumor cells detectable by immune cells [1]. Nonetheless, tumor cells usually escape from the immune cells and thereby gain the opportunity to develop and invade. Major mechanisms involved in immune evasion include reduced immune recognition through the loss of tumor antigens and expression of cytokines (e.g., VEGF, IL-10, TGFβ) or immunoregulatory molecules (e.g., IDO and B7 family checkpoint molecules), that lead to the induction of an immunosuppressive tumor environment and enhance tumor resistance or survival via elevated expression of STAT3 or of BCL-2 [2, 3].
Engineered T cells with selective antigen receptors are highly potent to identify and destroy tumor cells in efficient manner [3]. In this context, the main approaches include chimeric antigen receptors (CARs), T cell receptor (TCR) engineered T cells (TCR-Ts), TCR-like CARs, and TCR-CARs. CARs are chimeric molecules engineered to recognize a tumor antigen, leading to the MHC-independent activation of CAR T cell. Since most of the tumor mutations, including neoepitopes, are presented on the cell surface through MHC class I molecules, there is a limitation of target selection for CAR design. This limitation can be overcome by using approaches based on the TCRs such as TCR-like CARs, TCR-CARs [4, 5]. Unlike CARs, TCRs are not restricted to the cell surface antigens, but can detect and bind to the peptides presented by MHC molecules (pMHCs). This feature provides a wide range of potential targets for TCRs such as tumor-specific neoepitopes. Of note, redirection of TCR-based CARs on the highly tumor-specific neoepitopes can prevent “off-tumor” toxicities that are commonly associated with CAR therapies [3]. However, rearrangement of the native and transduced TCR’s αβ chains may cause autoimmune reactions because of their unknown specificity [6]. Thus, the combination of TCR and CAR features will apparently result in more benefits. TCR-like CARs comprise an extracellular single-chain variable fragment (scFv) arising from TCR-like antibodies which recognize tumor-associated pMHCs in the same way as full TCRs do [7]. This approach does not suffer from the problem of competition and rearrangement with endogenous TCRs, and possess many potential pMHC targets. In TCR-CARs, the variable domains of TCR (TCRv) recognizing pMHC are linked to intracellular domains of CARs [4].
Neoepitopes
Over the recent years, tumor-specific neoepitopes lay the path forward for the personalized immunotherapy approaches. Almost all cancers display various genetic alterations (such as single-nucleotide variants (SNVs), insertions and deletions (indel), gene fusions, frameshift mutations, and structural variants (SVs)) [8], a minority of which may result in somatic tumor mutations harboring elegant tumor specificity. These “neoepitopes” are foreign in nature and presented on MHC class I or II molecules, and in case they can provoke immune reactivities through putative mechanisms, are referred to as “neoantigens” [9, 10]. Here, we actually refer to the antigenic neoepitopes when discussing neoepitopes. Identification of different neoepitope repertoires in cancer patients who have reaped the therapeutic benefit compared to the patients with immunotherapy resistance consequences could provide beacons for an efficient choice of ideal neoepitopes for therapy. Of note, neoepitopes’ qualitative characteristics, which may result in a vigorous and sustained immunity, is of remarkable importance. In the first instance, untranslated mutations could obviously not evoke an immune response, therefore, any signs of transcript downregulation including, promoter methylation, exon skipping as well as chromatin remodeling should take into account during the characterization of prospective mutations as they might open the opportunity of immune evasion [11]. HLA loss is another well-reported mechanism of immune escape and subsequent resistance during tumor progression. Thus, it would be of high value to classify multiple HLA-binding neoepitopes as a high priority since it may prevent the resistance evolvement to neoepitope-targeted therapies due to the HLA loss [12]. The clonal fraction is the other key feature of neoepitopes that should be considered. Evidence supports the fact that effective responses to checkpoint inhibitor therapy are correlated with the burden and fraction of clonal neoepitopes [13]. By their nature, clonal neoepitopes are expressed by a higher cancerous cell fraction compared to sub-clonal neoepitopes. Moreover, every cancer cell does not express sub-clonal neoepitopes, hence, they potentially possess less chance for efficient immune control within all parts of the disease [12]. Not to mention that similarity to the self or known antigens is also worth to be noted. Due to the fact that the immune system is equipped with the capability to recognize non-native antigens, “non-self” degree of a peptide can affect the probability of efficient immune control [12]. Importantly, the vast majority of potential mutations are typically accounted for passenger rather than driver events and their loss via chromosomal instability during tumor growth could be commonly tolerated. However, mutations in cancer driver and cell survival-associated genes are considered as essential neoepitopes. According to the available evidence, multiple HLA-binding clonal neoepitopes expressing in crucial genes that could not be deleted or repressed owing to their position in the genome might be considered as the high-quality neoepitopes. Such targets could apparently serve as potential means of immune surveillance and immunotherapy approaches such as adoptive T cell therapy.
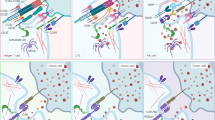
To distinguish neoepitopes, TCRv undergoes affinity maturation and subsequent selections. Therefore, in addition to neoepitopes, it is also critical to characterize neoepitope-reactive TCRv that can be used in the TCR-based therapies (Fig. 1). Neoepitope identification is technically complicated and affected by the current approaches. Using tumor and normal DNA, whole-exome sequencing (WES) is recruited to characterize tumor-specific non-synonymous mutations (NSM). When possible, RNA-seq is also employed to choose expressed mutations [14]. After the identification of NSMs, some strategies are exploited to select the list of candidate neoepitopes that will be subsequently evaluated for immunogenicity. In the WES data-based strategy, unfiltered candidate neoepitopes are listed and identified. However, there is a high success rate for tumors with a high mutation load [15]. Mass spectrometry (MS)-based immunopeptidomics is another technique which not only provides the characterization of post-translational modified peptides and non-canonical neoepitopes but also directly identifies naturally presented HLA-bound peptides [16, 17]. Nonetheless, the MS-based method is less sensitive and depends on the expression of HLA in cancerous cells. Besides, it requires a large number of samples from tumor tissue. In silico peptide prediction is also used to increase the reliability of candidate neoepitopes. However, it is not ideal for HLA class II-presented peptides, and also some peptides might be imprecisely predicted [18]. Although prompt and accurate identification of authentic neoepitopes in any given patient remains an obstacle, the technological advances and innovative screening assays might be promising for effective translation of neoepitopes targeting into more beneficial treatment strategies for cancer patients [18].

Conventional TCRs can recognize cognate pMHC complexes, but there is a need for further co-stimulatory signaling to fully activate T cells. When a mutation occurs in the tumor antigen, the previous tumor epitope-reacting TCRs have no/low affinity to the generated neoepitope. In order to recognize the neoepitope/MHC complex, the variable region of TCR undergoes affinity maturation process, and further TCR clonal selection is occurred. Neoepitope-reactive TCR variable domains (TCRv) can be further used in CAR structure. Although CAR T cells (here a 3rd generation CAR) are rapidly and strongly activated when stimulated by the target antigen, they can only target cell membrane surface proteins which are ~1% of total proteins expressed in the cell. Therefore, CARs are unable to target neoepitopes in the context of major histocompatibility complex (MHC) molecules. However, TCR-like CARs (with a scFv targeting pMHC) or TCR-CARs (with a TCRv targeting pMHC) can be developed to have the advantages of both TCRs (for targeting neoepitopes) and CARs (for rapid and robust activation).
Adoptive therapies targeting neoepitopes
Recent technological advances in genome sequencing and T cell engineering have paved the way for the development of adoptive cell therapy (ACT) targeting multiple cancer-specific mutations. Avoiding vital healthy tissue damage, neoepitope-reactive T cell administration has revealed clinical benefits for patients suffering from advanced solid tumors [19].
In this strategy, tumor specimens are used to provide tumor-infiltrating lymphocytes (TILs) containing CD8 and CD4 T cells, applying to recognize tumor-associated antigens and TCR sequences. The designed experiment of Stevanovic et al., indicated that the application of tumor-infiltrating adoptive T cell therapy in patients with papillomavirus-associated metastatic cervical cancer led to the entire cancer regression. Interestingly, mutated neoepitopes or a cancer germline antigen (KK-LC-1) caused the prominent T cell reactivities compared to the common viral antigens, suggesting a novel landscape of directing nonviral targets in immunotherapy of virally mediated cancers. Importantly, PD-1 expression was significantly observed in both viral and nonviral tumor antigen-specific T cells, indicating that antitumor T cell reactivities might be unleashed by PD-1 blockade strategies [20]. In a study of chemo-refractory metastatic breast cancer, TILs were directed against neoepitopes formed by non-synonymous mutation of four proteins: SLC3A2, KIAA0368, CADPS2, and CTSB. Administration of the neoepitope-specific TILs in combination with IL-2 and a brief course of checkpoint blockade (pembrolizumab) substantially provoked mutation-specific polyclonal responses and consequently led to the complete regression of cancer for more than 22 months [21]. Efforts are more recently underway to employ oncogenes and tumor suppressor gene mutations, so-called “hotspot” mutations, for validation of potential neoepitopes. For instance, objective tumor regression in a patient with metastatic colorectal cancer was recorded following polyclonal CD8 T cell reactivities against mutant-type K-Ras (K-RasG12D). However, the lack of expression of the HLA-C*08:02 molecule in a single lesion mediated tumor immune escape and resulted in tumor progression nine months after the treatment [22]. Furthermore, in addition to characterization of T cell responses targeting mutant-type K-Ras variants (G12D and G12V), Cafri and colleagues [23] identified CD8 memory T cells recognizing mutant-type SMAD5 and MUC4 proteins in the peripheral blood of patients with metastatic colon cancer, which could be potentially recruited to develop efficient personalized cancer immunotherapy based on neoepitope-reactive T cells. The most common mutated gene in cancer, TP53, is also an ideal candidate for assessment of targeted cancer immunotherapy due to its immunogenic potential. The most common hotspot mutations in TP53 (eight positions) were detected in 24% of common epithelial cancers (133 patients). T cell reactivities against the p53 neoepitopes presented through both intracellular and extracellular pathways were systematically and thoroughly analyzed using autologous antigen-presenting cells (APCs) expressing entire HLA class I and II molecules. This study laid the foundations for ongoing clinical trials evaluating the capability of TP53 mutation-specific TILs and TCRs to eradicate metastatic cancers [24].
Despite perceptible developments, targeting neoepitopes through adoptive therapy approaches yet is facing serious challenges regarding tumor cells and their microenvironment. Heterogeneity is a critical issue, as antigen processing and target presentation by tumor cells are variable. In other words, it must be taken into special consideration that not every “neoepitope” creates “neoantigen” that can be practically distinguished and trigger efficient T cell reactivities when presented by APCs with sufficient MHC-peptide expression [25]. In addition, infiltration and trafficking of cytotoxic T cells (CTL) into tumor is essential for their function and there are different approaches to enhance the homing of genetically modified T cells into the tumor microenvironment. For instance, the level of CXCL8/IL-8 is increased in the melanoma tumor microenvironment, and engineered MAGR-A3 TCR-T cells expressing CXCR2 have shown higher infiltration in xenograft murine models [26]. Immune checkpoint inhibitors can also increase CAR T cells’ efficacy and hinder the immune-suppressive tumor niche [27]. Besides that, the suppressive extracellular matrix (ECM) of tumors can be overcome by the expression of degrading enzymes such as Heparanase in CAR T cells [28], which can be applicable for TCR-CAR-based adoptive therapies.
TCR-like CAR and TCR-CAR
Genetically manipulation of TCRs has provided the basis of CARs development. CARs are synthetic receptors that typically contain CD3ζ as the intracellular domain, responsible for downstream signaling plus a co-stimulatory domain(s) which is commonly CD28 or 4-1BB, and an extracellular antigen-binding domain that is an scFv derived from an antibody interacting with unprocessed antigens including cell surface-expressed proteins, glycolipids, and carbohydrates. However, this recognition is in a MHC-independent manner, which limits CAR T cells’ recognition capability only to cell surface antigens (~1% of the whole cell’s expressed proteins). Nonetheless, TCRs are composed of an αβ transmembrane heterodimer and CD3 subdomains and possess the advantage of MHC-dependency. Thus, they are able to target any peptide derived from cellular protein degradation. In other words, the whole proteome can be recognized by TCRs [29]. Although TCR-T cells with defined specificity have exhibited treatment efficiency, the competition of exogenous therapeutic TCR with the endogenous TCR for CD3 signaling will augment the possibility of formation of mispaired dimers, which may subsequently lead to the unfavorable specificity and function. Importantly, TCR localization is restricted to T cells, as T cells prepare all substituents for effective TCR induction. While other immune cells like natural killers (NKs) and macrophages also can be engineered to express CAR construct [30, 31]. Moreover, compared to CARs, TCRs are contrarily characterized by almost lower affinity owing to the fact that high-affinity TCR-expressing T cells are subjected to negative selection in the thymus, as the majority of tumor antigens are “self” proteins but typically overexpressed. Accordingly, further advances in the area of immunotherapy depend in part on promoting the functional capacity of the engineered components. In this regard, the development of TCR-like CAR T cell is a novel strategy. In this approach, a new mode of epitope/MHC complex-specific antibodies (known as TCR mimic/TCR-like antibodies) are developed to bind MHC-bound targets. TCR-like antibodies were designed with almost 103–105 times stronger affinity compared to the natural TCRs. Furthermore, the CAR construct can convey its own activation signals through embedded transmembrane and intracellular domains without competition with endogenous TCR for signaling domains [5]. This approach primarily was introduced by Willemsen and coworkers in 2001 [32] when a CAR construct linked with a TCR-like antibody against MAGE-A1/HLA-A1 complex. On the other hand, affinity enhancement of TCRv through amino acid replacement can improve their MHC-bound target recognition and interaction. TCRv structure can also be linked to the intracellular signaling domains of CAR construct, generating TCR-CAR [4]. These novel structures are being improved to simultaneously represent favorable characteristics of both TCRs and CARs. Figure 2 shows a schematic illustration of the recent procedures of neoepitope characterization and development of neoepitope-reactive TCR-like antibodies or TCRv applied in TCR-like CAR or TCR-CAR platforms, respectively.

First, a tumor is surgically excised from the patient and tumor samples are used as the subject for performing whole-exome sequencing and identifying tumor-specific non-synonymous mutations that occurred in coding regions. In parallel, tumor-infiltrating lymphocytes (TILs) and tumor cell lines are also obtained from tumor samples to identify and validate neoepitope-reactive TCRs. The predicated neoepitope sequences surrounding each mutation by flanking regions of 12 codons on either side are synthesized as tandem minigenes (TMGs; constructs encoding multiple neoepitopes each encoding a specific mutation) or single long peptides with 25 amino acid lengths. TILs are co-cultured in the presence of an antigen-presenting cells (APC) expressing the TMG or pulsed with the long peptides. In parallel, the neoepitope/HLA monomers can be sued for isolation of TCR-like antibodies targeting the cognate neoepitope/HLA complex. Finally, characterized TCR variable domains (TCRv) or single-chain variable fragment (scFv) specific for neoepitope/MHC complex are used in a CAR structure to produce TCR-CARs or TCR-like CARs, respectively. The modified T cells can be further expanded and formulated in a proper buffer and infused to the patient by intravenous (IV) injection to specifically destroy tumor cells. CAR ICDs CAR intracellular domains.
TCR-based CARs against tumor epitopes/neoepitopes
In contrast to the “self” epitopes derived from shared or overexpressed antigens, neoepitopes can be potentially expressed at far higher rates and be recognized by highly tumor-specific TCRs [33]. Accordingly, TCR-like CARs and TCR-CARs against MHC molecules bound to a tumor antigen-derived epitope/neoepitope have been proven to be very efficient. For instance, Wälchli and colleagues constructed two TCR-CARs against DMF5 (from MELAN-A antigen) and Radium-1 (from a mutated form of TGFβR2). Both TCR-CARs, particularly Radium-1 TCR-CAR, stably expressed and redirected their host cell on cognate pMHC, where the engineered cell could efficiently kill target cells [4].
Historically, the β-chain was the topic of interest for the majority of researchers studying in the TCR biology field. This is apparently due to the higher capacity of β-chain in generating diversity (because of D gene component) as well as its unique expression in each single cell, where the expression of two different α chains can be observed in the same cell [34, 35]. In an interesting approach, the single variable domain of the β-chain (Vβ) targeting NY-ESO-1 and MAGE-A3 antigens were constructed and assessed in the structure of TCR-CAR and TCR. The TCR-CAR Jurkat cells carrying only Vβ domains as extracellular compartment showed a dose-dependent activation in the co-culture of serially diluted peptides and APC. This confirms the feasibility of Vβ-only TCR-CAR constructs as β-chain can ideally mimic whole TCR [36].
Recently, TCR-like antibodies targeting pMHC complexes have shown strong antitumor effects and many of these compounds are now under development in pre-clinical settings [37]. It has been previously established that the application of antibody-derived moieties as the antigen-recognition domain of CARs can substantially eradicate tumor cells with down-regulated antigen expression (~200 copies/cell) [38]. Therefore, these structures can also be used in TCR-like CAR structure. In this context, Sadelain’s group generated TCR-like CAR T cells restricted to HLA-A2/NY-ESO-1157–165. However, they found that despite the specificity of high-affinity Fab fragments in soluble form, the TCR-like CAR T cells expressing the Fab extracellular domain showed only moderate lysis of HLA-A2/NY-ESO-1157–165 expressing targets. The authors hypothesized that this might be because of the strong binding affinity of the Fab to HLA-A2 molecules. Thus, they lowered the Fab’s HLA-A2 binding affinity to TCR level by a rational mutagenesis approach, and thereby, improved the specificity and efficacy of TCR-like CAR T cells [39]. In another antibody-based TCR-like CAR design, Akahori et al., designed a TCR-like CAR with low affinity (Kd = 741 nM) against HLA-A*2402/WT1235–243 complex. The study confirmed the therapeutic efficacy of this strategy. Importantly, the authors indicated that scFvs with higher binding affinity to WT1235–243/A*2402 (Kd = 34.4 nM) had lower effector function in WT1+/A*2402+ tumor cell lines (probably with low antigen load). Therefore, they suggested an optimum binding affinity of TCR-like CARs to the pMHC as a crucial criterion for the serial triggering of the target antigen (quick release of TCR-like CAR from the pMHC) and facilitating modified T cell activation [40]. In support of this notion, Oren et al. [41], found that TCR-like CAR T cells containing a high-affinity scFv recognizing HLA-A2-WT1Db126 exhibited lower effector functions and loss of specificity compared to the engineered cells expressing low-affinity αβ-TCRs. However, this is still controversial since previous evidence suggested that high-affinity TCR-like CAR, but not the low affinity one, demonstrated specific and potent cytotoxicity [42]. This is plausibly because of the fact that scFvs with high affinities to pMHC can decrease the threshold of antigen density required for T cell activation [43]. Petrausch and colleagues developed and used one of these high-affinity scFvs recognizing NY-ESO-1157–165/HLA-A*02:01. Intriguingly, the transduced T cells had a predominant effector memory phenotype, and showed an specific antitumor activity and cytokine secretion when stimulated with NY-ESO-1157–165 [44]. Overall, it seems that the researchers should ensure the optimum binding affinity of the antigen-recognition domain prior to designing TCR-like CAR T cells.
An expected advantage of TCR-like CARs or TCR-CARs is their capability in distinguishing neoepitopes from wild-type epitopes, as TCRs are able to specifically recognize tumor neoepitopes and spare wild-type peptides [45]. Thus, triggering tumor neoepitopes via effector cells expressing TCR-like CAR or TCR-CAR is increasingly garnering considerable attention. A close example is targeting of minor histocompatibility antigens (mHAgs), known as neoantigens’ therapeutic equivalents [46]. mHAgs are a group of immunogenic peptides that are originated from polymorphic genes and presented on the cell surface in association with class I or class II MHC molecules evoking strong alloreactivity [5]. Like neoepitopes, a subset of mHAgs can be strictly expressed on the hematopoietic malignant cells introducing them as tumor-specific antigens [46]. Inaguma et al., produced TCR-like CARs by introducing scFvs targeting the mHAg HA-1H presented by HLA-A2. Similar to previous reports [40, 41], the authors showed that high-affinity scFvs (Kd = 19.9 nM) exerted lower cytotoxicity against target cells with low-density peptide/MHC complexes (~100 per cell) than the scFvs with moderate to low affinity (Kd = 446 nM) [47].
Another encouraging sign for the development of TCR-based CARs against neoepitopes is the feasibility of neoepitope-reactive TCR-T cells. In this regard, accumulating evidence suggests that TCR-Ts against neoepitopes such as those arising from K-Ras (NCT03190941) or TP53 mutations [48] have clinical benefit. Thus, it is anticipated that the benefits of neoepitope-reactive TCR-T cells will be extended to the TCR-like CAR/TCR-CAR T cells. Table 1 summarizes some examples of TCR-like CARs or TCR-CARs targeting the complex of epitope/neoepitope and HLA molecule.
Limitations and concluding remarks
Multiple groups have found that patient-derived T cell repertoire recognizing neoepitopes are remarkably efficacious for personalized cancer immunotherapy [49, 50]. Following these encouraging outcomes, a concerted effort is currently in progress to develop endogenous TCR alternatives such as TCR-like CARs or TCR-CARs to extend the TCR specificity into the CAR platform while garnering the advantages of CAR platforms [4]. To this end, current studies have mainly focused on the improvement of methodologies used for neoepitopes identification in order to precisely identify tumor-specific neoepitopes. However, some limitations might hinder the success of neoepitope-directed TCR-like CAR or TCR-CAR therapies. First, the feasibility of this approach might be restricted to the tumors which tend to have a high mutational burden such as melanoma and lung cancer, but not common epithelial tumors that have much lower mutational loads [51]. Second, during tumor recurrence or relapse, it is plausible to observe a different profile and rate of neoepitopes expression [52]. Third, neoepitopes with the highest expression level and putative binding affinity to MHC molecules are not always suitable candidates, because they might not sufficiently exert neoepitope-reactive T cell response [53]. Therefore, in parallel with neoepitopes characterization in tumor cells, there is a need to validate neoepitope-reactive TCRs. As fourth limitation, TCR-CARs, especially those with higher binding affinity to pMHC, are potent in the redundant recognition of alternative targets and may cause subsequent toxicities owing to the “off-target” cross-reactivity [54]. Fifth, the majority of neoepitopes differ from one patient to another that limits their use to solely personalized therapies. However, this is being addressed by the identification of “shared neoepitopes” observed in a large subset of patients [55]. And finally, immune pressure by TCR-like CAR or TCR-CAR T cells can drive tumor cells to activate escape pathways and modulate the expression of target antigens [3]. Consequently, the ubiquitously tumor-expressed mutations and generation of novel predominant neoepitopes following the TCR-based CAR T therapies might further enhance the complexity of this treatment modality.
Collectively, while neoepitope targeting with engineered T cells carrying TCR-based chimeric receptors is an appealing strategy, candidate neoepitope or neoepitope-targeting domains should be selected with considerable caution. Moreover, the safety and tolerability of these modified cells are still speculative and further improvements are still needed to drive impressive clinical outcomes.
Change history
24 March 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41417-021-00323-7
References
Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. 2017;8:1679.
Muenst S, Läubli H, Soysal S, Zippelius A, Tzankov A, Hoeller S. The immune system and cancer evasion strategies: therapeutic concepts. J Intern Med. 2016;279:541–62.
Poorebrahim M, Abazari MF, Sadeghi S, Mahmoudi R, Kheirollahi A, Askari H, et al. Genetically modified immune cells targeting tumor antigens. Pharmacol Therap. 2020;214:107603.
Walseng E, Köksal H, Sektioglu IM, Fåne A, Skorstad G, Kvalheim G, et al. A TCR-based chimeric antigen receptor. Sci Rep. 2017;7:1–10.
Akatsuka Y. TCR-like CAR-T cells targeting MHC-bound minor histocompatibility antigens. Front Immunol. 2020;11:257.
Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16:565–70.
Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571.
Schumacher TN, Scheper W, Kvistborg P. Cancer neoantigens. Annu Rev Immunol. 2019;37:173–200.
Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12:93.
Vormehr M, Diken M, Boegel S, Kreiter S, Sahin U. Mutanome directed cancer immunotherapy. Curr Opin Immunol. 2016;39:14–22.
Rosenthal R, Cadieux EL, Salgado R, Al Bakir M, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–85.
McGranahan N, Swanton C. Neoantigen quality, not quantity. Sci Transl Med. 2019;11:eaax7918.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J. 2018;16:15–24.
Jurtz VI, Olsen LR. Computational methods for identification of T cell neoepitopes in tumors. Methods Mol Biol. 2019;1878:157–72.
van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RC, Kester MG, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Investig. 2019;129:774–85.
Stern LJ, Santambrogio L. The melting pot of the MHC II peptidome. Curr Opin Immunol. 2016;40:70–7.
Gros A, Garcia-Garijo A, Fajardo CA. Determinants for neoantigen identification. Front Immunol. 2019;10:1392.
Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat Med. 2019;25:1488–99.
Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356:200–5.
Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24:724–30.
Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl J Med. 2016;375:2255–62.
Cafri G, Yossef R, Pasetto A, Deniger DC, Lu Y-C, Parkhurst M, et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun. 2019;10:1–9.
Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Investig. 2019;129:1109–14.
Schappert A, Schneck JP, Suarez L, Oelke M, Schütz C. Soluble MHC class I complexes for targeted immunotherapy. Life Sci. 2018;209:255–8.
Idorn M, Skadborg SK, Kellermann L, Halldorsdottir HR, Holmen Olofsson G, Met O, et al. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology. 2018;7:e1450715.
Poorebrahim M, Melief J, Pico de Coaña Y, L Wickström S, Cid-Arregui A, Kiessling R. Counteracting CAR T cell dysfunction. Oncogene. 2020;40:421–35.
Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–9.
Poorebrahim M, Sadeghi S, Fakhr E, Abazari MF, Poortahmasebi V, Kheirollahi A, et al. Production of CAR T-cells by GMP-grade lentiviral vectors: latest advances and future prospects. Crit Rev Clin Lab Sci. 2019;56:393–419.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–53.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947–53.
Willemsen R, Debets R, Hart E, Hoogenboom H, Bolhuis R, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8:1601–8.
Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med. 2019;25:1488–99.
Woodsworth DJ, Castellarin M, Holt RA. Sequence analysis of T-cell repertoires in health and disease. Genome Med. 2013;5:98.
Padovan E, Casorati G, Dellabona P, Meyer S, Brockhaus M, Lanzavecchia A. Expression of two T cell receptor alpha chains: dual receptor T cells. Science. 1993;262:422–4.
Oh J, Warshaviak DT, Mkrtichyan M, Munguia ML, Lin A, Chai F, et al. Single variable domains from the T cell receptor β chain function as mono-and bifunctional CARs and TCRs. Sci Rep. 2019;9:1–12.
He Q, Liu Z, Liu Z, Lai Y, Zhou X, Weng J. TCR-like antibodies in cancer immunotherapy. J Hematol Oncol. 2019;12:99.
Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194:911–20.
Maus MV, Plotkin J, Jakka G, Stewart-Jones G, Rivière I, Merghoub T, et al. An MHC-restricted antibody-based chimeric antigen receptor requires TCR-like affinity to maintain antigen specificity. Mol Ther Oncol. 2016;3:1–9.
Akahori Y, Wang L, Yoneyama M, Seo N, Okumura S, Miyahara Y, et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood. 2018;132:1134–45.
Oren R, Hod-Marco M, Haus-Cohen M, Thomas S, Blat D, Duvshani N, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol. 2014;193:5733–43.
Zhao Q, Ahmed M, Tassev DV, Hasan A, Kuo TY, Guo HF, et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015;29:2238–47.
Stewart-Jones G, Wadle A, Hombach A, Shenderov E, Held G, Fischer E, et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci USA. 2009;106:5784–8.
Schuberth PC, Jakka G, Jensen SM, Wadle A, Gautschi F, Haley D, et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther. 2013;20:386–95.
Parkhurst M, Gros A, Pasetto A, Prickett T, Crystal JS, Robbins P, et al. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin Cancer Res. 2017;23:2491–505.
Mutis T, Xagara A, Spaapen RM. The connection between minor H antigens and neoantigens and the missing link in their prediction. Front Immunol. 2020;11:1162.
Inaguma Y, Akahori Y, Murayama Y, Shiraishi K, Tsuzuki-Iba S, Endoh A, et al. Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specific for minor histocompatibility antigen HA-1H. Gene Ther. 2014;21:575–84.
Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-cell responses to TP53 “Hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. 2018;24:5562–73.
Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. 2018;3:e122467.
Liu S, Matsuzaki J, Wei L, Tsuji T, Battaglia S, Hu Q, et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J Immunother Cancer. 2019;7:156.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8.
Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, Tanaka S, et al. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res. 2019;7:1148–61.
Peng S, Zaretsky JM, Ng AHC, Chour W, Bethune MT, Choi J, et al. Sensitive detection and analysis of neoantigen-specific T cell populations from tumors and blood. Cell Rep. 2019;28:2728–2738.e7.
Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–71.
Lee KL, Schlom J, Hamilton DH. Combination therapies utilizing neoepitope-targeted vaccines. Cancer Immunol Immunother. 2020. ahead of print.
Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. 2017;23:478–88.
Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. 2016;18:985–94.
Acknowledgements
M.P. is supported by a postdoctoral research fellowship from Alexander von Humboldt Foundation. E.F. is the recipient of a PhD fellowship from the German Academic Exchange Service (DAAD). A.C.-A. gratefully acknowledges Prof. Harald zur Hausen for his continuous scientific support and encouragement.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poorebrahim, M., Mohammadkhani, N., Mahmoudi, R. et al. TCR-like CARs and TCR-CARs targeting neoepitopes: an emerging potential. Cancer Gene Ther 28, 581–589 (2021). https://doi.org/10.1038/s41417-021-00307-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-021-00307-7
This article is cited by
-
Computational immunogenomic approaches to predict response to cancer immunotherapies
Nature Reviews Clinical Oncology (2024)
-
Neoantigens: promising targets for cancer therapy
Signal Transduction and Targeted Therapy (2023)
-
Antibody-based binding domain fused to TCRγ chain facilitates T cell cytotoxicity for potent anti-tumor response
Oncogenesis (2023)
-
The landscape of T cell antigens for cancer immunotherapy
Nature Cancer (2023)
-
Characterizations of a neutralizing antibody broadly reactive to multiple gluten peptide:HLA-DQ2.5 complexes in the context of celiac disease
Nature Communications (2023)
