
Abstract
Immune cell infiltration into solid tumors, their movement within the tumor microenvironment (TME), and interaction with other immune cells are controlled by their directed migration towards gradients of chemokines. Dysregulated chemokine signaling in TME favors the growth of tumors, exclusion of effector immune cells, and abundance of immunosuppressive cells. Key chemokines directing the migration of immune cells into tumor tissue have been identified. In this review, we discuss well-studied chemokine receptors that regulate migration of effector and immunosuppressive immune cells in the context of cancer immunology. We discuss preclinical models that have described the role of respective chemokine receptors in immune cell migration into TME and review preclinical and clinical studies that target chemokine signaling as standalone or combination therapies.
Similar content being viewed by others


Introduction
The immune system is a dynamic and complex, yet highly organized, network of cells. Immune cells are motile and migrate to specific organs in a context-specific manner. Moreover, they need to come into spatial proximity with other cells to exchange information and function. Diverse immune responses in different situations are a result of these interactions. When they reach their destined organs, immune cells move relatively shorter distances to reach the appropriate microanatomical niche. This intra-organ movement is critical and determinative of immune outcomes [1, 2]. The inter- and intra-organ movement of immune cells is guided by a set of secreted molecules called chemokines. Immune cells that express the cognate chemokine receptor migrate based upon gradients of the respective ligands in a process called chemotaxis. Chemokines were originally discovered as inflammatory cytokines that could guide leukocytes to sites of inflammation, but it is now known that chemokines have additional roles even in the absence of inflammation [3]. For example, chemokines play a major role in the development of lymphoid organs [1, 4]. Defects in the expression of chemokines or chemokine receptors have been associated with dysfunctional lymphoid organ development and defective, aberrant, or exacerbated immune response [4,5,6,7,8,9]. Thus far, 50 chemokine ligands and 20 chemokine receptors have been described, and all but 6 chemokine receptors respond to multiple chemokines. Chemokines are grouped into four main classes depending on the location of the first two cysteine (C) residues in their protein sequence: namely, CC, CXC, C, and CX3C chemokines. Most chemokine receptors are transmembrane-spanning heterotrimeric G-protein-coupled receptors [1]. Cognate chemokine receptor binding induces G-protein coupling and subsequent activation of downstream signaling proteins involved in cell migration such as Rac, Rho, and Cdc42. The net effect is movement of the cells towards the chemotactic gradient [1].
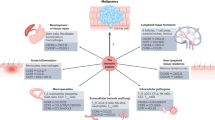
The infiltration of immune cells in the TME is a key factor in cancer prognosis, and chemokines play an essential role in guiding the migration of both activating and suppressive immune cell types [10,11,12,13]. The migration of immune cells into tumor tissue is more unpredictable than homeostatic migration of immune cells into lymphoid organs, as solid tumors are ectopic, heterogeneous, and do not have a defined anatomy. Even among tumors of the same type, the migration patterns of immune cells vary by case and time. Nonetheless, understanding the chemotactic environment of solid tumors and identifying chemokines that regulate immune cell entry into solid tumors is imperative in improving current immunotherapeutic interventions, including immune checkpoint blockade (ICB) (Fig. 1). Chemokines also play a role in the intrinsic growth and metastasis of tumor cells, mechanisms of which have been reviewed previously [14]. In the current review, we discuss chemokines that can guide anti-tumor effector and immunosuppressive immune cells into solid tumors and how these chemotactic axes have been targeted to develop immunotherapies.

Illustration depicts chemokines that have been commonly found to guide different types of immune cells into solid TME.
Chemokines recruit effector anti-tumor immune cells to the TME
CXCR3 and CXCR4 direct the migration of T cells and NK cells in solid tumors
Effector immune cells such as activated CD8+ T cells and NK cells can recognize and lyse tumor cells [15, 16]. In addition to their cytotoxic function, they secrete the major stimulatory cytokine interferon gamma (IFNγ) to induce and maintain interferon-induced anti-tumor immunity [17]. To mediate anti-tumor effects, effector cells must first enter the tumor by engaging chemokine receptors expressed on their cell surfaces. Lack of intratumoral T cell infiltration is associated with resistance to widely applied therapies including ICB [18]. Upon activation, T cells and NK cells upregulate expression of the chemokine receptor, C–X–C chemokine receptor (CXCR) 3. CXCR3-expressing cells follow gradients of the interferon-inducible ligands, C–X–C chemokine ligand (CXCL) 9 (MIG), CXCL10 (IP-10), and CXCL11 (I-TAC). CXCR3 guides the migration of NK cells into lymph nodes (LNs) and tumors [19, 20]. CXCR3-dependent T cell infiltration has been demonstrated in murine models of lymphoma, renal cell carcinoma, melanoma, and breast cancer [21,22,23,24,25], and is a prerequisite to the success of programmed cell death protein-1 (PD-1)/programmed cell death protein ligand 1 (PD-L1) blockade therapy [26]. In line with the above murine models, increased expression of CXCL9 and CXCL10 has been associated with increased infiltration of activated T cells in many human cancers, including melanoma, ovarian, and colon cancer [27,28,29,30]. Indeed, a study correlating chemokine expression and CD8+ T cell infiltration in human solid tumors showed that chemokine C–C motif ligand (CCL) 5 and CXCL9 had the highest correlation with CD8+ T cell infiltration across different cancer types and also demonstrated cooperation between CXCL9/10 and CCL5 in recruiting effector T cells into tumors [31].
IFNγ is the major driver of CXCR3–CXCL9/10/11 axis. CXCR3 is inducible by IFNγ; CXCL9 is inducible by IFNγ but not by IFNα/β (type-1 interferon); CXCL10 is inducible by both IFNγ and IFNα/β and tumor necrosis factor alpha [32,33,34,35,36]. Indeed, systemic administration of IFNγ increased levels of CXCL10 and intratumoral T cell infiltration in a phase 0 clinical trial for sarcoma, and intratumoral injection of IFNγ increased tumor CXCL10 and CXCL11 in melanoma patients [37, 38]. Strategies to directly increase the level of CXCR3 ligands have been described in preclinical models. These include plasmid-borne CXCL9 [39], intra-tumor injection of CXCL9 [22], recombinant CXCL10 protein with adoptive cell therapy (ACT) [40], intra-tumor injection of CXCL10 [41], retroviral transduction tumor cells with CXCL10 [42, 43], and intraperitoneal injection of oncolytic vaccinia virus expressing CXCL11 [44]. All the above strategies were effective in increasing T cell infiltration and reducing tumor growth in animal models but have not yet been investigated in clinical trials. Several challenges in taking the above therapeutics into the clinic can be envisaged. Injection of naked protein cannot guarantee their lasting bioavailability. Gene therapy can overcome this but would rely on site-specific and high transduction rate within the tumor, which would be difficult to achieve and evaluate.
Alternate interventions including ICB indirectly modulate levels of CXCR3 ligands, presumably through increased production of IFNγ from activated T cells. PD-1 blockade increased tumor expression of CXCL10 in melanoma-bearing mice treated with ACT [45]. Higher levels of CXCL9, CXCL10, and CXCL11 were also detected in melanoma patients’ tumors following treatment with ipilimumab, an antagonistic antibody against cytotoxic T-lymphocyte-associated protein 4 (CTLA4), another inhibitory receptor expressed by T cells [46]. Interleukin-7 increased the expression of CXCL9 and CXCL10 in a murine lung cancer model [47]. Cyclooxygenase-2 (COX-2) is expressed by many cancer types and confers resistance to chemo- and radiotherapy [48]. The COX-2 inhibitor indomethacin enhanced IFNγ-induced expression of CXCL9/10 by ovarian cancer cell lines. In the same study, expression of COX-2 was also shown to be negatively correlated with CXCL10 expression [49]. Indomethacin and acetylsalicylic acid (aspirin), alternative COX inhibitors, similarly enhanced IFN-γ-mediated CXCL9 and CXCL10 in breast cancer cell lines [50]. Celecoxib, another COX-2 inhibitor, together with anti-PD-1 synergistically increased the production of CXCL9/10 [51]. Therapeutics that target toll-like receptors also induced expression of CXCL9-11 at the tumor site [52, 53]. Thus, various immunotherapies may rely on IFNγ-stimulated CXCR3/CXCL9-11 signaling and associated effector cell recruitment for maximal therapeutic effect.
CXCR4 is important for homing of naïve and memory CD8+ T cells and NK cells into bone marrow (BM) [54, 55]. CXCR4 also regulates homeostatic proliferation and memory maintenance of T cells, and the development of NK cells [56, 57]. CXCR4 can guide NK cells to tumors. NK cells genetically engineered with a chimeric antigen receptor (CAR) to over-express CXCR4 along with the target antigen receptor demonstrated enhanced mobilization towards CXCL12 (stromal cell-derived factor 1, SDF-1), the exclusive cognate ligand for CXCR4, and increased infiltration in a mouse model of glioblastoma [58]. However, CXCR4 can also prevent adequate infiltration of T cells into tumors, as the surrounding CXCL12-rich stroma can arrest CXCR4-expressing T cells from directly reaching carcinoma cells [59]. Additionally, a phenomenon where high concentration of chemokines repelled cells expressing the cognate receptor was demonstrated for CXCR4/CXCL12 axis in a murine melanoma model. This was termed chemorepulsion or fugetaxis. Tumor expressing high levels of CXCL12 repelled CXCR4-expressing T cells and evaded an anti-tumor response [60].
Pharmacological agents targeting the CXCR4/CXCL12 axis were developed to inhibit tumor growth and metastasis [61] and also mobilize hematopoietic stem cells to the periphery for subsequent transplantation [62]. AMD3100 is a small-molecule CXCR4 antagonist commonly used in the clinic in the latter context [63]. Two independent studies demonstrated that AMD3100 promotes T cell infiltration into the tumor, by releasing them from the CXCL12-rich surrounding stroma [59, 64]. Another CXCR4 antagonist, BL-8040, released T cells from the BM to infiltrate tumors in a mouse lung tumor model [65]. Moreover, in a phase I/II trial of patients with chemotherapy-refractory pancreatic ductal adenocarcinoma (PDA), administration of the CXCR4 inhibitor BL-8040 with a PD-1 blocking antibody led to increased effector T cell infiltration, as well as substantial disease control rates when combined with systemic chemotherapy [66]. Thus, targeting the CXCR4/CXCL12 axis can optimize intratumoral T cell localization, promote T cell anti-tumor activity, and enhance the efficacy of ICB and cytotoxic chemotherapy.
CCR5 and CCR6 orchestrate dendritic cell migration into tumors for maximal anti-tumor immunity
Antigen-presenting cells (APCs) are crucial to anti-tumor immunity [67]. Dendritic cells (DCs) are the most proficient at antigen presentation, and a particular subset of DCs, i.e., cDC1s, are specialized at cross-priming CD8+ T cells and have been shown to play a non-redundant role in eliciting anti-tumor immune responses [68, 69]. Moreover, tumor-infiltrating DCs, especially CD103+ DCs, boost the infiltration of effector T cells into the tumor by producing CXCL9/10 and hence recruiting CXCR3+ T cells [70]. The mechanisms of intratumoral DC migration, and the chemokines involved, have been less extensively studied in comparison to T cells. However, chemokine receptors involved in DC migration in other disease settings have been described previously [71]. A few reports have implicated the role of CCR5 and CCR6 in the recruitment of DCs into the TME. CCR5 is a key receptor through which HIV enters target cells, but it also mediates physiologic functions of immune cells [72, 73]. CCR5 expression by DCs was required for their response in a parasite infection model [74]. CCR5 was also elevated in DCs in multiple sclerosis and acute monosymptomatic optic neuritis patients [75]. CCR5 expression on DCs has not been clearly demonstrated in tumor models or patients. However, in a genetically engineered mouse model of melanoma, defective expression of CCL4 (macrophage inflammatory protein-1-beta), a ligand for CCR5, led to reduced DC recruitment to tumors [76]. Using the same model, another study showed that CCL5 (RANTES), another ligand for CCR5, was crucial for DC recruitment into tumors. In this study, NK cell infiltration was required for production of CCL5 and subsequent recruitment of DCs [77]. Such NK cell-mediated recruitment of DCs was also demonstrated in a murine colon tumor model [78]. The above two studies suggest that immune cells can sequentially enter tumors and guide the migration of additional incoming cells by specific chemokine secretion.
CCR6 guides DCs into sites of inflammation [79, 80]. The expression of CCL20, a ligand for CCR6, is associated with higher infiltration of immature DCs in breast carcinoma patients [81]. Furumoto et al. [82] showed that CCL20 increased DC infiltration into murine melanomas by using either CCL20-transduced tumor cells or intratumoral CCL20 protein injections.
In the anti-tumor immunity cycle, DCs have to migrate into the tumor, capture tumor antigens, then exit the tumor and migrate to the draining LN to prime naïve tumor-specific T cells [67]. This means that unlike other immune cells, DCs must travel into and egress out of the tumor. Activation-induced CCR7 expression on DCs is crucial for their migration from peripheral tissues into the LN following the gradients of cognate ligands CCL19/21 [5]. The level of CCR7 expression correlates with their capacity to migrate into LNs and also from the LN periphery to the deep T cell zone of the LN [83]. It is reasonable to hypothesize that once DCs are in the tumor parenchyma, they use the same CCR7-dependent migration to make their way into the draining LN; however, this has not been demonstrated. Whether intratumoral DCs can access terminal lymphatics in tumors for their CCR7-dependent migration towards the draining LNs needs remains to be investigated. DCs are crucial to anti-tumor immunity, but much works remains to be done to identify the dominant chemokine(s) affecting their migration to and from the TME.
Chemokines recruit immunosuppressive cells into the TME
CCR4 regulates migration of regulatory T cells into TME
Regulatory T cells (Tregs) are a specialized subset of CD4+ T cells that are highly immunosuppressive and play a crucial role in maintaining immune tolerance during homeostasis and suppressing exacerbated immune responses in various pathological conditions [84]. In cancer models, they have been shown to suppress the anti-tumor immune response and promote tumor growth [85]. CCR4 expression has consistently been shown to mediate Treg migration in various cancer models. Primary tumor cells and tumor-associated macrophages (TAMs) from ovarian tumors produced CCL22, a ligand for CCR4, which was crucial for the migration Tregs, whose CCR4 expression was higher than other CD4+ T cells [86]. The group of Christine Ménétrier-Caux in two different studies demonstrated CCL22-mediated recruitment of Tregs in breast cancer [87, 88]. Elevated expression CCL22 and CCL17, an alternate ligand for CCR4, was associated with higher Treg infiltration in gastric cancer and esophageal squamous cell carcinoma [89, 90].
Several strategies to abrogate CCR4–CCL22/17 axis-mediated Treg recruitment, including blocking antibodies, siRNAs, and antagonists, have been effective in preclinical cancer models [91,92,93,94]. Many therapeutics targeting this axis are currently being investigated in clinical trials. For example, mogamulizumab (KW-0761) is a humanized, glycoengineered IgG1κ monoclonal antibody that targets CCR4-expressing cells by antibody-dependent, cell-mediated cytotoxicity (ADCC) [95]. Although it was originally developed to target malignant T cells in lymphomas and leukemias, clinical trial results show that it abrogates Treg accumulation in lymphomas [96, 97]. Objective responses were noted for 13 of 37 lymphoma patients (35%; 95% CI, 20–53%), including five patients (14%) with complete response; however, it is difficult to ascertain whether the effect was caused by depletion of Tregs or antibody-dependent cellular toxicity to CCR4-expressing T cell lymphomas [97]. A clinical trial of mogamulizumab in lung and esophageal cancer patients demonstrated that the antibody was well tolerated and led to efficient depletion of Tregs [98]. Allosteric antagonists that bind to the intracellular domain of CCR4 (class II antagonists) have been developed by pharmaceutical companies, including GlaxoSmithKline (GSK2239633) and AstraZeneca (AZD-1678, AZD-2098) [99, 100]. RAPT pharmaceuticals have developed small-molecule inhibitors of CCR4, including FLX475 and RPT193. Phase 1/2 trials with FLX475 in combination with pembrolizumab for advanced or metastatic cancer have been recently completed but not yet reported (NCT03674567). The full therapeutic potential of CCR4 inhibition and Treg depletion remains to be defined.
CCR2 and CCR5 regulate the migration of tumor-associated macrophages and myeloid-derived suppressor cells
Monocytes are pliable cells that develop in the BM but can further differentiate in peripheral tissues. Depending on the environment, they differentiate into immunostimulatory or immunosuppressive cells [101]. Monocytes bear resemblance to DC-precursors in ontogeny and can function as inflammatory APCs. However, in the context of cancer, monocyte-derived TAMs have frequently been shown to contribute to tumor progression and are associated with poor clinical prognosis [102, 103]. Myeloid-derived suppressor cells (MDSCs) are closely related to monocytes but are believed to arise from precursors distinct from monocytes [104, 105]. Owing to their close resemblance with monocyte-derived macrophages, MDSCs are often described interchangeably with TAMs, especially in the context of human tumors, and have similar immunosuppressive functions. In homeostatic conditions, CCR2 is exclusively expressed by myeloid cells, especially monocytes, and guides migration from BM to peripheral sites during both homeostatic and inflammatory conditions [106]. CCR2 is pivotal for the migration of monocytes and MDSCs into solid tumors [107]. Tumors in Ccr2 knockout mice are minimally infiltrated by MDSCs, which results in reduced tumor growth and metastasis [108,109,110]. Once myeloid cells infiltrate the tumor, they can further produce the cognate ligand CCL2 and maintain or even augment monocyte trafficking into tumors [111, 112]. Generally, tumor cells and tumor-associated stroma are rich sources of CCL2, the ligand for CCR2 [113, 114]. Elevated CCL2 expression in tumors was shown to recruit high numbers of monocyte or monocyte-derived cells in mouse models of glioma, renal tumors, lung cancer, prostate cancer, and melanoma [115,116,117,118,119,120]. High levels of CCL2 correlated with increased monocyte/macrophage recruitment and was an indicator of adverse prognosis in patients with breast, ovarian, gastric, and esophageal carcinomas [108, 121,122,123,124]. A study compared the levels of CCL2 in tumor tissue and adjacent healthy tissues from patients with breast, gastric, and ovarian cancer, and consistently observed an increased level of CCL2 in tumor samples [125].
Therapeutics that disrupt the CCR2/CCL2 axis have been effective in blocking macrophage infiltration and reducing tumor growth and metastasis in preclinical models of cancer, with variable activity demonstrated in early phase clinical trials. The therapeutic benefit of anti-CCL2 antibodies was seen in a mouse model of renal cell carcinoma [126]. Inhibition of CCR2/CCL2 with a CCL2 blocking antibody inhibited the infiltration of monocytes and metastatic seeding of breast cancer cells in mice [127]. In a phase II clinical trial for patients with cancer metastatic to the bone, an anti-CCR2 antibody (MLN1202) caused considerable reduction in urinary N-telopeptide (uNTX) values after 43 days of treatment in for 14 out of 43 patients with bone metastasis [128] (ClinicalTrials.gov ID: NCT01015560). A CCR2 inhibitor (PF-04136309) reduced TAM infiltration and tumor growth in a syngeneic PDA tumor mouse model [129]. A phase 1b trail of PF-04136309 in combination with nab-paclitaxel/gemcitabine in metastatic PDA patients demonstrated a decrease in inflammatory monocytes (IM) in the peripheral blood, without accumulation in the bone marrow. Unfortunately, CCR2 inhibition also caused increased pulmonary toxicity compared with standard chemotherapy [130]. In another phase 1b trial, a combination of PF-04136309 and FOLFIRINOX, a different standard chemotherapy regimen, was safe, reduced monocyte egress from BM, TAM infiltrate in the primary tumor, and resulted in a higher than expected objective response rate (49%) in patients with borderline resectable and locally advanced pancreatic cancer [131]. Another CCR2 antagonist, CCX872, in combination with FOLFIRINOX was safe and increased overall survival compared to FOLFIRINOX alone (29% versus 18.6% at 18 months) in pancreatic cancer patients [132]. CCR2 is the most common target of therapeutics developed to block monocyte infiltration into tumors.
Although the CCR2/CCL2 axis has been consistently found to mediate the recruitment of TAMs/MDSCs, other chemokines and receptors have also been shown to contribute to the process. In a transgenic mouse melanoma model, CCR5-expressing MDSCs induced immunosuppression in tumors. The same study also showed an enrichment of CCR5+ MDSCs and increased concentration of the cognate ligands, i.e., CCL3, CCL4, and CCL5, in melanoma specimens compared to matched serum [133]. In mice injected with TRAMP prostate cancer cell line, CCR5 guided CD11b+Ly6G+Ly6Clow polymorphonuclear granulocytic MDSCs from BM to tumors where they induced immunosuppression. The cognate ligands produced in the tumor, i.e., CCL3/4/5, not only recruited MDSCs but also induced the proliferation of MDSC-progenitors in the BM [134]. In the above two studies, it was shown that MDSCs that expressed CCR5 were more immunosuppressive than their CCR5− counterparts. In a mouse model of Her2-driven breast cancer, CCL5 over-expression promoted tumor recurrence by recruiting CCR5-expressing macrophages [135]. Elevated CCL5 expression is also associated with disease progression in breast [136], ovarian [137], gastric [138], and pancreatic cancer [139]. It remains to be determined what contextual factors drive CCR5-mediated suppressive versus stimulatory myeloid infiltration into the TME.
Inhibition of the CCR5 axis has been tested in preclinical models and early phase clinical trials. Maraviroc, a CCR5 antagonist, repolarized macrophages to an immune-active phenotype in organotypic explants derived from patients with metastatic colorectal cancer (CRC). In the same study, it was reported that a clinical trial of Maraviroc in patients with metastatic CRC demonstrated partial responses in patients with previously treatment-refractory disease. The effects included marked reductions in key cytokines and growth factors promoting tumor growth, chemotherapy resistance, or angiogenesis [140]. CCR5 expression on many different types of cancer cells promotes metastatic phenotype, metabolic and cell survival pathways, angiogenesis, and DNA repair [73]. Thus, CCR5 antagonists, developed for HIV treatment, have also been tested as cancer therapeutics. Some of these antagonists could inhibit tumor growth through dual mechanisms, i.e., inhibiting intrinsic tumor cell activity and reducing suppressive macrophage infiltration in the tumor. For example, Met-CCL5, an antagonist of CCR5, reduced the frequency of infiltrating macrophages in a murine model of breast cancer [141]. mCCR5–Ig fusion antibody, which sequesters CCR5 ligands, reduced MDSCs and tumor growth in a transgenic melanoma model [133]. Maraviroc inhibited TAM infiltration and tumor growth in mice inoculated with BM1 tumor cells [142]. Because CCR5 expression can guide both immunosuppressive or immune-stimulatory myeloid cells, its effects on both should be carefully considered before targeting this receptor.
CXCR1 and CXCR2 direct migration of neutrophils
Neutrophils, immune cells of the myeloid lineage, act as first responders to inflammation. At inflammatory sites, neutrophils play a role in phagocytosis, intracellular lysis of pathogens, and wound healing. Neutrophil infiltration has been observed in many cancer types and implicated in both anti- and pro-tumor roles [143]. Tumor-associated neutrophil have been generally classified into “N1” versus “N2” depending on if they exhibit anti-tumor or pro-tumor features respectively [144]. However the consensus leans towards the pro-tumorigenic role of neutrophils and many lines of evidence show neutrophils contribute to neovascularization in tumors, much akin to their role in wound healing, and thus promote tumor growth and metastasis [145]. Neutrophils express CXCR1 and CXCR2, which guide them towards their dominant ligand, CXCL8, as well as alternate ligands CXCL1, CXCL2, CXCL5, and CXCL6 [146]. CXCL8 expression is elevated in many cancers, including melanoma, colon, lung, prostate, and ovarian carcinoma [146]. CXCL5 and CXCL2 levels were high in tumor cells and tumor-associated stroma in a mouse model of PDA. Genetic ablation of CXCR2 resulted in lower neutrophil infiltration and suppression of tumor growth [147]. CXCR1–CXCL8 axis was essential in recruiting neutrophils to the tumor site in a zebrafish model of glioblastoma [148]. CXCL1 and CXCL2 chemokine gradients in melanoma-bearing mice induced neutrophil recruitment and tumor angiogenesis [149]. Hepatocellular carcinomas (HCC) have high expression of CXCL5, which attracts neutrophils, and is an indicator of poor prognosis [150]. CXCL6 over-expression in a mouse model of melanoma resulted in high neutrophil infiltration and increased angiogenesis [151]. Even in reports where neutrophils have been shown to exert an anti-tumor effect, CXCL5 and CXCL8 were shown to positively correlate with neutrophil infiltration [152].
Therapeutic agents that disrupt CXCR1/CXCR2 receptor binding with cognate ligands have been efficacious in suppressing tumor growth as shown in preclinical studies. Reparixin, a clinical grade CXCR1/2 inhibitor, suppresses breast cancer growth in vitro [153]. Small-molecule antagonists of CXCR1/2, including SCH479833 and SCH527123, exerted anti-tumor activity in xenograft models of breast cancer, CRC, spontaneous colon cancer liver metastasis, and melanoma [154,155,156,157]. ABX-CXCL8, a CXCL8 blocking antibody, inhibited melanoma growth in a xenograft model [157]. However, in the above studies the effect of therapeutics on migration of neutrophils was not demonstrated and remains to be evaluated in the clinical setting.
Concluding remarks
The bioavailability of chemokines in a solid tumor microenvironment critically influences the immune cell composition therein. However, research to understand the regulation of chemokine expression in tumors can be challenging due to heterogeneity of solid tumors and time-dependent changes in chemokine expression. More progress has been made on our understanding of the relevant chemokine receptors guiding immune cell migration into the TME. Although multiple chemokine receptors have been implicated in the migration of each immune cell type into the TME, one or two dominant receptors for each cell type have been consistently found to play a role for each cell type across various preclinical models (Table 1). While some strategies targeting these receptors have shown preliminary success in clinical trials, either alone or in combination with other interventions such as ICB and ACT, many strategies effective in mouse models, e.g., intratumoral injection of CXCR3 ligands, remain to be studied in patients. Therapies that block chemokine signaling could be easier to develop and monitor than strategies to constitutively increase chemokine expression in tumors. Pharmacological agents developed for other purposes have also been shown to manipulate immune cell trafficking into cancer. For example, antagonists to CXCR4, CCR2, CCR5, and CXCR1/2 were developed to disrupt tumor invasiveness, but now have been observed to influence immune cell migration in the TME. Targeting chemokines and receptors to improve cancer treatment can also be challenging, as expression is not exclusive to one immune cell type, and thus inhibiting one could have pleiotropic effects on several types of immune cells. Thus, the selection of candidate chemokine axes to target requires consideration of collateral effects. Biomarker assays to measure chemokine levels, receptor expression on cancer cells and immune cells, and immune cell infiltrate will be necessary to identify ideal pathways to target in an individual patient’s tumor. Understanding and manipulating intratumoral chemokine signaling offers a promising avenue to improve responses to current immunotherapies.
References
Schulz O, Hammerschmidt SI, Moschovakis GL, Förster R. Chemokines and chemokine receptors in lymphoid tissue dynamics. Annu Rev Immunol. 2016;34:203–42.
Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702.
Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992;307:97–101.
Eckert N, Permanyer M, Yu K, Werth K, Förster R. Chemokines and other mediators in the development and functional organization of lymph nodes. Immunol Rev. 2019;289:62–83.
Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8:362–71.
Müller G, Lipp M. Shaping up adaptive immunity: the impact of CCR7 and CXCR5 on lymphocyte trafficking. Microcirculation. 2003;10:325–34.
Kunkel SL, Godessart N. Chemokines in autoimmunity: from pathology to therapeutics. Autoimmun Rev. 2002;1:313–20.
Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7. PMID: 25635046.
Viola A, Luster AD. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharm Toxicol. 2008;48:171–97.
Ladányi A. Prognostic and predictive significance of immune cells infiltrating cutaneous melanoma. Pigment Cell Melanoma Res. 2015;28:490–500.
Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. 2017;117:451–60.
Di Caro G, Marchesi F, Laghi L, Grizzi F. Immune cells: plastic players along colorectal cancer progression. J Cell Mol Med. 2013;17:1088–95.
Burugu S, Asleh-Aburaya K, Nielsen TO. Immune infiltrates in the breast cancer microenvironment: detection, characterization and clinical implication. Breast Cancer. 2017;24:3–15.
Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. 2014;2:1125–31.
Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234:8509–21.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–36.
Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. 2018;9:847.
Georganaki M, van Hooren L, Dimberg A. Vascular targeting to increase the efficiency of immune checkpoint blockade in cancer. Front Immunol. 2018;9. PMID: 30627131.
Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-γ and CXCR3 ligands. Cancer Res. 2008;68:8437–45.
Martín-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat Immunol. 2004;5:1260–5.
Hensbergen PJ, Wijnands PG, Schreurs MW, Scheper RJ, Willemze R, Tensen CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in vivo involving attraction of CD8+ T lymphocytes but not inhibition of angiogenesis. J Immunother. 2005;28:343–51.
Pan J, Burdick MD, Belperio JA, Xue YY, Gerard C, Sharma S, et al. CXCR3/CXCR3 ligand biological axis impairs RENCA tumor growth by a mechanism of immunoangiostasis. J Immunol. 2006;176:1456–64.
Reckamp KL, Figlin RA, Moldawer N, Pantuck AJ, Belldegrun AS, Burdick MD, et al. Expression of CXCR3 on mononuclear cells and CXCR3 ligands in patients with metastatic renal cell carcinoma in response to systemic IL-2 therapy. J Immunother. 2007;30:417–24.
Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458.
Chheda ZS, Sharma RK, Jala VR, Luster AD, Haribabu B. Chemoattractant receptors BLT1 and CXCR3 tegulate sntitumor immunity by gacilitating CD8+ T cell migration into tumors. J Immunol. 2016;197:2016.
Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50:1498–512.e5.
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85.
Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138:1429–40.
Mullins IM, Slingluff CL, Lee JK, Garbee CF, Shu J, Anderson SG, et al. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res. 2004;64:7697–701.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl J Med. 2003;348:203–13.
Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell. 2019;35:885–900.e10.
Farber JM. Mig and IP‐10: CXC chemokines that target lymphocytes. J Leukoc Biol. 1997;61:246–57.
Qian C, An H, Yu Y, Liu S, Cao X. TLR agonists induce regulatory dendritic cells to recruit Th1 cells via preferential IP-10 secretion and inhibit Th1 proliferation. Blood. 2007;109:3308–15.
Ohmori Y, Wyner L, Narumi S, Armstrong D, Stoler M, Hamilton TA. Tumor necrosis factor-alpha induces cell type and tissue-specific expression of chemoattractant cytokines in vivo. Am J Pathol. 1993;142:861–70.
Rani MS, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM. Characterization of β-R1, a gene that is selectively induced by interferon β (IFN-β) compared with IFN-α. J Biol Chem 1996;271:22878–84.
Nakajima C, Mukai T, Yamaguchi N, Morimoto Y, Park WR, Iwasaki M, et al. Induction of the chemokine receptor CXCR3 on TCR‐stimulated T cells: dependence on the release from persistent TCR‐triggering and requirement for IFN‐γ stimulation. Eur J Immunol. 2002;32:1792–801.
Zhang S, Kohli K, Black RG, Yao L, Spadinger SM, He Q, et al. Systemic Interferon-gamma increases MHC Class I expression and T-cell infiltration in cold tumors: results of a phase 0 clinical trial. Cancer Immunol Res. 2019;7:1237–43.
Mauldin IS, Wages NA, Stowman AM, Wang E, Smolkin ME, Olson WC, et al. Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases. Cancer Immunol Immunother. 2016;65:1189–99.
Zhang R, Tian L, Chen LJ, Xiao F, Hou JM, Zhao X, et al. Combination of MIG (CXCL9) chemokine gene therapy with low-dose cisplatin improves therapeutic efficacy against murine carcinoma. Gene Ther. 2006;13:1263–71.
Wang X, Lu XL, Zhao HY, Zhang FC, Jiang XB. A novel recombinant protein of IP10-EGFRvIIIscFv and CD8(+) cytotoxic T lymphocytes synergistically inhibits the growth of implanted glioma in mice. Cancer Immunol Immunother. 2013;62:1261–72.
Arenberg DA, White ES, Burdick MD, Strom SR, Strieter RM. Improved survival in tumor-bearing SCID mice treated with interferon-gamma-inducible protein 10 (IP-10/CXCL10). Cancer Immunol Immunother. 2001;50:533–8.
Feldman AL, Friedl J, Lans TE, Libutti SK, Lorang D, Miller MS, et al. Retroviral gene transfer of interferon-inducible protein 10 inhibits growth of human melanoma xenografts. Int J Cancer. 2002;99:149–53.
Sun Y, Finger C, Alvarez-Vallina L, Cichutek K, Buchholz CJ. Chronic gene delivery of interferon-inducible protein 10 through replication-competent retrovirus vectors suppresses tumor growth. Cancer Gene Ther. 2005;12:900–12.
Liu Z, Ravindranathan R, Li J, Kalinski P, Guo ZS, Bartlett DL. CXCL11-armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. 2016;5:e1091554.
Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Res. 2012;72:5209–18.
Ji R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–31.
Andersson A, Srivastava MK, Harris-White M, Huang M, Zhu L, Elashoff D, et al. Role of CXCR3 ligands in IL-7/IL-7R alpha-Fc-mediated antitumor activity in lung cancer. Clin Cancer Res. 2011;17:3660–72.
Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234:5683–99.
Bronger H, Singer J, Windmüller C, Reuning U, Zech D, Delbridge C, et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer. 2016;115:553–63.
Bronger H, Kraeft S, Schwarz-Boeger U, Cerny C, Stöckel A, Avril S, et al. Modulation of CXCR3 ligand secretion by prostaglandin E2 and cyclooxygenase inhibitors in human breast cancer. Breast Cancer Res. 2012;14:R30.
Li Y, Fang M, Zhang J, Wang J, Song Y, Shi J, et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. OncoImmunology 2016;5:e1074374.
Soong R-S, Song L, Trieu J, Knoff J, He L, Tsai Y-C, et al. Toll-like receptor agonist imiquimod facilitates antigen-specific CD8+ T-cell accumulation in the genital tract leading to tumor control through IFNγ. Clin Cancer Res. 2014;20:5456–67.
Brackett CM, Kojouharov B, Veith J, Greene KF, Burdelya LG, Gollnick SO, et al. Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induces immunity by stimulating an NK-dendritic-CD8+ T-cell axis. Proc Natl Acad Sci USA 2016;113:E874–E83.
Goedhart M, Gessel S, van der Voort R, Slot E, Lucas B, Gielen E, et al. CXCR4, but not CXCR3, drives CD8(+) T-cell entry into and migration through the murine bone marrow. Eur J Immunol. 2019;49:576–89.
Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mrna transfection with gain-of-function variant CXCR4R334X. Front Immunol. 2019;10. PMID: 31231387.
Chaix J, Nish SA, Lin W-HW, Rothman NJ, Ding L, Wherry EJ, et al. Cutting edge: CXCR4 is critical for CD8+ memory T cell homeostatic self-renewal but not rechallenge self-renewal. J Immunol. 2014;193:1013–6.
Noda M, Omatsu Y, Sugiyama T, Oishi S, Fujii N, Nagasawa T. CXCL12-CXCR4 chemokine signaling is essential for NK-cell development in adult mice. Blood. 2011;117:451–8.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 2015;38:197–210.
Seo YD, Jiang X, Sullivan KM, Jalikis FG, Smythe KS, Abbasi A, et al. Mobilization of CD8+ T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic cancer. Clin Cancer Res. 2019;25:clincanres.0081.2019.
Vianello F, Papeta N, Chen T, Kraft P, White N, Hart WK, et al. Murine B16 melanomas expressing high levels of the chemokine stromal-derived factor-1/CXCL12 induce tumor-specific T cell chemorepulsion and escape from immune control. J Immunol. 2006;176:2902–14.
Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EGE, et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219–30.
Fiorina P, Jurewicz M, Vergani A, Petrelli A, Carvello M, D’Addio F, et al. Targeting the CXCR4-CXCL12 axis mobilizes autologous hematopoietic stem cells and prolongs islet allograft survival via programmed death ligand 1. J Immunol. 2011;186:121–31.
Susek KH, Karvouni M, Alici E, Lundqvist A. The role of CXC chemokine receptors 1-4 on immune cells in the tumor microenvironment. Front Immunol. 2018;9:2159.
Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7.
Gaur P, Verma V, Gupta S, Sorani E, Vainstein Haras A, Oberkovitz G, et al. CXCR4 antagonist (BL-8040) to enhance antitumor effects by increasing tumor infiltration of antigen-specific effector T-cells. J Clin Oncol. 2018;36:73.
Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat. Med. 2020;26.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39:1–10.
Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L, et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J Immunother Cancer. 2018;6:139.
Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014;74:705–15.
Pfirschke C, Siwicki M, Liao H-W, Pittet MJ. Tumor microenvironment: no effector T cells without dendritic cells. Cancer Cell. 2017;31:614–5.
Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30–48.
Vangelista L, Vento S. The expanding therapeutic perspective of CCR5 blockade. Front Immunol. 2018;8. PMID: 29375583.
Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 2019;79:4801–7.
Aliberti J, Reis e Sousa C, Schito M, Hieny S, Wells T, Huffnagle GB, et al. CCR5 provides a signal for microbial induced production of IL-12 by CD8α+ dendritic cells. Nat Immunol. 2000;1:83–7.
Pashenkov M, Teleshova N, Kouwenhoven M, Kostulas V, Huang Y-M, Söderström M, et al. Elevated expression of CCR5 by myeloid (CD11c+) blood dendritic cells in multiple sclerosis and acute optic neuritis. Clin Exp Immunol. 2002;127:519–26.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–37.e14.
Allen F, Bobanga ID, Rauhe P, Barkauskas D, Teich N, Tong C, et al. CCL3 augments tumor rejection and enhances CD8+ T cell infiltration through NK and CD103+ dendritic cell recruitment via IFNγ. OncoImmunology. 2018;7:e1393598.
Rescigno M. CCR6+ dendritic cells: the gut tactical-response unit. Immunity 2006;24:508–10.
Caux C, Ait-Yahia S, Chemin K, de Bouteiller O, Dieu-Nosjean MC, Homey B, et al. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Semin Immunopathol. 2000;22:345–69.
Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In Breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. 1999;190:1417–26.
Furumoto K, Soares L, Engleman EG, Merad M. Induction of potent antitumor immunity by in situ targeting of intratumoral DCs. J Clin Invest. 2004;113:774–83.
Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019;19:89–103.
Khor B. Regulatory T cells: central concepts from ontogeny to therapy. Transfus Med Rev. 2017;31:36–44.
Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356–71.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9.
Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69:2000–9.
Faget J, Biota C, Bachelot T, Gobert M, Treilleux I, Goutagny N, et al. Early detection of tumor cells by innate immune cells leads to Treg recruitment through CCL22 production by tumor cells. Cancer Res. 2011;71:6143–52.
Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122:2286–93.
Maruyama T, Kono K, Izawa S, Mizukami Y, Kawaguchi Y, Mimura K, et al. CCL17 and CCL22 chemokines within tumor microenvironment are related to infiltration of regulatory T cells in esophageal squamous cell carcinoma. Dis Esophagus. 2010;23:422–9.
Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. 2006;66:5716–22.
Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA. 2013;110:17945–50.
Maeda S, Murakami K, Inoue A, Yonezawa T, Matsuki N. CCR4 blockade depletes regulatory T cells and prolongs survival in a canine model of bladder cancer. Cancer Immunol. Res. 2019;7:1175–87.
Pere H, Montier Y, Bayry J, Quintin-Colonna F, Merillon N, Dransart E, et al. A CCR4 antagonist combined with vaccines induces antigen-specific CD8+ T cells and tumor immunity against self antigens. Blood. 2011;118:4853–62.
Wilcox RA. Mogamulizumab: 2 birds, 1 stone. Blood. 2015;125:1847–8.
Ni X, Jorgensen JL, Goswami M, Challagundla P, Decker WK, Kim YH, et al. Reduction of regulatory T cells by Mogamulizumab, a defucosylated anti-CC chemokine receptor 4 antibody, in patients with aggressive/refractory mycosis fungoides and sézary syndrome. Clin Cancer Res. 2015;21:274–85.
Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J Clin Oncol. 2014;32:1157–63.
Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia study of FoxP3+ CD4 Treg fepletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. 2015;21:4327–36.
Cahn A, Hodgson S, Wilson R, Robertson J, Watson J, Beerahee M, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of GSK2239633, a CC-chemokine receptor 4 antagonist, in healthy male subjects: results from an open-label and from a randomised study. BMC Pharm Toxicol. 2013;14:14.
Kindon N, Andrews G, Baxter A, Cheshire D, Hemsley P, Johnson T, et al. Discovery of AZD-2098 and AZD-1678, two potent and bioavailable ccr4 receptor antagonists. ACS Med Chem Lett. 2017;8:981–6.
Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–62.
Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–62.
Noy R, Pollard Jeffrey W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8.
Yamasaki R, Liu L, Lin J, Ransohoff RM. Role of CCR2 in immunobiology and neurobiology. Clin Exp Neuroimmunol. 2012;3:16–29.
Jeong J, Suh Y, Jung K. Context Drives diversification of monocytes and neutrophils in orchestrating the tumor microenvironment. Front Immunol. 2019;10. PMID: 31474975.
Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. 2020;19:41.
Chen X, Wang Y, Nelson D, Tian S, Mulvey E, Patel B, et al. CCL2/CCR2 regulates the tumor microenvironment in HER-2/neu-driven mammary carcinomas in mice. PLoS ONE. 2016;11:e0165595
Ren G, Zhao X, Wang Y, Zhang X, Chen X, Xu C, et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell. 2012;11:812–24.
Li D, Ji H, Niu X, Yin L, Wang Y, Gu Y, et al. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2020;111:47–58.
Lee S, Lee E, Ko E, Ham M, Lee HM, Kim E-S, et al. Tumor-associated macrophages secrete CCL2 and induce the invasive phenotype of human breast epithelial cells through upregulation of ERO1-α and MMP-9. Cancer Lett. 2018;437:25–34.
Li B-H, Garstka MA, Li Z-F. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol Immunol. 2020;117:201–15.
Yoshimura T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine. 2017;98:71–8.
Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76:5671–82.
Hale M, Itani F, Buchta CM, Wald G, Bing M, Norian LA. Obesity triggers enhanced MDSC accumulation in murine renal tumors via elevated local production of CCL2. PLoS ONE. 2015;10:e0118784.
Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, et al. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol Cell. 2017;65:730–42.e5.
Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72:876–86.
Mizutani K, Sud S, McGregor NA, Martinovski G, Rice BT, Craig MJ, et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia. 2009;11:1235–42.
Loberg RD, Ying C, Craig M, Yan L, Snyder LA, Pienta KJ. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9:556–62.
Dutta P, Sarkissyan M, Paico K, Wu Y, Vadgama JV. MCP-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res Treat. 2018;170:477–86.
Heiskala M, Leidenius M, Joensuu K, Heikkilä P. High expression of CCL2 in tumor cells and abundant infiltration with CD14 positive macrophages predict early relapse in breast cancer. Virchows Arch. 2019;474:3–12.
Ohta M, Kitadai Y, Tanaka S, Yoshihara M, Yasui W, Mukaida N, et al. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human gastric carcinomas. Int J Oncol. 2003;22:773–8.
Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol. 1997;150:1723–34.
Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007;252:86–92.
Arakaki R, Yamasaki T, Kanno T, Shibasaki N, Sakamoto H, Utsunomiya N, et al. CCL2 as a potential therapeutic target for clear cell renal cell carcinoma. Cancer Med. 2016;5:2920–33.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5.
Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7:28697–710.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–41.
Noel M, O’Reilly EM, Wolpin BM, Ryan DP, Bullock AJ, Britten CD, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest N. Drugs 2020;38:800–11.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62.
Linehan D, Noel MS, Hezel AF, Wang-Gillam A, Eskens F, Sleijfer S, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: correlation with blood monocyte counts. J Clin Oncol. 2018;36:92
Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018;78:157–67.
Hawila E, Razon H, Wildbaum G, Blattner C, Sapir Y, Shaked Y, et al. CCR5 blood to support tumor development. Cell Rep. 2017;21:2212–22.
Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife. 2019;8:e43653
Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008;267:271–85.
Tsukishiro S, Suzumori N, Nishikawa H, Arakawa A, Suzumori K. Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecologic Oncol. 2006;102:542–5.
Sima AR, Sima HR, Rafatpanah H, Hosseinnezhad H, Ghaffarzadehgan K, Valizadeh N, et al. Serum chemokine ligand 5 (CCL5/RANTES) level might be utilized as a predictive marker of tumor behavior and disease prognosis in patients with gastric adenocarcinoma. J Gastrointest Cancer. 2014;45:476–80.
Singh SK, Mishra MK, Eltoum I-EA, Bae S, Lillard JW, Singh R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep. 2018;8:1323.
Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell. 2016;29:587–601.
Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AEI, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003;63:8360–5.
Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T, et al. Metastasis suppressors regulate the tumor microenvironment by blocking recruitment of prometastatic tumor-associated macrophages. Cancer Res. 2015;75:4063–73.
Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 2019;9. PMID: 31799175.
Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe?. Carcinogenesis. 2012;33:949–55.
Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016;37:41–52.
Xie K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001;12:375–91.
Chao T, Furth EE, Vonderheide RH. CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol Res. 2016;4:968–82.
Powell D, Lou M, Barros Becker F, Huttenlocher A. Cxcr1 mediates recruitment of neutrophils and supports proliferation of tumor-initiating astrocytes in vivo. Sci Rep. 2018;8:13285.
Jablonska J, Wu C-F, Andzinski L, Leschner S, Weiss S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int J Cancer. 2014;134:1346–58.
Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology. 2012;56:2242–54.
Van Coillie E, Van Aelst I, Wuyts A, Vercauteren R, Devos R, De Wolf-Peeters C, et al. Tumor angiogenesis induced by granulocyte chemotactic protein-2 as a countercurrent principle. Am J Pathol. 2001;159:1405–14.
López-Lago MA, Posner S, Thodima VJ, Molina AM, Motzer RJ, Chaganti RSK. Neutrophil chemokines secreted by tumor cells mount a lung antimetastatic response during renal cell carcinoma progression. Oncogene. 2013;32:1752–60.
Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest. 2010;120:485–97.
Brandolini L, Cristiano L, Fidoamore A, De Pizzol M, Di Giacomo E, Florio TM, et al. Targeting CXCR1 on breast cancer stem cells: signaling pathways and clinical application modelling. Oncotarget. 2015;6:43375–94.
Ning Y, Labonte MJ, Zhang W, Bohanes PO, Gerger A, Yang D, et al. The CXCR2 antagonist, SCH-527123, shows antitumor activity and sensitizes cells to oxaliplatin in preclinical colon cancer models. Mol Cancer Ther.2012;11:1353–64.
Varney ML, Singh S, Li A, Mayer-Ezell R, Bond R, Singh RK. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011;300:180–8.
Singh S, Sadanandam A, Nannuru KC, Varney ML, Mayer-Ezell R, Bond R, et al. Small-molecule antagonists for CXCR2 and CXCR1 inhibit human melanoma growth by decreasing tumor cell proliferation, survival, and angiogenesis. Clin Cancer Res. 2009;15:2380–6.
Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol, Immunother. 2015;64:225–35.
Kim J, Kim JS, Lee HK, Kim HS, Park EJ, Choi JE, et al. CXCR3-deficient natural killer cells fail to migrate to B16F10 melanoma cells. Int Immunopharmacol. 2018;63:66–73.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
VGP receives research funding from Merck, Ipsen, and AstraZeneca. He currently has a consulting role at Takeda and has consulted Merck, Imvax, and GlaxoSmithKline in the past.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kohli, K., Pillarisetty, V.G. & Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther 29, 10–21 (2022). https://doi.org/10.1038/s41417-021-00303-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-021-00303-x
This article is cited by
-
Tumor-derived GLI1 promotes remodeling of the immune tumor microenvironment in melanoma
Journal of Experimental & Clinical Cancer Research (2024)
-
Tumor immune dysfunction and exclusion subtypes in bladder cancer and pan-cancer: a novel molecular subtyping strategy and immunotherapeutic prediction model
Journal of Translational Medicine (2024)
-
Acellular scaffold-based approach for in situ genetic engineering of host T-cells in solid tumor immunotherapy
Military Medical Research (2024)
-
Spatially fractionated GRID radiation potentiates immune-mediated tumor control
Radiation Oncology (2024)
-
Neoadjuvant lutetium PSMA, the TIME and immune response in high-risk localized prostate cancer
Nature Reviews Urology (2024)
