
Abstract
It is of great clinical significance to develop potential novel strategies to prevent diabetic cardiovascular complications. Endothelial progenitor cell (EPC) dysfunction is a key contributor to diabetic vascular complications. In the present study we evaluated whether low-dose nifedipine could rescue impaired EPC-mediated angiogenesis and prevent cardiovascular complications in diabetic mice. Diabetes was induced in mice by five consecutive injections of streptozotocin (STZ, 60 mg·kg−1·d−1, i.p.). Diabetic mice were treated with low-dose nifedipine (1.5 mg·kg−1·d−1, i.g.) for six weeks. Then, circulating EPCs in the peripheral blood were quantified, and bone marrow-derived EPCs (BM-EPCs) were prepared. We showed that administration of low-dose nifedipine significantly increased circulating EPCs, improved BM-EPCs function, promoted angiogenesis, and reduced the cerebral ischemic injury in diabetic mice. Furthermore, we found that low-dose nifedipine significantly increased endothelial nitric oxide synthase (eNOS) expression and intracellular NO levels, and decreased the levels of intracellular O2.− and thrombospondin-1/2 (TSP-1/2, a potent angiogenesis inhibitor) in BM-EPCs of diabetic mice. In cultured BM-EPCs, co-treatment with nifedipine (0.1, 1 μM) dose-dependently protected against high-glucose-induced impairment of migration, and suppressed high-glucose-induced TSP-1 secretion and superoxide overproduction. In mice with middle cerebral artery occlusion, intravenous injection of diabetic BM-EPCs treated with nifedipine displayed a greater ability to promote local angiogenesis and reduce cerebral ischemic injury compared to injection of diabetic BM-EPCs treated with vehicle, and the donor-derived BM-EPCs homed to the recipient ischemic brain. In conclusion, low-dose nifedipine can enhance EPCs’ angiogenic potential and protect against cerebral ischemic injury in diabetic mice. It is implied that chronic treatment with low-dose nifedipine may be a safe and economic manner to prevent ischemic diseases (including stroke) in diabetes.
Similar content being viewed by others


Introduction
Diabetes is a major risk factor for morbidity and mortality worldwide [1, 2]. It is associated with a reduced lifespan, largely as a consequence of its cardiovascular complications (including stroke) [2,3,4]. Compared with nondiabetic patients, diabetic patients have at least twice the risk for stroke, and approximately 20% of diabetic patients will die from stroke, making it one of the leading causes of death in this population [5]. However, recent randomized controlled trials (the ACCORD and ADVANCE trials) demonstrated that, as compared with standard therapy, the use of intensive therapy to target near-normal glycemic control for a median of 3.5 to 5 years does not significantly reduce cardiovascular events within that time frame, but increased mortality in the ACCORD trial [2,3,4]. Thus, it should be of great clinical importance to develop new therapeutic strategies except glycemic control to prevent diabetic cardiovascular complications.
Diabetic cardiovascular complications are frequently associated with endothelial progenitor cells (EPCs) dysfunction and reduced EPC-mediated angiogenesis in response to ischemia [6, 7]. EPCs are a population of new cells released into peripheral blood by bone marrow to promote endothelial repair and neovasculogenesis in response to an ischaemic injury [8,9,10]. Thus, it is postulated that promoting EPC function and EPC-mediated ischemic angiogenesis may serve as a potential strategy to prevent cardiovascular complications (such as stroke, coronary heart disease, heart failure, and so on) in diabetes.
Nifedipine, a classic calcium channel blocker, has been widely used in the treatment of hypertension for several decades, and it has been reported that nifedipine could enhance EPCs number and activity in hypertensive patients [11]. Based on these findings, we hypothesized that low-dose nifedipine may serve as a safe and economic option to prevent ischemic diseases by rescuing impaired EPC-mediated angiogenesis in diabetic mice. Here, we demonstrate that chronic low-dose nifedipine can rescue impaired angiogenic potential of diabetic EPCs and lead to an improved response to cerebral ischemic injury in diabetic mice.
Materials and methods
Animals and chronic low-dose nifedipine exposure
Male C57BL/6 mice at 10–12 weeks of age (20–25 g, purchased from the Sino-British SIPPR/BK Laboratory Animal Co., Ltd.) were rendered diabetic by intraperitoneal (i.p.) injection of 60 mg/kg STZ (Sigma-Aldrich, St Louis, MO, USA) in 0.05 M sodium citrate (pH 4.5) daily for five consecutive days during the first week of the study [12, 13]. A 5-day low-dose STZ injection regimen was used to ensure sustained hyperglycemia [13]. Half of the mice treated with STZ were also co-treated daily by oral gavage with low-dose nifedipine (1.5 mg·kg−1·d−1; Sigma-Aldrich, St Louis, MO, USA), and the other half were also co-treated daily with the same amount of vehicle (saline solution) via gavage for 6 consecutive weeks [12, 13]. The control mice were treated with intraperitoneal injection of citrate buffer and subsequently with saline solution by oral gavage. Previous studies reported that estrogens exert neuroprotective effects in an animal model of ischemia [14, 15]. To avoid the interference of estrogen on ischemic stroke, only male mice were selected in this study. Fasting blood glucose level from tail blood was measured from the beginning of the STZ injection and monitored weekly thereafter. Mice with tail vein blood glucose levels greater than 15 mM 1 week after STZ (Sigma-Aldrich) injection were deemed diabetic [16]. The body weight and blood pressure level were measured before and after 6 weeks of nifedipine treatment.
After 6 weeks of low-dose nifedipine (Sigma-Aldrich) treatment, mice were used for EPCs isolation and assessment, or were subjected to permanent focal cerebral ischemia. Administration of nifedipine was discontinued 3 days before EPCs isolation or cerebral ischemia to decrease potential off-target effects. Experiments were carried out in a random and blinded fashion. Experimental procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (USA). All animal experiments complied with the ARRIVE guidelines. The animal protocol was planned in compliance with the animal protection, animal welfare, and ethical principles and was approved by the IACUC of the Naval Medical University.
Quantification of circulating EPCs
After 6 weeks of low-dose nifedipine treatment, the circulating EPCs in the peripheral blood were quantified by flow cytometry using a previously reported technique [17,18,19]. Briefly, peripheral blood was obtained by cardiac puncture after the mice were anesthetized. Peripheral blood mononuclear cells (PB-MNCs) were isolated by Histopaque-1083 (Sigma-Aldrich, St Louis, MO, USA) density gradient centrifugation at 400 × g for 30 min. The mononuclear fraction was collected and washed in phosphate buffer saline (PBS) (pH 7.4), red blood cells were lysed with ammonium chloride solution (Stemcell Technologies, Vancouver, BC, Canada), then washed twice with PBS and once with 5% albumin bovine serum (BSA)/PBS (PBS-A; Sigma-Aldrich, St Louis, MO, USA). Freshly isolated PB-MNCs were re-suspended in 100 μL of l5% PBS-A and incubated with antibodies to Sca-1-PE (BD Pharmingen, San Jose, CA, USA) and Flk-1-FITC (BD Pharmingen) for 1 h on ice. The isotype-specific conjugated anti-immunoglobulin G (IgG; Sigma-Aldrich, St Louis, MO, USA) was used as a negative control. After washing and centrifugation, the cell pellets were suspended in 300 µL of 2% paraformaldehyde (Beyotime Biotechnology, Shanghai, China). Sca-1/Flk-1 double-positive cells were quantified using a BD Flow cytometer (FACScan; Becton Dickenson Biosciences, San Jose, CA, USA). Quantitative analysis of flow cytometry data was expressed as the percentage of Sca-1+/Flk-1+ cells in peripheral blood from Control, Diabetic and chronic low-dose nifedipine-treated diabetic mice [17,18,19].
Bone marrow-derived EPCs (BM-EPCs) isolation and culture
BM-EPCs were isolated and cultured according to the published methods [19, 20]. Bone marrow-derived mononuclear cells (BM-MNCs) were isolated from the murine tibia and femur, seeded in six-well cell culture plates coated with rat vitronectin (1 mg/mL; Sigma-Aldrich, St Louis, MO, USA) at a density of 5 × 106 cells/well, and cultured in endothelial growth medium-2 (EGM-2; Lonza, Walkersville, MD, USA). After four days of culture, nonadherent cells were removed and the adherent cells were further cultivated for 3 days. And then EPCs were either used for in vitro studies (including function assays and Western blot analysis) or for EPC transplantation.
EPC adhesion assay
1 × 104 cells were plated per well of a 96-well plates precoated with vitronectin (1 µg/mL; Sigma-Aldrich, St Louis, MO, USA). After 4-h incubation time, nonadherent BM-EPCs were removed, and residual adherent BM-EPCs were stained with 10 μg/mL of Hoechst (Hoechst 33258; Sigma-Aldrich, St Louis, MO, USA) and then fixed with 2% PFA. Adherent cells were counted at five random fields at magnification of ×100 per sample. Three wells were measured for each cell sample [21].
EPC migration assay
BM-EPCs were plated at a number of 5 × 104 per well in the upper Boyden’s chamber with M199 (Hyclone, Shanghai, China), while M199 supplemented with 50 ng/mL of VEGF (R&D systems, Minneapolis, MN, USA) and 10% FBS (Bioind, Kibbutz Beit Haemek, Israel) was placed in the lower chamber. After 24-h incubation time, migrated BM-EPCs adhering to the lower membrane were fixed and then stained with 10 μg/mL of Hoechst 33258 (Sigma-Aldrich). Then, the number of migrated BM-EPCs were determined by counting the BM-EPCs on the lower side of the membrane under a microscope at magnification ×100. In blinded analyses, for each sample, five images were taken from random fields [21].
EPC tube formation assay
A number of 5 × 104 BM-EPCs were plated per well of a 96-well plate precoated with growth factor-reduced Matrigel-Matrix (BD Biosciences, San Jose, CA, USA). After 6 h of incubation, images of tube morphology were taken by inverted microscope (Leica), and tube numbers were counted at five random fields at magnification of ×100 per sample [17, 21].
Western blot analysis
Western blot analysis was performed as previously described [19, 22]. Briefly, collected BM-EPCs culture media were concentrated with an Amicon Ultra 4 centrifugal filter device with a 10,000 molecular weight cutoff (Merck Millipore, Darmstadt, Germany) according to the manufacturer’s recommendations. For loading control, protein concentrations were determined by the BCA method using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and samples containing equal amounts of protein were subjected to 8% SDS/PAGE. Gels were transferred to nitrocellulose membranes (Millipore) and incubated with antibodies specific to TSP-1 (Abcam, Cambridge, MA, USA; ab1823) and TSP-2 (BD Transduction Laboratories™; 611150). Secondary antibodies included IR Dye 800 conjugated anti-mouse IgG (1:5000, Rockland Immunochemicals, Gilbertsville, PA, USA).
BM-EPCs were extracted in lysis buffer containing protease inhibitors. Protein concentrations was determined by the BCA method using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal quantities of proteins from total cell lysates were fractionated by electrophoresis under denaturing conditions on 8% polyacrylamide gel and transferred to nitrocellulose membranes (Millipore). Proteins were detected by probing Western blots with primary antibodies which were purified mouse anti-eNOS (BD Transduction Laboratories™; 610296), rabbit anti-p-eNOS (Cell Signaling Technology; 9571) and mouse anti-GAPDH (Abcam; ab8245) overnight, and then conjugated using anti-mouse IgG secondary antibody (1:5000, Cell Signaling Technology) (eNOS, beta Actin) or anti-rabbit IgG secondary antibody (1:1000, Cell Signaling Technology) (p-eNOS).
Expression levels of the proteins were detected by Odyssey infrared image system (LI-COR), and the band intensities were quantified with the NIH ImageJ software.
Intracellular superoxide (O2 .−) measurement
O2.− levels were estimated using the fluorescent probe dihydroethidium (DHE) (Sigma-Aldrich, St Louis, MO, USA), a commonly used cell-permeable dye which is sensitive to O2.− and may be oxidized to the red fluorescent molecule ethidium. BM-EPCs were washed, collected and resuspended in M199 medium, and then labeled with 1 µM DHE for 40 min at 37 °C in dark. After that, labeled BM-EPCs were washed three times with 5% BSA/PBS and then fixed in 2% PFA. Finally, the labeled BM-EPCs were analyzed by flow cytometry [19].
Intracellular nitric oxide (NO) measurement
NO levels were visualized using DAF-FM diacetate (4-amino-5-methylamine-2’,7’-difluorofluorescein diacetate, Molecular Probes, USA). BM-EPCs were collected, washed and resuspended in PBS, and then labeled with 1 µM DAF-FM diacetate in dark. After incubation at 37 °C for 40 min, labeled BM-EPCs were washed three times with PBS and then fixed in 2% PFA. Finally, the labeled BM-EPCs were analyzed by flow cytometry [19].
Small interfering RNA transfection in EPCs
After 6-day culture, BM-EPCs were replanted into 6-well plates at a density of 2.5 × 105 cells/well. After 24 h, the TSP-1 or TSP-2 siRNA SMARTpool (si-TSP, Dharmacon, Lafayette, CO, USA) was diluted to a 2 μM working solution and delivered to cells at 100 nM final concentration for 72 h through a lipid-mediated DharmaFECT Transfection Reagent (Dharmacon) according to the manufacturer’s protocol [13, 17]. The nonrelated scramble oligonucleotides (Dharmacon) were used as negative controls. After 72 h of transfection, the cells and medium were harvested for the other experiments.
Animal model of stroke
Permanent focal cerebral ischemia was induced in mice according to previously described methods [18, 21]. Briefly, mice were anesthetized with 3.5% chloral hydrate at 0.1 mL/10 g body weight by intraperitoneal injection. A 0.5-cm skin incision was made between the left orbit and ear. Then, the left distal middle cerebral artery was exposed through a craniotomy and permanently cauterized above the rhinal fissure. The body temperature was maintained at 37 ± 0.5 °C throughout the surgical procedure using a heating lamp and pad. Behavioral assessment (including Body Asymmetry Test and Beam Test) was performed three days after middle cerebral artery occlusion, then mice were euthanized, and their brains were immediately removed. Infarct volume was determined by staining with 2,3,5-triphenyltetrazolium chloride (TTC, Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) and was analyzed with Image J software [21, 23]. The experiments were performed in a blind and random fashion.
Additionally, on day 3 after middle cerebral artery occlusion, the mice were anesthetized and the ischemic brains were flushed with PBS and perfused in 4% PFA, before being dehydrated and embedded in paraffin. Serial sections (6 µm thick) were cut in the coronal plane from paraffin-embedded blocks. Every 10th section was processed for immunohistochemical staining. For quantification of angiogenesis, the area of ischemic boundary was immunostained with CD31 antibody [21, 24, 25].
Neurological behavior analysis: Beam Test and Body Asymmetry Test
-
(1)
Beam Test—Beam walking across a bridge was used to assess motor coordination and balance after stroke injury. The mice were trained for five days before the MCA occlusion to traverse a narrow round beam (5 mm in diameter, 900 mm in length) to reach an enclosed escape platform. They were placed on one end of the beam and the latency to traverse the central 80% of the beam toward the enclosed escape platform at the other end was recorded. Data are expressed as mean latency to cross the beam in three trials [18, 21].
-
(2)
Body Asymmetry Test—To measure motor asymmetry, the mice were examined using the elevated body swing test as described previously [18, 21]. The mice were examined for head swings while being suspended by their tails. The direction of the swing (right or left) was recorded when the mouse turned its head sideways by approximately at a 10° angle to the body’s midline. After each swing, each mouse was allowed to move freely in a Plexiglas box for at least 30 s before undergoing the next test; the trials were repeated 20 times for each animal. The frequency of the head swings toward the contralateral side was counted and normalized as follows:
EPC transplantation and animal stroke model in diabetic mice
To further confirm whether the EPC-mediated ischemic angiogenesis might relate to the protection of nifedipine against cerebral ischemic injury in diabetic mice, a total of 1×106 bone marrow-derived diabetic EPCs and nifedipine (1 μmol/L for 1 day) incubated diabetic EPCs in 200 μL PBS were respectively systemically injected into diabetic mice via a tail vein just after middle cerebral artery occlusion, and equivalent volume of PBS was administered to control diabetic mice [10, 24]. On day 3 after stroke, behavioral tests were performed, and then the ischemic brains were serially cut and stained with 2% TTC for 5 min (55 °C) to determine the infarct area [21, 23].
Additionally, on day 3 after middle cerebral artery occlusion, the mice were anesthetized and the ischemic brains were flushed with PBS and perfused in 4% PFA, before being dehydrated and embedded in paraffin. Serial sections (6 µm thick) were cut in the coronal plane from paraffin-embedded blocks. Every 10th section was processed for immunohistochemical staining. For quantification of angiogenesis, the area of ischemic boundary was immunostained with CD31 antibody [18, 24, 25].
In vivo EPC integration in diabetic mice
5-Bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich, St Louis, MO, USA) labeling of EPCs was performed as published methods [17]. On day 5 of culture, BrdU (1:100) was added to the media, and new medium supplemented with BrdU was replenished daily until day 7. Thereafter, the labeled EPCs were collected and resuspended in PBS and were then transplanted into mice (1 × 106 cells/mouse) via the tail vein just after middle cerebral artery occlusion.
On day 3 after stroke, the ischemic brains were exsanguinated by transcardial perfusion with PBS and perfusion fixed with 4% PFA at 4 °C overnight. After being washed in fresh PBS, the ischemic brains were dehydrated, cleared and embedded in paraffin. Serial sections (6 µm thick) were cut in the coronal plane from the paraffin-embedded block. Every 10th section was chosen for immunohistochemical staining (a total of three ischemic brain sections each) [21, 24, 25]. These sections were stained with anti-CD31 antibody at 4 °C overnight, followed by BrdU antibody. Secondary antibodies applied were conjugates of Alexa Fluor 488 or Cy3. Finally, DAPI was employed to stain nuclei [10, 17].
Statistical analysis
Data were expressed as mean ± SEM. An unpaired Student’s t-test was used for comparisons between two groups and one-way ANOVA followed by Newman-Keuls post hoc analysis was used for comparison of variables in more than two groups. A value of P < 0.05 was considered statistically significant.
Results
Chronic low-dose nifedipine administration had no effect on fasting blood glucose levels, body weights and blood pressure in diabetic mice
A 5-day low-dose STZ injection regimen was used to ensure sustained hyperglycemia [13]. Five consecutive intraperitoneal daily injections of STZ (60 mg/kg) were administered to male C57BL/6 mice that were monitored for six weeks, in comparison with a group of vehicle (citrate buffer)-treated control mice. Half of the STZ-injected mice were also co-treated daily by oral gavage with low-dose nifedipine (1.5 mg·kg−1·d−1) for six consecutive weeks, and the other half were also co-treated daily by oral gavage with vehicle (saline solution) for six consecutive weeks [12]. As shown in Fig. 1a, b, a significant increase in fasting blood glucose levels and a considerable reduction in body weight were observed in the STZ-treated mice. However, 6-week treatment with low-dose nifedipine had no effect on fasting blood glucose levels and body weight in diabetic mice. Blood pressure levels did not differ among all the groups before or after 6 weeks of low-dose nifedipine administration (Fig. 1c).

a Fasting blood glucose levels. b Body weights. c Blood pressure. Throughout, error bars represent mean ± SEM. n = 4–12 per group. **P < 0.01 vs. Diabetes, Diabetes+Nif. *P < 0.05. Control, vehicle (citrate buffer and saline solution)-treated mice; Diabetes, vehicle (saline solution)-treated diabetic mice; Diabetes+Nif, chronic low-dose nifedipine-treated diabetic mice.
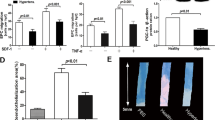
Chronic low-dose nifedipine administration increased circulating EPC number and rescued BM-EPC dysfunction in diabetic mice
After six weeks of low-dose nifedipine treatment, the circulating EPC numbers and BM-EPC functions were determined in diabetic mice (Fig. 2a). Flow cytometry analysis revealed a 60% decrease in circulating Sca-1+/Flk-1+ EPCs in diabetic mice. However, compared with diabetic mice, a 6-week low-dose nifedipine treatment increased the number of circulating EPCs (P < 0.05; Fig. 2b). Results showed that the adhesion, migration and tube formation functions were significantly impaired in diabetic BM-EPCs, which were rescued by chronic low-dose nifedipine treatment (P < 0.01; Fig. 2c–e).

a Experimental protocols: five consecutive intraperitoneal daily injections of STZ (60 mg/kg) were administered to male C57BL/6 mice that were monitored for 6 weeks. Half of the STZ-injected mice were also co-treated daily by oral gavage with low-dose nifedipine (1.5 mg·kg−1·d−1), and the other half were also co-treated daily with the same amount of vehicle (saline solution) via gavage for 6 consecutive weeks. The control mice were treated with intraperitoneal injection of citrate buffer and subsequently with saline solution by oral gavage. Then, EPCs were enumerated in peripheral blood by flow cytometry, and BM-EPCs were isolated, cultured and examined. b Circulating EPCs were quantified by enumerating Sca-1+/Flk-1+ cells. Quantitative analysis of flow cytometry data was expressed as the percentage of Sca-1+/Flk-1+ cells in peripheral blood from Control, diabetic and chronic low-dose nifedipine-treated diabetic mice (n = 6–8). c EPCs adhesion assay (n = 5). d EPCs migration assay (n = 5–6). e EPCs tube formation assay (n = 5–7). Values were normalized to Control. *P < 0.05, **P < 0.01. Scale bar, 100 μm. Control, vehicle (citrate buffer and saline solution)-treated mice; Diabetes, vehicle (saline solution)-treated diabetic mice; Diabetes+Nif, chronic low-dose nifedipine-treated diabetic mice.
To investigate the potential mechanisms underlying chronic low-dose nifedipine administration protecting BM-EPCs functions, the expression of eNOS, P-eNOS, TSP-1, TSP-2 and the levels of intracellular O2.− and intracellular NO of BM-EPCs were examined in diabetic mice after 6-week low-dose nifedipine administration (Fig. 3a). Compared to control, both eNOS and P-eNOS were significantly decreased (−32.8%, P < 0.01 and −32.8%, P < 0.01, respectively) in BM-EPCs from diabetic mice. BM-EPCs from chronic low-dose nifedipine treatment diabetic mice showed higher levels of both total and phosphorylated eNOS (+41.2%, P < 0.01 and +48.0%, P < 0.01, respectively) compared to diabetic mice (Fig. 3b–d). No difference was found between control group and chronic low-dose nifedipine treated diabetic mice. In addition, intracellular NO level of BM-EPCs from diabetic mice was significantly attenuated compared to that in control mice, which was rescued in BM-EPCs from chronic low-dose nifedipine-treated diabetic mice (P < 0.01; Fig. 3e). As shown in Fig. 3f, intracellular O2.− level of BM-EPCs from diabetic mice was increased ~4.8-fold compared to control group. However, it was significantly attenuated in BM-EPCs from chronic low-dose nifedipine treated diabetic mice when compared with that of the diabetic mice (P < 0.01). Since TSP-1 and TSP-2 are secreted proteins and their levels in BM-EPCs were too low to be detected (data not shown), we examined the TSP-1 and TSP-2 levels in BM-EPCs culture media using Western blot analysis [19, 22]. TSP-1 expression level in BM-EPCs media from diabetic mice was increased ~1.7-fold compared with that of the control group, which was reduced in chronic low-dose nifedipine treated diabetic mice (−52.2%, P < 0.01 vs. Diabetes) (Fig. 3g, h). TSP-2 expression level in BM-EPCs media was also enhanced significantly in diabetic mice (+36.1%, P < 0.01 vs. Control), which was decreased in chronic low-dose nifedipine-treated diabetic mice (−29.7%, P < 0.01 vs. Diabetes) (Fig. 3g, i).

a Experimental protocols: five consecutive intraperitoneal daily injections of STZ (60 mg/kg) were administered to C57 male mice that were monitored for 6 weeks, in comparison with a group of vehicle (citrate buffer and saline solution)-treated control mice. Half of STZ-injected mice were also co-treated daily by oral gavage with low-dose nifedipine (1.5 mg·kg−1·d−1) and the other half were also co-treated daily by oral gavage with vehicle (saline solution) for 6 consecutive weeks. Then, BM-EPCs were isolated, cultured and examined. b, c, d The representative images and the protein expression levels of total eNOS (n = 5) and phosphorylated eNOS (n = 5). e Intracellular NO levels of BM-EPCs (n = 4–7). f Intracellular O2.− levels of BM-EPCs (n = 6–11). g, h, i Western blot analysis of secreted TSP-1 (n = 4–6) and TSP-2 (n = 3–4) levels in BM-EPCs culture media. Values were normalized to Control. **P < 0.01. Control, vehicle (citrate buffer and saline solution)-treated mice; Diabetes, vehicle (saline solution)-treated diabetic mice; Diabetes+Nif, chronic low-dose nifedipine-treated diabetic mice.
Chronic low-dose nifedipine administration promoted local angiogenesis in ischemic brain in diabetic mice
EPCs have been shown to promote angiogenesis by secreting various angiogenic growth factors [10]. Therefore, local angiogenesis was assessed by immunostaining at three days after cerebral ischemia using an antibody against the angiogenesis marker, i.e., CD31 (Fig. 4a). It was found that the number of CD31+ microvessels in the boundary area of ischemic brains was reduced in diabetic mice compared with control (−40.7%, P < 0.05), which was significantly increased in 6-week low-dose nifedipine treated diabetic mice (+171.5%, P < 0.01 vs. Diabetes) (Fig. 4b).

a Experimental protocols: five consecutive intraperitoneal daily injections of STZ (60 mg/kg) were administered to male C57BL/6 mice that were monitored for 6 weeks, in comparison with a group of vehicle (citrate buffer and saline solution)-treated control mice. Half of the STZ-injected mice were also co-treated daily by oral gavage with low-dose nifedipine (1.5 mg·kg−1·d−1) for 6 consecutive weeks, and the other half were also co-treated daily by oral gavage with vehicle (saline solution) for 6 consecutive weeks. Then the mice were subjected to focal cerebral ischemia by permanent occlusion. Infarct volumes, functional outcome and the local angiogenesis in ischemic brain 3 days after focal cerebral ischemia were investigated in a murine model of ischemic stroke. b Microvessels (CD31+) in the boundary area of ischemic brains. The bar graph shows that the number of microvessels was reduced in diabetic mice compared with control, which was significantly increased in chronic low-dose nifedipine treated diabetic mice. n = 6. Scale bar, 100 μm (left); 50 μm (right). c Images are representative of 2,3,5-triphenyltetrazolium chloride-stained ischemic brain sections, cerebral infarct volumes (d) and neurobehavioral outcomes (e, Beam Test; f, Body Asymmetry Test) on day 3 after middle cerebral artery occlusion. n = 7–12. Values were normalized to Control. *P < 0.05, **P < 0.01. Control, vehicle (citrate buffer and saline solution)-treated mice; Diabetes, vehicle (saline solution)-treated diabetic mice; Diabetes+Nif, chronic low-dose nifedipine-treated diabetic mice.
Chronic low-dose nifedipine administration protected diabetic mice against cerebral ischemic injury
Next, we attempted to study whether the promotion of EPC functions and local angiogenesis in ischemic brain produced by chronic low-dose nifedipine treatment would lead to a decrease of ischemic brain injury in diabetic mice. After 6-week of treatment, mice were subjected to cerebral ischemia (Fig. 4a). Compared to control mice, diabetic mice exhibited more severe cerebral ischemic injury (+88.3%, P < 0.01) and higher neurological deficits at three days after cerebral ischemia. However, the infarct volume was significantly reduced (−45.7%, P < 0.01) and the neurological deficits were markedly improved in chronic low-dose nifedipine treated diabetic mice compared with diabetic mice (Fig. 4c–f). These results indicate that chronic low-dose nifedipine treatment could protect diabetic mice against cerebral ischemic injury.
Incubation with nifedipine protected BM-EPCs against high-glucose-induced impairment of migration function
To determine whether nifedipine exerted direct beneficial effects in protecting BM-EPCs functions, we further performed cell functions, TSP-1 level, TSP-2 level, intracellular O2.− and intracellular NO assays in high-glucose-treated BM-EPCs (Fig. 5a). BM-EPCs were treated with vehicle (EBM-2), high glucose (33 mM) or high glucose with additional nifedipine (0.1 μM and 1 μM) respectively for 1 day. Equimolar mannitol addition controlled for any potential effect of increased osmolarity under high-glucose conditions. Results showed that the migration function was significantly impaired in high-glucose-treated BM-EPCs, which was rescued by nifedipine treatment (1 µM for 1 day, P < 0.01). As a control for potential osmotic effects of added high glucose, equimolar mannitol produced no impairment of migration function in BM-EPCs (Fig. 5b). In addition, incubation of BM-EPCs with high glucose led to a reduction in intracellular NO level and an increase in intracellular O2.− level, TSP-1 and TSP-2 secretion, which were rescued by co-incubation with nifedipine (P < 0.01) (Fig. 5c–e).

a Experimental protocols: BM-EPCs from male young C57BL/6 mice were cultured for 6 days. Then, cells were treated with vehicle (EBM-2), high glucose (33 mM), high glucose with additional nifedipine (0.1 μΜ, 1 μΜ) or mannitol respectively, and 1 day later, the cells functions, intracellular NO level, intracellular O2.− level and TSP-1 and TSP-2 expression levels were examined. Equimolar mannitol was used as an osmotic control of high glucose. b migration assay of BM-EPCs (n = 11–17). Scale bar, 100 μm. c Intracellular NO levels of BM-EPCs (n = 5). d Intracellular O2.− levels of BM-EPCs (n = 6–8). e, f, g Western blot analysis of secreted TSP-1 (n = 3) and TSP-2 (n = 3) levels in BM-EPCs culture media. Values were normalized to EPC. *P < 0.05, **P < 0.01. EPC, vehicle (EBM-2)-treated BM-EPCs; eHG, high glucose-treated BM-EPCs; eHG+0.1, BM-EPCs treated with high glucose and additional nifedipine (0.1 μΜ, 1 day); eHG+1, BM-EPCs treated with high glucose and additional nifedipine (1 μΜ, 1 day); eMan, mannitol-treated BM-EPCs.
Silencing TSP-1 or TSP-2 blunted diabetic BM-EPCs function improvement induced by nifedipine treatment
To further investigate whether TSP-1 and TSP-2 play a causal role in nifedipine-induced improvement of diabetic BM-EPCs functions, TSP-1 siRNA or TSP-2 siRNA was used to knock down TSP-1 or TSP-2 in high-glucose-treated BM-EPCs before nifedipine treatment. An ON-TARGETplus non-targeting siRNA pool from Dharmacon served as a control, and all targeting siRNA were compared to this scramble control (Fig. 6a). Western blot analysis confirmed that TSP-1 siRNA decreased TSP-1 levels by 49.7% compared with scramble control transfection in BM-EPCs. Meanwhile, TSP-2 siRNA decreased TSP-2 levels by 31.9% compared with scramble control transfection in BM-EPCs. High-glucose-induced increase of TSP-1 or TSP-2 level in BM-EPCs was blunted after TSP-1 or TSP-2 silencing. Meanwhile, nifedipine-induced reduction of TSP-1 or TSP-2 level in high-glucose-treated BM-EPCs was blunted after TSP-1 or TSP-2 silencing (Fig. 6b, f). The results of in vitro function assays showed that, when TSP-1 or TSP-2 expression was knocked down by its siRNA, BM-EPCs functions were increased compared with scramble control BM-EPCs. Nifedipine-induced improvement of adhesion and migration functions of high-glucose-incubated BM-EPCs were blunted after TSP-1 silencing (Fig. 6c, d). However, silencing TSP-1 did not alter the increased tube formation function of high-glucose-treated BM-EPCs following nifedipine treatment (Fig. 6e). Meanwhile, nifedipine-induced improvement of migration and tube formation functions of high-glucose-incubated BM-EPCs were blunted after TSP-2 silencing (Fig. 6h, i). However, silencing TSP-2 did not alter the increased adhesion function of high-glucose-treated BM-EPCs following nifedipine treatment (Fig. 6g).

a Experimental protocols: BM-EPCs from male young C57BL/6 mice were cultured for 6 days. Then, cells were treated with scramble control siRNA or TSP-1/TSP-2 siRNA respectively, and 2 days later, TSP-1/TSP-2 siRNA-treated EPCs were co-treated with HG or HG with additional nifedipine (1 μΜ), then, after one day, secreted TSP-1/TSP-2 expression levels and the cells functions were examined. b Western blot analysis of secreted TSP-1 level in BM-EPCs culture media (n = 4). c EPCs adhesion assay (n = 4). d EPCs migration assay (n = 6). e EPCs tube formation assay (n = 4). f Western blot analysis of secreted TSP-2 level in BM-EPCs culture media (n = 3). g Adhesion assay of EPCs (n = 5). h Migration assay of EPCs (n = 5). i Tube formation assay of EPCs (n = 4–5). Values were normalized to scramble control siRNA. *P < 0.05, **P < 0.01.
Nifedipine enhanced the therapeutic effect of diabetic BM-EPCs on cerebral ischemic injury reduction and angiogenesis promotion in diabetic mice
The beneficial effects of incubation with nifedipine (1 µM for one day) on diabetic BM-EPCs functions were firstly determined (Fig. 7a). Results showed that diabetic BM-EPCs exhibited impaired migration, tube formation and adhesion functions. However, incubation with nifedipine (1 µM for one day) significantly rescued the impaired functions of diabetic BM-EPCs (P < 0.01; Fig. 7b–d).

a Experimental protocols: EPCs from control mice (10–12 weeks) or diabetic mice were cultured for 6 days. Then, diabetic EPCs were incubated with vehicle (PBS) or nifedipine (1 μΜ) respectively, and 1 day later, the EPC functions were examined. b EPCs adhesion assay (n = 5). c EPCs migration assay (n = 6). d EPCs tube formation assay (n = 6). Values were normalized to Control. **P < 0.01 Scale bar, 100 μm. Control, control EPC; Diabetes, diabetic EPC; Diabetes + Nif 1d, Nifedipine (1 μΜ, 1 day)-incubated diabetic EPC.
To determine whether nifedipine can enhance the therapeutic effect of diabetic BM-EPCs on cerebral ischemic injury reduction and whether the EPC-mediated ischemic angiogenesis may contribute to the protection of nifedipine against cerebral ischemic injury in diabetic mice, 1×106 bone marrow-derived EPCs from diabetic mice with or without nifedipine (1 µM for 1 day) incubation were injected via tail vein into diabetic mice immediately after middle cerebral artery occlusion, and control diabetic mice received equal volume of vehicle (PBS) (Fig. 8a). As shown in Fig. 8b–e, diabetic BM-EPCs or nifedipine-incubated diabetic BM-EPCs transplantation attenuated infarct volume in diabetic mice (−18.3%, P < 0.01 or −44.5%, P < 0.01) and the corresponding neurobehavioral outcomes were significantly improved in the two groups of EPC-treated mice compared with controls. What is more, nifedipine-incubated diabetic BM-EPCs showed a stronger therapeutic efficacy against cerebral ischemic injury than diabetic BM-EPCs (P < 0.01). Furthermore, the angiogenesis in ischemic brain was assessed on day 3 after middle cerebral artery occlusion. It was found that the number of CD31+ microvessels in the boundary area of ischemic brains was significantly increased in the two groups of EPC-treated diabetic mice. However, nifedipine-incubated diabetic BM-EPCs exerted a markedly stronger effect on local angiogenesis promotion compared to the diabetic BM-EPCs (P < 0.01) (Fig. 8f).

a Surgical protocols: male diabetic mice were randomly allocated to 3 groups. Each group of animals respectively received a single injection of vehicle (PBS), diabetic EPCs or nifedipine-incubated diabetic EPC just after middle cerebral artery occlusion. Infarct volumes, functional outcome and the local angiogenesis in ischemic brain three days after focal cerebral ischemia were investigated in a murine model of ischemic stroke. b Images are representative of TTC-stained ischemic brain sections and cerebral infarct volumes (c). d, e Neurobehavioral outcomes (d, Beam Test; e, Body Asymmetry Test) on day 3 after middle cerebral artery occlusion. n = 5–12. Values were normalized to Vehicle. f CD31 immunostaining shows microvessels in ischemic brains of diabetic mice. The bar graph shows the number of CD31+ microvessels was increased in the two groups of EPCs-treated mice. Moreover, nifedipine-incubated EPCs exerted a more positive effect on angiogenesis promotion compared to the EPCs without nifedipine incubation. n = 3. Scale bar, 100 μm (left); 50 μm (right). Values were normalized to EPC. g Typical photographs (taken from the ischemic boundary area) indicate that some BrdU-positive cells (red fluorescence) migrated into the ischemic area of brains and were found near the CD31-positive vessels (green fluorescence) after 3 days of injection. The nucleus was counterstained with DAPI (blue fluorescence). Scale bar, 25 μm. **P < 0.01. Vehicle, diabetic mice treated with vehicle (PBS); EPC, diabetic mice treated with diabetic EPCs; EPC-Nif 1d, diabetic mice treated with nifedipine (1 μΜ, 1 day)-incubated diabetic EPC.
To further validate that donor derived (BrdU-positive) EPCs already successfully home to the area of ischemic boundary and participate in angiogenesis in diabetic mice, the sections were stained for CD31 and BrdU. As shown in Fig. 8g, inspection of microvessels (labeled with CD31 immunostaining) proximal to the ischemic brain in the mice with transplantation of EPCs revealed a conspicuous accumulation of BrdU-labeled cells (red). Previous studies have found that donor-derived BM-EPCs participate in angiogenesis in ischemic hindlimb and ischemic brain [10, 17]. Our finding demonstrated that the donor derived EPCs homed to the area of ischemic boundary and participated in angiogenesis in diabetic mice.
These findings collectively demonstrated that the transplanted EPCs could incorporate into the area of ischemic boundary, promote local neovascularization capacity and protect against the cerebral ischemic injury in diabetic mice, and the nifedipine increased the therapeutic effect of EPCs from diabetic mice on angiogenesis promotion and cerebral ischemic injury reduction.
Discussion
The present study showed that chronic low-dose nifedipine rescued impaired EPC-mediated angiogenesis and prevented against cerebral ischemic injury in diabetic mice, independently of blood pressure.
EPCs, as important precursors of endothelial cells, have been shown to participate in vascular formation, and have been used to successfully restore endothelial function and enhance neovascularization capacity in ischemic brain [10, 14, 17]. However, ample studies indicate that EPCs are dysfunctional in diabetic patients [26]. The present study demonstrated that chronic low-dose nifedipine could improve EPC function, enhance neovascularization capacity in the ischemic brain and reduce the cerebral ischemic injury in diabetic mice. In addition, it has been shown that EPC functions are fundamentally impaired in diabetic patients [26]. Here we found that incubation with nifedipine significantly rescued EPC dysfunctions in diabetic mice. Furthermore, we found that nifedipine increased the therapeutic effect of EPCs from diabetic mice on angiogenesis promotion and cerebral ischemic injury reduction in diabetic mice, and the donor-derived EPCs homed to the recipient ischemic brain. Taken together, these findings demonstrate that the promotion of EPC-mediated ischemic brain angiogenesis may partly contribute to the protection of chronic low-dose nifedipine against cerebral ischemic injury in diabetic mice.
It has been reported that nifedipine has a number of blood pressure-independent effects, including improving the endothelial function and decreasing oxidative stress [27]. To avoid the potential adverse effects induced by a significant reduction in blood pressure levels in normotensive patients, the present work aim to choose a low-dose of nifedipine that shows no effect on blood pressure but can rescue impaired EPC-mediated angiogenesis in diabetes. It has been reported that 14-day treatment with a dose of nifedipine (5 mg·kg−1·d−1) had no effect on blood pressure in angiotensin II infused hyperphenylalaninemia (hph-1) mice [28]. Based on this study [28] and our preliminary experiments, the dose of continuous oral administration of nifedipine (1.5 mg·kg−1·d−1) was determined. In addition, chronic low-dose nifedipine possesses some obvious advantages: (1) low-dose nifedipine may be considered as a safe and feasible candidate to promote EPC-mediated ischemic angiogenesis in diabetes; (2) since EPCs has been demonstrated to participate in vascular repair and revascularization in various ischemic organs besides brain tissue [10, 17], chronic low-dose nifedipine treatment might also be a strategy to prevent other diabetic cardiovascular complications except cerebral ischemia, such as amputation and myocardial infarction, which remains to be comprehensively tested.
It has been demonstrated that eNOS critically regulate EPC function [29]. In vascular endothelial cells, loss of eNOS activity leads to increased O2.− level [30, 31]. The increase in intracellular O2.− and reduction in intracellular NO represent a major mechanism underlying EPC dysfunction in diabetic and hypertensive mice [13, 17, 19]. In addition, the decreased NO production or increased O2.− production may upregulate TSP-1 and TSP-2 expression in cultured EPC [19, 32], and the upregulation of TSP-1 and TSP-2 may impair EPC function, inhibit angiogenesis and reendothelialization in mice [19, 32, 33]. Thus, the increased eNOS, p-eNOS and intracellular NO levels together with the decrease of TSP-1, TSP-2 and intracellular O2.− levels, as observed in the present study, might partly contribute to the promotion of EPC functions in chronic low-dose nifedipine-treated diabetic mice (Fig. 9).

The decreased expression of eNOS and p-eNOS enhances TSP-1 and TSP-2 expression levels in diabetic EPCs by increasing intracellular O2.− level and lowering intracellular NO level, resulting in impaired EPC functions in diabetes mice. Diabetic mice have impaired endothelial progenitor cell function, decreased ischemic angiogenesis, increased cerebral infarct volumes, and reduced corresponding neurobehavioral outcomes. Low-dose nifedipine reduces cerebral ischemic injury via rescuing impaired EPC-mediated angiogenesis in diabetic mice. STZ streptozotocin, TSP-1/2 thrombospondin-1/2, EPC Endothelial progenitor cell.
Incubation with nifedipine suppressed TSP-1 secretion, which protected EPCs against high-glucose-induced impairment of migration function, suggesting that downregulation of TSP-1 may play a crucial role in mediating the protective effect of nifedipine on diabetic EPC functions. This point was further confirmed by the evidence that TSP-1 silencing blunted the improvement of adhesion and migration function caused by nifedipine treatment in high-glucose-incubated EPCs. Meanwhile, nifedipine-induced improvement of migration and tube formation function of high-glucose-incubated EPCs were blunted after TSP-2 silencing. However, silencing TSP-1 did not alter the increased tube formation function of high-glucose-treated EPCs following nifedipine treatment. Meanwhile, silencing TSP-2 did not alter the increased adhesion function of high-glucose-treated EPCs following nifedipine treatment. These may be elucidated by other angiogenesis-related mechanisms, which are supposed to be regulated by nifedipine treatment.
In conclusion, chronic low-dose nifedipine could prevent cerebral ischemic injury in diabetic mice, which might be partly attributed to the improvement of EPC-mediated ischemic brain angiogenesis (Fig. 9). This result implies that chronic treatment with low-dose nifedipine, may be a safe and economic manner to prevent ischemic diseases (including stroke) in diabetes, and priming with nifedipine may be a feasible option to improve the efficacy of EPCs-based therapy for ischemic diseases.
References
Xu Y, Wang LM, He J, Bi YF, Li M, Wang TG, et al. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310:948–59.
Dluhy RG, McMahon GT. Intensive glycemic control in the ACCORD and ADVANCE trials. N Engl J Med. 2008;358:2630–3.
Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59.
ADVANCE Collaborative Group, Patel A, MacMahon S, Chalmers J, Neal B, Billot L, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–72.
Phipps MS, Jastreboff AM, Furie K, Kernan WN. The diagnosis and management of cerebrovascular disease in diabetes. Curr Diab Rep. 2012;12:314–23.
Loomans CJM, Koning EJPde, Staal FJT, Rookmaaker MB, Verseyden C, Boer HCde, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–9.
Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–6.
Hill JM, Zalos G, Halcox JPJ, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600.
Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007.
Fan YF, Shen FX, Frenzel T, Zhu W, Ye JQ, Liu JR, et al. Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann Neurol. 2010;67:488–97.
Sugiura T, Kondo T, Kureishi-Bando Y, Numaguchi Y, Yoshida O, Dohi Y. et al. Nifedipine improves endothelial function: role of endothelial progenitor cells. Hypertension. 2008;52:491–8.
Zauli G, Toffoli B, Iasio MGdi, Celeghini C, Fabris B, Secchiero P. Treatment with recombinant tumor necrosis factor-related apoptosis-inducing ligand alleviates the severity of streptozotocin-induced diabetes. Diabetes. 2010;59:1261–5.
Wang XR, Zhang MW, Chen DD, Zhang Y, Chen AF. AMP-activated protein kinase rescues the angiogenic functions of endothelial progenitor cells via manganese superoxide dismutase induction in type 1 diabetes. Am J Physiol Endocrinol Metab. 2011;300:E1135–45.
Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, et al. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J Neurosurg. 1997;87:724–30.
Suzuki S, Brown CM, Wise PM. Neuroprotective effects of estrogens following ischemic stroke. Front Neuroendocrinol. 2009;30:201–11.
Ju LN, McFadyen JD, Al-Daher S, Alwis I, Chen YF, Tønnesen LL, et al. Compression force sensing regulates integrin αIIbβ3 adhesive function on diabetic platelets. Nat Commun. 2018;9:1087.
Marrotte EJ, Chen DD, Hakim JS, Chen AF. Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing indiabetic mice. J Clin Invest. 2010;120:4207–19.
Dong XH, Sun X, Jiang GJ, Chen AF, Xie HH. Dietary intake of sugar substitutes aggravates cerebral ischemic injury and impairs endothelial progenitor cells in mice. Stroke. 2015;46:1714–8.
Xie HH, Zhou S, Chen DD, Channon KM, Su DF, Chen AF. GTP cyclohydrolase I/BH4 pathway protects EPCs via suppressing oxidative stress and thrombospondin-1 in salt-sensitive hypertension. Hypertension. 2010;56:1137–44.
Dong XH, Peng C, Zhang YY, Jiang Y, Yang LJ, He JB, et al. Low-dose piperlongumine rescues impaired function of endothelial progenitor cells and reduces cerebral ischemic injury in high-fat diet-fed mice. Front Pharmacol. 2021;12:689880.
Dong XH, Peng C, Zhang YY, Tao YL, Tao X, Zhang C. et al. Chronic exposure to subtherapeutic antibiotics aggravates ischemic stroke outcome in mice. EBioMedicine. 2017;24:116–26.
Tran MD, Neary JT. Purinergic signaling induces thrombospondin-1 expression in astrocytes. Proc Natl Acad Sci USA. 2006;103:9321–6.
Zhu W, Fan YF, Frenzel T, Gasmi M, Bartus RT, Young WL, et al. Insulin growth factor−1 gene transfer enhances neurovascular remodeling and improves long-term stroke outcome in mice. Stroke. 2008;39:1254–61.
Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res. 2002;90:284–8.
Chen JL, Zhang CL, Jiang H, Li Y, Zhang LJ, Robin A, et al. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–90.
Awal HB, Nandula SR, Domingues CC, Dore FJ, Kundu N, Brichacek B, et al. Linagliptin, when compared to placebo, improves CD34+ve endothelial progenitor cells in type 2 diabetes subjects with chronic kidney disease taking metformin and/or insulin: a randomized controlled trial. Cardiovasc Diabetol. 2020;19:72.
Kito T, Shibata R, Kondo M, Yamamoto T, Suzuki H, Ishii M, et al. Nifedipine ameliorates ischemia-induced revascularization in diet-induced obese mice. Am J Hypertens. 2012;25:401–6.
Miao XN, Siu KL, Cai H. Nifedipine attenuation of abdominal aortic aneurysm in hypertensive and non-hypertensive mice: mechanisms and implications. J Mol Cell Cardiol. 2015;87:152–9.
Thum T, Fraccarollo D, Schultheiss M, Froese S, Galuppo P, Widder JD, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666–74.
Adams GN, LaRusch GA, Stavrou E, Zhou YH, Nieman MT, Jacobs GH, et al. Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood. 2011;117:3929–37.
Sato N, Kaneko M, Tamura M, Kurumatani H. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese Zucker (fatty) rats. Diabetes. 2010;59:1092–100.
Bae O, Wang JM, Baek SH, Wang QD, Yuan H, Chen AF. Oxidative stress-mediated thrombospondin-2 upregulation impairs bone marrow-derived angiogenic cell function in diabetes mellitus. Arterioscler Thromb Vasc Biol. 2013;33:1920–7.
Li M, Takenaka H, Asai J, Ibusuki K, Mizukami Y, Maruyama K, et al. Endothelial progenitor thrombospondin-1 mediates diabetes-induced delay in reendothelialization following arterial injury. Circ Res. 2006;98:697–704.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology of China (2021YFA0804803), the National Natural Science Foundation of China (81870189, 82070266, 81900243, 81930012, 81730013, 92168120 and 81974506), and the Beijing Natural Science Foundation (Z200019).
Author information
Authors and Affiliations
Contributions
HHX conceived and designed the experiments. CP, LJY, YJ, LWXS, JBH and ZWZ performed the experiments. HHX, LT, CZ and AFC analyzed and interpreted the data. XT provided intellectual contribution and useful discussion. HHX and CP wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Peng, C., Yang, Lj., Zhang, C. et al. Low-dose nifedipine rescues impaired endothelial progenitor cell-mediated angiogenesis in diabetic mice. Acta Pharmacol Sin 44, 44–57 (2023). https://doi.org/10.1038/s41401-022-00948-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00948-w
