
Abstract
Cholestasis is a common feature of liver injury, which manifests as bile acid excretion and/or enterohepatic circulation disorders. However, very few effective therapies exist for cholestasis. Recently, 18β-Glycyrrhetinic acid (18b-GA), a major metabolic component of glycyrrhizin, which is the main ingredient of licorice, was reported to protect against alpha-naphthylisothiocyanate (ANIT)-induced cholestasis. However, its protective mechanism remains unclear. We hypothesized that 18b-GA may stimulate the signaling pathway of bile acid (BA) transportation in hepatocytes, resulting its hepatoprotective effect. According to the results, 18b-GA markedly attenuated ANIT-induced liver injury as indicated the hepatic plasma chemistry index and histopathology examination. In addition, the expression levels of nuclear factors, including Sirt1, FXR and Nrf2, and their target efflux transporters in the liver, which mainly mediate bile acid homeostasis in hepatocytes, significantly increased. Furthermore, we first revealed that 18b-GA treatment significantly activated FXR, and which can be significantly reduced by EX-527 (a potent and selective Sirt1 inhibitor), indicating that 18b-GA activates FXR through Sirt1. Taken together, 18b-GA confers hepatoprotection against ANIT-induced cholestasis by activating FXR through Sirt1, which promotes gene expression of the efflux transporter, and consequently attenuates dysregulation of bile acid homeostasis in hepatocyte compartments.
Similar content being viewed by others


Introduction
Cholestasis is clinically associated with a variety of liver diseases, has a very high incidence, and mainly manifests as a bile secretion disorder and excessive bile acid (BA) accumulation in the liver. Without effective intervention, cholestasis will eventually evolve into hepatic fibrosis and cirrhosis [1, 2]. The etiology of cholestasis is more complex, but cholestatic disease due to any cause can lead to the retention of toxic substances, such as BA, leading to liver damage and cholestatic liver disease [3,4,5,6]. Currently, obeticholic acid and ursodeoxycholic acid (UDCA) are two FDA-approved therapeutic drugs used as single agents or in combination for primary biliary cholangitis (PBC) treatment in adults, while there are no approved drugs for other cholesteric diseases that are found throughout the world [7, 8]. Therefore, novel therapeutic strategies for the treatment of cholestasis are needed.
Many traditional Chinese medicines have been shown to protect against liver diseases, such as hepatitis, cholestasis and liver fibrosis [9]. Glycyrrhizin, which is the principal triterpene component of licorice root, has been used to treat patients with chronic hepatitis B in China and Japan for many years [10]. 18β-Glycyrrhetinic acid (18b-GA) is a product of glycyrrhizin metabolism through the intestinal flora [11]. 18b-GA has recently been reported to have anti-inflammatory [12], anticancer [13, 14], and hepatoprotective [15] effects. In addition, recent evidence indicates that 18b-GA exerts a powerful protective effect in multiple liver injury models, including models of exposure to free fatty acids [16], BA [9], a choline-deficient l-amino acid-defined diet [17], and carbon tetrachloride [18]. Although 18b-GA has been reported to protect against alpha-naphthylisothiocyanate (ANIT)-induced hepatotoxicity [10, 19], the mechanism remains unclear.
Interrupting BA metabolism and transportation in hepatocyte compartments is widely believed to play a role in cholestasis. BAs are steroid acids that are mainly synthesized in the liver via cholesterol oxidation and are subsequently secreted to help digest fats [20]. When BA metabolism is impaired, BAs accumulate in the liver at high concentrations, which may cause hepatocyte apoptosis and necrosis [6]. Additionally, abnormal BA transporter and enzyme expression and function may cause BA retention in the liver, ultimately leading to cholestasis [21, 22]. The expression and function of BA transporters and enzymes can be regulated by nuclear receptors and transcription factors. Farnesoid X receptor (FXR) plays important regulatory roles in repressing BA synthetic enzymes, inhibiting the hepatic uptake transporter, inducing bile efflux transporters and increasing BA metabolism in the liver [23]. Nuclear factor-E2-related factor-2 (Nrf2), which is known to be mainly involved in the body’s antioxidant response [24], also participates in mediating phase II drug metabolism enzymes and the efflux transporters [25]. Recent studies have reported that Sirt1, a nuclear factor family receptor, can regulate the activities of FXR and Nrf2, indicating that Sirt1 is a transcription factor and plays an important role in BA homeostasis [8, 26, 27]. Therefore, we hypothesized that the therapeutic effect of 18b-GA on hepatic cholestasis may be related to its regulatory effect on Sirt1 to affect downstream nuclear receptors, consequently attenuating the imbalance of metabolism and transport of BA.
In this paper, based on an assessment of the protective effect of 18b-GA on ANIT-induced intrahepatic cholestasis, we further systematically studied the effect of 18b-GA on BA transporter expression and the ability of this compound to reverse ANIT-induced toxicity. Therefore, we investigated key nuclear receptors, which are crucial in regulating BA homeostasis. Subsequent studies using luciferase assays on transfected cells were performed to explore the effect of 18b-GA on signaling pathways of Sirt1-FXR.
Materials and Methods
Animals and experimental design
All animal experiments were performed in accordance with the Institutional Ethics Committee of Shanghai Institute of Materia Medica. Eighteen 6- to 8-week-old male Sprague-Dawley specific pathogen-free (SPF)-grade rats were purchased from Shanghai Laboratory Animal Co. (Shanghai, China). They were housed under conditions of 55% humidity at 20-25 °C, provided a standard diet with water available ad libitum and were kept on a 12-h light/dark cycle. The rats were fed for one week and allowed to acclimate to the environment before the experiment. The rats were then randomly divided into three groups. Control group rats were injected intraperitoneally with 5% dimethyl sulfoxide (DMSO) in olive oil for 7 days, and olive oil was orally administered on the 5th day. ANIT-treated rats were injected intraperitoneally with 5% DMSO in olive oil for 7 days and, on the 5th day, were orally administered ANIT (dissolved in olive oil) at 100 mg/kg body weight, which was selected according to the supplemental data (Supplementary Figure S1). ANIT and 18b-GA cotreated rats were injected intraperitoneally with 18b-GA (60 mg/kg, which was selected according to preliminary data (Supplementary Figure S2) for 7 days and given ANIT (100 mg/kg) via oral administration 4 h after the 5th 18b-GA injection. On the 7th day, 48 h after ANIT (100 mg/kg) treatment, the rats were sacrificed to collect the livers and blood. Blood samples were centrifuged at 3000 rpm for 10 min and stored at −80 °C. Liver samples were immediately frozen and stored at −80 °C. ANIT, olive oil, and 18b-GA were purchased from Sigma-Aldrich (St Louis, MO, USA).
Plasma biochemistry
Reagents for measuring plasma aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), total bilirubin (TBIL), and γ-glutamyl transpeptidase (GGT) were obtained from Roche, Germany and were tested using a biochemical analyzer (Roche, Germany).
Histopathological analysis
After rats were euthanatized, the livers were fixed in 10% paraformaldehyde. Tissue samples were then dehydrated and embedded in paraffin, sectioned (4 μm thickness), and stained with hematoxylin and eosin (H&E). Histological lesions were scored by a pathologist . We evaluated tissue damage by a semi-quantitative method according to the literature [28]. For semiquantitative analyses, the area of necrosis or lesions in the detected area was scored as +1 for <25%, +2 for 25–50%, +3 for 50–75%, and +4 for >75%.
RNA isolation and qRT-PCR analysis of mRNA expression
Total RNA was extracted from 30 mg liver samples using TRIzol (Invitrogen, USA) according to the manufacturer’s instructions. cDNA was synthesized using a PrimeScript RT kit (Takara, Japan), and quantitative real-time polymerase chain reactions (qRT-PCR) were analyzed on a Rotor-Gene Q 2plex HRM system (Qiagen, USA) using a SYBR® premix Ex Taq™ II kit (Takara, Japan). The primer sequences are listed in Table 1. All results were normalized to GAPDH expression and calculated using the ΔΔCt method.
Western blotting
Rat livers (30 mg) were lysed using radio immunoprecipitation assay buffer (Beyotime, China). Protein lysates were separated on 10% SDS-PAGE gels and then transferred to PVDF membranes (Millipore, USA). The membranes were incubated overnight at 4 °C with antibodies against FXR (sc-1204, Santa Cruz Biotechnology, USA), sirtuin 1 (Sirt1) (8469, Cell Signaling Technology, USA), p-glycoprotein (P-gp; ab170904, Abcam, USA), bile salt export pump (Bsep; ab112494, Abcam, USA), multidrug resistance-associated protein 3 (Mrp3; sc-5774, Santa Cruz Biotechnology, USA) and GAPDH (ab9485, Abcam, USA). Membranes were then washed three times and incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., USA). Protein band chemiluminescence was detected using an ECL Plus immunoblot detection system (Millipore, Billerica, MA, USA), and protein band density was quantified by ImageQuant software (GE Healthcare, UK).
Primary rat hepatocyte isolation and sandwich culture
Primary hepatocytes were isolated from adult male Sprague-Dawley rats by a two-step collagenase perfusion method and purified by 45% isotonic Percoll [29]. The cell treatments and culture methods have been described previously [30].
Under Mrp2 transport in cells, 5(and 6)-carboxy-2′,7′-dichlorofluorescein (CDF), which is a probe substrate for Mrp2, is hydrolyzed from a fluorescent CDF diacetate promoiety (CDF-DA). CDF fluorescence intensity is indicative of Mrp2 transport function. CDF-DA was obtained from Sigma-Aldrich (St Louis, MO, USA).
FXR luciferase assay
The luciferase reporter expression plasmids pcDNA3.1-FXRα, pcDNA3.1-RXRα, and pGL3-FXRE-Luc and the Renilla luciferase gene-containing plasmid pRL-SV40 were kind gifts from Prof. Xu Shen (School of Medicine and Life Sciences, Nanjing University of Chinese Medicine, Nanjing, China). We seeded Huh7 cells (ATCC, USA) in 48-well plates for luciferase assay experiments. The experimental method was consistent with that of Prof. Shen’s laboratory [31]. Cell extracts were collected at 24 h after transfection, and luciferase activity was measured using a Dual-Luciferase reporter assay system (Promega, Madison, WI, USA). Renilla luciferase was used for normalization.
Statistical analysis
Data were statistically analyzed and graphed using GraphPad Prism software (version 5.03; GraphPad Software, Inc., CA, USA). All cell experiments were performed independently at least three times. The data are expressed as the mean ± SEM, and Student’s t-tests were used to analyze the significance of differences. P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
18b-GA has anti-cholestatic and hepatoprotective effects in vivo
To estimate the protective effect of 18b-GA on ANIT-induced hepatotoxicity and cholestasis, rats were intraperitoneally injected with 18b-GA (60 mg/kg) for 7 days, and ANIT was administered orally (100 mg/kg) 48 h before euthanasia. Then, plasma biochemistry was analyzed. Plasma ALT, AST, ALP, GGT, and TBIL levels were increased by 31.2-, 33.4-, 5.1-, 5.0-, and 91.3-fold, respectively, in ANIT-induced rats (P < 0.0001), and all levels were significantly decreased by 62.0%, 38.5%, 45.7%, 51.6%, and 39.7%, respectively, after 18b-GA treatment (P < 0.05) (Fig. 1a–e).

Protective effects of 18b-GA against ANIT-induced cholestasis in rats. Rats were intraperitoneally administered 18b-GA (60 mg/kg) or vehicle for 7 days and were orally provided ANIT (100 mg/kg, ig) or vehicle after the 5th 18b-GA dose. The levels of biochemical indicators (a) ALT, (b) AST, (c) ALP, (d) GGT, and (e) TBIL were measured. Blood samples were collected and analyzed, as described in the Materials and Methods. Data are shown as the mean ± SEM. (n = 6). *P < 0.05, ** P < 0.01, ***P < 0.001 compared with the ANIT-treated group
H&E staining of liver sections showed that compared with the control group, the ANIT-induced group displayed multifocal portal vein degeneration/coagulation necrosis, fibrosis, neutrophil and monocyte accumulation, and bile duct epithelial cell apoptosis and hypertrophy. 18b-GA and ANIT coadministration significantly improved the above symptoms in bile duct epithelial cells, as shown in Fig. 2a. Histological scores revealed a significant improvement in the animals treated with 18b-GA (Fig. 2b).

Liver histology of ANIT-treated rats and 18b-GA-attenuated liver injury induced by ANIT. a Liver tissues were fixed, followed by H&E staining (×200). No histological change was observed in the control group, large areas of necrosis and inflammation were observed in the ANIT-treated group, and a few spotty areas of necrosis and inflammation were observed in the ANIT and 18b-GA cotreated group. b The graph shows the semi-quantitative analysis of cholangiocyte inflammation, necrosis, hypertrophy, and fibrosis. Data are shown as the mean ± SEM. (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the ANIT-treated group
18b-GA altered receptor factors involved in BA homeostasis
To elucidate the protective effects of 18b-GA in ANIT-induced cholestasis, we measured the gene expression of several nuclear receptors and transcription factors associated with BA homeostasis. As shown in Fig. 3a, 18b-GA and ANIT cotreatment significantly increased FXR, Nrf2, and Sirt1 mRNA levels and had little effect on NF-ƙB, Ahr, and Hnf4α levels. These data demonstrated that FXR, Nrf2, and Sirt1, but not NF-ƙB, Ahr, and Hnf4α, are key factors in the anti-cholestatic effect of 18b-GA. According to the literature, 18b-GA may protect the liver through Nrf2-induced Mrp3/4 upregulation (Fig. 3b). Next, FXR target genes in rats were studied by real-time PCR, which revealed involvement of Shp, Bsep, and Mrp2 (Fig. 3c). These data revealed that 18b-GA regulates the BA balance through FXR.

Effects of 18b-GA on bile acid nuclear receptor and transcription factor homeostasis in vivo. Quantitative real-time PCR analysis was performed to measure (a) the BA transcription factor; (b) the expression levels of Nrf2 target genes, including Mrp3 and Mrp4; and (c) the expression levels of FXR target genes, including Shp, Bsep, and Mrp2. d Western blot was used to measure transcription factor and transporter levels. Data are reported as the mean ± SEM. (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the ANIT-treated group
To verify the accuracy of the qRT-PCR results on the induction of transporters and nuclear factors by 18b-GA, we measured protein levels using western blot analysis. The results revealed that ANIT treatment marginally changed Bsep and FXR protein expression and slightly increased P-gp, Mrp3 and Sirt1 protein expression. 18b-GA cotreatment dramatically increased Bsep, P-gp, Mrp3, FXR, and Sirt1 protein levels (Fig. 3d). Therefore, 18b-GA and ANIT coadministration can increase Bsep, Mrp3, P-gp, FXR, and Sirt1 mRNA and protein expression. This finding suggested that the protective effect of 18b-GA against cholestasis may be a result of enhanced BA output, which is possibly mediated via FXR or Sirt1 activation.
18b-GA activated FXR through Sirt1 in vitro
As FXR plays an important role in regulating BA homeostasis, we tested whether 18b-GA exerted protective effects against cholestasis through modulating FXR activities. Interestingly, 18b-GA exposure resulted in dose-dependent FXR activation when the FXR ligand chenodeoxycholic acid (CDCA) was present, and 18b-GA exposure did not activate FXR when CDCA was absent. (Fig. 4a, b), as indicated by the FXR luciferase assay in Huh7 cells (SRT1720, a selective activator of human Sirt1, as a positive control). Furthermore, as Sirt1 is a mediator of FXR, we investigated whether 18b-GA activated FXR through Sirt1. The results revealed that 18b-GA treatment significantly activated FXR, which was significantly reduced by EX-527 (a potent and selective Sirt1 inhibitor) (Fig. 4c), indicating that 18b-GA-mediated FXR activation is associated with Sirt1. Similar results were obtained when another tool, HEK293T cells, were used in the FXR luciferase assay (Supplementary Figure S3).

18b-GA activates FXR in vitro. After transfection with FXR plasmid DNA for 6 h, the dose-dependent response of Huh7 cells to 18b-GA exposure for 24 h with (a) and without (b) CDCA (50 μM), an effective FXR endogenous ligand, was measured. (c) Luciferase reporter assays confirmed that FXR is regulated by Sirt1. 18b-GA treatment significantly activated FXR, and FXR activity was significantly reduced by EX-527. SRT1720, a selective activator of human Sirt1, and EX-527, a potent and selective Sirt1 inhibitor, were used. Data are reported as the mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 compared with DMSO, #P < 0.05 compared with 18b-GA
18b-GA regulated FXR activity and FXR target gene expression via Sirt1
Since 18b-GA activated FXR through Sirt1 in vitro, we further evaluated the protective effect of 18b-GA against EX-527 by transporters and nuclear factors in sandwich-cultured rat hepatocytes (SCRHs). As illustrated in Fig. 5a, 18b-GA can significantly increase Sirt1 and FXR mRNA levels, which are significantly reduced by EX-527. Then, we examined the mRNA levels of downstream FXR genes, such as Shp, Bsep and Mrp2. The results revealed that 18b-GA significantly increased Shp, Bsep and Mrp2 mRNA expression, which was inhibited by EX-527 (Fig. 5b). In addition, cells were pretreated with 18b-GA for 1 day and then re-treated with ANIT for 24 h, as shown in Fig. 5c. After treatment with ANIT, the fluorescence substrate of Mrp2, CDF, was decreased. Compared with ANIT treatment, 18b-GA and SRT1720 (a selective activator of human Sirt1) cotreatment increased CDF fluorescence intensity. ANIT treatment reduced the fluorescence intensity of CDF, which was reversed when the cells were cotreated with 18b-GA and ANIT, thus increasing the intensity of the fluorescent substrate of Mrp2. These results indicated that 18b-GA can protect against ANIT-induced impairment of the efflux function of Mrp2. Taken together, these observations indicated that 18b-GA ameliorated ANIT-induced cholestasis through the Sirt1/FXR pathway.

18b-GA upregulates the expression of FXR and FXR target genes, and 18b-GA promotes Mrp2 function in SCRHs. Rat primary hepatocytes were treated with 18b-GA (30 μM) and/or EX-527 (10 μM) for 3 days and with ANIT (40 μM) on the 4th day. RNA isolation and quantitative real-time PCR analysis were performed to measure the levels of the transcription factors Sirt1 and FXR (a) and FXR target genes (b). Data are reported as the mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 compared with ANIT and 18b-GA cotreatment). c Intracellular amounts of CDF were determined by spectrofluorimetry. Rat primary hepatocytes were treated with ANIT (40 μM) and 18b-GA (7.5, 15, 30 μM); 18b-GA pretreatment lasted for 1 day, and cells were treated with ANIT the following day. Then, a fluorescent substrate of Mrp2, CDF, was added. The fluorescence represents the function of the Mrp2-related canalicular efflux pump. Scale bar = 100 μm
Discussion
Our data highlight the protective effect of 18b-GA against ANIT-induced liver injury by stimulating the expression of Sirt1, FXR and Nrf2 nuclear factors. Nrf2 activation stimulated the expression of its targeted efflux transporter gene (Mrp3, Mrp4) and regulated bile acid homeostasis. In addition, 18b-GA treatment significantly activated FXR, which promotes the expression of its targeted efflux transporter gene (Mrp2, Bsep), and this effect was prevented by the selective Sirt1 inhibitor EX-527. Therefore, 18b-GA activated FXR through activation of Sirt1 and exerted a hepatoprotective effect against ANIT-induced cholestasis, which promotes the expression of efflux transporter genes, thus impairing bile acid homeostasis disorders in hepatocytes.
ANIT, a hepatotoxic agent that is widely used to induce intrahepatic cholestasis in rodents, leads to cholestasis by impairing parenchymal hepatic cell polarization, directly injuring biliary epithelial cells and inhibiting BA transporter function and expression [32]. According to the literature, treating rats with ANIT significantly decreases bile flow, and cotreatment with ANIT and 18b-GA significantly increases bile flow [10]. We speculated that a close relationship exists between increased bile flow and BA efflux. Since BA content is regulated by various nuclear factors, we investigated whether the expression of these genes was changed in the liver by 18b-GA cotreatment. The results revealed that compared with the ANIT treatment group, 18b-GA cotreatment significantly increased FXR, Nrf2 and Sirt1 gene expression levels. However, changes in BA-related nuclear receptors induced by ANIT are inconsistent across studies [21, 23, 33,34,35,36,37,38,39]. Our results show that compared with the control rats, the levels of FXR, Sirt1, and Nrf2 mRNA in the rats treated with ANIT did not change. The diversity of this effect may be due to the different experimental conditions and the complex regulation of nuclear receptors, i.e., the negative feedback of BA balance, the body’s stress response [35, 36], cross-regulation of BA in the body [39], and even intestinal flora [40, 41].
Activating Nrf2 may ameliorate bile duct ligation (BDL)- or ANIT-induced cholestasis [7, 42, 43]. Recent studies have reported that Mrp2, Mrp3, and Mrp4 are direct Nrf2 target genes whose expression was increased by prototypical Nrf2 chemical activators in rodent livers and decreased to basal expression levels in Nrf2-knockout mice [44,45,46]. Our results revealed that 18b-GA significantly increased Mrp2, Mrp3, and Mrp4 mRNA expression (Fig. 3c). The protective effect of 18b-GA in ANIT-induced cholestasis has been indicated to be due to increased bile efflux, which is closely related to Nrf2-mediated Mrp2, Mrp3, and Mrp4 upregulation. Under baseline conditions, FXR-knockout (FXR−/−) mice develop plasma BA, and when fed cholic acid, these mice develop severe liver toxicity [47, 48]. Previous studies have demonstrated that the synthetic FXR agonist GW4064 improves cholestatic symptoms induced by ANIT [49] and BDL [50]. To further confirm the effect of 18b-GA on the FXR pathway, the expression of FXR and FXR target genes, including Bsep, Mrp2, and Shp, were quantified in rats treated with 18b-GA. The results demonstrated that 18b-GA improved FXR, Shp, Bsep and Mrp2 expression, as shown in Fig. 3b, d, indicating that 18b-GA alleviates liver injury via activation of the FXR/Shp signaling pathway.
Sirt1, a class III NAD-dependent histone deacetylase, plays a key role in lipid, glucose, and BA metabolism. Sirt1 and FXR can form an interactive regulatory network: acetylation stabilizes FXR but reduces its activity by decreasing FXR/RXR heterodimerization and the ability of FXR to bind FXRE and transactivate other genes [51, 52]. Liver-specific Sirt1 deletion can lead to BA metabolic dysfunction by downregulating FXR signaling and affects cholesterol gallstone development in lithogenic diets, which can be reversed by Sirt1 overexpression [53]. To analyze the influence of 18b-GA on Sirt1/FXR signaling, we first tested whether 18b-GA can activate FXR in Huh7 cells. In the presence of CDCA, an endogenous FXR ligand, 18b-GA activated FXR in a dose-dependent manner; this effect did not occur in the absence of CDCA (Fig. 4b). Reports in the literature have also demonstrated that when no ligand is present, FXR remains in a silent state [54]. Subsequently, we found that 18b-GA activated FXR, and this effect was prevented by EX-527, a potent and selective Sirt1 inhibitor, indicating that 18b-GA-mediated FXR activation is associated with Sirt1.
To further analyze the effect of 18b-GA on the Sirt1/FXR pathway in vitro, Sirt1 and FXR target gene expression levels were quantified in SCRHs. The results demonstrated that 18b-GA increased Sirt1, FXR, Shp, Mrp2, and Bsep expression levels, which were significantly inhibited by EX-527 (Fig. 5a, b). In addition, we found that CDF fluorescence intensity was markedly increased in 18b-GA-treated SCRHs compared with that in ANIT-treated SCRHs (Fig. 5c), indicating that 18b-GA can improve BA efflux into bile. As CDF is a substrate of efflux transporter Mrp2, its fluorescence in bile pockets of SCRHs may reflect the function of efflux transporters at the bile canalicular side of hepatocytes.
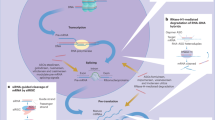
In conclusion, the current study indicates that 18b-GA protects against ANIT-induced cholestasis through activation of the Sirt1/FXR signaling pathway and the Sirt1/Nrf2 signaling pathway (Fig. 6). However, how 18b-GA activates FXR through Sirt1 is still unclear. Further studies focusing on molecular structure may help determine their interactive mechanism.

The possible mechanism of Sirt1/FXR activation by 18b-GA in an ANIT-induced cholestasis rat model. 18b-GA can activate Sirt1 and promote the binding of FXR and RXR, which promotes upregulation of canalicular efflux transporters and entry into the bile. On the other hand, activation of Sirt1 promotes the expression of Nrf2, leading to marked upregulation of the basolateral efflux transporters, thereby promoting bile into the blood. ARE: antioxidant response element, RXR: retinoid X receptor, FXRE: FXR-responsive element
References
Chawla A, Saez E, Evans RM. Don’t know much bile-ology. Cell. 2000;103:1–4.
Chiang JY. Recent advances in understanding bile acid homeostasis. F1000Res. 2017;6:2029.
Kang YZ, Sun XY, Liu YH, Shen ZY. Autoimmune hepatitis-primary biliary cirrhosis concurrent with biliary stricture after liver transplantation. World J Gastroenterol. 2015;21:2236–41.
Liu J, Wu KC, Lu YF, Ekuase E, Klaassen CD. Nrf2 protection against liver injury produced by various hepatotoxicants. Oxid Med Cell Longev. 2013;2013:305861.
Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4:25–36.
Wu JS, Li YF, Li YY, Dai Y, Li WK, Zheng M, et al. Huangqi decoction alleviates alpha-naphthylisothiocyanate induced intrahepatic cholestasis by reversing disordered bile acid and glutathione homeostasis in mice. Front Pharmacol. 2017;8:938.
Tanaka A, Gershwin ME. Finding the cure for primary biliary cholangitis - still waiting. Liver Int. 2017;37:500–2.
Yu L, Liu X, Yuan Z, Li X, Yang H, Yuan Z, et al. SRT1720 alleviates ANIT-induced cholestasis in a mouse model. Front Pharmacol. 2017;8:256.
Gumpricht E, Dahl R, Devereaux MW, Sokol RJ. Licorice compounds glycyrrhizin and 18beta-glycyrrhetinic acid are potent modulators of bile acid-induced cytotoxicity in rat hepatocytes. J Biol Chem. 2005;280:10556–63.
Zhai D, Zhao Y, Chen X, Guo J, He H, Yu Q, et al. Protective effect of glycyrrhizin, glycyrrhetic acid and matrine on acute cholestasis induced by alpha-naphthyl isothiocyanate in rats. Planta Med. 2007;73:128–33.
Akao T, Akao T, Hattori M, Kanaoka M, Yamamoto K, Namba T, et al. Hydrolysis of glycyrrhizin to 18 beta-glycyrrhetyl monoglucuronide by lysosomal beta-D-glucuronidase of animal livers. Biochem Pharmacol. 1991;41:1025–9.
Huang QC, Wang MJ, Chen XM, Yu WL, Chu YL, He XH, et al. Can active components of licorice, glycyrrhizin and glycyrrhetinic acid, lick rheumatoid arthritis? Oncotarget. 2016;7:1193–202.
Fiore C, Eisenhut M, Krausse R, Ragazzi E, Pellati D, Armanini D, et al. Antiviral effects of Glycyrrhiza species. Phytother Res. 2008;22:141–8.
Kong SZ, Chen HM, Yu XT, Zhang X, Feng XX, Kang XH, et al. The protective effect of 18beta-Glycyrrhetinic acid against UV irradiation induced photoaging in mice. Exp Gerontol. 2015;61:147–55.
Jeong HG, You HJ, Park SJ, Moon AR, Chung YC, Kang SK, et al. Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon tetrachloride-induced liver injury: inhibition of cytochrome P450 2E1 expression. Pharmacol Res. 2002;46:221–7.
Wu X, Zhang L, Gurley E, Studer E, Shang J, Wang T, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47:1905–15.
Makino T, Ohtake N, Watanabe A, Tsuchiya N, Imamura S, Iizuka S, et al. Down-regulation of a hepatic transporter multidrug resistance-associated protein 2 is involved in alteration of pharmacokinetics of glycyrrhizin and its metabolites in a rat model of chronic liver injury. Drug Metab Dispos. 2008;36:1438–43.
Chen S, Zou L, Li L, Wu T. The protective effect of glycyrrhetinic acid on carbon tetrachloride-induced chronic liver fibrosis in mice via upregulation of Nrf2. PLoS One. 2013;8:e53662.
Wang H, Fang ZZ, Meng R, Cao YF, Tanaka N, Krausz KW, et al. Glycyrrhizin and glycyrrhetinic acid inhibits alpha-naphthyl isothiocyanate-induced liver injury and bile acid cycle disruption. Toxicology. 2017;386:133–42.
Marin JJG, Macias RIR, Briz O, Banales JM, Monte MJ. Bile acids in physiology, pathology and pharmacology. Curr Drug Metab. 2016;17:4–29.
Xu L, Sheng T, Liu X, Zhang T, Wang Z, Han H. Analyzing the hepatoprotective effect of the Swertia cincta Burkillextract against ANIT-induced cholestasis in rats by modulating the expression of transporters and metabolic enzymes. J Ethnopharmacol. 2017;209:91–9.
Cui YJ, Aleksunes LM, Tanaka Y, Goedken MJ, Klaassen CD. Compensatory induction of liver efflux transporters in response to ANIT-induced liver injury is impaired in FXR-null mice. Toxicol Sci. 2009;110:47–60.
Gao X, Fu T, Wang C, Ning C, Liu K, Liu Z, et al. Yangonin protects against cholestasis and hepatotoxity via activation of farnesoid X receptor in vivo and in vitro. Toxicol Appl Pharmacol. 2018;348:105–16.
Wang X, Li C, Xu S, Ishfaq M, Zhang X. NF-E2-related factor 2 deletion facilitates hepatic fatty acids metabolism disorder induced by high-fat diet via regulating related genes in mice. Food Chem Toxicol. 2016;94:186–96.
Shen G, Kong AN. Nrf2 plays an important role in coordinated regulation of phase II drug metabolism enzymes and phase III drug transporters. Biopharm Drug Dispos. 2009;30:345–55.
Tang W, Jiang YF, Ponnusamy M, Diallo M. Role of Nrf2 in chronic liver disease. World J Gastroenterol. 2014;20:13079–87.
You M, Jogasuria A, Taylor C, Wu J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg Nutr. 2015;4:88–100.
Quaglia A, Duarte R, Patch D, Ngianga-Bakwin K, Dhillon AP. Histopathology of graft versus host disease of the liver. Histopathology. 2007;50:727–38.
Kotani N, Maeda K, Watanabe T, Hiramatsu M, Gong LK, Bi YA, et al. Culture period-dependent changes in the uptake of transporter substrates in sandwich-cultured rat and human hepatocytes. Drug Metab Dispos. 2011;39:1503–10.
Guo C, He L, Yao D, A J, Cao B, Ren J, et al. Alpha-naphthylisothiocyanate modulates hepatobiliary transporters in sandwich-cultured rat hepatocytes. Toxicol Lett. 2014;224:93–100.
Xu X, Xu X, Liu P, Zhu ZY, Chen J, Fu HA, et al. Structural basis for small molecule NDB (N-benzyl-N-(3-(tert-butyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide) as a selective antagonist of farnesoid X receptor alpha (FXR alpha) in stabilizing the homodimerization of the receptor. J Biol Chem. 2015;290:19888–99.
Yang T, Mei H, Xu D, Zhou W, Zhu X, Sun L, et al. Early indications of ANIT-induced cholestatic liver injury: Alteration of hepatocyte polarization and bile acid homeostasis. Food Chem Toxicol. 2017;110:1–12.
Yan JY, Xie GX, Liang CG, Hu YY, Zhao AH, Huang FJ, et al. Herbal medicine Yinchenhaotang protects against alpha-naphthylisothiocyanate-induced cholestasis in rats. Sci Rep. 2017;7:4211.
Meng Q, Chen XL, Wang CY, Liu Q, Sun HJ, Sun PY, et al. Alisol B 23-acetate protects against ANIT-induced hepatotoxity and cholestasis, due to FXR-mediated regulation of transporters and enzymes involved in bile acid homeostasis. Toxicol Appl Pharmacol. 2015;283:178–86.
Tanaka Y, Aleksunes LM, Cui YJ, Klaassen CD. ANIT-induced intrahepatic cholestasis alters hepatobiliary transporter expression via Nrf2-dependent and independent signaling. Toxicol Sci. 2009;108:247–57.
Aleksunes LM, Slitt AM, Cherrington NJ, Thibodeau MS, Klaassen CD, Manautou JE. Differential expression of mouse hepatic transporter genes in response to acetaminophen and carbon tetrachloride. Toxicol Sci. 2005;83:44–52.
Wang Y, Jiang YM, Fan XM, Tan HS, Zeng H, Wang YT, et al. Hepato-protective effect of resveratrol against acetaminophen-induced liver injury is associated with inhibition of CYP-mediated bioactivation and regulation of SIRT1-p53 signaling pathways. Toxicol Lett. 2015;236:82–9.
Hu XD, Jogasuria A, Wang JY, Kim C, Han Y, Shen H, et al. MitoNEET deficiency alleviates experimental alcoholic steatohepatitis in mice by stimulating endocrine adiponectin-Fgf15 axis. J Biol Chem. 2016;291:22482–95.
Fu ZD, Cui JY, Klaassen CD. The role of Sirt1 in bile acid regulation during calorie restriction in mice. PLoS One. 2015;10:e0138307.
Kazgan N, Metukuri MR, Purushotham A, Lu J, Rao A, Lee S, et al. Intestine-specific deletion of SIRT1 in mice impairs DCoH2-HNF-1 alpha-FXR signaling and alters systemic bile acid homeostasis. Gastroenterology. 2014;146:1006–16.
Clark A, Mach N. The crosstalk between the gut microbiota and mitochondria during exercise. Front Physiol. 2017;8:319.
Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, et al. Nrf2 counteracts cholestatic liver injury via stimulation of hepatic defense systems. Biochem Biophys Res Commun. 2009;389:431–6.
Aleksunes LM, Slitt AL, Maher JM, Dieter MZ, Knight TR, Goedken M, et al. Nuclear factor-E2-related factor 2 expression in liver is critical for induction of NAD(P)H: quinone oxidoreductase 1 during cholestasis. Cell Stress Chaperon. 2006;11:356–63.
Maher JM, Dieter MZ, Aleksunes LM, Slitt AL, Guo G, Tanaka Y, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46:1597–610.
Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G735–47.
Anwar-Mohamed A, Degenhardt OS, El Gendy MA, Seubert JM, Kleeberger SR, El-Kadi AO. The effect of Nrf2 knockout on the constitutive expression of drug metabolizing enzymes and transporters in C57Bl/6 mice livers. Toxicol Vitr. 2011;25:785–95.
Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care. 2009;32(Suppl 2):S237–45.
Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;328:116–22.
Yang F, Tang XW, Ding LL, Zhou Y, Yang QL, Gong JT, et al. Curcumin protects ANIT-induced cholestasis through signaling pathway of FXR-regulated bile acid and inflammation. Sci Rep. 2016;6:33052.
Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. 2003;112:1678–87.
Kulkarni SR, Soroka CJ, Hagey LR, Boyer JL. Sirtuin 1 activation alleviates cholestatic liver injury in a cholic acid-fed mouse model of cholestasis. Hepatology. 2016;64:2151–64.
Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, et al. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 2009;10:392–404.
Purushotham A, Xu Q, Lu J, Foley JF, Yan XJ, Kim DH, et al. Hepatic deletion of SIRT1 decreases hepatocyte nuclear factor 1 alpha/Farnesoid X receptor signaling and induces formation of cholesterol gallstones in mice. Mol Cell Biol. 2012;32:1226–36.
Kanaya E, Shiraki T, Jingami H. The nuclear bile acid receptor FXR is activated by PGC-1 alpha in a ligand-dependent manner. Biochem J. 2004;382:913–21.
Acknowledgements
This work was supported by the National New Drug Creation Program of China (No. 2018ZX09201017-004) and the “Strategic Priority Research Program” of the Chinese Academy of Sciences (No. XDA12050305) and the National Natural Science Foundation of China (No. 81403028)
Author contributions
SW, LG, GX, and JR designed the research project; SW, SC, LW, YZ, and XY performed the experiments; SW, SC, and LG analyzed the data; HL, as a pathologist, scored the histological lesions; and SW and LG wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wu, Sy., Cui, Sc., Wang, L. et al. 18β-Glycyrrhetinic acid protects against alpha-naphthylisothiocyanate-induced cholestasis through activation of the Sirt1/FXR signaling pathway. Acta Pharmacol Sin 39, 1865–1873 (2018). https://doi.org/10.1038/s41401-018-0110-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0110-y
Keywords
This article is cited by
-
18β-Glycyrrhetinic Acid Ameliorates Neuroinflammation Linked Depressive Behavior Instigated by Chronic Unpredictable Mild Stress via Triggering BDNF/TrkB Signaling Pathway in Rats
Neurochemical Research (2023)
-
Influence Mechanism of New-type Urbanization on Urban Land Use Efficiency in the Yangtze River Delta, China
Chinese Geographical Science (2023)
-
Furmonertinib (Alflutinib, AST2818) is a potential positive control drug comparable to rifampin for evaluation of CYP3A4 induction in sandwich-cultured primary human hepatocytes
Acta Pharmacologica Sinica (2022)
-
Research progress on the protective effects of licorice-derived 18β-glycyrrhetinic acid against liver injury
Acta Pharmacologica Sinica (2021)
-
Prevention of D-GalN/LPS-induced ALI by 18β-glycyrrhetinic acid through PXR-mediated inhibition of autophagy degradation
Cell Death & Disease (2021)

{kind=link}
{kind=link}
{kind=link}