
Abstract
Cellular metabolism is an intricate network satisfying bioenergetic and biosynthesis requirements of cells. Relevant studies have been constantly making inroads in our understanding of pathophysiology, and inspiring development of therapeutics. As a crucial component of epigenetics at post-transcription level, RNA modification significantly determines RNA fates, further affecting various biological processes and cellular phenotypes. To be noted, immunometabolism defines the metabolic alterations occur on immune cells in different stages and immunological contexts. In this review, we characterize the distribution features, modifying mechanisms and biological functions of 8 RNA modifications, including N6-methyladenosine (m6A), N6,2′-O-dimethyladenosine (m6Am), N1-methyladenosine (m1A), 5-methylcytosine (m5C), N4-acetylcytosine (ac4C), N7-methylguanosine (m7G), Pseudouridine (Ψ), adenosine-to-inosine (A-to-I) editing, which are relatively the most studied types. Then regulatory roles of these RNA modification on metabolism in diverse health and disease contexts are comprehensively described, categorized as glucose, lipid, amino acid, and mitochondrial metabolism. And we highlight the regulation of RNA modifications on immunometabolism, further influencing immune responses. Above all, we provide a thorough discussion about clinical implications of RNA modification in metabolism-targeted therapy and immunotherapy, progression of RNA modification-targeted agents, and its potential in RNA-targeted therapeutics. Eventually, we give legitimate perspectives for future researches in this field from methodological requirements, mechanistic insights, to therapeutic applications.
Similar content being viewed by others
Introduction
Since the first documentation of RNA modification as early as 1950s, over 170 types have been identified, ubiquitously existing in coding RNAs and non-coding RNAs.1 The contributions of nucleoside base modifications in developing mRNA vaccine against COVID-19, which is awarded the Nobel Prize for 2023, immensely refresh the biologists studying RNA-based therapeutics. In most situations, the RNA modifications we talked about are reversible type, similar to DNA methylation. These modifications are deposited, removed and recognized by dedicated machineries, composed of writers, erasers and readers. These post-transcriptional modifications alter the canonical ribose and base structure to determine RNA fates, including splicing, trafficking, degradation, translation, and so on. Via regulating gene expression and cellular phenotypes, RNA modifications are extensively involved in various cellular processes.2
Cellular metabolism, a sophisticated network involving multitudes of biochemical reactions, continuously invigorates scientific researches. “Metabolism reprogramming” was originally proposed in cancer research, and gradually expanded to other non-tumor diseases and normal physiological processes. Used to be defined as “changes of tumor cellular bioenergetics”, the current perception tends to regard it as an inherent adaptive capacity of all cells, which is strengthened in tumor cells via abnormally activated pre-existing processes.3 Such metabolic adaptability is based on the interaction between cells and environment. During these biochemical processes, epigenetic modifications adjust the cell-environment relationship in a context-dependent manner.
According to the alarming statistics of several recent public health researches, metabolic diseases appear as an increasingly severe burden in human society. There have been more than 1.9 billion adults and over 650 million adults qualified as obese and overweight globally in 2016.4 According to International Diabetes Federation, 537 million adults had diabetes in 2021, which has become the ninth major cause of death worldwide.5 NAFLD is the most common chronic liver disease worldwide, the global prevalence of which was 25%.6 On the other hand, recent clinical trials targeting cancer metabolism come out with unsatisfactory efficacy and frustrating adverse reactions.7,8,9 Leaving dietary interventions alone, metabolic therapy is divided into agents targeting nucleotide metabolism and non-nucleotide metabolism. Not a few metabolic drugs targeting nucleotide metabolism, mostly nucleotide analogs, have been commonly employed in clinical practice. But development of the non-nucleotide metabolism-targeted drug remains in its nascent stages.
Therefore, for better insights into pathophysiology and optimized therapeutic strategies, integrated multi-omics analysis of metabolism is imperative. Not a few excellent studies have discussed the regulatory roles of RNA modifications on metabolism in specified pathological situations, with a particular focus on cancers.10,11,12 However, there is a deficiency of a comprehensive and wide-scale review on the epigenetic-metabolic interaction covering health and disease context.
Notably, immunometabolism is emerging field, expected to provide novel therapeutic strategies in cancer, autoimmunity, and metabolic diseases. The concept illustrates metabolic changes occurring in immune cells during their differentiation and activation processes. Studies have confirmed that various immune cells, including T cells, macrophages, NK cells, and DCs, proceeded metabolism remodeling to fulfill their specific functions in discrete contexts.13,14,15,16 According to current knowledge, RNA modification could exert influences on immunometabolism through cell-intrinsic and extrinsic mechanisms. The former is intrinsic programs, including mTORC1 signaling and metabolic-related genes expression. The latter refers to tissue microenvironment and nutrient availability.
In this review, we first introduce the history and current understanding of RNA modifications, and focus on their regulatory roles in cellular metabolism to construct epigenetic-metabolic landscape in physiological and pathological situations. And the influences of RNA modifications on immunometabolism in different immune responses is discussed separately. Eventually, we highlight the clinical implications of RNA modifications and provide perspectives for further studies.
Overview of RNA modifications
Brief history of RNA modification research
Modified nucleosides in RNA, beyond the canonical A, U, C and G, have been recognized for more than half a century. Figure 1 illustrates the historical milestones of RNA modifications research. Pseudouridine (Ψ) is the first RNA modification type to be identified in 1950s.17 In 1965, sequencing of the first biological RNA, alanine tRNA derived from yeast, confirmed 10 modification types.18 Due to technological advancement, over 170 RNA modifications have been discovered, ubiquitously existing in various coding and non-coding RNAs. However, it was not until last decade that the functional significance of RNA modifications gradually got recognized, prominently the widespread prevalence and biological functions of N6-methyladenosine (m6A).19 Following 5′ cap and 3′ poly(A) tail of messenger RNA (mRNA), internal modifications on mRNAs were identified, represented by the most common methylation m6A.20,21 These modifications were observed to exert significant roles in every link of mRNA fate, including pre-mRNA splicing, nuclear export, translation, stabilization and degradation. Transfer RNA (tRNA) modification is renowned for the largest number, with an average of 13 modifications per molecule.22 Their biological roles could be generalized in two aspects, which are maintaining the tertiary structure and facilitating codon–anticodon recognition.23 For ribosomal RNA (rRNA), RNA modifications are especially indispensable, as rRNA biogenesis is interrupted without pseudouridines and 2′-O-methyls. Modifications of long noncoding RNA (lncRNA) are mainly methyl nucleotide derivatives, including m6A and m5C.24 Though far from being elucidated, lncRNAs modifications have been revealed to influence the stability, protein interactions, and subcellular distribution of lncRNAs.25 Human small nuclear RNA (snRNA) contains 2′-O-methyls, pseudouridines, and base methylations, participating in RNA splicing reaction. At present, mainstream RNA-seq methods are incapable for comprehensive and quantitative mapping of modifications on small non-coding RNAs. Here we summarize the current knowledge of RNA modifications, focusing on the regulatory mechanisms and biological consequences of several well-learned types.

The milestone events in RNA modification field. The first RNA modification type Pseudouridine (Ψ) was discovered in 1951. Since then, other RNA modification, including m6A, m1A, m6Am, etc were discovered. Along with the accumulation of epi-transcriptomic knowledge, comprehensive databases like RNAMDB and MODOMICS were incepted. Since Liquid Chromatograph-Mass Spectrometer (LC-MS) technique was utilized for the quantitative analysis of modified ribonucleosides in 2015, more specific high-throughput mapping methods gradually emerged. Recent years have witnessed the application of single-cell sequencing technologies in mapping RNA modification. The figure is generated with BioRender (https://biorender.com)
Main types of RNA modification
To build a general intuition of RNA modifications, we first sketch the distinctions between reversible and non-reversible modifications. Reversible types are usually smaller-scale modifications on chemical side chains, spanning from simple methylation to some appendages of large-molecular mass. These plastic and reversible RNA modifications extensively exist in gene regulation and cellular states. The extensive catalog of nonreversible RNA modifications includes RNA editing, splicing, and transcript-content modification (such as intron retention). Contrary to the reversible type, these modifications directly alter the sequence information, magnifying plasticity and diversity of transcriptome. The chemical structure and distribution of eight RNA modifications were showed in Fig. 2 and Table 1.

The chemical structure, distribution, and molecular functions of eight RNA modifications. Chemical modification occurs on many types of RNA and modulate every links of RNA metabolism. m6A N6-methyladenosine, m6Am N6,2′-O-dimethyladenosine, m5C 5-methylcytosine, m1A N1-methyladenosine, m7G 7-methylguanosine, ac4C N4-acetylcytidine, ψ pseudouridine, A-to-I editing adenosine-to-inosine RNA editing, CDS coding sequence, UTR untranslated regions, pri-miRNA primary microRNA, pre-miRNA precursor microRNA. The figure is generated with BioRender (https://biorender.com)
Adenosine modification
N6-methyladenosine (m6A)
m6A modification refers to the methylation of the adenosine base at the N-6 position. m6A targeted at consensus sequences DRACH (D = G, A, or U; R = G or A; H = A, C, or U), which are mainly enriched in CDS and 3’UTR region of mRNA,26 as well as most non-coding RNAs, including rRNAs, lncRNAs, circular RNAs (circRNAs), microRNAs (miRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs).27 Growing studies have confirmed that m6A could exert significant impacts on various biological processes in mammals, including DNA damage response, cell cycle, circadian rhythm, heat shock response, meiotic progression, development of hematopoietic, central nervous and reproductive systems, myogenesis, and fat differentiation.28,29,30,31,32,33
m6A deposition in mRNA is dependent on methyltransferase complex (MTC), of which the methyltransferase-like 3/14 (METTL3/14) heterodimer is the key component.34 Therein, METTL3 exerts catalytic role via transferring methyl group of S-adenosyl methionine (SAM) and METTL14 provides structural support. In METTL3, two methyltransferase domains (MTD) bind to methyl donors, CCH-type zinc finger domain (ZFD) recognizes targets, while nuclear localization signal (NLS) domain and leading helix structure (LH) domain coordinately mediate the interaction between METTL3 and METTL4.35,36 There are several auxiliary subunits for localizing and initiating methylation, including Wilms’ tumor 1-associating protein (WTAP), RNA-binding motif protein 15/15B (RBM15/15B), zinc finger CCCH-type containing 13 (ZC3H13) and vir-like m6A methyltransferase-associated (VIRMA, also known as KIAA1429).37,38,39 METTL16 is responsible for m6A formation in U6 snRNA, targeting a conserved UACAGAGAA sequence.40 METTL16 also participates in maintaining homeostasis of SAM in a m6A-dependent manner.41
Zinc Finger CCHC-Type Containing 4 (ZCCHC4) and METTL5 mediate m6A modification of 28 S and 18 S rRNA at A4220 and A1832 region, respectively.42,43 Both m6A demethylases, Fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5), belong to AlkB family of the Fe (II)/α-ketoglutarate-dependent dioxygenase superfamily. FTO is the first discovered m6A eraser for mRNA and snRNA, which also mediates demethylation of N6,2′-O-dimethyladenosine (m6Am) and N1-methyladenosine (m1A).44,45 Which one of m6A and m6Am is the principal substrate of FTO remains controversial. It was reported that FTO catalyzed m6A demethylation at a concentration at least twice that of m6Am.46,47 But Zhang et al. proposed that FTO equivalently demethylated m6A and m6Am deposited on the same RNA sequence.48 Significantly, Wei et al. discovered that nuclear FTO showed an affinity bias towards m6A, which tends to be inconspicuous in cytoplasm, due to altered abundance of m6A.49 Whereas, ALKBH5 exclusively catalyzes m6A demethylation in mRNA.50
The most studied readers are the YT521-B homology (YTH) domain family members, including YTHDF1/2/3 and YTHDC1/2, most of which localize to cytoplasm except for YTHDC1 in nucleus.51,52 The prevailing idea is that YTHDFs bind to different m6A-modified RNAs, but Zaccara et al. hold that all m6A-modified RNAs are subjected to YTHDFs and they act redundantly in mediating RNA degradation.53 YTHDC1 contributes to RNA splicing, nuclear export and degradation, while YTHDC2 promotes translation efficacy and decay.54,55 YTHDF1 could stabilize transcripts and initiate translation via interacting with eIF3, YTHDF3 not only facilitates translation but works in synergism with YTHDF2 in inducing mRNA degradation.56,57 The insulin-like growth factor 2 mRNA-binding protein family, IGF2BP1/2/3, is another group of readers. IGF2BPs possess 4 repetitive KH domains and bind to m6A sites with KH3/4 to stabilize transcripts and facilitate translation.58,59 The heterogeneous nuclear ribonucleoprotein (HNRNP) family includes HNRNPC, HNRNPG, and HNRNPA2B1. HNRNPs can mediate splicing of precursor (pre)-mRNAs and/or primary (pri)-miRNAs through ‘the m6A-switch’ mechanism, in which m6A alters the local structure of mRNA or lncRNA to facilitate the binding of HNRNPs.60 HNRNPA2B1 directly binds to pri-miRNAs to mediate alternative splicing. Meanwhile, its interaction with the miRNA microprocessor complex protein DGCR8 promoted primary miRNA processing.61 And HNRNPG could elicit co-transcriptional m6A-dependent alternative splicing regulation via directly binding to RNA polymerase II (RNAPII).62 Besides, proline rich coiled-coil 2 A (PRRC2A) and Staphylococcal nuclease and tudor domain-containing 1 (SND1) could serve as readers to stabilize m6A-modified RNAs.63,64
To sum up, m6A modification extensively influences fate of different RNA classes, consequently regulates various cellular processes. In mRNAs, m6A methylation can affect splicing, exportation, stabilization, degradation, and translation.65 In rRNAs, the A1832 methylation in 18 S rRNA and A4220 methylation in 28 S rRNA are essential for translation.42,43 In miRNAs, m6A could facilitate pri-miRNA processing via recruiting DGCR8,61 or downregulate several miRNAs via some exclusive mechanism.66 In lncRNAs, m6A modification could serve as a structural switch to regulate RNA-protein interactions,67 or stabilize lncRNAs to ensure its function.68 In cirRNAs, m6A could facilitate cytoplasmic export,69 translation70 and degradation.71 Moreover, m6A participates in modulating splicing and biogenesis of snRNA.72 Although m6A methylation has been widely investigated, the underlying rationales are far from clarified. For example, m6A modification could modulate RNA life via diverse mechanisms, but how these selective effects are determined in different cellular contexts remains unclear. While previous studies notably focus in mRNAs, the interplay between m6A and non-coding RNAs deserves more attention. The same is true for m6A readers, which are unheeded compared to writers and erasers. And the significance of methodology development cannot be stressed enough, as bona fide m6A mapping and elaborate edition on specific m6A sites will provide a wide scope for future researches.
N6,2′-O-dimethyladenosine (m6Am)
m6Am is produced at a 2′-O-methylated adenosine which is methylated co-transcriptionally at the N6 position. It is discovered in the first position adjacent to the 5′ cap structure in many mRNAs and snRNAs in mammals, and also found as internal modification in the snRNA U2.73 According to quantification studies, m6Am content ranges from 10% to almost 50% in mRNAs of different organisms and cell types.74 Previous studies have shown that m6Am installed by host PCIF1 on viral RNA mediated immune evasion, while host m6Am exhibited both anti-viral and pro-viral roles.75,76
The enzyme catalyzes m6Am next to the 5′ cap of mRNAs and in snRNAs is “phosphorylated CTD-interacting factor 1” (PCIF1), also known as “cap-specific adenosine methyltransferase” (CAPAM).77 The core region of PCIF1 contains the methyltransferase domain and helical domain that functions as the RNA-binding surface,78 and a specific site (m7Gsite) located between the two domains mediated the specific recognition of the m7G cap.79 It was revealed that knockout of PCIF1 altered cell proliferation under oxidative stress conditions in human HEK293T cell line.79 Another m6Am writer, METTL4 methylated the internal 2′-O methylated adenine, at position 30 in human U2 snRNA.80 METTL4 contains a C-terminal domain that is similar to METTL3, a middle domain (MID) and a N-terminal domain (NTD), which enables METTL4 works as a monomer with no need for METTL14.81 It was indicated that METTL4 was highly conserved and exclusive for U2 snRNA.82 However, overexpressed METTL4 tends to modify A instead of Am in mRNAs with consensus HMAGKD (H = A/C/U, M = A/C, K = G/U, D = A/G/U).83 Also, METTL4 was found to catalyze mt-DNA m6A in human cell line.84 Ablation of METTL4 did not influence viability of HEK293T cell line, but altered adipocyte differentiation of mouse 3T3-L1 cells.83,85
To date, FTO is the only known demethylase for m6Am, which, as mentioned above, show a substrate preference between m6A and m6Am depending on its cellular localization.49 In cytoplasm, FTO preferentially demethylates cap-adjacent m6Am and internal m6A on mRNAs, while nuclear FTO acts on m6Am in RNA Pol II-transcribed snRNAs, and internal m6Am and m6A in the snRNAs U2 and U6.49 Studies have identified that FTO distribution was correlated with cell cycle phase and regulated by casein kinase II-mediated phosphorylation.86 To be mentioned, structural analysis demonstrated that the catalytic activity of FTO was mediated by recognizing N6-methyl of adenine rather than the 2′-O methyl group of the ribose.48
There are discrepancies exist in present studies on influences of m6Am modification on gene expression, as an inherited issue from the past immature m6Am mapping methodologies. For instance, m6Am methylation was initially suggested to play a positive role in mRNA stability in a cell-type-specific manner.45 However, a recent study, developing the specific sequencing method m6Am-seq, has clarified that PCIF1 was not required for stabilization of m6Am-modified mRNAs.77 There are other studies implicated that m6Am did not have direct effects on mRNA stability.77,79 As for translation, the current cognition is that m6Am modifications in mRNA cap exert a cell-specific influence on translation.79,87 And such effects are dependent on 2′-O-methylation modification in the second nucleotide of the cap-structure.88 Moreover, the effect of m6Am modification in splicing need more verification. It was suggested that METTL4 had no direct influence on U2 snRNA expression levels but rather altered splicing regulation.80,82
To sum up, the cap-adjacent location endows m6Am modification with potential to regulate stability and translation. The significant discovery of PCIF1, specifically catalyzes m6Am in the cap structure, drives relevant exploration. However, methodological deficiency is the major problems in m6Am researches. Most of previous studies adopted m6A mapping protocols instead of specific m6Am mapping methods, which led to poor reproducibility and controversial results. Thus, more specific and efficient methods are in urgent need to clarify the regulatory roles of m6Am modification in gene expression.
N1-methyladenosine (m1A)
m1A, the methylation of adenosine at position N1 identified in 1960s, has been found in tRNA, rRNA, lncRNA, and mRNA, among which tRNA is the most heavy-modified class.89,90 Particularly, m1A can transfer to m6A after “Dimroth rearrangement” under alkaline conditions and they also share some regulators.49 m1A has been identified and enriched in specific regions of viral RNA, but its influences in innate immunity is not yet clear.91
In mitochondrial tRNA, m1A methylation is catalyzed by tRNA methyltransferase (TRMT61B) and TRMT10C at positions 58 (m1A58) and 9 (m1A9), respectively.92,93 TRMT61A and TRMT6 form a heterotetrameric complex to methylate both cytoplasmic tRNA at A58 and mRNAs with GUUCRA tRNA-like motifs, as TRMT61A functions as the catalytic subunit.94 TRMT61B mediates m1A in mitochondrial 16 S rRNA,95 and nucleomethylin (NML, also known as RRP8) methylates 28 S rRNA in nuclei.96 And no specific m1A writer for mRNA has been reported yet. ALKBH1 catalyzes demethylation of most m1A in cyto-tRNAs, while m1A58 is the major substrate.97 ALKBH3 demethylates m1A in both tRNAs and mRNAs.98,99 ALKBH7 can demethylate m1A within mitochondrial Leu1 pre-tRNA regions in the nascent polycistronic mitochondrial RNAs.100 And FTO was also proved to demethylated m1A in tRNA.49 YTHDF1/2/3 and YTHDC1 have been confirmed to directly bind to m1A marks, with weaker affinity than that of m6A.101,102 The evolutionarily conversed YTH domain was suggested to be the key to methyl recognition, but the mechanistic research remains deficient.102
The methyl group of m1A carries a positive electrostatic charge, which affects RNA base pairing, and subsequently influences molecule structure and function of modified RNAs. Notably, the electro-chemical interaction of m1A is supposed to play roles in maintaining or stabilizing the T-loop-like structure, and further strengthening the structure.103 As for translation, m1A modification has effects on initiation or elongation process via regulating tRNA, mRNA and rRNA. Several studies have indicated that m1A on either tRNA or mt-tRNA could facilitate translation.97,104 Whereas, m1A modification on mRNA plays diverse roles in protein synthesis, as m1A in 5’UTR correlates with enhanced translation initiation and efficiency,105 but m1A in the CDS exerts inhibitory effects.92,106 In rRNA, m1A is likely associated with translation initiation, as loss of yeast RRP8-catalyzed m1A led to incompetent formation of the 80 S initiation complex.107 Moreover, m1A modification participates in the structural thermostability of tRNAs108 and the nascent polycistronic mt-RNA processing.100
As one of the most abundant internal RNA modifications, the machinery and biological functions of m1A remain largely unknown. The roles of YTH domain-containing proteins as m1A readers may provide novel scientific prospects. And whether its impact on RNA base pairing influences RNA interaction, such as miRNA with mRNA, lncRNA, and circRNA, requires more exploration.
Cytosine modification
5-methylcytosine (m5C)
For decades, methylation of cytosine residues at the position 5 in DNA have been quite familiar. Ever since it was identified in RNA in 1958, m5C has been revealed to distribute widely in RNAs, including tRNA, rRNA, mRNA, enhancer RNA (eRNA), and miRNA.109,110 Studies figured out that m5C modification extensively occurred on maternal mRNA in zygotes of different eukaryotic species, regulating embryogenesis in mouse, zebrafish and Drosophila.111,112,113
In eukaryotes, m5C modification is catalyzed by members of the NOL1/NOP2/SUN domain (NSUN) family of proteins, NSUN1-7 and DNA methyltransferase (DNMT) homolog DNMT2. For rRNA, NSUN1 and NSUN5 introduce m5C at position 4413 and 3761 of human 28 S rRNA, while their homologs in yeast methylate 25S-C2870/25S-C2278.114,115 For tRNA, NSUN2 could modify several sites in various tRNAs, including C34, C40, C48, C49, and C50.116 NSUN6 and DNMT2 methylate C72 and C38 in particular tRNAs, respectively.117,118 NSUN3 and NSUN4 are responsible for methylation of mitochondrial tRNA and 12 S rRNA.119,120 And NSUN4 forms a complex with the mitochondrial transcription factor MTERF4 for lack of RNA recognition motif.121 The m5C methyltransferase specific for mRNAs has not been confirmed yet, but NSUN2 was described to target mRNAs in several studies.122,123 Besides, m5C modifications of ncRNA and eRNA are modified by NSUN2 and NSUN7, respectively.124,125 The identified m5C erasers include ten-eleven translocation (TET) proteins (TET1–3) and ALKBH1. ALKBH1 can successively catalyze m5C into 5-hydroxymethylcytidine (hm5C), 5-formylcytosine (f5C), and 5-carboxylcytosine, at position 34 of cytoplasmic and mitochondrial tRNA,126,127 whereas TETs has been only reported to complete the first step for RNA m5C.128 Aly/REF Export Factor (ALYREF) is the first identified m5C reader in mRNA, a well-known complex that promotes the nuclear export.129 Y-box-binding protein 1 (YBX1) is located in cytoplasm and could recruit stability maintainer ELAV like RNA binding protein 1 (ELAVL1) to stabilize m5C-modified mRNAs.112 Also, YTHDF2 has been reported to modulate the maturation of m5C-modified rRNAs.130
Collectively, m5C modification plays a crucial role in RNA stabilization, exportation, and translation. m5C at C2278 of 25 S rRNA stabilizes the structural conformation of the ribosome.115 Hypermethylated mRNAs with m5C are stabilized via YBX1-dependent manner.131 NSUN2-mediated m5C modifications in vault RNA are significant for its processing into derived small RNAs and protect eRNAs from degradation.132 Also, NSUN2 modified cyclin-dependent kinase inhibitor 1 A (CDKN1A) mRNA and promoted its nuclear export and translation.133
As mentioned above, the dizzying matchup between m5C modifiers and their specific targets brings out challenges as well as opportunities. Targeting certain writers or manipulating specific modification sites reserve great therapeutic potential.
N4-acetylcytosine (ac4C)
ac4C, acetylation of the N4 position of cytosine, is the first acetylation event described. As initially found in tRNA and rRNA, ac4C was also confirmed to be widely present on mRNAs.134 In tRNA, ac4C is located at the wobble of tRNAMet and the D-arm of tRNASer/Leu.135 In eukaryotic 18 S rRNA, ac4C is deposited in helix 34 and helix 45 near the decoding site.136 In mRNA, ac4C is detected in the CDS region and 5ʹUTR, enriched in the third codon encoding amino acid.134 Advances in the study of RNA ac4C modification in cell cycle, inflammatory stress, tumors, premature diseases and viral infection have been reported.91,137,138
Currently, N-acetyltransferase 10 (NAT10) is the only identified ac4C writer, with acetyl-CoA providing acetyl and ATP/GTP hydrolysis supplying energy.139 When modifying tRNA, the assistance of THUMP domain containing 1 (THUMPD1) is necessary,140 while box C/D snoRNPs act as antisense to guide 18 S rRNA acetylation.141 For now, no ac4C eraser has been identified and it remains unknown whether ac4C modification is reversible.
The presence of ac4C on tRNA helps maintain the thermal stability of tRNA and a high heat tolerance of cells, and improves fidelity and efficiency of translation.134,142 ac4C on mRNA CDS region significantly enhance mRNA stability and facilitate translation, probably by preserving codon-anticodon interaction.143 However, ac4C on 5ʹUTR mainly regulates translation initiation in a location-specific manner, as ac4C downstream a weak translation initiation site could promote translation, but the one adjacent to a strong AUG start codon disturbs translation.144 In 18 S rRNA, ac4C modification is crucial for maintaining translation accuracy, pre-rRNA processing and ribosome synthesis.140
The cognition of ac4C modifiers and molecular functions remains largely unknown. Since cofactors of NAT10 have been identified during ac4C formation in human rRNA or tRNA, whether novel cofactors exist in catalyzing mRNA ac4C is noteworthy. Particularly, no erasers or readers has been found yet, whether a deacetylation mechanism exist require more validation.
Guanosine modification
N7-methylguanosine
m7G, referring to the RNA methylation of guanine at position N7, was first found at the 5′ cap (m7GPPPN) of mRNA, stabilizing transcripts and further mediating cap-related biological functions.145 Until now, m7G has been discovered at internal position within mRNA, tRNA, and rRNA,146,147 and tRNA nucleotide position 46 (m7G46) in the variable loop region is the most prevalent m7G methylation site.148
The most well-characterized m7G writer is METTL1, which forms a functional complex with WD repeat domain 4 (WDR4) to install m7G on tRNA, miRNA, and mRNA.147 RNA guanine-7 methyltransferase (RNMT) is responsible for m7G on recapped mRNAs, cooperated with RNMT-activating mini-protein (RAM).149 Williams–Beuren syndrome chromosome region 22 (WBSCR22) methylate G1639 in human 18 S rRNA, requiring tRNA methyltransferase activator subunit 112 (TRMT112).150 Trimethylguanosine synthase 1 (TGS1) might also function as a modifier, catalyzing hypermethylation of m7G caps into m2,2,7 G in snRNAs and snoRNAs.151 The eukaryotic translation initiation factor eIF4E and the cap-binding complex (CBC) can recognize m7G cap and further affect RNA maturation, nuclear export, and translation.152
Notably, m7G modification is extensively involved in various biological processes. For mRNA, the m7G cap could regulate pre-mRNA slicing, nuclear export, translation,152 and indirectly enhance translational capacity by driving ribosome biogenesis.153 And internal m7G also influences translation.154 For tRNA, METTL1/WDR4-mediated m7G methylome plays pivotal roles in maintaining tRNA structural integrity, thereby facilitating translation and reducing ribosome pausing.155 For rRNA, m7G modification participates in 18 S rRNA precursor biogenesis and nuclear export of the 40 S rRNA.150,156 Moreover, m7G on G-quadruplex structures in pri-miRNA could promote miRNA processing.157
At present, our understanding of m7G regulators is apparently limited. No specific demethylase has been identified to regulate the global balance of m7G. And whether m7G modification regulating gene expression via affecting the secondary structure of RNA or recruiting RNA binding proteins remains unclear. Furthermore, the interplay among m7G and other post-transcription attracts growing attention, more explorations are imperative to unravel the underlying mechanism.
Uridine modification
Pseudouridine (Ψ)
Ψ, the 5–riboside isomer of uridine, is the first discovered and most abundant RNA modification.17,158 The C5 atom, instead of N1, forms a new carbon-carbon bond (C5–C1′) with pentose at its non-Watson-Crick edge, endowing Ψ with unique chemical properties. Ψ is present in a wide range of RNAs, including tRNA, rRNA, and various snRNAs, which is highly conserved among species.158,159 The widespread distribution determines its importance in regulating gene expression, steering cellular programs both in development and disease.
The pseudouridylation is mainly catalyzed by pseudouridine synthases (PUSs), via RNA-dependent or -independent manner. The RNA-dependent mechanism involves Dyskerin pseudouridine synthase 1 (DKC1), which forms a complex with box H/ACA snRNA to pseudouridylates rRNA.160 The RNA-independent PUSs includes PUS1, PUSL1, PUS3, TRUB1, TRUB2, PUS7, PUS7L, RPUSD1–4, and PUS10.161,162,163 Regrettably, no Ψ eraser or reader has been documented. And it was speculated that C5–C1′ bond render pseudouridylation irreversible.164
Ψ on tRNA is critical for stabilizing tRNA structure and tRNA codon–anticodon base pairing, further affecting translation processes. Also, Ψ-modified tRNA-derived fragments could restrain aberrant protein synthesis.165 Besides, Ψ is also involved in pre-mRNA processing, structure and stability of mRNA, translational fidelity and termination.166,167 The rRNA Ψ plays a functional role in rRNA processing and protein synthesis.168 It was demonstrated that hypo-pseudouridylated rRNAs decreased affinity for tRNA of ribosomes, impairing translational fidelity.169 snRNP Ψ participates in its biogenesis and splicing.170 Ψ35 in the 5′ end of the U2 snRNA was considered as necessary for early spliceosome formation.171
Although discovered 70 years ago, there are still plenty of vacancies in knowledge on the mechanisms and functions of Ψ. Elucidating whether pseudouridylation is reversible will be one of the key directions in the future. Since efforts to approach inducible pseudouridylation have generated exciting results, which open up new avenues for exploring potential therapeutics. Remarkably, Ψ has already been validated to make critical contribution to COVID-19 mRNA vaccines.172
RNA editing
A-to-I editing
RNA editing modifies primary mRNA and miRNA in posttranscriptional level, altering coding information of DNA. It was first discovered in trypanosome mitochondrial mRNA in 1986.173 So far, RNA editing has been found in tRNA, rRNA and miRNA.174,175,176 The most prevalent type is conversion of adenosine into inosine (A-to-I editing),177 and then inosine is recognized as guanine by the translational machinery. It has been implicated that ADAR1-mediated A-to-I editing was involved in stem cell pluripotency and maintenance, neurological development and function, and immune response.178,179
A-to-I editing only occurs in the double-stranded regions of RNAs made from inverted Alu repetitive elements (Alu dsRNAs), and is far less frequent in coding sequences than noncoding sequences such as UTRs and introns.180 Precursors of certain miRNAs are also common targets.181 The editing levels dramatically vary in cell and tissue type of different origins and development stages, ranging from 2%-100%.182,183 The conversion is catalyzed by adenosine deaminase acting on RNA (ADAR) protein.184 In vertebrates, the isoforms of ADAR protein, ADAR1-3 have identified. These ADAR enzymes possess a C-terminal conserved catalytic deaminase domain, and double-stranded RNA binding domain (dsRBD) at the N-terminus, three for ADAR1 and two for ADAR2-3.185 Functionally, ADAR1 and ADAR2 are responsible for all known A-to-I editing events, while ADAR3 has no documented deaminase activity.186 The mechanism of ADAR substrate specificity remains unclear, in which length and structure of dsRNA was suggested to play an important role,187 and editor modulators like snoRNAs also participated in.188 The consequences of A-to-I editing in coding sequences includes alternative splicing, nonsynonymous amino acid substitutions, nuclear retention and degradation of mRNA. Also, these editing could regulate gene expression via influencing splicing enhancers/silencers recognition sites of ncRNAs in non-coding sequence.189 For several miRNAs, A-to-I editing negatively affects the expression and function of the mature miRNAs.181 In opposition, ADAR1 could facilitate miRNA processing and RNA interference (RNAi) efficacy via forming a complex with Dicer.190
Recently, RNA editing, represented by A-to-I editing, has emerged as a powerful tool to correct pathogenetic mutations, modulate gene expression and protein function. And its transient pharmacodynamic effects could be applied in treatment of several diseases like viral infection, obesity, inflammation, and acute pain. In addition, the transient modulation of protein functions opens up new avenues for oncology and regenerative drugs.
Main database of RNA modifications
To our knowledge, there have been 15 databases established for RNA modifications, two of which are concentrated on biochemical features of RNA modifications, and the rest aimed at elucidating the biological roles. The latter part includes reversible RNA modification database, which can be further classified as comprehensive and type-specific, and nonreversible RNA modification database, namely RNA editing database (Table 2).
Sequencing methods of RNA modification profiling
With the advances in next-generation sequencing (NGS) technologies, many experimental methods have been designed to profile RNA modifications. Generally, the principles of sequencing methods could be classified as two types. The first type is based on antibody or chemical label to capture modified RNA fragments, such as MeRIP-seq for m6A profiling. Another strategy is using enzyme-assisted reaction or a specific chemical reaction on the modified bases, such as Pseudo-seq for Ψ. And these reactions bring about base deletions, substitutions, or truncations, either before or after the modified bases. Here we briefly introduce characteristics of current sequencing methods in Table 3.
RNA modifications and cellular metabolism
Cellular metabolism is a flexible network that allows cells to satisfy their bioenergetic and biosynthesis requirements. In malignant cells, metabolism reprogramming is implicated in tumorigenesis, progression, metastasis and chemoresistance. Aside from the well-concerned cancer metabolism, metabolic adoptions extensively exist in various diseases, including diabetes, obesity, nonalcoholic fatty liver disease (NAFLD), and atherosclerosis. In these pathologies, dysregulated RNA modifiers significantly participate in metabolic alterations via targeting metabolic enzymes, transporters, metabolism-related transcription factors or pathways. Here we summarize current knowledge of how dysregulated RNA modifiers influence glucose, lipid, amino acid, and mitochondrial metabolism, and then, discuss the metabolic effects on RNA modifications.
Glucose metabolism
Glucose is the main energy source of cells, the metabolic pathways principally include aerobic oxidation, anaerobic digestion, pentose phosphate pathway (PPP), glycogen synthesis and gluconeogenesis. Glycolysis is the fundamental energy-producing process in organisms, in which glucose is decomposed into pyruvate with free energy released into ATP.191 Normally, glycolysis in the cytosol is followed by mitochondrial oxidative phosphorylation (OXPHOS) to produce a large amount of ATP under aerobic condition. While in cancer cells, glycolysis had priority over mitochondrial respiration even with sufficient oxygen supply, known as Warburg effect or aerobic glycolysis.192 The key glycolytic enzymes, such as hexokinase (HK), enolase (ENO), Aldolase A (ALDOA), pyruvate kinase isozyme M1/2 (PKM1/2), pyruvate dehydrogenase kinase (PDK), lactate dehydrogenase (LDH) and glucose transporter (GLUT) are crucial targets of RNA modifications in various pathological processes. The pentose phosphate pathway (PPP) is next to glycolysis and the tricarboxylic acid (Krebs) cycle, subdivided into two branches, known as the oxidative and non-oxidative PPP. The non-oxidative PPP is virtually ubiquitous and can occur non-enzymatically, supporting biosynthesis of aromatic amino acid and RNA backbone with ribose 5-phosphate and erythrose 4-phosphate.193 The oxidative branch depends on glucose-6-phosphate (G-6-P) to produce ribulose-5-phosphate, carbon dioxide, and nicotinamide adenine dinucleotide phosphate (NADPH), absent in many aerobic and thermophilic organisms.194 Glycogen synthesis is catalyzed by glycogen synthase under balanced phosphorylation/de-phosphorylation of various kinases, exemplified by glycogen synthase kinase 3 (GSK-3). In fasting state, GSK-3 is activated through de-phosphorylation, thus inhibits glycogen synthesis and facilitates glycogenolysis. While normal feeding inactivated GSK-3 and promotes glycogen synthesis.195 Gluconeogenesis refers to the process that cells synthesize glucose or glycogen from non-sugar precursors such as lactic acid, glycerol, and amino acids. The liver gluconeogenesis is enhanced by decreased insulin and increased glucagon. Remarkably, RNA modifications have been confirmed to play crucial roles in glucose metabolic pathways via directly or indirectly regulating expression of glycolytic-related genes (Fig. 3 and Table 4).

The roles of RNA modifications in glucose metabolism. The schematic diagram shows the direct regulation of RNA modification on glucose metabolism pathways. The key glycolytic enzymes, such as hexokinase (HK), enolase (ENO), Aldolase A (ALDOA), pyruvate kinase isozyme M1/2 (PKM1/2), pyruvate dehydrogenase kinase (PDK), lactate dehydrogenase (LDH) and glucose transporter (GLUT) are crucial targets of dysregulated RNA modifiers, and are generally upregulated in various pathologies. The figure is generated with BioRender (https://biorender.com)
Diabetes mellitus
Type 2 diabetes (T2D) is characterized by insulin resistance and hyperglycemia. And functional integrity of β-cell in pancreatic islet is indispensable for glucose homeostasis. It has been demonstrated that high glucose concentrations reduce m6A level in human and mouse islets.196 Notably, m6A modification played a vital role in pancreatic beta-cell biology. In β-cell specific METTL14-knockout mice, dysfunction of islet, manifested as reduced β-cell proliferation and insulin degranulation, was observed, accelerating the occurrence of diabetes.197 Accordingly, Wang et al. revealed the essential role of METTL3/14 in beta-cell functional maturity. Depletion of METTL3/14 in endocrine progenitors implicated that METTL3/14 were dispensable for beta-cell differentiation but modulated expression of an essential transcription factor MAFA, leading to hypo-insulinemia and hyperglycemia.198 The m6A reader IGF2BP2 is identified as crucial for β-cell proliferation, PDX1 expression level, insulin secretion, and further related with T2DM susceptibility. Mechanistically, IGF2BP2 could stimulate PDX1 translation in an m6A dependent manner and orchestrate IGF2-AKT-GSK3beta-PDX1 signaling to stabilize PDX1 polypeptides.199 And IGF2BP2 is involved in restraining cardiac fibrosis in diabetic heart through LncRNA Airn /IGF2BP2/p53 axis in an m6A-dependent manner.200 Sun et al. figured out that YTHDF2-mediated m6A modification suppress the expression of poly (ADP-ribose) polymerase 1 (PARP1), which is indispensable in the progression of diabetic retinopathy (DR).196 Moreover, METTL14-mediated m6A mitigates diabetic cardiomyopathy via promoting the degradation of LncRNA TINCR dependent on YTHDF2.201 Particularly, FTO gene polymorphism rs9939609 and rs9940128 are closely associated with hyperglycemia, insulin resistance and diabetes mellitus in different populations.202,203,204 In T2D patients, FTO, METTL3, METTL14, and WTAP are upregulated and global m6A level was reduced. And FTO was positively correlated with serum glucose and expression level of several glucose-metabolic genes, such as forkhead box protein O1 (FOXO1), glucose-6-phosphate (G6P) and diacylglycerol O-acyltransferase 2 (DGAT2).205 Thereinto, FOXO1, as an essential transcription factor in gluconeogenesis, has been verified as a direct substrate of FTO. And the potential FTO inhibitor entacapone elicits glucose-lowering function in vivo.206 Moreover, unregulated activating transcription factor 4 (ATF4) was found in FTO-overexpressed transgenic mice, which could augment glucose production by modulating G6P.207,208 Moreover, m6A modification exerts regulatory roles in insulin resistance (IR). Hu et al. proposed that inhibition of FTO aggravates the insulin resistance and adipose tissue inflammation in T2D mice.209
Cancer
Abnormal glucose metabolism, manifested as enhanced glycolytic activity and lactic acid fermentation, is a fundamental part of tumor metabolic reprogramming. Numerous studies have revealed that METTL3-induced m6A directly upregulated expression of various glycolytic enzymes in different cancers. In cervical cancer (CC) cells, METTL3 promotes the translation elongation and mRNA stability of PDK4 depending on YTHDF1/eEF-2 complex and IGF2BP3, respectively.210,211 In lung adenocarcinoma (LUAD), METTL3/m6A/YTHDF1 augment the stability of ENO1 mRNA.212 In colorectal cancer (CRC), METTL3 catalyzes m6A on 5’/3’UTR of HK2 and 3’UTR of GLUT1 (SLC2A1), further stabilizing the transcripts through IGF2BP2 or IGF2BP2/3, respectively.213 Consistently, Chen et al. identified that METTL3/m6A/GLUT1/mTORC1 axis, and overexpression of METTL3 could predict poor survival of CRC patients.214 In esophageal carcinoma (ESCA), the multivariate analysis confirmed the positive association between METTL3 level and expression of GLUT1 and HK2.215 And Li et al. have verified its enhancement on HK2 expression in PDAC cells.216 In 5-FU resistant CRC cells, overexpressed METTL3 not only promoted the transcription of LDHA via stabilizing mRNA of HIF-1α, but also triggered its translation in a YTHDF1-dependent manner.217 And METTTL3 could indirectly activate expression of GLUT1, ALDOA, PKM2, and LDHA in CRC cells via IGF2BP2-mediated stabilization of PTTG3P mRNA.218 Similar indirect activation on GLUT4 and ENO2 was achieved via IGF2BP3-mediated stabilization of HDGF mRNA in GC cells.219
Aside from above glycolytic-related key enzymes or transporters, METTL3 exerts extensive regulation on other metabolic-related targets to motivate glycolysis. Known as a wide-ranging oncogenic determinant, c-MYC was found to be upregulated by METTL3 via m6A/DLGAP1-AS2/YTHDF1 in non-small cell lung cancer (NSCLC)220 and m6A/YTHDF/APC/β-catenin in ESCA, further advancing glycolytic metabolism.221 HIF-1α is responsible for hypoxia conditions in tumor environment, which form mutual feedback with tumor growth. METTL3-induced m6A modification positively regulates HIF-1α level, leading to enhanced aerobic glycolysis.222 METTL3 could also regulate glycolysis and tumorigenesis of breast cancer (BRCA) via YAP, the downstream of Hippo pathway. In mechanism, YTHDF2 accelerated degradation of m6A-modified LATS1 mRNA, thus reduced phosphorylation of YAP/TAZ and activated it.223 In addition to these compelling transcription factors (TFs), METTL3 boosted expression of NDUFA4 in GC,224 AKR1B10 in cholangiocarcinoma (CCA),225 NCAPH in clear cell renal cell carcinomas (ccRCC),226 thus promoted glycolysis and malignant phenotypes. Notably, METTL3-induced m6A interacted with ncRNAs to improve glycolysis, such as stabilizing effects on lncRNA ABHD11-AS1 in NSCLC,227 lncRNA SNHG7 in prostate cancer (PC),228 circQSOX1 in CRC,229 and linc-UROD in PC,230 which are generally mediated by IGF2BPs.
Interestingly, METTL14 seems to exert negative influences on tumor glucose metabolism. In CRC, METTL14 repressed glycolysis via YTHDF2-dependent processing of pri-miR-6769b and pri-miR-499a, which attenuated SLC2A3 and PGAM1 expression, respectively.231 In HCC, METTL14 stabilizes USP48 mRNA, which mediated deubiquitination at the K33 and K128 sites of SIRT6, thus hindered glycolytic reprogramming.232 In RCC, METTL14 attenuated stability of BPTF mRNA, which constituted super-enhancers that activated downstream glycolysis-related genes like ENO2 and SRC.233 Lin et al. proposed that METTL14 positively regulated LHPP expression to restrain aerobic glycolysis of GC.234
WTAP, another m6A writer, was identified to promote Warburg effect in several cancers. WTAP targets 3′-UTR of HK2 mRNA and increased its stability in GC,235 while it indirectly upregulates HK2 via interacting with DGCR8 to boost miR-200 maturation in Ovarian Cancer (OVC).236 Ou et al. supplemented that WTAP-induced m6A methylation could facilitate expression of ENO1 in BRCA.237 In Colon adenocarcinoma (COAD), WTAP stabilizes FOXP3 mRNA via YTHDF1, and FOXP3 bound to SMARCE1 promoter to exert transcriptional activation.238 In CRC, WTAP modifies NT5DC3 to suppress the tumorigenesis under hyperglycemia via repressing Hexokinase domain component 1 (HKDC1).239 Besides, writer KIAA1429 upregulates HK2 and GLUT1 level in methyltransferase activity-dependent manner, facilitating glycolytic process of CRC and GC,240,241 and RBM15 catalyzes m6A modification to accelerate expression of HK2, glucose-6-phosphate isomerase (GPI) and phosphoglycerate kinase1 (PGK1) in OS,242 while ZC3H13 significantly destabilizes PKM2 mRNA to weaken glycolytic reprogramming and enhance cisplatin sensitivity of HCC.243
FTO, the m6A eraser, demonstrates an ambiguous role in regulating glycolytic metabolism. FTO-triggered demethylation was found to enhance glycolysis of HCC and GBM via directly facilitating expression of key enzymes PKM2 and PDK1.244,245 Especially, studies have confirmed the suppressive effect on glycolysis of some selective FTO inhibitors. In SCLC cell line, meclofenamic acid (MA) treatment significantly induced attenuated glycolysis and enhanced mitochondrial metabolism.246 R-2-hydroxyglutarate (R-2HG) represses aerobic glycolysis of leukemia cells via abrogating m6A/YTHDF2-mediated upregulation of PFKP and LDHB, thus inhibiting leukemogenesis in vivo.247 Besides, FTO elevates TFs c-Jun, JunB, and C/EBPβ to upregulate glycolysis-related genes in melanoma, contributing to escaping immune surveillance. Targeting FTO with a small compound Dac51 successfully stimulated therapeutic benefit of anti-PD-L1 blockade.248 Nevertheless, several researchers proposed opposite conclusions. Recently, Liu et al. reported that overexpression of FTO could partially reverse E6E7-induced improvement on HK2 in CC.249 In LUAD, Wnt signaling induces downregulation of FTO, thus increased m6A level leads to enhanced YTHDF1-mediated translation of c-MYC and subsequently increases glycolysis.250 FTO diminishes IGF2BP2-dependent stabilization of APOE mRNA, thus restrains glycolysis and growth of papillary thyroid cancer (PTC).251
Likewise, another m6A eraser, ALKBH5 also have dual regulatory effects. ALKBH5 enhanced stability of G6PD mRNA, thereby activating PPP and promoting proliferation of glioma cells.252 In HER2 resistant BRCA cells, ALKBH5 stimulated glycolysis via protecting GLUT4 mRNA from YTHDF2-mediated decay.253 However, in PTC, knockdown of ALKBH5 accelerates glycolysis through upregulating circNRIP1 and consequently increased PKM2 expression.254 Zhao et al. discovered that overexpressed ALKBH5 elevated expression level of UBR7, which inhibited glycolysis by indirectly suppressing HK2 expression through Nrf2/Bach1 axis.255
Particular attention has been given to m6A readers, which recognize m6A marks and mediate highly context-specific regulation on glycolytic process. Generally, YTHDF1 positively controls glycolysis through stabilizing transcripts or initiating translation in a wide range of cancers.210,212,256 Consistent to its binary regulation on gene expression, YTHDF2 indeed plays diverse roles in reprogramming glycolytic metabolism, with underlying rationale to be further elucidated. For instance, YTHDF2 accelerates decay of GLUT4 mRNA in BRCA,253 while facilitates expression of PFKP and LDHB in leukemia,247 leading to opposite effects. Meanwhile, YTHDF2 participates in modulating other glucose metabolic pathways of glucose like PPP and glycogen synthesis. In LC, overexpressed YTHDF2 binds to m6A sites on 3′-UTR of G6PD mRNA to promotes its translation, enhancing PPP flux,257 and Chen et al. proposed that YTHDF2 enhanced PPP via reducing G6PD ubiquitination by circ_0003215/miR-663b/DLG4 axis.258 In CRC, YTHDF2 is capable to stabilize mRNA of GSK3 to inhibit glycogen synthesis and facilitate glycogenolysis.259 Also, YTHDF2-mediated degradation of STEAP3 mRNA attenuated STEAP3-induced phosphorylation and inactivation of GSK3β in CRC.260
YTHDF3 facilitated aerobic glycolysis of HCC cells by elevating PFKL expression, and PFKL in turn upregulated YTHDF3 through reducing its ubiquitination.261 In PDAC, YTHDF3-mediated destabilization of lncRNA DICER1-AS1 contributes to enhancing expression of glycolytic genes like LDHA, HK2, PGK1, and SLC2A1,262 while YTHDF3 targeted m6A-modified PGK1 mRNA to exert a stabilizing role in OS.263 IGF2BP1 was highly expressed in GC tissue and associated with poor prognosis for GC patients. IGF2BP1 promoted the migration and aerobic glycolysis of GC cells via directly interacting with c-MYC mRNA to stabilize it.264 The gain/loss functional assays proved IGF2BP1-mediated stabilization of LDHA mRNA in ccRCC.265 Moreover, upregulated IGF2BP2 has been found as a predictor of poor prognosis in CC and OSCC, which improved stability of c-MYC and HK2 mRNA, respectively.266,267 Recently, Ye et al. suggested that miR4458HG interacted with IGF2BP2 and activated the improvement of HK2 and GLUT1 expression in HCC.268 In OSCC, IGF2BP3 interacted with circFOXK2 to stabilize GLUT1 mRNA.269 And LOC101929709 bound to LIN28B and IGF2BP3, facilitating LIN28B to stabilize m6A-modified c-MYC mRNA in GC.270
Recently, Wu et al. demonstrated the interplay between m1A modification and tumor glycolytic metabolism. In HeLa cells, ALKBH3 promoted glycolysis by upregulating ATP5D, a subunit of mitochondrial ATP synthase. Mechanistically, the m1A marks on ATP5D mRNA hinders its translation elongation via recruiting YTHDF1/eRF1 complex, and m1A modification destabilizes E2F1 mRNA to block the initiation of ATP5D transcription. ALKBH3-/- HeLa cells displayed reduced glycolysis and weakened growth, both depletion m1A of ATP5D by dm1ACRISPR and overexpression of ATP5D could recede the suppression effect.271 In bladder cancer (BLCA), m5C reader ALYREF bound to 3′-UTR of PKM2 mRNA to stabilize it, and HIF-1α exerted indirect activation on ALYREF in this process.272 Bioinformatics studies have preliminarily implicated that m7G modification participated in glycolytic metabolism. In adrenocortical carcinoma (ACC), a novel m7G risk signature consisted of METTL1, NCBP1, NUDT1 and NUDT5 was constructed, and the risk score presented significant correlation with enrichment of glycolysis. Especially, METTL1, was found to positively regulate the expression of HK1.273
Other diseases
Epigenetic influence of RNA modification on dysregulated glycolysis has been noted in several other pathological processes. Zhang et al. first investigated the role of FTO as a m6A eraser in cardiac metabolism and suggested that FTO could attenuate cardiac dysfunction by regulating glucose uptake and glycolysis with pressure overload-induced heart failure (HF) in mice. Future studies are warranted to systematically assess the potential of FTO for HF prevention and treatment.274 For cardiac fibrosis, METTL3 could repress androgen receptor (AR) expression in a YTHDF2-dependent manner, which activates HIF-1α signaling, thus enhancing glycolysis and cardiac fibroblast proliferation.275 Cai et al. revealed the potential metabolic-related regulation of RNA modification in osteogenic differentiation, inspiring future clinical applications in metabolic bone diseases and stem cell therapy. The mechanistic study showed that METTL3 enhanced stability of ATP citrate lyase (ACLY) and a mitochondrial citrate transporter (SLC25A1) mRNA mediated by IGF2BP2 and IGF2BP2/3, respectively.276 In palmitate (PA)-induced IR C2C12 cells and high-fat diet (HFD)-fed mice model, Quercetin downregulated METTL3, lead to decreased phosphorylated insulin receptor substrate 1 (p-IRS1) levels, increased serine-threonine kinase protein kinase D2 (PRKD2), GLUT4 and p-AKT, further enhancing glucose uptake and alleviating oxidative stress.277
Lipid metabolism
Lipids are essential components of biological membranes, building blocks of biosynthesis, and significant energy storage. According to the comprehensive classification system, lipids are categorized into fatty acyls (FA), glycerolipids (GL), glycerophospholipids (GP), sphingolipids (SP), sterol lipids (ST), prenol lipids (PR), saccharolipids (SL), polyketides (PK).278 FA could be esterified and stored in lipid droplets during high nutrient availability, while hydrolyzed to generate ATP by FA oxidation (FAO), also called β-oxidation, under energy stress conditions. FA synthesis is under control of sterol regulatory element-binding protein 1c (SREBP1c). Stimulated by growth factors, the precursor is processed into mature SREBP1c, and then translocated into nucleus to improve the transcription of target genes, including fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC), stearoyl-CoA desaturase1 (SCD1), and ACLY.279,280 Cholesterol is the material for synthesis of fat-soluble vitamins and steroid hormones, and also the constitution of membranes, together with GL, GP and SP.281 Dysregulated lipid metabolism is implicated in several pathologies, with RNA modifications participating in various metabolic links (Fig. 4 and Table 5).

The roles of RNA modifications in lipid and amino acid metabolism. For lipid metabolism, key enzymes in FA synthesis, including fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC), stearoyl-CoA desaturase1 (SCD1), and ACLY, are significant targets of RNA modifications. Relevant studies on amino acid metabolism are limited. The figure is generated with BioRender (https://biorender.com)
Obesity
In current cognition, obesity is the result of genetic and environmental factors, thereinto, epigenetic regulation such as RNA modifications play significant roles.
Transcriptome profile of human adipose tissue displayed that several m6A modifiers, including WTAP, VIRMA, ALKBH5, and YTHDC1, are associated with obesity and clinical variables, while single nucleotide polymorphisms of METTL3 correlates with body mass index (BMI).282 In brown adipose tissue (BAT), METTL3 is essential for the postnatal maturation and BAT-specific depletion of METTL3 accelerated development of HFD-induced obesity.283 Hepatocyte-specific ablation of METTL3 could promote fatty acid metabolism in mice fed with HFD through regulating fatty acid synthase (FASN), enhancing insulin sensitivity.284 METTL3/m6A/YTHDF2 mediate decay of cyclin D1 (CCND1) mRNA to block cell-cycle progression and inhibit adipogenesis.285
Since FTO was initially discovered as an obesity-related protein before as an eraser, its correlation with obesity has been widely reported in different populations.33,286,287 Significantly, FTO plays a critical role in lipogenesis and obesity susceptibility dependent on m6A demethylase activity. FTO could adjust exonic splicing of adipogenic regulatory factor runt-related transcription factor 1 (RUNX1T1) through eliciting m6A modifications around splice sites, further induces the differentiation of mouse 3T3-L1 preadipocytes.288 FTO could restrain cell cycle progression of preadipocytes and adipogenesis via YTHDF2-dependent decay of cyclin A2 (CCNA2) and cyclin dependent kinase 2 (CDK2).289,290 Also, Zinc finger protein (ZFP217) regulate adipogenesis via FTO/m6A/YTHDF2 axis.291
FTO-mediated demethylation facilitates the expression of peroxisome proliferator-activated receptor gamma (PPARG) mRNA, which promotes the differentiation of bone marrow stem cells (BMSCs) into adipocytes.292 Although depletion of endothelial FTO has no effect on the development of obesity and dyslipidemia, it could promote AKT (protein kinase B) phosphorylation in endothelial cells and skeletal muscle to preserve myogenic tone in resistance arteries, which ultimately alleviates obesity-induced hypertension.293 In accordance, AMP-activated protein kinase (AMPK) was found to regulate lipid metabolism of skeletal muscle via FTO-dependent m6A demethylation.294 Moreover, betaine-mediated downregulation of FTO contributes to dysfunctional adipose tissue induced by high-fat diet.295 YTHDF1 was identified to enhance translation of mitochondrial carrier homology 2 (MTCH2) mRNA and in an m6A-dependent way, which promoting lipogenesis.296 And YTHDF2 facilitated degradation of cyclin D1 mRNA to mediate adipogenesis inhibition.297
NAFLD
Metabolic disorders, manifested as dysregulated de novo lipogenesis, fatty acid uptake, fatty acid oxidation, and triglycerides export, are essential part of pathological mechanism of NAFLD.298 In NAFLD mice model, targeting METTL3/14 upregulated level of ACLY and SCD1, promoting cholesterol production and lipid droplet deposition.299 METTL3/m6A/YTHDF1 exerts a stabilizing effect of Rubicon mRNA and promotes its expression, leading to hepatic lipid deposition.300 m6A hypomethylation state and increased FTO level are detected in HFD-induced NAFLD mice, and FTO participates in the hepatoprotective effects of betaine.301 Overexpressed FTO significantly enhanced lipogenesis and oxidative stress in vitro.302 In the glucocorticoid (GC)-induced NAFLD model, glucocorticoid receptor (GR)-mediated FTO transactivation promotes hepatocyte adipogenesis and lipid accumulation via m6A demethylation of SREBF1 and SCD1.303 Exposure of endocrine disrupting chemicals (EDCs) was confirmed to induce NAFLD, in which process decreased global m6A level and altered expression of m6A modulators was observed.304 Inhibition of FTO effectively alleviates progression of dexamethasone-induced fatty liver in mice.305 Reduced FTO expression mediates the ameliorating effect of exenatide therapy on lipid accumulation and inflammatory responses in NAFLD.306 Moreover, FTO-mediated m6A demethylation increases interleukin-17 receptor A (IL-17RA) level in tumor adjacent tissues with chronic inflammation, suggesting the potential role of FTO in inflammation-carcinogenesis transformation of HCC.307
And ALKBH5-dependent demethylation drives lipogenesis and NAFLD-HCC progression via stabilizing LINC01468, which accelerates cullin4A (CUL4A)-linked degradation of inositol polyphosphate phosphatase-like 1 (INPPL1, SHIP2).308 Nevertheless, another study demonstrated that overexpressed ALKBH5 could ameliorate liver fibrosis and inactivate Hepatic stellate cells (HSCs) via upregulating Patched 1 (PTCH1).309 Moreover, YTHDF3 was also reported to restrain liver fibrosis and HSC activation via facilitating peroxiredoxin 3 (PRDX3) translation in an m6A-dependent manner.310
Atherosclerosis
During the development and progression of atherosclerosis (AS), RNA modifications play critical roles in lipid deposition and fiber cap formation.311 Growing evidences showed that RNA modifications participate in development of AS via modulating inflammatory cell infiltration and immune response, including vascular endothelial cells, macrophages, and vascular smooth muscle cells (VSMCs).312
In an ox-LDL-induced AS model, highly expressed METTL3 in VSMCs facilitates the binding of DGCR8 to pri-miR-375 and further improved miR-375-3p expression, which targets PDK1 transcription, inducing phenotypic transformation of VSMCs and rendering AS plaques more vulnerable.313 In ox-LDL-treated human umbilical vein endothelial cells (HUVEC), upregulated METTL3 and METTL14 were detected, and METTL14 modified p65 mRNA to facilitate lipoprotein synthesis and AS development.314 METTL14 was found to reduce cholesterol efflux and enhanced atherosclerotic plaque inflammation via modifying lncRNA ZFAS1.315 FTO exerts various effects on vascular homeostasis properties via influencing lipid metabolism. Researchers have reported the anti-atherosclerotic properties of FTO. In-vivo experiments showed that overexpressed FTO induced by Adeno-associated virus serotype 9 (AAV9) obviously decreased the lipidic profiles including plasma total cholesterol and LDL cholesterol, and mitigated the formation of atherosclerotic plaques.316 Yang et al. demonstrated that FTO inhibits macrophage lipid influx by downregulating PPARγ expression and facilitating cholesterol efflux via phosphorylation of AMPK, thereby meliorating foam cell formation and AS development.317 Also, Kruger et al. found that endothelial-specific knockdown of FTO could prevent obesity-induced vascular dysfunction.293
Cancer
Activated de novo synthesis of fatty acids serves as an essential energy source, while enhanced FAO contributes to ATP production, intracellular ROS reduction. Except for bioenergetic demand, remodeled lipid metabolism could assist tumor development through modulating ferroptosis, enabling metastasis and invasion, and crosstalk with other hallmarks in TME.318,319,320
In BLCA, METTL14-mediated m6A elevates level of lncDBETm, which interacts with Fatty acid-binding protein 5 (FABP5) to activate peroxisome proliferator-activated receptors (PPARs), markers of lipid metabolism-related signaling pathways.321 And previous study suggested that METTL3 could recruit YTHDF2 to stabilize PPARα mRNA, regulating circadian rhythms of hepatic lipid metabolism.322 In breast and liver cancer cell, elevated m6A modification upregulated ERRγ by triggering the splicing of precursor ESRRG mRNA, subsequently improved expression of carnitine palmitoyl transferase 1B (CPT1B), a rate-limiting enzyme of FAO, conferring to chemoresistance.323 Peng et al. confirmed that METTL5 promoted de novo lipogenesis and HCC progression via ACSL4-mediated FAO. Targeting ACSL4 and METTL5 cooperatively suppresses HCC tumorigenesis in vivo.324
Overexpressed FTO enhances lipogenesis and lipid droplet enlargement in liver, and inhibits CPT1-mediated FAO via the SREBP1c pathway. FTO-dependent m6A demethylation indirectly elevates SREBP1c expression, thus activating downstream lipogenic genes.303 Knockdown of FTO markedly enhanced m6A abundance of FASN mRNA and promoted YTHDF2-mediated decay, further reduced protein levels of ACC1 and ACLY, which suppressed de novo lipogenesis in HepG2 cells.325 In EC, mechanism study revealed that FTO promoted the formation of lipid droplets by enhancing HSD17B11 expression.326 ALKBH5 was downregulated in cervical squamous cell carcinoma (CESC) and predicted an unfavorable prognosis. ALKBH5 attenuated stability of SIRT3 mRNA in an IGF2BP1-dependent manner, further reduced ACC1 level repress its deacetylation, thus suppresses fatty acid synthesis to modulate CESC lipid metabolism.327
YTHDF2 was reported to targets m6A-marked transcripts of key lipogenic genes to induce their degradation, thus suppressing liver steatosis.328 In GBM, YTHDF2 facilitates decay of LXRA and HIVEP2 mRNA, negatively regulating cholesterol synthesis, efflux, and uptake.329 In CRC, reduced m6A methylation promoted DEGS2 expression via attenuating YTHDF2-mediated decay, which contributed to dysregulated lipid metabolism, especially suppressed ceramide synthesis.330 In AML, IGF2BP2 stabilizes PRMT6 mRNA through m6A-dependemt manner, which catalyzes H3R2me2a and suppresses lipid transporter MFSD2A expression, thus decreasing docosahexaenoic acid levels and promoting LSC maintenance.331 HNRNPA2B1 participates in enhancing fatty acid metabolism of GC via stabilizing RPRD1B mRNA, which facilitated uptake and synthesis of FA by transcriptionally activating c-Jun/c-Fos, further upregulated SREBP1.332 Abrogation of HNRNPA2B1 inhibits de novo fatty acid synthesis in ESCA cells though downregulating expression of ACLY and ACC1.333
Several studies have dictated the regulatory functions of m5C modification in lipid metabolism. Function analysis demonstrated that highly m5C-marked genes were enriched in pathways correlated with decreased adipogenesis and improved myogenesis. Particularly, reader ALYREF recognized m5C targets on YBX2 and SMO and mediated the shuttling from nucleus to cytoplasm, thereby regulating adipogenesis and myogenesis, implicating a novel therapeutic approach for metabolic disorder diseases.334 The m5C writer NSUN2 has been found to advance adipogenesis through targeting CDKN1A mRNA and recruiting ALYREF to facilitate its nuclear export, thus accelerating cell cycle progression to promote lipid accumulation in preadipocytes.133 In OS, NSUN2-induced m5C modification stabilized FABP5 mRNA to positively regulated FA metabolism, further enhancing OS progression.335 In silico analysis identified that two clusters OVC samples with different m5C modification pattern exhibited distinct metabolic characteristics, with distinct expression profile of lipid metabolism-related pathways.336 In HCC, the m1A methyltransferase complex, TRMT6/TRMT61A facilitate PPARδ translation, which augmented cholesterol synthesis to initiate Hedgehog signaling, eventually driving self-renewal of liver CSCs and tumorigenesis.337
Mitochondrial metabolism
Mitochondrion, the metabolic center in cells, plays an indispensable role in oncogenesis. Although aerobic glycolysis occupies an essential position in tumor bioenergetic metabolism, the OXPHOS and mitochondria-dependent energy supply were considered as key to maintain the stemness of some tumor cells.338 Besides, providing materials for anabolism, producing ROS, and maintaining regulated cell death (RCD) signaling significantly conduce to tumor progression.339
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is an essential cofactor for mitochondrial biogenesis and energy metabolism, which play significant parts in pathologies of monocyte-macrophage inflammation-associated diseases, such as atherosclerosis and rheumatoid arthritis. METTL3/m6A/YTHDF2 mediated degradation of PGC-1α, as well as cytochrome c (CYCS) and NADH ubiquinone oxidoreductase subunit C2 (NDUFC2), inducing mitochondrial dysfunction and oxLDL-induced inflammation in monocytes.340 During myoblasts differentiation, FTO positively modulates mTOR-PGC-1α pathway-mediated mitochondria biogenesis depending on its m6A demethylase activity.32 Consistently, ectopic expression of FTO in VHL-deficient ccRCC cells upregulated expression of PGC-1α to regain mitochondrial activity, exhibiting anti-proliferation effect.341
In NSCLC, METTL3-induced m6A methylation on DCP2 mRNA accelerates its degradation, which activates mitochondrial autophagy through the Pink1-Parkin pathway, inducing resistance to cisplatin and etoposide.342 In amoxifen-resistant BRCA cells, METTL3 was overexpressed and contributed to upregulation of AK4, which stimulating ROS production and p38 phosphorylation, further suppressing mitochondrial apoptosis to desensitize tamoxifen. Additionally, METTL3 knockdown abrogated AK4 expression and drug resistance.343 Also, METTL3 exerts inhibitive role in mitochondrial apoptosis via AKT signaling pathway in EC.344 METTL14 plays a pivotal part in regulating mitochondrial homeostasis in CRC via m6A/YTHDC2/miR-17-5p/MFN2 axis. Low expression of METTL14 ultimately led to less apoptosis and 5-FU chemoresistance in CRC.345 METTL14-catalyzed m6A increases expression of Fission 1 (FIS1), contributing to cadmium (Cd)-induced mitochondrial fission and dysfunction.346
FTO-mediated demethylation exerts protective roles in progression of hepatic ischemia-reperfusion injury (HIRI) via targeting dynamin-related protein 1 (Drp1) to alleviate liver oxidative stress and mitochondrial fragmentation in vivo and in vitro.347 Moreover, in silico analysis showed that overexpressed FTO and METTL5 were significantly associated with OXPHOS in NSCLC and GC, respectively.348,349 IGF2BP1 augmented stability of m6A-modified NDUFA4, and upregulated NDUFA4 enhanced oxidative metabolism in GC cells, whereas suppression of mitochondrial fission could switch the NDUFA4-induced mitochondrial activities and tumor growth of GC.224 In GBM, IGF2BP2 was involved in maintaining stability of SHMT2 mRNA, which played a crucial role in OXPHOS activities, as a significant driving force of GBM tumorigenesis.350 Elevated m6A level and IGF2BP2 expression conduced to maintenance of hematopoietic stem cells (HSCs) via restricting mitochondrial activity. IGF2BP2 deficiency accelerated degradation of Bmi1 mRNA, relieving its depression on mitochondria-related genes, thus impaired quiescence state and functions of HSCs.351 A novel RNA-binding protein, RALY, a member of HNRNPC subfamily, augmented pri-miRNA processing via the METTL3-mediated m6A modification, further reprogrammed mitochondrial metabolism by downregulating the electron transport chain (ETC) genes in CRC cells. Depletion of RALY demonstrated effective inhibition of tumor growth and development in vivo and in organoid models.352 In glioma, YTHDF1 participated in c-MYC-induced restraint on mitochondrial autophagy by directly interacting with FDX1 and upregulated its expression, which was closely correlated with malignant phenotypes and clinical prognosis.353 Highly-expressed HNRNPA2B1 served as an adverse prognostic factor in MM. HNRNPA2B1 recognized m6A sites at TLR4 and elevated its expression in post-transcriptional level, which protecting mitochondria under proteasome inhibitor exposure.354
Amino acid metabolism
Except for serving as substrates for synthesizing proteins or peptides, amino acids are decomposed through deamination or transamination to generate building blocks for anabolism, like α-ketoacids utilized to release energy in TCA cycle. Particularly, glutamine has multiple biofunctions beyond a metabolic fuel or protein precursor. Glutamine decomposition is another significant feature of tumor metabolism remodeling.355 Given that cancers are normally auxotrophic for some non-essential amino acids, targeting the supply of these amino acids have been validated as an effective therapeutic intervention.356
Previous studies have substantiated the regulatory effects of m6A modification in glutamine metabolism. FTO-mediated m6A demethylation upregulated expression of the glutamine transporter SLC1A5, and FTO inhibition exclusively suppressed the proliferation and vitality of VHL-deficient ccRCC cells independent of HIF.357 Han et al. suggested that m6A modification exerted a significant role in the antitumor efficacy of glutaminolysis inhibition in CRC cells. FTO was upregulated upon glutaminolysis inhibition to promote expression of ATF4 via abrogating m6A/YTHDF2-mediated RNA degradation, and ATF4 activated pro-survival autophagy via transcriptionally upregulating DDIT4 to suppress mTOR signaling.358 Actually, it was found earlier that m6A modifications in the 5′ UTR of ATF4 mRNA modulated its alternative translation to mediate re-initiation independent of eIF2α signaling pathway, in response to amino acid starvation, implicating the translation regulation of m6A in integrated stress response.359 In CRC, YTHDF1 bound to 3′ UTR of glutaminase (GLS) mRNA and induced translational promotion, leading to enhanced glutamine uptake, further mediating chemoresistance, and targeting YTHDF1 effectively re-sensitizes CRC cells to cisplatin.360 IGF2BP2 positively modulated glutamine metabolism in AML cells via advancing mRNA stability and translation of several metabolic-related genes, such as GPT2, SLC1A5, and MYC, thereby accelerating AML development.361
Recently, Chen et al. have proposed that m6A methylation was involved in WZ35-mediated enhanced radiotherapeutic sensitivity via Glutathione (GSH) exhaustion. Mechanistically, WZ35 consumed GSH through the ROS-YAP-AXL-ALKBH5-GLS2 loop, inducing metabolic remodeling and further repressing GC cell metastasis.362 Moreover, METTL16 participated in regulating branched-chain amino acid (BCAA) metabolism by facilitating expression of BCAT1 and BCAT2 in an m6A-dependent manner, serving as an oncogene in leukemogenesis and LSC maintenance of AML.363
In summary, as shown in Fig. 5, Numerous proteins and RNA modification mediated metabolism are implicated in the progression of various diseases. To put into practice the preventive and treatment options for these diseases, a thorough understanding of RNA modification and metabolic disorders is needed.

Epigenetic regulation of RNA modification on metabolism in diseases. RNA modifications broadly regulated metabolic pathways in diverse diseases, covering glucose (green box), lipid (blue box), amino acid and mitochondrial (yellow box) metabolism. As intuitively shown in the picture, Gastric cancer (GC), Colorectal Cancer (CRC), Hepatocellular Carcinoma (HCC), Acute Myeloid Leukemia (AML), and Breast Cancer (BRCA) are especially related to RNA modifications-mediated metabolic deregulations. The figure is generated with BioRender (https://biorender.com)
RNA modification and immunometabolism
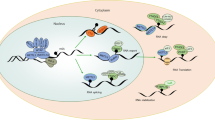
Generally speaking, the immunometabolism concerns distinctions between activated and resting immune cells. The former metabolizes in a manner similar to malignant cells, Warburg effect without obvious OXPHOS, while the latter obtains energy from FAO and the Krebs cycle. Corresponding metabolic patterns of different immune cell subgroups have been described.364 Herein, we focus on the contributions of RNA modifications on immunometabolism in diverse immune responses (Fig. 6).

Effects of RNA modification on immunometabolism. This figure shows current findings about how RNA modifications regulate immunometabolism. On the one hand, RNA modifications are involved in the intrinsic metabolic adaptation of immune cells, further affecting function and state of immunocytes. On the other hand, m6A modification could mediate several phenotype alterations of immunocytes induced by glucose deficiency and high lactate in TME. The figure is generated with BioRender (https://biorender.com)
Anti-tumor immunity
The concept of tumor immune microenvironment (TIME) emphasizes the interplay between immune cells, tumor cells, and other components in immunity system, which profoundly influences the immune responses, via nutrients depletion and metabolites release. To sustain the rapid proliferation, tumor cells consume large amounts of glucose, glutamine and other amino acids like arginine, generally constituting an adverse residential environment for immune cells. The universal recognition is that glucose exhaustion induced by tumor cells contributes to immunosuppressive TME.
Nevertheless, the pioneering work of Reinfeld et al. showed that glucose is not a limiting factor in the TME, and resident immunocytes were capable to enhance glucose uptake in compensation for depleted glutamine. Indeed, multiple pathways are conducive to the impaired glucose metabolism of T cells from TME, but the effects are likely to be context-dependent. Mechanism study about epigenetic regulation on glycolytic reprogramming of T cells remains deficient, but the glycolytic-epigenetic interplay has been found in development of Tfh cells. VHL deficiency induced expression of glycolytic enzyme GAPDH, which acted as an epigenetic regulator to enhance METTL3/METTL14-catalyzed m6A modification on ICOS mRNA, thereby suppressed ICOS expression led to attenuated Tfh cell differentiation.365
Glucose depletion in TME predisposes the differentiation of macrophages to M2-like TAM, which preferentially employs OXPHOS for ATP synthesis rather than consuming glucose.366 Recently, METTL3 was considered as the top candidates for regulating M1 macrophages activation via targeting mTOR/NF-κB-mediated metabolic adaptation.367 However, Ning et al. proposed that m6A modification was responsible for inhibited glycolysis and M1 macrophage polarization, through YTHDF2-mediated degradation of m6A-modified STAT1 mRNA, further attenuating glycolysis-related genes expression.368
Distinct glutamine acquisition is another essential aspect of nutrient partitioning. Tumor cells tend to have advantage in glutamine consumption over those immune cells, as tumor cells have over-expressed methionine transporter Slc43a2, thereby restricts methionine metabolism and the antitumor function of T cells.369 However, whether RNA modifications participate in glutamine metabolism of immune cells remains to be explored.
Except for nutrient competition, metabolites secreted by tumor cells also exert immunosuppressive effects on anti-tumor immunity. Tumor cells produce large amounts of lactic acid to generate highly acidic regions, as a hallmark of immunosuppressive TME. High concentration of extracellular lactic acid significantly inhibits the survival and activation of T and NK Cells, further blocks the immunosurveillance.370 m6A-mediated stabilization of circQSOX1 enhance lactic acid accumulation in CRC, thus supports Treg cells and facilitate immune escape, which further impacts efficacy of anti-CTLA-4 therapy in vivo.229 As mentioned above, the intratumoral myeloid cells have vigorous glycolytic activity, but whether myeloid-derived lactate limits T cell effector functions, remains to be explored.371
Lactate also negatively influences functions of macrophage and skews the differentiation of macrophages toward the M2 phenotype. M2 macrophage infiltration in endometriosis was positively correlated with lactate accumulation. Mechanism study showed that lactate promoted M2 macrophage polarization via METTL3-mediated m6A modification on Trib1 mRNA, which enhanced its stability.372
Tumor cell is a significant source of intratumoral lipids, including cholesterol, fatty acid or oxidized lipids, which have a deleterious effect on T cells, DCs, and macrophages. Several studies have confirmed that m6A methylation was involved in maintaining the functional homeostasis of macrophages via targeting the balance between lipid uptake and cholesterol efflux. Elevated METTL3 in oxidized low-density lipoprotein (oxLDL)-treated macrophages facilitates lipid uptake via interacting with DDX5 to target MSR1 mRNA and stabilize it in m6A-dependent manner.373
FTO is upregulated in macrophages loaded with ox-LDL, which enhances cholesterol efflux via motivating AMPK/ACC phosphorylation to promote ABCA1/G1-mediated efflux, and attenuates cholesterol ester accumulation through restricting PPARγ to reduce CD36 expression.316 More direct and convincing evidences are expected to elucidate the epigenetic regulation on TAMs. Besides, cholesterol uptake activates PD-1 expression in tumor-infiltrating CD8 + T cells, which in turn facilitates FAO and lipolysis.374 Extracellular fatty acids are more effectively consumed by Treg cells than effector T cells, which eventually supports Treg accumulation.375 Oxidized lipids restrain cross-representation in DCs376 and enhanced uptake of fatty acids and peroxidation lead to dysfunctional state of tumor-derived DCs.377 But the role of RNA modifications in these cellular processes has not been identified yet.
Antiviral immunity
The pathogenesis of infectious diseases is consisted of two parts, deficiency of immune system itself and immune escape of pathogens. On one hand, specific RNA modifications on viral RNAs have been described, including m6A, m5C ac4C, Ψ, and RNA editing, thus affecting viral RNA sensing and signaling.378 On the other hand, RNA modifications influence host responses to viral infection via regulating immune cell functions. The interferon pathway is the major target of m6A modification to modulate antiviral innate immunity. METTL3/14 enhances the turnover rate of IFNB mRNAs via YTHDF2-mediated manner and accelerates viral propagation.379,380 The coordination between m6A methylation and other RBPs also mediate the negative effects on immunity. DEAD-box helicase 5 (DDX5), hijacked by viruses to promote replication, could interact with METTL3 to facilitate formation of the METTL3/14 complex during vesicular stomatitis virus infection.381 In addition to interferon pathway, ALKBH5 could promote viral propagation relying on metabolic rewiring. It was showed that viral infection impaired the enzymatic activity of ALKBH5 in posttranslational level and thus downregulated expression of α-ketoglutarate dehydrogenase (OGDH), leading to reduced itaconate production, a metabolite that inhibits viral replication.382 Beyond that, the association between RNA modification and metabolic processes remains largely unknown.
Inflammation and autoimmune disorders
Inflammatory response is achieved through a coordinately regulated gene expression program, including acute and chronic type.383 In response to microorganisms, autoimmunity, allergies, dysregulated metabolism, and physical damage, different types of inflammation are produced.384 Until recently, regulatory roles of RNA modification in inflammation and anti-inflammation gene expression have been verified. Previous studies have shown that m6A modification is involved in pathogenesis of autoimmune diseases. For instance, METTL3 is significantly upregulated in RA patients and positively associated with CRP and ESR, the two common markers of RA disease activity.385 In systemic lupus erythematosus (SLE), decreased m5C level and low NSUN2 expression are found in CD4 + T cells, and hypermethylated m5C-modified upregulated genes in SLE are enriched in inflammatory pathways.386 Significantly, in DC-dependent inflammatory response, m6A-mediated glycolytic reprogramming is critical for feedback-control of DC migration. Mechanistically, in response to microbial products or inflammatory signals, upregulated CC-chemokine receptor 7 (CCR7) stimulated lnc-Dpf3 via removing its m6A modification to prevent degradation, and lnc-Dpf3 could negatively modulate HIF-1α pathway via binding to HIF-1α and suppressing HIF-1α-dependent transcription of the glycolytic gene Ldha.387 Also, m6A modification modulates macrophage phenotype in inflammatory responses. Previous study has reported that METTL3 was notably elevated in M1 macrophages and modulated polarization via metabolism reprogramming. In mechanism, m6A methylation contributes to enhanced expression of HDGF, which increases glycolysis and lipids accumulation in M1, therefore aggravating the progression of atherosclerosis.388 And METTL3-meidated m6A of PGC-1α mRNA is involved in mitochondrial dysfunction and oxLDL-induced inflammation in monocytes.340 Although there are few studies on RNA modifications regulating inflammatory and autoimmune diseases in the aspect of immunometabolism.
Clinical implications of RNA modifications
RNA modifications and therapeutic responses of metabolic therapy
For the currently approved metabolic drugs, an impending challenge of clinic application is development of chemo-resistance owing to rewiring or compensatory metabolic pathways. Thus, the multiple pathways blockade or combined therapy may have superiority over the single-agent therapy. Notably, multiple studies have supported that combined utilization of targeting RNA modifications could improve chemo-resistance to some metabolism-targeted drugs.
The influences of m6A modification on CRC resistance to 5-FU is a representative example. Mechanism studies have demonstrated that METTL3 could induce 5-FU resistance of CRC cells via m6A/DGCR8/miR181d/NCALD axis,389 m6A/IGF2BP1/SEC62/β-catenin axis.390 Also, m6A methylation facilitates preferential splicing of p53 pre-mRNA to produce p53 R273H mutant protein, leading to multidrug resistance in CRC cells.391 Moreover, suppressing c-Myc-driven YTHDF1 transactivation was revealed to re-sensitize CRC cells to some anticancer drugs, including 5-FU.392 Consistently, Jiang et al. found that miR-136-5p could downregulated YTHDF1 to suppress tumor progression and chemoresistance to 5-FU, while miR-136-5p was declined in CRC cell lines and tissues.393
Moreover, METTL3 was identified to positively modulate gemcitabine (GEM) sensitivity of PC via DBH-AS1/miR-3163/USP44, and low expression level of METTL3 was closely related with GEM resistance.394 Upregulated METTL14 was observed in GEM-resistant PC cells, which was induced by p65 and downstream facilitated cytidine deaminase (CDA) expression to inactivate GEM in PC. Inhibition of METTL14 effectively re-sensitized GEM in vitro and in vivo, indicating a promising approach for circumvent chemo-resistance.395 Intriguingly, ALKBH5-mediated demethylation also exerts a positive role in GEM sensitivity of pancreatic ductal adenocarcinoma (PDAC) through suppressing Wnt pathway.396
RNA modifications and therapeutic responses of immunotherapy
Growing researches revealed that m6A regulators markedly affected therapeutic responses against checkpoint blockade. Wang et al. reported that depletion of METTL3/14 enhanced infiltration and cytokines secretion of CTL, augmenting anti-PD-1 therapy efficacy of CRC in vivo, through m6A/YTHDF2/STAT1/IRF1 axis.397 However, METTL14 could sensitize cholangiocarcinoma to ICB via YTHDF1-mediated degradation of SIAH2 mRNA.398 Knockdown of YTHDF1 enhances cross-presentation of DCs to CD8 + T cells by suppressing cathepsins expression, further increased IFN-γ secretion of T cells upregulates PD-L1 level in tumor cells.399 FTO was identified to negatively regulate ICB therapeutic efficacy in melanoma. Ablating FTO decreases expression of several significant melanoma-promoting genes and sensitized anti-PD-1 treatment in vivo.400 However, FTO was revealed to promote PD-L1 expression in an IFN-γ-independent manner of CRC cells, thus improving ICB treatment.401 Su et al. demonstrated that inhibition of FTO obviously downregulated immune check point gene LILRB4 in AML cells, with superiority over PD-L1/2, further repressing leukemia stem cell maintenance and immune evasion.402 In melanoma, FTO participates in rewiring tumor glycolysis metabolism to suppress T cell effector functions, and FTO inhibition synergizes with anti-PD-L1 therapy.248 Additionally, the silico analysis identified that high FTO level was associated with poor prognosis and unfavorable immunotherapy effect of GC patients.403 And ALKBH5 was supposed to be a potential predictor for anti-PD-1 blockade efficacy in melanoma. Deficiency of ALKBH5 induced downregulation of MCT4 expression and intra-tumoral lactate content, which negatively influenced polymorphonuclear myeloid derived cells and Tregs.404 Several bioinformatic studies have underlined the intimate connection between m6A modification and immunotherapy resistance.405 Moreover, loss of A-to-I editor ADAR1 significantly augmented anti-PD-1 treatment in melanoma and CRC. In mechanism, the interaction of tumor intrinsic type I and type II IFN signaling contribute to sensitize ADAR1-null cells to ICB.406
Except for checkpoint blockade therapy, targeting m6A modification has demonstrated promising potential in improving adoptive cell therapy. Marvelous progresses have been made in modulating METTL3 and YTHDF2 to enhance the proliferation and cytotoxicity of NK cells in vitro, which might inspire future protocols for NK cell-based immunotherapy.407,408 No attempt to modulate m6A methylation in CAR T cells has been reported yet, but considering the significant roles of m6A regulators in determining functions and fate of T cell, novel therapeutic strategies are expected.
Development of RNA modification-targeted agents
Targeting the dysregulated m6A regulators, which are overexpressed in tumor on most occasions, plentiful specific inhibitors have demonstrated exciting anti-tumor effects in vitro and in vivo (Table 6). FTO is considered as the most promising target. Within a decade, a series of selective inhibitors have come out, ranging from natural substance to small-molecule compound. The first natural inhibitor Rhein displayed therapeutic activity in leukemia mice,409 meclofenamic acid 2 (MA2) was observed to suppress glioblatoma progression.410 Small-molecule compounds CHTB and N-CDPCB were identified with novel binding sites by crystal structure screening.411 R-2-hydroxyglutarate (R-2HG) exerts anti-leukemia and anti-glioma effects, synergizing with current first-line chemotherapy agents.412 FB23-2 also significantly attenuates the progression of AML in vitro and in xeno-transplanted mice.413
Subsequently, more potent inhibitors with potential to improve anti-tumor immunity was developed. CS1/2-induced FTO inhibition not only attenuates leukemia stem cell self-renewal, but reprograms immune responses by downregulating expression of immune checkpoint gene, which overcomes HMA-induced immune evasion and sensitizes leukemia cells to T cell cytotoxicity.402 Also, Dac15 restores functions of CD8 + T cells, blocks FTO-mediated immune evasion, and synergizes with anti-PD-1 blockade.248 Recently, progresses have been made in more tumor types rather than AML and glioma. A small molecule inhibitor 18097 significantly restrained in vivo growth and lung colonization of breast cancer cells.414 Oxetanyl class demonstrated antiproliferative effects in GC, glioblastoma and AML models, while FTO-43 has potency comparable to 5-FU.415 Compound C6, a 1,2,3-triazole analogs, was suggested as a potential orally antitumor agent for esophageal cancer.416
Exploitation of inhibitors against other regulators, including METTL3 and ALKBH5, is also proceeding steadily. The ALKBH5 inhibitor ALK-04 effectively sensitized melanoma cells to anti-PD-1 blockade, as ALKBH5 attenuated immunotherapy responses via regulating lactate content and immunosuppressive cell infiltration in the TME.404 Selberg et al. discovred two compouds, 2-[1-hydroxy-2-oxo-2-phenylethyl]sulfanyl acetic acid and 4-[furan-2-yl]methyl amino-1,2-diazinane-3,6-dione, demonstrated cancer-cell-type-selective antiproliferative effects in selected leukemia cell lines.417 Targeting SAM binding sites, adenosine was first identified as METTL3 inhibitors. Non-nucleoside inhibitors with higher selectivity and permeability have been developed, such as UZH1a and UZH2.418,419 A novel METTL3 inhibitor STM2457 effectively blocked AML progression and prolonged survival in AML mouse models, without disturbing normal hematopoiesis.420
Furthermore, RNA m1A methylation is also a potential therapeutic target. The m1A methyltransferase complex, TRMT6/TRMT61A is highly expressed in HCC and correlated with poor prognosis. Wang et al. screened out three potential drugs targeting the interaction of TRMT6 and TRMT61A, thimerosal, phenylmercuric acetate (PMA), and thiram. Among them, the administration of thiram significantly attenuated HCC growth in preclinical models.421
RNA modifications in RNA-based therapeutics
Though RNA medicine has been facing challenges like efficacy and immunogenicity since from birth, the most recent hit of mRNA vaccines against COVID-19 provide new momentum to this field and bring RNA modifications back into to spotlight. Chemical modification of RNA could protect RNA from hydrolysis and nucleases, and decrease off-target cytotoxic effects. Once therapeutic RNAs form duplexes with targeted sequences, modifications lowering the melting temperature could destabilize the complex and improve target specificity via reducing base-paring with non-target RNAs. Moreover, RNA modifications are utilized for RNA delivery and strengthen the pharmaceutical activity of RNA.422
Base modifications have been successfully applied in improving the performance of therapeutic RNA, for example, replacement of uridine with the modified base 1-methylpseudouridine (N1-Me) in COVID-19 vaccines (Pfizer’s Comirnaty and Moderna’s Spikevac) effectively facilitates translation and reduces off-target side effects and immunogenicity of therapeutic mRNA.423 Additionally, m7G cap linked by a 5′-triphosphate to the 5′ end of the mRNA, which replicates the naturally occurring mRNA caps to prevent degradation of the 5′ end of mRNA, has been introduced into mRNA vaccines BNT162/Comirnaty and mRNA-1273/Spikevax.424 As for ribose modifications, modified hydroxyl group on the C-2′ position of the ribose could protect RNA against nuclease digestion and lower the thermal stability of duplexes. N-acetylgalactosamine (GalNAc) groups or lipophilic moieties attached cleavable linkers, including ester-based, peptide-based cleavable groups, could localize therapeutic RNA to target tissue.425 Moreover, modifications to the phosphate group in the sugar-phosphate backbone shelter RNAs from nucleases, represented by phosphorothioate. And eliminating the negative charge via replacing the oxygens on the phosphates with neutral groups or cations can assist the delivery into cell.426
Based on CAS Content Collection, a recent study summarized the modification content in approved RNA medicines, including antisense oligonucleotide (ASO), siRNA, aptamer, and mRNA.427 Thereinto, N1-Me is prominently abundant in two mRNA vaccines, along with 2′-O-methyl, 3′-methyl, m7G, 5′-5′-triphosphate. And 2′ -oxy-methoxyethylguanosine (2′-MOE) is exclusive in ASOs, which protects ASOs from degradation. The approved siRNAs have 2′-fluoro and 2′-O-methyl modification of the ribose, and three of them are 3′-glycosylated with the GalNAc conjugate, which specifically targets siRNAs to hepatocytes.428
Combination of targeting RNA modification and current therapy
Some gratifying results have been acquired in combined application of m6A regulators inhibitors with current anticancer therapy. The involvement of m6A modification in underlying mechanisms of resistance has been systematically summarized.429 Herein, we put emphasis on the combined utility of targeting RNA modification to circumvent resistance and improve individualized cancer treatment.
A mass of evidences showed that overexpressed METTL3 widely participated in the acquisition of various therapeutic resistance in many cancer types. Knockdown of METTL3 using short hairpin RNA improved sensitivity to anticancer reagents such as gemcitabine, 5-fluorouracil, cisplatin and irradiation in pancreas cancer (PC).430 Suppression of METTL3 restored chemosensitivity and attenuated CML cells viability.431 Nevertheless, few studies have ever investigated the utility of METTL3 inhibitors in overcoming chemoresistance. Targeting FTO also shed new light on improving chemoresistance. Upregulated FTO in oral squamous cell carcinoma played a pivotal part in arecoline-induced stemness and chemoresistance to cisplatin.432 Depletion of FTO sensitized breast cancer to doxorubicin via suppressing de novo synthesis of fatty acid.433 Specifically, FTO was revealed to facilitate GBM resistance to temozolomide (TMZ), and the inhibitor R-2HG demonstrated a synergistic effect with TMZ in suppressing proliferation of FTO-high glioma cells.412
Moreover, the feasibility of administrating m6A regulators inhibitors to improve immunotherapy effectiveness needs further investigation. Depletion of METTL3/14 was found to augment ICB therapeutic responses in mismatch-repair-proficient or microsatellite instability-low (pMMR-MSI-L) CRC and melanoma.434 In accordance, a recent study confirmed that targeting METTL3 by inhibitor STM2457 potentiate ICB efficacy in various CRC mouse models.435 Knockdown of FTO reduced PD1 expression in melanoma via m6A/YTHDF2-dependent manner, thus sensitized anti-PD-1 blockade.400 FTO was supposed to enhanced PD-L1 expression independent on IFN-γ in CRC.436 In AML, FTO inhibition induced by small-molecule compounds CS1/2 leads to downregulation of checkpoint gene LILRB4, reigniting the interest of introducing ICB to AML.402 Besides, Li et al. reported that the specific inhibitor to ALKBH5, ALK-04 markedly enhanced the efficacy of anti-PD-1 blockade in CRC model.404
Clinical trials targeting RNA modifications
We surveyed ClinicalTrials.gov as of October 20, 2023, to keep up-to-date with clinical implications of RNA modifications, basically including therapeutic effectiveness of agents targeting modifiers, potential as predictive biomarkers, and combined application with current treatment. However, hardly any above-mentioned specific inhibitor has progressed into clinical stage, in spite of the encouraging antitumor results of FTO-targeted agents in various cancers. Most recently, a phase 1, first-in-human study is designed to systematically evaluate the pharmacokinetics, pharmacodynamics and clinical activity of STC-15 in adult subjects with advanced malignancies (NCT05584111).
Several studies have evaluated the association between FTO polymorphisms (rs9939609 and rs1558902) and obesity in different populations, including Turkish population (NCT04205318), Indonesian obesity women (NCT04740528), as well as weight loss in overweight carriers induced by calorie restriction (NCT02940197), and intermittent or moderate continuous high intensity training programs (NCT03568773). Furthermore, there are projects aim at assess the correlation between FTO polymorphisms and risk of developing diabetes in Mexican adolescents with overweight and obesity (NCT02886013), and features of metabolic syndrome in children with T1D (NCT01279161). Considering that variants in FTO showed high correlation with body weight and also interact with dopamine signaling in the brain, a clinical trial was designed to develop a genotype-specific and individualized therapy approach for obesity targeting FTO (rs8050136) variant (NCT03525002). Genotyping for FTO was also incorporated into tailored therapeutic model for azathioprine-induced myelosuppression in inflammatory bowel disease patients (NCT03719118).
Conclusion and future perspectives
State-of-the-art methods for RNA modification sequencing
With advent of high-throughput methodologies, precision and sensitivity of RNA modification sequencing invented in an unprecedented space. Currently, mainstream MeRIP- and miCLIP-based methods have been widely accepted, yet with several disadvantages to be overcome. The poor sensitivity of antibody-based methods is first limitation, and chemical-assisted labeling is recognized as a promising approach. On account of the strong affinity of biotin-streptavidin binding, the m6A seal (m6A selective chemical labeling) method dramatically enhance enrichment efficiency via introducing a biotin tag to modified bases.437 In addition, to solve the incapability of quantifying modification ratio, m6A-LAIC-seq (m6A-level and isoform-characterization sequencing), originated from MeRIP-seq, could quantify m6A levels for all isoforms of transcripts for each gene via isolating m6A-positive and m6A-negative post-RIP fractions and sequencing full-length transcripts.438 Adding synthetic modification-free RNA molecules as internal reference is another strategy to realize quantitative sequencing.439 To be noted, single-cell sequencing technologies is an emerging hotspot in tumor immunology, which could effectively profile the intricate immune landscape in tumor TME. For instance, DART-seq (deamination adjacent to RNA modification target sequencing) is designed to monitor m6A at the single-cell level, which successfully reveal the heterogeneity in m6A scenarios across individual cells and identify differentially methylated mRNAs across the cell cycle.440 However, further application of DART-seq in clinics is limited by its dependency on overexpression of the APOBEC1-YTH fusion protein in cells. Hence, a free of genetic manipulation single-cell method for deciphering RNA modification is warranted.
Advancement of RNA modification databases
With advanced methodologies of detecting and profiling RNA modifications, rapid-accumulated enormous epi-transcriptome data call for centralized bioinformatics platforms to mine the underestimated treasure. For both experimental and computational studies of RNA modifications, such valuable resources will be of great help. Researchers focus on structural biology could take full advantage of comprehensive databases like RNAMDB and MODOMICS, while computational biologists perform their researches based on relational databases such as MetDBv2.0, m6A-Atlas, RMBase v2.0 etc. Exploration of novel types or modifiers of RNA modification fully relies on current knowledgebases. For clinics, these databases advance understanding of the role epi-transcriptomics plays in disease pathology. Not a few databases have provided information about relationship between disease-related varients with RNA modifications, such as m6A-Atlas, RMVar, and RMDisease.
Meanwhile, epitranscriptomics-disease links are highlighted in newest update of MODOMICS. The new section exhibits association between malfunction or misregulation of a given RNA-modifying enzymes with specific disease conditions. To better elucidate the post-transcriptional regulatory networks, multi-omics analysis is highly rated. Moreover, context-specificity of RNA modification should be taken into consideration via distinguishing species, cell type, and tissues. Finally, more user-friendly interface and webserver tools are significant for improving accessibility of these resources.
RNA modifications and immunology
During differentiation and development of immune cells, various clusters of functionally coordinated genes are under sophisticated control of RNA modifications. The highly selectivity and specificity of RNA modifying machinery still remains largely defined. The top priority is to distinguish different targeted transcripts according to a framework of classical immunological systems such as polarization of macrophages and CD4 + T cell differentiation. It was suggested that additional factors such as region-enriched cis-regulatory elements exerted a certain effect on selectivity of RNA marking. Besides, increasing evidences have implicated the crosstalk between RNA modifications on non-coding chromosome-associated regulatory RNA (carRNA) and chromatin modifications, thus RNA modifications may control immune responses to environmental stimuli via shaping the chromatin environment of immune cells.
In the aspect of tumor immunology, the discrepancy of epi-transcriptome between tumor and immune cells is acknowledged as an essential influencing factor of antitumor immune responses. However, relevant research is still in its infancy. Thus, rigorous dissection of RNA modification marks and regulators in tumor cells and immune cells is considered as a fundamental and crucial for developing effective interventions. Following marker-informed sorting of cell populations of interest, methods like mass spectrometry are used for profiling dynamic RNA modifications. Furthermore, integration of single-cell scale and transcriptome methods with RNA modification sequencing may provide valuable insights into dysregulated RNA modification in the TME. Aside from highly-specific RNA modification-targeted inhibitors, modification editing in immune cells is another promising direction for treating immune-related diseases.
Established on the understanding of metabolism, the application prospects of targeting m6A methylation in immunotherapy mainly consisted of two possibilities. One is to circumvent therapeutic resistance mediated by the metabolic antagonism in TME, the other is to potentiate proliferation efficiency and effector functions of immune cells for adoptive cell therapy. Recent advances clued some potential strategies: 1) a programmable m6A editing machinery to fine-tune RNA modifications of specific genes with minimal off-target alterations, 2) effective manners to manipulate m6A system ex vivo for optimal generation of NK cells and T cells, 3) efficient targeted delivery of m6A editors into cells, like nanoparticles, 4) inhibitors against m6A regulators with potential to modulate anti-tumor immunity.
RNA modifications and cancer metabolism, metabolic diseases
The current understanding is that metabolic phenotypes evolve as cancers process from premalignant lesions, localized invasive malignancies to metastatic cancers, even therapy-resistant states. The dynamic RNA modification along with emerging metabolic vulnerabilities in evolutionary process provide attractive clinical opportunities. In some cases, tumors exhibited stereotyped metabolic alterations without detectable mutations or DNA methylation abnormities,441 implicating the presence of other epigenetic regulation like RNA modifications. Delineating the evolving genetic, epigenetic, immune-metabolic landscape is quite necessary for designing effective strategies to preclude metastasis. Progresses in spatial-omics techniques and system biology research may help to address it.
Hyperactive metabolic pathways lead to brisk adaptation to nutrient deprivation, contributing to resistance to antimetabolic chemotherapy agents like antifolates. Metabolic coupling, characterized by catabolites transfer, is common in tumor for overcoming nutrient deficiency. Thus, combination of targeting glycolysis and OXPHOS was proposed as a promising strategy. Considering the underlying toxicity, the alternative is suppressing dysfunctional signals to indirectly target glycolysis, while directly targeting OXPHOS.
Increasing studies have indicated the significance of epigenetic regulation in metabolic diseases. Up to now, none of epigenetic drugs have been approved for metabolic diseases, and the efficacy of RNA modification-targeted agents have not been verified in metabolic diseases. Thus, investigation whether inhibition of RNA modifiers can be used for treatment of metabolic diseases is requested. Given that environmental factors shed influences on epi-transcriptome via intracellular metabolic changes, molecular insights of RNA modification in development of metabolic diseases remains largely unknown.
Superiority and challenges of targeting RNA modifications
Indeed, the universal distribution and broad functionality of RNA modification is a double-edged sword. For anticancer treatment, targeting a single identified driver sometimes turns out an unfavorable result, as a consequence of various reasons including the development of resistance and intra- or intertumoral heterogeneity. From this point, targeting RNA modification is advantageous to cover a network of targets. Especially, these RNA modifiers tend to be overexpressed or more active in cancerous tissue compared to matched normal control tissue. However, the essentiality and specificity of these RNA modifiers remain significant concerns.
For currently developed agents targeting RNA modifying enzymes, poor specificity and selectivity remain the main obstacle in their progression into clinical researches. And such deficiency is anticipated to be improved via optimized bioinformatic prediction models and high-throughput enzymatic tests. Here are several other outstanding questions to be further investigated. If pharmacologic inhibition of RNA modification enzyme is capable to reproduce the phenotypic activity induced by genetic deletion? If redundance in modifying enzymes, like METTL3/14 complex and METTL6, potentially induce resistance to pharmacological inhibition. Given that RNA modifiers tend to be overexpressed in tumor tissue but still present in normal tissues, an appropriate therapeutic window in a certain therapeutic context may be necessary. Aside from specific inhibitors or activators to those modifiers, RNA modifications have been applied to improve the stability, efficacy and target specificity of RNA-based therapies. Common strategies include utilizing synthetic chemical or naturally occurring modifications, and modulating sequence context or location of these modifications. For all current therapeutic RNA, RNA modifications are extensively present and poised to further enhance their effectiveness.
In summary, epigenetic regulation of RNA modifications exerts a crucial role in cellular metabolism in diverse physiological and pathological situations. Growing evidences suggest that such metabolic-epigenetic interplay significantly affects immune responses, via modulating biological activities of immune cells and remodeling immune context. Thus, delineating the evolving genetic, epigenetic, immune-metabolic landscape is quite necessary for designing effective strategies to preclude pathogenesis, including various metabolic disorders, immune-related diseases, and cancer. Recent years have witnessed remarkable advancements in methods for detecting and profiling RNA modifications, accompanied with a series of serviceable databases and tools springing up. At present, attempts to targeting RNA modification for improving current therapy have obtained some inspiring advances, but relevant researches are still in its infancy. And we can count on further in-depth exploration to accelerate the development of RNA modification-targeted therapy, metabolism-targeted therapy and immunotherapy.
References
Boccaletto, P. et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 50, D231–D235 (2022).
Yue, Y., Liu, J. & He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355 (2015).
Xiao, Z., Dai, Z. & Locasale, J. W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 10, 3763 (2019).
Koenen, M., Hill, M. A., Cohen, P. & Sowers, J. R. Obesity, adipose tissue and vascular dysfunction. Circ. Res. 128, 951–968 (2021).
Boulton, A. Strengthening the International Diabetes Federation (IDF). Diabetes Res Clin. Pract. 160, 108029 (2020).
Powell, E. E., Wong, V. W. & Rinella, M. Non-alcoholic fatty liver disease. Lancet 397, 2212–2224 (2021).
Long, G. V. et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 20, 1083–1097 (2019).
Tannir, N. M. et al. Efficacy and safety of telaglenastat plus cabozantinib vs placebo plus cabozantinib in patients with advanced renal cell carcinoma: the CANTATA randomized clinical trial. JAMA Oncol. 8, 1411–1418 (2022).
Goodwin, P. J. et al. Effect of metformin vs placebo on invasive disease-free survival in patients with breast cancer: the MA.32 randomized clinical trial. JAMA 327, 1963–1973 (2022).
An, Y. & Duan, H. The role of m6A RNA methylation in cancer metabolism. Mol. Cancer 21, 14 (2022).
Cai, X. et al. N6-methyladenosine modification and metabolic reprogramming of digestive system malignancies. Cancer Lett. 544, 215815 (2022).
Yue, S. W. et al. m6A-regulated tumor glycolysis: new advances in epigenetics and metabolism. Mol. Cancer 22, 137 (2023).
Everts, B. et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431 (2012).
Akkaya, M. et al. Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat. Immunol. 19, 871 (2018). -+.
Lauterbach, M. A. et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity 51, 997 (2019).
Wang, R. N. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Cohn, W. E. & Volkin, E. Nucleoside-5′-phosphates from ribonucleic acid. Nature 167, 483–484 (1951).
Holley, R. W., Everett, G. A., Madison, J. T. & Zamir, A. Nucleotide sequences in the yeast alanine transfer ribonucleic acid. J. Biol. Chem. 240, 2122–2128 (1965).
Jia, G. F. et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Desrosiers, R., Friderici, K. & Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl Acad. Sci. USA 71, 3971–3975 (1974).
Adams, J. M. & Cory, S. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature 255, 28–33 (1975).
Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 28, 395–404 (2018).
Lyons, S. M., Fay, M. M. & Ivanov, P. The role of RNA modifications in the regulation of tRNA cleavage. FEBS Lett. 592, 2828–2844 (2018).
Jacob, R., Zander, S. & Gutschner, T. The dark side of the epitranscriptome: chemical modifications in long non-coding RNAs. Int J. Mol. Sci. 18, 2387 (2017).
Sun, Z. et al. Aberrant NSUN2-mediated m(5)C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene 39, 6906–6919 (2020).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Huang, H., Weng, H. & Chen, J. m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37, 270–288 (2020).
Wang, Y. et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014).
Fustin, J. M. et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806 (2013).
Aguilo, F. et al. Coordination of m(6)A mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell 17, 689–704 (2015).
Schwartz, S. et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155, 1409–1421 (2013).
Wang, X. et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1alpha pathway-mediated mitochondria biogenesis. Cell Death Dis. 8, e2702 (2017).
Dina, C. et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet 39, 724–726 (2007).
Liu, J. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Wang, P., Doxtader, K. A. & Nam, Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63, 306–317 (2016).
Huang, J. et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N(6)-methyladenosine methyltransferase. Protein Cell 10, 272–284 (2019).
Ping, X. L. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 (2014).
Patil, D. P. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
Bokar, J. A. et al. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 269, 17697–17704 (1994).
Pendleton, K. E. et al. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169, 824 (2017). -+.
Shima, H. et al. S-adenosylmethionine synthesis is regulated by selective N-6-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 21, 3354–3363 (2017).
Ma, H. H. et al. N-6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation (vol 15, pg 88, 2019). Nat. Chem. Biol. 15, 549–549 (2019).
Ignatova, V. V. et al. The rRNA m(6)A methyltransferase METTL5 is involved in pluripotency and developmental programs. Gene Dev. 34, 715–729 (2020).
Jia, G. F. et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO (vol 7, pg 885, 2011). Nat. Chem. Biol. 8, 1008–1008 (2012).
Mauer, J. et al. Reversible methylation of m(6)A(m) in the 5′ cap controls mRNA stability. Nature 541, 371–375 (2017).
Mauer, J. & Jaffrey, S. R. FTO, m(6) Am, and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett. 592, 2012–2022 (2018).
Mauer, J. et al. Reversible methylation of m(6)A(m) in the 5′ cap controls mRNA stability. Nature 541, 371 (2017).
Zhang, X. et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl Acad. Sci. USA 116, 2919–2924 (2019).
Wei, J. et al. Differential m(6)A, m(6)A(m), and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell 71, 973–985.e975 (2018).
Zheng, G. Q. et al. ALKBH5 Is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Liao, S., Sun, H., Xu, C. & YTH Domain: a family of N(6)-methyladenosine (m(6)A) readers. Genom. Proteom. Bioinform. 16, 99–107 (2018).
Jain, D. et al. ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. Elife 7, e30919 (2018).
Zaccara, S. & Jaffrey, S. R. A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell 181, 1582–1595.e1518 (2020).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6, e31311 (2017).
Mao, Y. et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 10, 5332 (2019).
Shi, H. L. et al. YTHDF3 facilitates translation and decay of N-6-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Orouji, E. et al. Oncogenic role of an epigenetic reader of m(6)A RNA modification: YTHDF1 in merkel cell carcinoma. Cancers 12, 202 (2020).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Feng, M. D. et al. YBX1 is required for maintaining myeloid leukemia cell survival by regulating BCL2 stability in an m(6)A-dependent manner. Blood 138, 71–85 (2021).
Liu, N. et al. N-6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015).
Alarcon, C. R. et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162, 1299–1308 (2015).
Zhou, K. I. et al. Regulation of co-transcriptional Pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol. Cell 76, 70–81.e79 (2019).
Wu, R. et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 29, 23–41 (2019).
Baquero-Perez, B. et al. The Tudor SND1 protein is an m(6)A RNA reader essential for replication of Kaposi’s sarcoma-associated herpesvirus. Elife 8, e47261 (2019).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624 (2019).
Yang, L. et al. Proteinase-activated receptor 2 promotes cancer cell migration through RNA methylation-mediated repression of miR-125b. J. Biol. Chem. 290, 26627–26637 (2015).
Liu, N. et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015).
Zheng, Z. Q. et al. Long noncoding RNA FAM225A promotes nasopharyngeal carcinoma tumorigenesis and metastasis by acting as ceRNA to sponge miR-590-3p/miR-1275 and upregulate ITGB3. Cancer Res. 79, 4612–4626 (2019).
Chen, R. X. et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat. Commun. 10, 4695 (2019).
Yang, Y. et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 27, 626–641 (2017).
Paramasivam, A. & Vijayashree Priyadharsini, J. Novel insights into m6A modification in circular RNA and implications for immunity. Cell Mol. Immunol. 17, 668–669 (2020).
Charenton, C., Wilkinson, M. E. & Nagai, K. Mechanism of 5′ splice site transfer for human spliceosome activation. Science 364, 362–367 (2019).
Wei, C., Gershowitz, A. & Moss, B. N6, O2′-dimethyladenosine a novel methylated ribonucleoside next to the 5′ terminal of animal cell and virus mRNAs. Nature 257, 251–253 (1975).
Wang, J. et al. Quantifying the RNA cap epitranscriptome reveals novel caps in cellular and viral RNA. Nucleic Acids Res. 47, e130 (2019).
Zhang, Q. et al. HIV reprograms host m(6)Am RNA methylome by viral Vpr protein-mediated degradation of PCIF1. Nat. Commun. 12, 5543 (2021).
Wang, L. et al. PCIF1-mediated deposition of 5′-cap N(6),2′-O-dimethyladenosine in ACE2 and TMPRSS2 mRNA regulates susceptibility to SARS-CoV-2 infection. Proc. Natl Acad. Sci. USA 120, e2210361120 (2023).
Sendinc, E. et al. PCIF1 catalyzes m6Am mRNA methylation to regulate gene expression. Mol. Cell 75, 620–630.e629 (2019).
Keith, J. M., Ensinger, M. J. & Moss, B. HeLa cell RNA (2′-O-methyladenosine-N6-)-methyltransferase specific for the capped 5′-end of messenger RNA. J. Biol. Chem. 253, 5033–5039 (1978).
Akichika, S. et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 363, eaav0080 (2019).
Goh, Y. T. et al. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 48, 9250–9261 (2020).
Luo, Q. et al. Structural insights into molecular mechanism for N(6)-adenosine methylation by MT-A70 family methyltransferase METTL4. Nat. Commun. 13, 5636 (2022).
Chen, H. et al. METTL4 is an snRNA m(6)Am methyltransferase that regulates RNA splicing. Cell Res. 30, 544–547 (2020).
Goh, Y. T. et al. METTL4 catalyzes m(6)Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 48, 9250–9261 (2020).
Hao, Z. Y. et al. N-6-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol. Cell 78, 382 (2020).
Zhang, Z. X. et al. Regulation of adipocyte differentiation by METTL4, a 6mA methylase. Sci Rep. 10, 1 (2020).
Hirayama, M. et al. FTO demethylates cyclin D1 mRNA and controls cell-cycle progression. Cell Rep. 31, 107464 (2020).
Boulias, K. et al. Identification of the m(6)Am methyltransferase PCIF1 reveals the location and functions of m(6)Am in the transcriptome. Mol. Cell 75, 631–643.e638 (2019).
Drazkowska, K. et al. 2′-O-Methylation of the second transcribed nucleotide within the mRNA 5′ cap impacts the protein production level in a cell-specific manner and contributes to RNA immune evasion. Nucleic Acids Res. 50, 9051–9071 (2022).
Zhou, H. et al. Evolution of a reverse transcriptase to map N(1)-methyladenosine in human messenger RNA. Nat. Methods 16, 1281–1288 (2019).
Dunn, D. B. The occurrence of 1-methyladenine in ribonucleic acid. Biochim. Biophys. Acta 46, 198–200 (1961).
Furuse, Y. RNA modifications in genomic RNA of influenza A virus and the relationship between RNA modifications and viral infection. Int J. Mol. Sci. 22, 9127 (2021).
Chujo, T. & Suzuki, T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 18, 2269–2276 (2012).
Vilardo, E. et al. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase - extensive moonlighting in mitochondrial tRNA biogenesis (vol 40, pg 11583, 2012). Nucleic Acids Res. 46, 11126–11127 (2018).
Wang, M. X. et al. Crystal structure of the two-subunit tRNA m(1)A58 methyltransferase TRM6-TRM61 from Saccharomyces cerevisiae. Sci. Rep. 6, 32562 (2016).
Bar-Yaacov, D. et al. Mitochondrial 16S rRNA is methylated by tRNA methyltransferase TRMT61B in all vertebrates. PloS Biol. 14, e1002557 (2016).
Waku, T. et al. NML-mediated rRNA base methylation links ribosomal subunit formation to cell proliferation in a p53-dependent manner. J. Cell Sci. 129, 2382–2393 (2016).
Liu, F. et al. ALKBH1-mediated tRNA demethylation regulates translation. Cell 167, 816–828.e816 (2016).
Li, X. Y. et al. Transcriptome-wide mapping reveals reversible and dynamic N-1-methyladenosine methylome. Nat. Chem. Biol. 12, 311 (2016).
Chen, Z. et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 47, 2533–2545 (2019).
Zhang, L. S. et al. ALKBH7-mediated demethylation regulates mitochondrial polycistronic RNA processing. Nat. Cell Biol. 23, 684–691 (2021).
Safra, M. et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551, 251–255 (2017).
Dai, X. X., Wang, T. L., Gonzalez, G. & Wang, Y. S. Identification of YTH domain-containing proteins as the readers for N1-methyladenosine in RNA. Anal. Chem. 90, 6380–6384 (2018).
Voigts-Hoffmann, F. et al. A methyl group controls conformational equilibrium in human mitochondrial tRNA(Lys). J. Am. Chem. Soc. 129, 13382–13383 (2007).
Ueda, Y. et al. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci. Rep. 7, 42271 (2017).
Dominissini, D. et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446 (2016).
Zhou, H. Q. et al. m(1)A and m(1)G disrupt A-RNA structure through the intrinsic instability of Hoogsteen base pairs. Nat. Struct. Mol. Biol. 23, 803–810 (2016).
Sharma, S. et al. A single N-1-methyladenosine on the large ribosomal subunit rRNA impacts locally its structure and the translation of key metabolic enzymes. Sci. Rep. 8, 11904 (2018).
Oerum, S., Degut, C., Barraud, P. & Tisne, C. m1A post-transcriptional modification in tRNAs. Biomolecules 7, 20 (2017).
Bohnsack, K. E., Hobartner, C. & Bohnsack, M. T. Eukaryotic 5-methylcytosine (m(5)C) RNA methyltransferases: mechanisms, cellular functions, and links to disease. Genes 10, 102 (2019).
Amos, H. & Korn, M. 5-Methyl cytosine in the RNA of Escherichia coli. Biochim. Biophys. Acta 29, 444–445 (1958).
Chi, L. & Delgado-Olguin, P. Expression of NOL1/NOP2/sun domain (Nsun) RNA methyltransferase family genes in early mouse embryogenesis. Gene Expr. Patterns 13, 319–327 (2013).
Yang, Y. et al. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol. Cell 75, 1188–1202 e1111 (2019).
Zou, F. et al. Drosophila YBX1 homolog YPS promotes ovarian germ line stem cell development by preferentially recognizing 5-methylcytosine RNAs. Proc. Natl Acad. Sci. USA 117, 3603–3609 (2020).
Sharma, S. et al. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 41, 9062–9076 (2013).
Schosserer, M. et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun 7, 11530 (2016).
Brzezicha, B. et al. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucleic Acids Res. 34, 6034–6043 (2006).
Haag, S. et al. NSUN6 is a human RNA methyltransferase that catalyzes formation of m(5)C72 in specific tRNAs. RNA 21, 1532–1543 (2015).
Goll, M. G. et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398 (2006).
Nakano, S. et al. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNA(Met). Nat. Chem. Biol. 12, 546 (2016). -+.
Metodiev, M. D. et al. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. Plos Genet. 10, e1004110 (2014).
Camara, Y. et al. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 13, 527–539 (2011).
Khoddami, V., Yerra, A. & Cairns, B. R. Experimental approaches for target profiling of RNA cytosine methyltransferases. Method Enzymol. 560, 273–296 (2015).
Hussain, S. et al. The mouse cytosine-5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol. Cell Biol. 33, 1561–1570 (2013).
Aguilo, F. et al. Deposition of 5-methylcytosine on enhancer RNAs enables the coactivator function of PGC-1alpha. Cell Rep. 14, 479–492 (2016).
Nombela, P., Miguel-Lopez, B. & Blanco, S. The role of m(6)A, m(5)C and Psi RNA modifications in cancer: novel therapeutic opportunities. Mol. Cancer 20, 18 (2021).
Arguello, A. E. et al. Reactivity-dependent profiling of RNA 5-methylcytidine dioxygenases. Nat. Commun. 13, 4176 (2022).
Kawarada, L. et al. ALKBH1 is an RNA dioxygenase responsible for cytoplasmic and mitochondrial tRNA modifications. Nucleic Acids Res. 45, 7401–7415 (2017).
Fu, L. et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585 (2014).
Dominissini, D. & Rechavi, G. 5-methylcytosine mediates nuclear export of mRNA. Cell Res. 27, 717–719 (2017).
Dai, X. et al. YTHDF2 binds to 5-methylcytosine in RNA and modulates the maturation of ribosomal RNA. Anal. Chem. 92, 1346–1354 (2020).
Zhang, X. et al. The tRNA methyltransferase NSun2 stabilizes p16INK(4) mRNA by methylating the 3′-untranslated region of p16. Nat. Commun. 3, 712 (2012).
Hussain, S. et al. NSun2-mediated cytosine-5 methylation of vault noncoding Rna determines its processing into regulatory small RNAs. Cell Rep. 4, 255–261 (2013).
Liu, Y. H. et al. mRNA m5C controls adipogenesis by promoting CDKN1A mRNA export and translation. RNA Biol. 18, 711–721 (2021).
Arango, D. et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1872–1886.e1824 (2018).
Meier, J. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. J. Biol. Chem. 299, S654–S654 (2023).
Bortolin-Cavaille, M. L. et al. Probing small ribosomal subunit RNA helix 45 acetylation across eukaryotic evolution. Nucleic Acids Res. 50, 6284–6299 (2022).
Jin, C. et al. Acetyltransferase NAT10 regulates the Wnt/beta-catenin signaling pathway to promote colorectal cancer progression via ac(4)C acetylation of KIF23 mRNA. J. Exp. Clin. Cancer Res. 41, 345 (2022).
Cui, L. et al. RNA modifications: importance in immune cell biology and related diseases. Signal Transduct. Target Ther. 7, 334 (2022).
Ikeuchi, Y., Kitahara, K. & Suzuki, T. The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J. 27, 2194–2203 (2008).
Sharma, S. et al. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 43, 2242–2258 (2015).
Sharma, S. et al. Specialized box C/D snoRNPs act as antisense guides to target RNA base acetylation. Plos Genet. 13, e1006804 (2017).
Whipple, J. M. et al. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev. 25, 1173–1184 (2011).
Dominissini, D. & Rechavi, G. N(4)-acetylation of cytidine in mRNA by NAT10 regulates stability and translation. Cell 175, 1725–1727 (2018).
Arango, D. et al. Direct epitranscriptomic regulation of mammalian translation initiation through N4-acetylcytidine. Mol. Cell 82, 2797–2814.e2711 (2022).
Ramanathan, A., Robb, G. B. & Chan, S. H. mRNA capping: biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016).
Zhang, L. S. et al. Transcriptome-wide mapping of internal N-7-methylguanosine methylome in mammalian mRNA. Mol. Cell 74, 1304 (2019).
Alexandrov, A., Martzen, M. R. & Phizicky, E. M. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA 8, 1253–1266 (2002).
Alexandrov, A., Grayhack, E. J. & Phizicky, E. M. tRNA m(7)G methyltransferase Trm8p/Trm82p: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA 11, 821–830 (2005).
Trotman, J. B., Giltmier, A. J., Mukherjee, C. & Schoenberg, D. R. RNA guanine-7 methyltransferase catalyzes the methylation of cytoplasmically recapped RNAs. Nucleic Acids Res. 45, 10726–10739 (2017).
Haag, S., Kretschmer, J. & Bohnsack, M. T. WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N-7-methylation of G1639 in human 18S rRNA. RNA 21, 180–187 (2015).
Monecke, T., Dickmanns, A. & Ficner, R. Structural basis for m7G-cap hypermethylation of small nuclear, small nucleolar and telomerase RNA by the dimethyltransferase TGS1. Nucleic Acids Res. 37, 3865–3877 (2009).
Mars, J. C., Ghram, M., Culjkovic-Kraljacic, B. & Borden, K. L. B. The cap-binding complex CBC and the eukaryotic translation factor eIF4E: co-conspirators in cap-dependent RNA maturation and translation. Cancers 13, 24 (2021).
Galloway, A. et al. Upregulation of RNA cap methyltransferase RNMT drives ribosome biogenesis during T cell activation. Nucleic Acids Res. 49, 6722–6738 (2021).
Zhang, L. S. et al. Transcriptome-wide mapping of internal N(7)-methylguanosine methylome in mammalian mRNA. Mol. Cell 74, 1304–1316.e1308 (2019).
Lin, S. B. et al. Mettl1/Wdr4-mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell 71, 244 (2018).
Figaro, S. et al. Trm112 is required for Bud23-mediated methylation of the 18S rRNA at position G1575. Mol. Cell Biol. 32, 2254–2267 (2012).
Pandolfini, L. et al. METTL1 promotes let-7 microRNA processing via m7G methylation. Mol. Cell 74, 1278–1290.e1279 (2019).
Zhao, B. S. & He, C. Pseudouridine in a new era of RNA modifications. Cell Res. 25, 153–154 (2015).
Karijolich, J., Yi, C. Q. & Yu, Y. T. Transcriptome-wide dynamics of RNA pseudouridylation. Nat. Rev. Mol. Cell Biol. 16, 581–585 (2015).
Henras, A. et al. Accumulation of H/ACA snoRNPs depends on the integrity of the conserved central domain of the RNA-binding protein Nhp2p. Nucleic Acids Res. 29, 2733–2746 (2001).
Penzo, M. et al. RNA pseudouridylation in physiology and medicine: for better and for worse. Genes 8, 301 (2017).
Song, J. H. et al. Differential roles of human PUS10 in miRNA processing and tRNA pseudouridylation. Nat. Chem. Biol. 16, 160 (2020).
Purchal, M. K. et al. Pseudouridine synthase 7 is an opportunistic enzyme that binds and modifies substrates with diverse sequences and structures. Proc. Natl Acad. Sci. USA 119, e2109708119 (2022).
Xue, C. et al. Role of main RNA modifications in cancer: N-6-methyladenosine, 5-methylcytosine, and pseudouridine. Signal Transduct. Tar. 7, 142 (2022).
Guzzi, N. et al. Pseudouridine-modified tRNA fragments repress aberrant protein synthesis and predict leukaemic progression in myelodysplastic syndrome. Nat. Cell Biol. 24, 299 (2022).
Karikó, K. et al. Incorporation of pseudouridine Into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 16, 1833–1840 (2008).
Carlile, T. M. et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 (2014).
King, T. H., Liu, B., McCully, R. R. & Fournier, M. J. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell 11, 425–435 (2003).
Jack, K. et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell 44, 660–666 (2011).
Zhao, X. & Yu, Y. T. Pseudouridines in and near the branch site recognition region of U2 snRNA are required for snRNP biogenesis and pre-mRNA splicing in Xenopus oocytes. RNA 10, 681–690 (2004).
Yang, C., McPheeters, D. S. & Yu, Y. T. Psi35 in the branch site recognition region of U2 small nuclear RNA is important for pre-mRNA splicing in Saccharomyces cerevisiae. J. Biol. Chem. 280, 6655–6662 (2005).
Morais, P., Adachi, H. & Yu, Y. T. The critical contribution of pseudouridine to mRNA COVID-19 vaccines. Front. Cell Dev. Biol. 9, 789427 (2021).
Benne, R. et al. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46, 819–826 (1986).
Lavrov, D. V., Brown, W. M. & Boore, J. L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl Acad. Sci. USA 97, 13738–13742 (2000).
Decatur, W. A. & Fournier, M. J. RNA-guided nucleotide modification of ribosomal and other RNAs. J. Biol. Chem. 278, 695–698 (2003).
Choudhury, Y. et al. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Investig. 122, 4059–4076 (2012).
Wagner, R. W., Smith, J. E., Cooperman, B. S. & Nishikura, K. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc. Natl Acad. Sci. USA 86, 2647–2651 (1989).
Yang, Y., Okada, S. & Sakurai, M. Adenosine-to-inosine RNA editing in neurological development and disease. RNA Biol. 18, 999–1013 (2021).
Mannion, N. M. et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494 (2014).
Ramaswami, G. & Li, J. B. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–D113 (2014).
Yang, W. D. et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 13, 13–21 (2006).
Nishikura, K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat. Rev. Mol. Cell Biol. 7, 919–931 (2006).
Bass, B. L. RNA editing by adenosine deaminases that act on RNA. Annu Rev. Biochem. 71, 817–846 (2002).
Bass, B. L. & Weintraub, H. A developmentally regulated activity that unwinds RNA duplexes. Cell 48, 607–613 (1987).
Ryter, J. M. & Schultz, S. C. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 17, 7505–7513 (1998).
Chen, C. X. et al. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6, 755–767 (2000).
Nishikura, K. et al. Substrate specificity of the dsRNA unwinding/modifying activity. EMBO J. 10, 3523–3532 (1991).
Vitali, P. et al. ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J. Cell Biol. 169, 745–753 (2005).
Farajollahi, S. & Maas, S. Molecular diversity through RNA editing: a balancing act. Trends Genet 26, 221–230 (2010).
Ota, H. et al. ADAR1 forms a complex with dicer to promote MicroRNA processing and RNA-induced gene silencing. Cell 153, 575–589 (2013).
Chandel, N. S. Glycolysis. Cold Spring Harb. Perspect. Biol. 13, 5 (2021).
Warburg, O., Wind, F. & Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 (1927).
Stincone, A. et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 90, 927–963 (2015).
Grochowski, L. L., Xu, H. & White, R. H. Ribose-5-phosphate biosynthesis in Methanocaldococcus jannaschii occurs in the absence of a pentose-phosphate pathway. J. Bacteriol. 187, 7382–7389 (2005).
Han, H. S. et al. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 48, e218 (2016).
Sun, J. et al. PARP1 is upregulated by hyperglycemia Via N6-methyladenosine modification and promotes diabetic retinopathy. Discov. Med. 34, 115–129 (2022).
De Jesus, D. F. et al. m6A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat. Metab. 1, 765–774 (2019).
Wang, Y. Q. et al. m6A mRNA methylation controls functional maturation in neonatal murine β-cells. Diabetes 69, 1708–1722 (2020).
Regue, L. et al. RNA m6A reader IMP2/IGF2BP2 promotes pancreatic beta-cell proliferation and insulin secretion by enhancing PDX1 expression. Mol. Metab. 48, 101209 (2021).
Peng, T. et al. LncRNA Airn alleviates diabetic cardiac fibrosis by inhibiting activation of cardiac fibroblasts via a m6A-IMP2-p53 axis. Biol. Direct. 17, 32 (2022).
Meng, L. et al. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis. 13, 38 (2022).
Khoshi, A. et al. Association of Omentin rs2274907 and FTO rs9939609 gene polymorphisms with insulin resistance in Iranian individuals with newly diagnosed type 2 diabetes. Lipids Health Dis. 18, 142 (2019).
Hjort, R. et al. Interaction between overweight and genotypes of HLA, TCF7L2, and FTO in relation to the risk of latent autoimmune diabetes in adults and type 2 diabetes. J. Clin. Endocr. Metab. 104, 4815–4826 (2019).
Nasser, F. A. et al. The association of the common fat mass and obesity associated gene polymorphisms with type 2 diabetes in obese Iraqi population. Diabetes Metab. Synd. 13, 2451–2455 (2019).
Yang, Y. et al. Glucose is involved in the dynamic regulation of m6a in patients with type 2 diabetes. J. Clin. Endocr. Metab. 104, 665–673 (2019).
Peng, S. M. et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci. Transl. Med. 11, eaau7116 (2019).
Zhou, J. et al. Methyladenosine guides mRNA alternative translation during integrated stress response. Mol. Cell 69, 636 (2018). -+.
Li, K. et al. MicroRNA-214 suppresses gluconeogenesis by targeting activating transcriptional factor 4. J. Biol. Chem. 290, 8185–8195 (2015).
Hu, F. et al. MiR-495 regulates macrophage M1/M2 polarization and insulin resistance in high-fat diet-fed mice via targeting FTO. Pflug. Arch. 471, 1529–1537 (2019).
Li, Z. H. et al. N-6-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun 11, 1 (2020).
Wang, Q. Q. et al. N6-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis. 11, 911 (2020).
Ma, L. et al. The essential roles of m(6)A RNA modification to stimulate ENO1-dependent glycolysis and tumorigenesis in lung adenocarcinoma. J. Exp. Clin. Cancer Res. 41, 36 (2022).
Shen, C. Q. et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 19, 1 (2020).
Chen, H. R. et al. RNA N-6-methyladenosine methyltransferase METTL3 facilitates colorectal cancer by activating the m(6)A-GLUT1-mTORC1 axis and is a therapeutic target. Gastroenterology 160, 1284 (2021).
Liu, X. S. et al. Overexpression of METTL3 associated with the metabolic status on (18)F-FDG PET/CT in patients with Esophageal Carcinoma. J. Cancer 11, 4851–4860 (2020).
Li, F. J. et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol. Res. 187, 106555 (2023).
Zhang, K. et al. N-6-methyladenosine-mediated LDHA induction potentiates chemoresistance of colorectal cancer cells through metabolic reprogramming. Theranostics 12, 4802–4817 (2022).
Zheng, Y. et al. N6-methyladenosine modification of PTTG3P contributes to colorectal cancer proliferation via YAP1. Front. Oncol. 11, 669731 (2021).
Wang, Q. et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 69, 1193–1205 (2020).
Zhang, Q. et al. METTL3-induced DLGAP1-AS2 promotes non-small cell lung cancer tumorigenesis through m(6)A/c-Myc-dependent aerobic glycolysis. Cell Cycle 21, 2602–2614 (2022).
Wang, W. et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N(6)-methyladenosine-dependent YTHDF binding. Nat. Commun. 12, 3803 (2021).
Yang, N. et al. HBXIP drives metabolic reprogramming in hepatocellular carcinoma cells via METTL3-mediated m6A modification of HIF-1alpha. J. Cell Physiol. 236, 3863–3880 (2021).
Xu, Y. et al. The N6-methyladenosine METTL3 regulates tumorigenesis and glycolysis by mediating m6A methylation of the tumor suppressor LATS1 in breast cancer. J. Exp. Clin. Cancer Res. 42, 10 (2023).
Xu, W. et al. m6A RNA methylation-mediated NDUFA4 promotes cell proliferation and metabolism in gastric cancer. Cell Death Dis. 13, 715 (2022).
Cai, J. et al. METTL3 promotes glycolysis and cholangiocarcinoma progression by mediating the m6A modification of AKR1B10. Cancer Cell Int. 22, 385 (2022).
Chen, Z. et al. Non-SMC condensin I complex subunit H participates in anti-programmed cell death-1 resistance of clear cell renal cell carcinomas. Cell Prolif. 56, e13400 (2023).
Xue, L. et al. m(6)A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J. Cell Physiol. 236, 2649–2658 (2021).
Liu, J., Yuan, J. F. & Wang, Y. Z. METTL3-stabilized lncRNA SNHG7 accelerates glycolysis in prostate cancer via SRSF1/c-Myc axis. Exp. Cell Res. 416, 113149 (2022).
Liu, Z. H. et al. N6-methyladenosine-modified circular RNA QSOX1 promotes colorectal cancer resistance to anti-CTLA-4 therapy through induction of intratumoral regulatory T cells. Drug Resist. Update 65, 100886 (2022).
He, Y. et al. Linc-UROD stabilizes ENO1 and PKM to strengthen glycolysis, proliferation and migration of pancreatic cancer cells. Transl. Oncol. 27, 101583 (2023).
Hou, Y. et al. METTL14 modulates glycolysis to inhibit colorectal tumorigenesis in p53-wild-type cells. EMBO Rep. 24, e56325 (2023).
Du, L. et al. USP48 is upregulated by Mettl14 to attenuate hepatocellular carcinoma via regulating SIRT6 stabilization. Cancer Res. 81, 3822–3834 (2021).
Zhang, C. J. et al. Downregulated METTL14 accumulates BPTF that reinforces super-enhancers and distal lung metastasis via glycolytic reprogramming in renal cell carcinoma. Theranostics 11, 3676–3693 (2021).
Lin, J. X. et al. m6A methylation mediates LHPP acetylation as a tumour aerobic glycolysis suppressor to improve the prognosis of gastric cancer. Cell Death Dis. 13, 463 (2022).
Yu, H. et al. N-6-methyladenosine (m(6)A) methyltransferase WTAP accelerates the Warburg effect of gastric cancer through regulating HK2 stability. Biomed. Pharmacother. 133, 111075 (2021).
Lyu, Y. Y. et al. HIF-1 alpha Regulated WTAP overexpression promoting the Warburg effect of ovarian cancer by m6A-dependent manner. J. Immunol. Res 2022, 6130806 (2022).
Ou, B. C. et al. C5aR1-positive neutrophils promote breast cancer glycolysis through WTAP-dependent m6A methylation of ENO1. Cell Death Dis. 12, 737 (2021).
Zhang, Y. et al. WTAP mediates FOXP3 mRNA stability to promote SMARCE1 expression and augment glycolysis in colon adenocarcinoma. Mamm. Genome 33, 654–671 (2022).
Li, H., Li, C., Zhang, B. & Jiang, H. Lactoferrin suppresses the progression of colon cancer under hyperglycemia by targeting WTAP/m(6)A/NT5DC3/HKDC1 axis. J. Transl. Med. 21, 156 (2023).
Li, Y. et al. N(6)-methyladenosine methyltransferase KIAA1429 elevates colorectal cancer aerobic glycolysis via HK2-dependent manner. Bioengineered 13, 11923–11932 (2022).
Yang, D. S. et al. m(6)A transferase KIAA1429-stabilized LINC00958 accelerates gastric cancer aerobic glycolysis through targeting GLUT1. IUBMB Life 73, 1325–1333 (2021).
Yang, F. et al. Circ-CTNNB1 drives aerobic glycolysis and osteosarcoma progression via m6A modification through interacting with RBM15. Cell Proliferat 56, 1 (2023).
Wang, Q. B. et al. ZC3H13 inhibits the progression of hepatocellular carcinoma through m(6)A-PKM2-mediated glycolysis and enhances chemosensitivity. J Oncol 2021, 1328444 (2021).
Li, J. et al. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am. J. Transl. Res. 11, 6084–6092 (2019).
Li, X. D. et al. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 112, 4543–4552 (2021).
Yanar, S. et al. Proteomics analysis of meclofenamic acid-treated small cell lung carcinoma cells revealed changes in cellular energy metabolism for cancer cell survival. J. Biochem. Mol. Toxic. 37, 4 (2023).
Qing, Y. et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol. Cell 81, 922 (2021).
Liu, Y. et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 33, 1221 (2021).
Liu, C. Y. et al. E6E7 regulates the HK2 expression in cervical cancer via GSK3B/FTO signal. Arch. Biochem. Biophys. 729, 109389 (2022).
Yang, X. et al. WNT/beta-catenin-suppressed FTO expression increases m(6)A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 12, 462 (2021).
Huang, J. et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J. Exp. Clin. Cancer Res. 41, 42 (2022).
Liu, Z., Chen, Y., Wang, L. & Ji, S. ALKBH5 promotes the proliferation of glioma cells via enhancing the mRNA stability of G6PD. Neurochem. Res. 46, 3003–3011 (2021).
Liu, H. et al. ALKBH5-mediated m6A demethylation of GLUT4 mRNA promotes glycolysis and resistance to HER2-targeted therapy in breast cancer. Cancer Res. 82, 3974–3986 (2022).
Ji, X. et al. ALKBH5-induced circular RNA NRIP1 promotes glycolysis in thyroid cancer cells by targeting PKM2. Cancer Sci. 114, 2318–2334 (2023).
Zhao, L. et al. UBR7 inhibits HCC tumorigenesis by targeting Keap1/Nrf2/Bach1/HK2 and glycolysis. J. Exp. Clin. Cancer Res. 41, 330 (2022).
Wang, Y. Q. et al. HLA complex P5 upregulation is correlated with poor prognosis and tumor progression in esophageal squamous cell carcinoma. Bioengineered 13, 9301–9311 (2022).
Sheng, H. et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis 41, 541–550 (2020).
Chen, B. X. et al. N6-methyladenosine modification of circ_0003215 suppresses the pentose phosphate pathway and malignancy of colorectal cancer through the miR-663b/DLG4/G6PD axis. Cell Death Dis. 13, 804 (2022).
Li, H. Y. Downregulation of microRNA-6125 promotes colorectal cancer growth through YTHDF2-dependent recognition of N6-methyladenosine-modified GSK3 beta. Clin. Transl. Med. 12, e1085 (2022).
Zhou, L. et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/beta-catenin signaling to promote colorectal cancer progression by preventing m(6)A-mediated degradation of STEAP3 mRNA. Mol. Cancer 21, 168 (2022).
Zhou, R. et al. A functional loop between YTH domain family protein YTHDF3 mediated m(6)A modification and phosphofructokinase PFKL in glycolysis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 41, 334 (2022).
Hu, Y. H. et al. A reciprocal feedback between N6-methyladenosine reader YTHDF3 and lncRNA DICER1-AS1 promotes glycolysis of pancreatic cancer through inhibiting maturation of miR-5586-5p. J. Exp. Clin. Cancer Res. 41, 62 (2022).
Liu, D. Y. et al. N-6-methyladenosine reader YTHDF3 contributes to the aerobic glycolysis of osteosarcoma through stabilizing PGK1 stability. J. Cancer Res. Clin. 149, 4601–4610 (2022).
Luo, F. & Lin, K. N-6-methyladenosine (m(6)A) reader IGF2BP1 accelerates gastric cancer aerobic glycolysis in c-Myc-dependent manner. Exp. Cell Res. 417, 113176 (2022).
Yuan, B. & Zhou, J. N6-methyladenosine (m6A) reader IGF2BP1 facilitates clear-cell renal cell carcinoma aerobic glycolysis. PeerJ. 11, e14591 (2023).
Xu, K., Dai, X. J., Wu, J. K. & Wen, K. N-6-methyladenosine (m(6)A) reader IGF2BP2 stabilizes HK2 stability to accelerate the Warburg effect of oral squamous cell carcinoma progression. J. Cancer Res. Clin. 148, 3375–3384 (2022).
Hu, C. C. et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m(6)A-MYC expression. Int. J. Biol. Sci. 18, 507–521 (2022).
Ye, Y. et al. lncRNA miR4458HG modulates hepatocellular carcinoma progression by activating m6A-dependent glycolysis and promoting the polarization of tumor-associated macrophages. Cell Mol. Life Sci. 80, 99 (2023).
Cui, Y. M. et al. m(6)A-modified circFOXK2 targets GLUT1 to accelerate oral squamous cell carcinoma aerobic glycolysis. Cancer Gene Ther. 30, 163–171 (2023).
Xu, T. P. et al. LOC101929709 promotes gastric cancer progression by aiding LIN28B to stabilize c-MYC mRNA. Gastr. Cancer 26, 169–186 (2023).
Wu, Y. M. et al. RNA m(1)A methylation regulates glycolysis of cancer cells through modulating ATP5D. Proc. Natl Acad. Sci. USA. 119, e2119038119 (2022).
Wang, J. Z. et al. The role of the HIF-1 alpha/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun. 41, 560–575 (2021).
Xu, F. S. et al. N7-methylguanosine regulatory genes well represented by METTL1 define vastly different prognostic, immune and therapy landscapes in adrenocortical carcinoma. Am. J. Cancer Res. 13, 538 (2023).
Zhang, B. J. et al. m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct. Tar 6, 377 (2021).
Zhou, Y. et al. METTL3 boosts glycolysis and cardiac fibroblast proliferation by increasing AR methylation. Int. J. Biol. Macromol. 223, 899–915 (2022).
Cai, W. et al. METTL3-dependent glycolysis regulates dental pulp stem cell differentiation. J. Dent. Res. 101, 580–589 (2022).
Jiao, Y., Williams, A. & Wei, N. Quercetin ameliorated insulin resistance via regulating METTL3-mediated N6-methyladenosine modification of PRKD2 mRNA in skeletal muscle and C2C12 myocyte cell line. Nutr. Metab. Cardiovasc. 32, 2655–2668 (2022).
Fahy, E. et al. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50, S9–14, (2009).
Ahmed, M. H. & Byrne, C. D. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD). Drug Discov. Today 12, 740–747 (2007).
Li, L. et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 63, 1900–1913 (2016).
Luo, J., Yang, H. & Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 21, 225–245 (2020).
Rønningen, T. et al. m6A regulators in human adipose tissue—depot-specificity and correlation with obesity. Front Endocrinol. 12, 778875 (2021).
Wang, Y. Q. et al. METTL3 is essential for postnatal development of brown adipose tissue and energy expenditure in mice. Nat. Commun. 11, 1648 (2020).
Xie, W. et al. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of Fasn mRNA and promoting fatty acid metabolism. Biochem. Biophys. Res. Commun. 518, 120–126 (2019).
Liu, Q. et al. ZFP217 regulates adipogenesis by controlling mitotic clonal expansion in a METTL3-m(6)A dependent manner. RNA Biol. 16, 1785–1793 (2019).
Ningombam, S. S. et al. Differential distribution and association of FTO gene polymorphism with obesity: a cross-sectional study among two tribal populations of India with East-Asian ancestry. Gene 647, 198–204 (2018).
Hebbar, P. et al. Variant rs1421085 associates with increased body weight, soft lean mass, and total body water through interaction with ghrelin and apolipoproteins in Arab population. Front. Genet. 10, 1411 (2020).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014).
Wu, R. et al. FTO regulates adipogenesis by controlling cell cycle progression via m(6)A-YTHDF2 dependent mechanism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 1323–1330 (2018).
Wu, R. et al. Epigallocatechin gallate targets FTO and inhibits adipogenesis in an mRNA m(6)A-YTHDF2-dependent manner. Int. J. Obes. 42, 1378–1388 (2018).
Song, T. et al. Zfp217 mediates m6A mRNA methylation to orchestrate transcriptional and post-transcriptional regulation to promote adipogenic differentiation. Nucleic Acids Res. 47, 6130–6144 (2019).
Shen, G. S. et al. The GDF11-FTO-PPARγ axis controls the shift of osteoporotic MSC fate to adipocyte and inhibits bone formation during osteoporosis. BBA-Mol. Basis Dis. 1864, 3644–3654 (2018).
Kruger, N. et al. Loss of endothelial FTO antagonizes obesity-induced metabolic and vascular dysfunction. Circ. Res. 126, 232–242 (2020).
Wu, W. C. et al. AMPK regulates lipid accumulation in skeletal muscle cells through FTO-dependent demethylation of N6-methyladenosine. Sci. Rep. 7, 41606 (2017).
Zhou, X. H. et al. The beneficial effects of betaine on dysfunctional adipose tissue and N6-methyladenosine mRNA methylation requires the AMP-activated protein kinase α1 subunit. J. Nutr. Biochem. 26, 1678–1684 (2015).
Jiang, Q. et al. promotes adipogenesis in intramuscular preadipocytes an m6A-YTHDF1-dependent mechanism. FASEB J. 33, 2971–2981 (2019).
Liu, Q. et al. ZFP217 regulates adipogenesis by controlling mitotic clonal expansion in a METTL3-m6A dependent manner. RNA Biol. 16, 1785–1793 (2019).
Liu, W. et al. ‘Micro-managers’ of hepatic lipid metabolism and NAFLD. Wiley Interdiscip. Rev. Rna 6, 581–593 (2015).
Yang, Y. et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol. Ther. 30, 2342–2353 (2022).
Peng, Z. et al. METTL3-m(6)A-Rubicon axis inhibits autophagy in nonalcoholic fatty liver disease. Mol. Ther. 30, 932–946 (2022).
Chen, J. et al. FTO-dependent function of N6-methyladenosine is involved in the hepatoprotective effects of betaine on adolescent mice. J. Physiol. Biochem. 71, 405–413 (2015).
Guo, J. et al. Fat mass and obesity-associated gene enhances oxidative stress and lipogenesis in nonalcoholic fatty liver disease. Dig. Dis. Sci. 58, 1004–1009 (2013).
Hu, Y. et al. GR-mediated FTO transactivation induces lipid accumulation in hepatocytes via demethylation of m(6)A on lipogenic mRNAs. RNA Biol. 17, 930–942 (2020).
Sun, L. M. et al. Differential mechanisms regarding triclosan vs. bisphenol A and fluorene-9-bisphenol induced zebrafish lipid-metabolism disorders by RNA-seq. Chemosphere. 251, 126318 (2020).
Hu, Y. et al. GR-mediated FTO transactivation induces lipid accumulation in hepatocytes via demethylation of m6A on lipogenic mRNAs. RNA Biol. 17, 930–942 (2020).
Li, S. et al. Exenatide ameliorates hepatic steatosis and attenuates fat mass and gene expression through PI3K signaling pathway in nonalcoholic fatty liver disease. Braz. J. Med. Biol. Res. 51, e7299 (2018).
Gan, X. et al. FTO promotes liver inflammation by suppressing m6A mRNA methylation of IL-17RA. Front. Oncol. 12, 989353 (2022).
Wang, H. Q. et al. LINC01468 drives NAFLD-HCC progression through CUL4A-linked degradation of SHIP2. Cell Death Discov. 8, 499 (2022).
Yang, J. J. et al. ALKBH5 ameliorated liver fibrosis and suppressed HSCs activation via triggering PTCH1 activation in an m6A dependent manner. Eur. J. Pharmacol. 922, 174900 (2022).
Sun, R. M. et al. The m6A reader YTHDF3-mediated PRDX3 translation alleviates liver fibrosis. Redox Biol. 54, 102378 (2022).
Libby, P., Ridker, P. M. & Hansson, G. K. Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 (2011).
Back, M. et al. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Chen, J. et al. Silencing METTL3 stabilizes atherosclerotic plaques by regulating the phenotypic transformation of vascular smooth muscle cells via the miR-375-3p/PDK1 axis. Cardiovasc. Drugs Ther. 37, 471–486 (2023).
Liu, Y. et al. Methyltransferase-like 14 silencing relieves the development of atherosclerosis via m(6)A modification of p65 mRNA. Bioengineered 13, 11832–11843 (2022).
Gong, C., Fan, Y. & Liu, J. METTL14 mediated m6A modification to LncRNA ZFAS1/ RAB22A: A novel therapeutic target for atherosclerosis. Int. J. Cardiol. 328, 177 (2021).
Mo, C. et al. Fat mass and obesity-associated protein attenuates lipid accumulation in macrophage foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. J. Hypertens. 35, 810–821 (2017).
Yang, Z. et al. Critical roles of FTO-mediated mRNA m6A demethylation in regulating adipogenesis and lipid metabolism: implications in lipid metabolic disorders. Genes Dis. 9, 51–61 (2022).
Ladanyi, A. et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301 (2018).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Hisano, Y. & Hla, T. Bioactive lysolipids in cancer and angiogenesis. Pharm. Ther. 193, 91–98 (2019).
Liu, P. H. et al. m(6)A-induced lncDBET promotes the malignant progression of bladder cancer through FABP5-mediated lipid metabolism. Theranostics 12, 6291–6307 (2022).
Zhong, X. et al. Circadian clock regulation of hepatic lipid metabolism by modulation of m(6)A mRNA methylation. Cell Rep. 25, 1816–1828 e1814 (2018).
Chen, Z. J. et al. N6-methyladenosine-induced ERR. triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics 10, 3382–3396 (2020).
Peng, H. et al. N(6)-methyladenosine (m(6)A) in 18S rRNA promotes fatty acid metabolism and oncogenic transformation. Nat. Metab. 4, 1041–1054 (2022).
Sun, D. L. et al. Fat mass and obesity-associated protein regulates lipogenesis via m(6)A modification in fatty acid synthase mRNA. Cell Biol. Int. 45, 334–344 (2021).
Duan, X. R. et al. m6A demethylase FTO promotes tumor progression via regulation of lipid metabolism in esophageal cancer. Cell Biosci. 12, 60 (2022).
Zhen, L. & Pan, W. Y. ALKBH5 inhibits the SIRT3/ACC1 axis to regulate fatty acid metabolism via an m6A-IGF2BP1-dependent manner in cervical squamous cell carcinoma. Clin. Exp. Pharm. Pract. 50, 380–392 (2023).
Zhou, B. et al. N-6-methyladenosine reader protein YT521-B homology domain-containing 2 suppresses liver steatosis by regulation of mRNA stability of lipogenic genes. Hepatology 73, 91–103 (2021).
Fang, R. P. et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholestÿerol dysregulation and invasive growth of glioblastoma. Nat. Commun. 12, 117 (2021).
Guo, W. et al. M6A methylation of DEGS2, a key ceramide-synthesizing enzyme, is involved in colorectal cancer progression through ceramide synthesis. Oncogene 40, 5913–5924 (2021).
Cheng, Y. et al. Decoding m(6)A RNA methylome identifies PRMT6-regulated lipid transport promoting AML stem cell maintenance. Cell Stem Cell 30, 69–85 e67 (2023).
Jia, Y. X. et al. Long non-coding RNA NEAT1 mediated RPRD1B stability facilitates fatty acid metabolism and lymph node metastasis via c-Jun/c-Fos/SREBP1 axis in gastric cancer. J. Exp. Clin. Cancer Res. 41, 287 (2022).
Guo, H. M. et al. m(6)A Reader HNRNPA2B1 promotes esophageal cancer progression via up-regulation of ACLY and ACC1. Front. Oncol. 10, 553045 (2020).
Liu, Y. et al. mRNA m(5)C inhibits adipogenesis and promotes myogenesis by respectively facilitating YBX2 and SMO mRNA export in ALYREF-m(5)C manner. Cell Mol. Life Sci. 79, 481 (2022).
Yang, M. et al. NSUN2 promotes osteosarcoma progression by enhancing the stability of FABP5 mRNA via m(5)C methylation. Cell Death Dis. 14, 125 (2023).
Xu, J. et al. RNA 5-methylcytosine regulators contribute to metabolism heterogeneity and predict prognosis in ovarian cancer. Front. Cell Dev. Biol. 10, 807786 (2022).
Wang, Y. et al. N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat. Commun. 12, 6314 (2021).
Courtney, K. D. et al. Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo. Cell Metab. 28, 793–800.e792 (2018).
Porporato, P. E. et al. Mitochondrial metabolism and cancer. Cell Res. 28, 265–280 (2018).
Zhang, X. et al. The m(6)A methyltransferase METTL3 modifies PGC-1alpha mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J. Biol. Chem. 297, 101058 (2021).
Zhuang, C. S. et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1 alpha signalling axis. J. Cell Mol. Med. 23, 2163–2173 (2019).
Sun, Y. et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J. Exp. Clin. Cancer Res. 42, 65 (2023).
Liu, X. C. et al. Adenylate kinase 4 modulates the resistance of breast cancer cells to tamoxifen through an m(6)A-based epitranscriptomic mechanism. Mol. Ther. 28, 2593–2604 (2020).
Hou, H. Y. METTL3 promotes the proliferation and invasion of esophageal cancer cells partly through AKT signaling pathway. Pathol. Res. Pract 216, 153087 (2020).
Sun, K. Y. et al. METTL14-dependent maturation of pri-miR-17 regulates mitochondrial homeostasis and induces chemoresistance in colorectal cancer. Cell Death Dis. 14, 148 (2023).
Deng, P. et al. Long-term cadmium exposure impairs cognitive function by activating lnc-Gm10532/m6A/FIS1 axis-mediated mitochondrial fission and dysfunction. Sci. Total Environ. 858, 159950 (2023).
Du, Y. D. et al. N6-methyladenosine demethylase FTO impairs hepatic ischemia-reperfusion injury via inhibiting Drp1-mediated mitochondrial fragmentation. Cell Death Dis. 12, 442 (2021).
Li, N. et al. Gene characteristics and prognostic values of m(6)A RNA methylation regulators in nonsmall cell lung cancer. J. Health. Eng. 2021, 2257066 (2021).
Wang, Z. S. et al. Expression and prognostic potential of ribosome 18S RNA m(6)A methyltransferase METTL5 in gastric cancer. Cancer Cell Int. 21, 569 (2021).
Wang, C. et al. Crosstalk of oxidative phosphorylation-related subtypes, establishment of a prognostic signature and immune infiltration characteristics in colorectal adenocarcinoma. Cancers 14, 4503 (2022).
Yin, R. et al. Differential m(6)A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell 29, 149–159.e147 (2022).
Sun, L. et al. RNA-binding protein RALY reprogrammes mitochondrial metabolism via mediating miRNA processing in colorectal cancer. Gut 70, 1698–1712 (2021).
Li, G. W., Lu, X. F., Xu, Q. Q. & Jin, Y. P. The FDX1 methylation regulatory mechanism in the malignant phenotype of glioma. Genomics 115, 110601 (2023).
Jia, C. et al. HNRNPA2B1-mediated m6A modification of TLR4 mRNA promotes progression of multiple myeloma. J. Transl. Med. 20, 537 (2022).
Curi, R. et al. Molecular mechanisms of glutamine action. J. Cell Physiol. 204, 392–401 (2005).
Vettore, L., Westbrook, R. L. & Tennant, D. A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 122, 150–156 (2020).
Xiao, Y. R. et al. The m(6)A RNA demethylase FTO is a HIF-independent synthetic lethal partner with the VHL tumor suppressor. Proc. Natl Acad. Sci. USA 117, 21441–21449 (2020).
Han, S. T. et al. Targeting ATF4-dependent pro-survival autophagy to synergize glutaminolysis inhibition. Theranostics 11, 8464–8479 (2021).
Zhou, J. et al. N(6)-methyladenosine guides mRNA alternative translation during integrated stress response. Mol. Cell 69, 636–647.e637 (2018).
Chen, P. et al. Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism. Mol. Ther.Oncol. 20, 228–239 (2021).
Weng, H. Y. et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell 40, 1566 (2022).
Chen, T. et al. WZ35 inhibits gastric cancer cell metastasis by depleting glutathione to promote cellular metabolic remodeling. Cancer Lett. 555, 216044 (2023).
Han, L. et al. METTL16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming BCAA metabolism. Cell Stem Cell 30, 52 (2023).
Traba, J., Sack, M. N., Waldmann, T. A. & Anton, O. M. Immunometabolism at the nexus of cancer therapeutic efficacy and resistance. Front. Immunol 12, 657293 (2021).
Zhu, Y. Y. et al. The E3 ligase VHL promotes follicular helper T cell differentiation via glycolytic-epigenetic control. J. Exp. Med. 216, 1664–1681 (2019).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015).
Zhu, X., Tang, H., Yang, M. & Yin, K. N6-methyladenosine in macrophage function: a novel target for metabolic diseases. Trends Endocrinol. Metab. 34, 66–84 (2023).
Ning, H. F. et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int. Immunopharmacol 83, 106432 (2020).
Bian, Y. et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 (2020).
Brand, A. et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 24, 657–671 (2016).
Reinfeld, B. I. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282 (2021).
Gou, Y. L. et al. Ectopic endometriotic stromal cells-derived lactate induces M2 macrophage polarization via Mettl3/Trib1/ERK/STAT3 signalling pathway in endometriosis. Immunology 168, 389–402 (2023).
Zhao, W. et al. RNA helicase DDX5 participates in oxLDL-induced macrophage scavenger receptor 1 expression by suppressing mRNA degradation. Exp. Cell Res. 366, 114–120 (2018).
Patsoukis, N. et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015).
Kumagai, S. et al. An oncogenic alteration creates a microenvironment that promotes tumor progression by conferring a metabolic advantage to regulatory T cells. Immunity 53, 187 (2020).
Cao, W. et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 192, 2920–2931 (2014).
Herber, D. L. et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–U857 (2010).
Gokhale, N. S. & Horner, S. M. RNA modifications go viral. Plos Pathog 13, e1006188 (2017).
Xue, M. G. et al. Viral RNA N6-methyladenosine modification modulates both innate and adaptive immune responses of human respiratory syncytial virus. PloS Pathog 17, e1010142 (2021).
Rubio, R. M. et al. RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 32, 1472–1484 (2018).
Xu, J. et al. The RNA helicase DDX5 promotes viral infection via regulating N6-methyladenosine levels on the DHX58 and NFkappaB transcripts to dampen antiviral innate immunity. Plos Pathog. 17, e1009530 (2021).
Liu, Y. et al. N (6)-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 365, 1171–1176 (2019).
Medzhitov, R. & Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 (2009).
Hawiger, J. & Zienkiewicz, J. Decoding inflammation, its causes, genomic responses, and emerging countermeasures. Scand. J. Immunol. 90, e12812 (2019).
Wang, J. et al. METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-kappaB signaling pathway. Mediators Inflamm. 2019, 3120391 (2019).
Guo, G. et al. Disease activity-associated alteration of mRNA m(5) C methylation in CD4(+) T cells of systemic lupus erythematosus. Front Cell Dev. Biol. 8, 430 (2020).
Liu, J. et al. CCR7 chemokine receptor-inducible lnc-Dpf3 restrains dendritic cell migration by inhibiting HIF-1alpha-mediated glycolysis. Immunity 50, 600–615.e615 (2019).
Zheng, L. B. et al. RNA-m6A modification of HDGF mediated by Mettl3 aggravates the progression of atherosclerosis by regulating macrophages polarization via energy metabolism reprogramming. Biochem. Biophys. Res. Commun. 635, 120–127 (2022).
Pan, S. et al. N6-methyladenosine upregulates miR-181d-5p in exosomes derived from cancer-associated fibroblasts to inhibit 5-FU sensitivity by targeting NCALD in colorectal cancer. Int. J. Oncol 60, 14 (2022).
Liu, X. et al. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/beta-catenin pathway. J. Exp. Clin. Cancer Res 40, 132 (2021).
Uddin, M. B. et al. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharm. 160, 134–145 (2019).
Nishizawa, Y. et al. Oncogene c-Myc promotes epitranscriptome m(6)A reader YTHDF1 expression in colorectal cancer. Oncotarget 9, 7476–7486 (2018).
Jiang, Z. et al. Circular RNA protein tyrosine kinase 2 (circPTK2) promotes colorectal cancer proliferation, migration, invasion and chemoresistance. Bioengineered 13, 810–823 (2022).
Ye, X. et al. Increased m(6)A modification of lncRNA DBH-AS1 suppresses pancreatic cancer growth and gemcitabine resistance via the miR-3163/USP44 axis. Ann. Transl. Med. 10, 304 (2022).
Zhang, C. et al. m(6)A methyltransferase METTL14-mediated upregulation of cytidine deaminase promoting gemcitabine resistance in pancreatic cancer. Front Oncol. 11, 696371 (2021).
Tang, B. et al. m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol. Cancer 19, 3 (2020).
Wang, L. et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 39, e104514 (2020).
Zheng, H. et al. Decreased expression of programmed death ligand-L1 by seven in absentia homolog 2 in cholangiocarcinoma enhances T-cell-mediated antitumor activity. Front Immunol. 13, 845193 (2022).
Han, D. et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274 (2019).
Yang, S. et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 10, 2782 (2019).
Mimura, K. et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 109, 43–53 (2018).
Su, R. et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell 38, 79 (2020).
Yuan, C. et al. Crosstalk of histone and RNA modifications identified a stromal-activated subtype with poor survival and resistance to immunotherapy in gastric cancer. Front. Pharm. 13, 868830 (2022).
Li, N. et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl Acad. Sci. USA 117, 20159–20170 (2020).
Zhu, J. et al. Integrative analysis of m6A RNA methylation regulators and the tumor immune microenvironment in non-small-cell lung cancer. Dis. Mark. 2022, 2989200 (2022).
Ishizuka, J. J. et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43 (2019).
Ma, S. et al. The RNA m6A reader YTHDF2 controls NK cell antitumor and antiviral immunity. J. Exp. Med. 218, e20210279 (2021).
Song, H. et al. METTL3-mediated m(6)A RNA methylation promotes the anti-tumour immunity of natural killer cells. Nat. Commun. 12, 5522 (2021).
Chen, B. et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J. Am. Chem. Soc. 134, 17963–17971 (2012).
Huang, Y. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 43, 373–384 (2015).
He, W. et al. Identification of a novel small-molecule binding site of the fat mass and obesity associated protein (FTO). J. Med. Chem. 58, 7341–7348 (2015).
Su, R. et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105.e123 (2018).
Huang, Y. et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35, 677–691.e610 (2019).
Xie, G. Y. et al. A novel inhibitor of N-6-methyladenosine demethylase FTO induces mRNA methylation and shows anti-cancer activities. Acta Pharm. Sin. B 12, 853–866 (2022).
Huff, S. et al. Rational design and optimization of m(6)A-RNA demethylase FTO inhibitors as anticancer agents. J. Med. Chem. 65, 10920–10937 (2022).
Qin, B. et al. Discovery of novel mRNA demethylase FTO inhibitors against esophageal cancer. J. Enzym Inhib. Med. Chem. 37, 1995–2003 (2022).
Selberg, S., Seli, N., Kankuri, E. & Karelson, M. Rational design of novel anticancer small-molecule RNA m6A demethylase ALKBH5 inhibitors. ACS Omega 6, 13310–13320 (2021).
Moroz-Omori, E. V. et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. Chemmedchem 16, 3035–3043 (2021).
Dolbois, A. et al. 1,4,9-Triazaspiro[5.5]undecan-2-one derivatives as potent and selective METTL3 inhibitors. J. Med. Chem. 64, 12738–12760 (2021).
Yankova, E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597 (2021).
Wang, Y. Y. et al. N-1-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat. Commun. 12, 6314 (2021).
Damase, T. R. et al. The limitless future of RNA therapeutics. Front. Bioeng. Biotech. 9, 628137 (2021).
Parr, C. J. C. et al. N 1-Methylpseudouridine substitution enhances the performance of synthetic mRNA switches in cells. Nucleic Acids Res. 48, e35 (2020).
Shatkin, A. J. Capping of eucaryotic mRNAs. Cell 9, 645–653 (1976).
Yan, Y. et al. Non-viral vectors for RNA delivery. J. Control Release 342, 241–279 (2022).
Putney, S. D., Benkovic, S. J. & Schimmel, P. R. A DNA fragment with an alpha-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease-iii and can be replicated invivo. Proc. Natl Acad. Sci. Biol. 78, 7350–7354 (1981).
Sasso, J. M. et al. The progress and promise of RNA medicine—an arsenal of targeted treatments. J. Med Chem. 65, 6975–7015 (2022).
Springer, A. D. & Dowdy, S. F. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 28, 109–118 (2018).
Liu, W. W., Zhang, Z. Y., Wang, F. & Wang, H. Emerging roles of m6A RNA modification in cancer therapeutic resistance. Exp. Hematol. Oncol. 12, 21 (2023).
Taketo, K. et al. The epitranscriptome m(6)A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J. Oncol. 52, 621–629 (2018).
Lai, X. et al. Dysregulation of LINC00470 and METTL3 promotes chemoresistance and suppresses autophagy of chronic myelocytic leukaemia cells. J. Cell Mol. Med. 25, 4248–4259 (2021).
Li, X. et al. Fat mass and obesity-associated protein regulates tumorigenesis of arecoline-promoted human oral carcinoma. Cancer Med. 10, 6402–6415 (2021).
Wang, X. et al. Fatty acid receptor GPR120 promotes breast cancer chemoresistance by upregulating ABC transporters expression and fatty acid synthesis. EBioMedicine 40, 251–262 (2019).
Wang, L. L. et al. m(6)A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 39, e104514 (2020).
Chen, H. et al. METTL3 inhibits antitumor immunity by targeting m(6)A-BHLHE41-CXCL1/CXCR2 axis to promote colorectal cancer. Gastroenterology 163, 891–907 (2022).
Tsuruta, N. et al. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem. Biophys. Res. Commun. 530, 235–239 (2020).
Wang, Y. et al. Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol. 16, 896 (2020).
Molinie, B. et al. m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat. Methods 13, 692 (2016).
Zhang, Z. et al. Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library. Nat. Methods 18, 1213 (2021).
Tegowski, M., Flamand, M. N. & Meyer, K. D. scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells. Mol. Cell 82, 868 (2022).
Xu, J. N. et al. Abnormal oxidative metabolism in a quiet genomic background underlies clear cell papillary renal cell carcinoma. Elife 8, e38986 (2019).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015).
Chen, K. et al. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) A sequencing. Angew. Chem. Int. Ed. Engl. 54, 1587–1590 (2015).
Zhang, Z. et al. Single-base mapping of m(6)A by an antibody-independent method. Sci. Adv. 5, eaax0250 (2019).
Meyer, K. D. DART-seq: an antibody-free method for global m(6)A detection. Nat. Methods 16, 1275–1280 (2019).
Koh, C. W. Q., Goh, Y. T. & Goh, W. S. S. Atlas of quantitative single-base-resolution N(6)-methyl-adenine methylomes. Nat. Commun. 10, 5636 (2019).
Wang, Y. et al. Antibody-free enzyme-assisted chemical approach for detection of N(6)-methyladenosine. Nat. Chem. Biol. 16, 896–903 (2020).
Shu, X. et al. A metabolic labeling method detects m(6)A transcriptome-wide at single base resolution. Nat. Chem. Biol. 16, 887–895 (2020).
Li, X. et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12, 311–316 (2016).
Li, X. et al. Base-resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 68, 993–1005 e1009 (2017).
Khoddami, V. & Cairns, B. R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 31, 458–464 (2013).
David, R. et al. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 29, 445–460 (2017).
Cui, X. et al. 5-methylcytosine RNA methylation in arabidopsis thaliana. Mol. Plant 10, 1387–1399 (2017).
Schwartz, S. et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162 (2014).
Li, X. et al. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597 (2015).
Enroth, C. et al. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. 47, e126 (2019).
Malbec, L. et al. Dynamic methylome of internal mRNA N(7)-methylguanosine and its regulatory role in translation. Cell Res. 29, 927–941 (2019).
Thalalla Gamage, S., Sas-Chen, A., Schwartz, S. & Meier, J. L. Quantitative nucleotide resolution profiling of RNA cytidine acetylation by ac4C-seq. Nat. Protoc. 16, 2286–2307 (2021).
Sakurai, M. & Suzuki, T. Biochemical identification of A-to-I RNA editing sites by the inosine chemical erasing (ICE) method. Methods Mol. Biol. 718, 89–99 (2011).
Acknowledgements
This study was supported by the Scientific Research Project of Anhui Provincial Education Department (2022AH020079), the Natural Science Foundation of Anhui Province (2008085MH241).
Author information
Authors and Affiliations
Contributions
Wei-Wei Liu, Si-Qing Zheng, Tian Li, Chen Wang and Shuang Zhang collected the related literature, wrote the manuscript and drew the figures. Hao Wang, Fei Wang, and Guan-Min Jiang participated in the design of the review and revised the manuscript. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Consent for publication Not Applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, WW., Zheng, SQ., Li, T. et al. RNA modifications in cellular metabolism: implications for metabolism-targeted therapy and immunotherapy. Sig Transduct Target Ther 9, 70 (2024). https://doi.org/10.1038/s41392-024-01777-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01777-5
