
Abstract
Cell and gene therapies hold tremendous promise for treating a range of difficult-to-treat diseases. However, concerns over the safety and efficacy require to be further addressed in order to realize their full potential. Synthetic receptors, a synthetic biology tool that can precisely control the function of therapeutic cells and genetic modules, have been rapidly developed and applied as a powerful solution. Delicately designed and engineered, they can be applied to finetune the therapeutic activities, i.e., to regulate production of dosed, bioactive payloads by sensing and processing user-defined signals or biomarkers. This review provides an overview of diverse synthetic receptor systems being used to reprogram therapeutic cells and their wide applications in biomedical research. With a special focus on four synthetic receptor systems at the forefront, including chimeric antigen receptors (CARs) and synthetic Notch (synNotch) receptors, we address the generalized strategies to design, construct and improve synthetic receptors. Meanwhile, we also highlight the expanding landscape of therapeutic applications of the synthetic receptor systems as well as current challenges in their clinical translation.
Similar content being viewed by others
Introduction
As the next milestone in fighting diseases, cell and gene therapies are transforming the field of medicine by offering targeted and personalized treatments to patients that are not achievable by conventional pharmaceutics.1,2,3 As of now, chimeric antigen receptor T (CAR T) cell therapies for blood cancers,4,5,6 genetically engineered hematopoietic stem cells for hematologic disorders7,8 and gene therapies to treat a range of rare diseases including inherited retinal dystrophy and spinal muscular atrophy (SMA)9,10,11,12 are already clinically approved products on the market. The research and development continue to grow at a fast rate, with more novel therapies advancing in clinical development. However, moving to the next stage, there are major issues to be addressed. For cell and gene therapies, as the basic safety and efficacy feature, to precisely adjust the activity levels of the therapeutic cells or genes by controlling the active dosage, timing and localization is crucial.13 But currently, the overactivity of therapeutic cells and off-target effects in gene therapies are still significant obstacles to overcome. Uncontrolled CAR T cell activity can lead to the development of cytokine release syndrome (CRS) and neurotoxicity when infused CAR T cells become overactivated, causing severe or even life-threatening adverse events.14,15 While gene therapies involving introducing genetic materials into patients’ cells might disrupt the function or regulation of non-targeted genes, therefore causing serious consequences. As an intriguing and rapidly evolving field, synthetic biology is offering new solutions to address these problems.13,16,17 Novel synthetic receptor platforms are established as powerful tools to precisely control the function of engineered cells.18,19 They can be applied to finetune the therapeutic activities like adjusting production of dosed, bioactive payloads by sensing and processing user-defined signals or biomarkers.13 The convergence of synthetic biology with therapeutic strategies might substantially accelerate the evolvement of cell and gene therapies to the next generation.
Here, we review the current knowledge of synthetic receptor systems, including their characteristics and applications, as well as strategies to engineer and improve synthetic receptors. We also discuss the challenges for developing and adapting synthetic receptor platforms to program novel gene and cell therapies.
Synthetic receptors: an overview
Receptors empower cells to timely sense and respond to extrinsic (extracellular) and intrinsic (intracellular) stimuli in the complex environment. Based on the in-depth studies of various natural receptors over the past decades, synthetic biologists have been able to deconstruct and reconstruct receptors, and rationally engineer synthetic receptors. For a functional synthetic receptor, there are at least two domains: a sensor domain for specific binding with input signals, and an actuator domain to transduce sensor activity into outputs.20 Synthetic receptors can be engineered using natural components and/or artificial ones in origin,20 which endows cells (termed as ‘designer cells’) with customized functionalities by rewiring cellular input-output relationships.20,21 Figure 1 summarizes the timeline of key discoveries in synthetic receptor research.

Landmark research achievements of the synthetic receptor over the past three decades. A timeline is shown with brief summaries of some of the key research milestones in the synthetic receptor field published in the past 30 years. CAR chimeric antigen receptor, RASSL receptor activated solely by a synthetic ligand, DREADD designer receptors exclusively activated by designer drug, TRUCK T-cells redirected towards universal cytokine killing, TEVp tobacco etch virus protease, synTF synthetic transcription factor, MESA modular extracellular sensor architecture, synNotch synthetic Notch, FDA the U.S. Food and Drug Administration, dCas9-synR dCas9 synthetic receptor, GPCR G protein-coupled receptor, iTango inducible Tango, Cal-Light calcium- and light-gated switch, FLARE fast light- and activity-regulated expression, SPARK specific protein associated tool giving transcriptional readout with rapid kinetics, SUPRA CAR split, universal and programmable CAR, RASER rewiring of aberrant signaling to effector release, LOCKR latching orthogonal cage-key protein, SPOC split-protease-cleavage orthogonal-coiled coil-based logic circuit, GEMS generalized extracellular molecule sensor, CHOMP circuits of hacked orthogonal modular protease, esNotch enhanced synNotch, TMD transmembrane domain, GEAR generalized engineered activation receptor, TCS two component system, POST phosphoregulated orthogonal signal transduction system, SNIPR synthetic intramembrane proteolysis receptor, AMBER advanced modular bispecific extracellular receptor, OCAR orthogonal chemically activated cell-surface receptor, DocTAR double-cut transcription activation receptor
Armed with synthetic receptors, designer cells are programmed to respond to multiple signals, achieving a spatiotemporal signaling and a subsequent behavior control13 (Fig. 2a). In a designer cell, there are three modules: the sensing module, the processing module and the response module.13,22,23 The sensing module includes but is not limited to various receptors that can detect a range of environmental cues, followed by signal transduction via downstream pathways.13,22,23 The processing module consists of rewired endogenous signaling pathways and orthogonal synthetic genetic circuits, which can process signals from multiple receptors.13,22,23 The response module are components generating measurable outputs, therefore employ user-defined changes (e.g., therapeutic effects)13,22,23 (Fig. 2a).

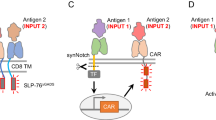
Programming cell and gene therapies using synthetic receptors. a Mammalian cells can be designed and engineered to sense and respond to a variety of stimuli such as chemicals and disease biomarkers, and subsequently trigger downstream signaling pathways, which can finetune customized therapeutic effects (e.g., gene expression, protein activity and secretion, etc.).13,22 b Engineering CAR T cell therapy.68,96 T cells are genetically engineered to express specific CAR proteins on their surface. When infused back into the body, CARs interact with the targeted antigens on cancer cells, causing the activation of CAR T cells for cancer-killing. PCMV cytomegalovirus promoter, scFv single-chain fragment variant, TMD transmembrane domain, CD costimulatory domain, CD3ζ CD3 zeta signaling domain, pA poly(A) signal. c Synthetic receptor applications in CAR T cell therapy.25,67 T cells can be engineered with a combination of CARs and synthetic receptors like synNotch for a more precise tumor recognition to reduce off-target toxicity. Synthetic receptors like MESA could also be used in combination with CAR to sense a soluble biomarker. In addition to driving CAR expression, synthetic receptors are also able to express additional beneficial payloads alongside the CAR, such as cytokines, chemokines, enzymes, single-chain fragment variants (scFvs), mono-antibodies (mAbs), ligands or receptors. TF transcription factor, SynP synthetic promoter, Prom promoter, synR synthetic receptor, pA poly(A) signal. d Therapeutic cell engineering.18,19 By incorporating synthetic receptors, therapeutic cells are designed to act as ‘smart drugs’ that can sense disease biomarkers or user-defined inputs, and trigger a therapeutic response, such as the release of a drug or a therapeutic protein. These engineered cells present clinical potentials as they were encapsulated and implanted in mouse to treat diseases in proof-of-concept studies.213,214 STAT3, signal transducer and activator of transcription 3; P, phosphorylation; shGLP-1, synthetic human glucagon-like peptide 1. e Rewiring of aberrant signaling to effector release (RASER).27,28 In RASER, a hepatitis C virus protease (HCVp) and an effector protein (e.g., OFP-Bid) are fused to two different domains that can sense overactive ErbB signaling. When ErbB activity is detected, the two domains are co-recruited together, causing HCVp to cleave and activate OFP-Bid. This leads to the induction of apoptosis specifically in ErbB hyperactive cancer cells, sparing normal cells. The compact size of RASER construct makes it suitable for AAV-delivered gene therapy. PCMV cytomegalovirus promoter, TMD transmembrane domain, SH2 Src homology 2 domain, CS cleavage site, Bid BH3 interacting domain death agonist, OFP orange fluorescent protein, P2A 2A peptide derived from porcine teschovirus-1, PTB phosphotyrosine-binding domain; NS3, hepatitis C virus nonstructural protein 3, pA poly(A) signal. f Engineering multicellular behaviors with synthetic receptor systems.33,34 (Left) To construct a three-layer structure, two separate cell lines are constructed using synNotch systems.31,32 CD19 ligands on the A-type cells can activate anti-CD19 synNotch receptors on the B-type cells, which induces the expression of Ecadhi (E-cadherin, high expression) and GFPlig (surface-bound GFP) in the B-type cells. Subsequently, these cells will form a two-layer structure with a green core and blue outer layer. Then, GFPlig on the B-type cells can send reciprocal signals to the A-type cells via anti-GFP synNotch, leading to the activation of Ecadlo (E-cadherin, low expression) and mCherry, which will induce the stepwise formation of the three-layer structure. (Right, Upper) Synthetic diffusive morphogen systems can be engineered using synNotch.35 In these systems, soluble ligands can form an artificial morphogen gradient and activate synthetic receptors on receiver cells. The gradient patterns can be tuned by modulating the expression level of synthetic morphogens (e.g., soluble GFP). The synNotch-based synthetic morphogen systems require an extra anchor protein to be expressed on the hybrid anchor/receiver cells (as shown here) or solely on the anchor cells. (Right, Lower) Another possible synthetic diffusive morphogen system using the synthetic receptor, such as MESA. In this supposed system, soluble ligands induce the dimerization of synthetic receptors, activating downstream gene transcription. GFP green fluorescent protein, mCherry a red fluorescent protein, BFP blue fluorescent protein, CD19 cluster of differentiation 19
In recent years, the modular synthetic receptors have been constantly engineered and evolved for biomedical applications. As a well-known example, CARs are synthetic receptors to interact with target cells with high specificity. CAR T cell therapies (Fig. 2b) are now market-approved pharmaceuticals,24 which has convincingly demonstrated the great potential of synthetic receptors to be applied in therapeutics. Another widely used synthetic receptor is synthetic Notch (synNotch), which is able to conditionally drive the expression of the CAR as well as additional payload in engineered T cells when targeting a secondary antigen25,26 (Fig. 2c). Besides, synthetic receptors have been used to engineer therapeutic cells, which can sense arbitrary inputs, such as small molecules and disease-associated biomarkers, and secrete therapeutic molecules in response in animal models19,23 (Fig. 2d). Also, synthetic receptors with compact sizes (e.g., RASER (rewiring of aberrant signaling to effector release) as shown in Fig. 2e) can be directly delivered in targeted gene therapies by the AAV vector to treat ErbB-hyperactive cancers.27,28
Apart from biomedical applications, synthetic receptors have been widely adopted in the fundamental research.29 They are powerful tools to investigate various aspects of cell signaling and behaviors, including cell differentiation, migration and morphogenesis, in a controlled and precise manner.30 For example, researches have used synNotch receptors to program engineered-cells to self-organize into multicellular structures in response to juxtacrine signaling31,32,33,34 (Fig. 2f). Meanwhile, synthetic receptors can also be engineered as part of synthetic orthogonal morphogen systems to pattern three-dimensional (3D) tissues in a tightly controlled manner34,35 (Fig. 2f).
Diverse types of synthetic receptors
Mammalian synthetic receptors can be classified based on different classification criteria. In this section, we provide an overview of diverse available synthetic receptors classified under four distinct principles. A summarization of the classified synthetic receptors is shown in Table 1, with their comparative advantages and limitations further discussed.
Cell-surface receptors and intracellular receptors
According to the location to bind ligands, synthetic receptors can be divided into cell-surface receptors (also known as transmembrane receptors) and intracellular receptors.36
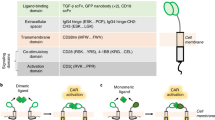
Cell-surface receptors generally comprise three types of transmembrane receptors (i.e., enzyme-linked receptors, G-protein-coupled receptors and ion channel-liked receptors), which span the plasma membrane and convert extracellular signals into intracellular signals.37,38 For this category, each synthetic receptor contains an extracellular ligand-binding domain, at least one transmembrane domain and an intracellular effector domain. Besides the best-known CAR,24,39 synNotch,31,40,41 SNIPR (synthetic intramembrane proteolysis receptor),42 RASSL (receptor activated solely by a synthetic ligand),43,44 Tango,45,46 MESA (modular extracellular sensor architecture),47,48 ChaCha,49 TCS (two-component system),50 chimeric cytokine receptor51,52 and GEMS (generalized extracellular molecule sensor)53 also fall into the category of cell-surface receptors (Table 1). This type of synthetic receptors can only sense external signals, making them suitable to detect cellular (e.g. CAR, synNotch and SNIPR) and systemic disease biomarkers (e.g. MESA and GMES).
Intracellular receptors can either locate in the cytoplasm or nucleus, or be anchored to the intracellular membrane of the cell. Cal-Light (calcium- and light-gated switch),54 CHOMP (circuits of hacked orthogonal modular protease),55 intrabody sensor,56 RASER (rewiring of aberrant signaling to effector release),27 LOCKR (latching orthogonal cage-key proteins),57,58 COMET (composable mammalian elements of transcription)59 and POST (phosphoregulated orthogonal signal transduction)60 belong to the category of intracellular receptors (Table 1). Notably, the synthetic receptors discussed here could also be referred to as synthetic protein switches.61 Since intracellular receptors can only be activated by intracellular input, many of them (e.g., LOCKR and COMET) are designed as a switch induced by chemical molecules that can cross the plasma membrane.
Natural signaling-based receptors and orthogonal signaling-based receptors
Activated receptors trigger signal transduction via multiple downstream pathways. According to the downstream pathway actuated, either natural or engineered, we divide synthetic receptors into natural signaling-based receptors and orthogonal signaling-based receptors.
Natural signaling-based receptors rewire endogenous signaling pathways to either original or customized outputs. This category of synthetic receptors includes RASSL,43,44 CAR,24,39 chimeric cytokine receptor,51,52 GEMS53 and GEAR62 (Table 1). They inevitably activate native pathways to actuate outputs. Among them, RASSL, CAR and chimeric cytokine receptor redirect endogenous pathways to user-defined ligands to execute desirable functions, and are no longer responsive to their original ligands.18,19,20 While GEMS and GEAR hijack endogenous pathways to generate additional customized outputs, they can simultaneously lead to activation of endogenous transcription networks.18,19,20 On the other hand, the capacity to modulate transcriptional networks endows natural signaling-based receptors with unique advantages over orthogonal signaling-based receptors, allowing for the execution of natural and highly complex programs. For example, CARs are invented to mimic the function of T-cell receptor (TCR) to achieve antitumor activity63 via modulating endogenous signaling pathways, but at the same time, additional transgenes can be induced by CAR activation (e.g., nuclear factor of activated T cells (NFAT)-driven cytokine expression), which could not be achieved by orthogonal systems.
By contrast, orthogonal signaling-based receptors are completely independent of endogenous signaling pathways. This attributes to the employment of orthogonal synthetic transcription factors (syn-TFs) or orthogonal signal transduction systems, as they cannot recognize endogenous regulatory elements and activate endogenous signaling cascades, respectively.18,19,20 Nevertheless, some syn-TFs like dCas9-based TFs and TALE-based TFs can program endogenous gene expression without the requirement of extra synthetic promoters.18,20 The currently available Tango,45,46 MESA,47,48 synNotch,31,40,41 ChaCha,49 Cal-Light,54 intrabody sensor,56 LOCKR57,58 and COMET59 employ syn-TFs for orthogonal signal pathway construction, while TCS50 and POST60 utilize orthogonal signaling systems derived from prokaryotes (Table 1).
Of note, the most widely used orthogonal TFs are of non-human origin (e.g., yeast and bacteria), therefore carrying high risks of immunogenicity in clinical applications. The use of humanized syn-TFs comprising both the DNA-binding domain and activation domain derived from human TFs will largely reduce the immunogenic risk. However, these fused TFs are still able to activate endogenous signaling, which reduces their orthogonality. Notably, Khalil and colleagues de novo designed a panel of fully humanized synthetic zinc finger transcription regulators (synZiFTRs) engineered by an array of zinc finger domains, which can specifically recognize their cognate short DNA-binding motifs, achieving genome-orthogonal specificities.64 This kind of pioneering work will accelerate the progress of humanization of orthogonal signaling-based receptors, which is pivotal to bridge the translational gap to the clinic.
Soluble ligand-binding receptors and surface ligand-binding receptors
Synthetic receptors can bind to a range of soluble and surface-bound ligands with a high specificity and sensitivity. In line with the features of ligands, the corresponding synthetic receptors can be categorized into soluble ligand-binding receptors and surface ligand-binding receptors.18,21
A large variety of ligands are in the soluble form, including most chemical molecules, hormones, cytokines, growth factors, intracellular soluble proteins, and some peptides cleaved from membrane proteins (e.g., carcinoembryonic antigen (CEA)65 and amyloid-beta (Aβ)66). The above listed soluble molecules can induce the activation of soluble ligand binding-receptors, such as Tango,45,46 ChaCha,49 MESA,47,48 chimeric cytokine receptor,51,52 CHOMP,55 GEMS53 LOCKR57,58 and POST60 (Table 1). By using small chemical molecules that can cross the plasma membrane, several intracellular synthetic receptors (e.g., POST, CHOMP and LOCKR) have been developed.20 Meanwhile, they can also act as extracellular ligands when the ligand-binding domain of a receptor locates outside the plasma membrane, scilicet cell-surface receptors (e.g., Tango, ChaCha, MESA and GEMS).20 For peptide/protein ligands, in most cases, they cannot enter the cell, and therefore act on cell-surface receptors to subsequently trigger downstream intracellular signaling cascades.
Surface-bound ligands fixed on the plasma membrane of sender cells can trans-activate cell-surface receptors on adjacent receiver cells.18,20 CAR and synNotch are typical surface ligand-sensing receptors which theoretically cannot be activated by soluble ligands.18,20 The different properties of these two types of synthetic receptors possess distinct mechanisms of action. Soluble ligand-binding receptors are usually activated by ligand-induced dimerization to trigger downstream signaling, whereas surface ligand-binding receptors require a mechanical force generated by ligand-receptor interaction to activate downstream signaling. Therefore, activation of soluble ligand-binding receptors (e.g., chimeric cytokine receptor and GEMS) can be induced by ligands after a long-distance transport, whereas surface ligand-binding receptors (CAR and synNotch) can only function in a juxtacrine manner.34
Partially modular receptors and fully modular receptors
Over the past decade, an increasing number of synthetic receptor systems have been engineered and refined. Based on whether they are made up entirely of reconfigurable components, we divide synthetic receptors into partially modular receptors and fully modular receptors.
Partially modular receptors can be constructed by engineering the artificial sensor domain while retaining the original actuator domain, or vice versa. The former synthetic receptors can rewire endogenous signaling pathways to new ligands (e.g., CAR, chimeric cytokine receptor and GEMS), while the latter ones can activate an alternative pathway by natural ligands (e.g., Tango, ChaCha and dCas-synR)18,19,20 (Table 1). Therefore, the former synthetic receptors can be categorized as natural signaling-based receptors and the latter ones as orthogonal signaling-based receptors (as discussed above).
Fully modular synthetic receptors, with both the sensor domain and actuator domain engineered, including synNotch,31,40,41 SNIPRs,42 MESA47,48 and LOCKR,57,58 can execute novel functions without disrupting the endogenous pathways in an orthogonal way (Table 1). It is worth noting that the ‘building brick’ for modular assembly of fully modular synthetic receptors can be either derived from either pre-exiting natural components (e.g., synNotch, SNIPRs and MESA) or de novo designed ones (e.g., LOCKR).20
Engineering of synthetic receptors
In recent years, the construction of evolved synthetic receptors has been facilitated by the rapid development of novel technologies and high-throughput methodologies like directed evolution, rational design and in silico design.67 Meanwhile, they are making the implementation of the most classical design-build-test-learn (DBTL) framework in the development process easier than ever. A better combination of the cutting-edge technologies and the DBTL cycle will surely advance a rapid prototyping and optimization of novel synthetic systems for various biomedical applications. Here, we highlight the development and evolution of four of the most advanced single-pass transmembrane synthetic receptors, CARs (Fig. 3a and Table 1), synNotch (Fig. 4a and Table 1), MESA (Fig. 5a and Table 1) and GEMS (Fig. 5b and Table 1).

Design and engineering of the chimeric antigen receptor (CAR). a The architecture of CARs comprises an extracellular sensor domain, a hinge, a transmembrane domain (TMD) and an intracellular signaling domain (actuator domain).330 The extracellular sensor domain, also known as antigen-binding domain, is usually a single-chain variable fragment (scFv) derived from a monoclonal antibody by fusing its light (VL) and heavy (VH)-chain variable domain with a flexible linker peptide. Other proteins like nanobodies, designed ankyrin repeat proteins (DARPins), natural ligands and small peptides can also function as the antigen-targeting moiety.99,101 The hinge derived from T cell proteins or immunoglobins can function as a flexible linker, providing sufficient conformational freedom to overcome steric hinderance to facilitate the access to the target antigen. T cell protein-derived or de novo designed TMD not only anchors the CAR in the cell membrane but also affects the stability and function of CAR. The intracellular signaling domain generally contains a CD3ζ signaling domain and CD28/4-1BB costimulatory domains (CDs), which facilitates T cell persistence and activity. Several other costimulatory domains including ICOS, OX-40, CD27, MyD88/CD40 and NKG2D are already underway.99,101 CAR architectures can be further engineered to express an ‘armor’, which aims to enhance the in vivo persistence and efficacy of CAR T cells. sbL, surface-bound ligand; ITAM, immunoreceptor tyrosine-based activation motif. b First-generation (1 G) CARs only contain a CD3ζ signaling domain in the intracellular domain (ICD), which outperforms the less popular FcεR1γ signaling domain. Second-generation (2 G) CARs harbor one CD, and third-generation (3 G) CARs contain more than one CDs in their intracellular signaling domain. Fourth-generation (4 G) CARs are based on 2 G CARs with additional expression of transgenic products (armor), such as cytokines, antibodies, enzymes, ligands or receptors.99 Fifth-generation (5 G) CARs are also based on 2 G CARs with the addition of a cytoplasmic domain derived from cytokine receptors (e.g., IL-2Rβ chain fragment)154 or synapse formation proteins (e.g., PDZbm scaffolding anchor domain).261 NFAT nuclear factor of activated T cells, IL-12 interleukin 12, IL-2Rβ interleukin 2 receptor beta-chain, JAK Janus kinase, STAT signal transducer and activator of transcription. c Numerous approaches to improve the safety and efficacy of CAR T cell therapy. Tandem CARs using bispecific single-chain variable fragments (scFvs) can operate an OR gate and overcome obstacles caused by tumor heterogeneity and antigen loss.79 Dual CAR engaging split CARs can perform AND gate to provide and enhance the specificity through targeting multiple antigens.165 Switch CARs with ON/OFF switches utilizing small molecule-triggered dimerization or degradation mechanisms can timely control CAR activity and overcome systemic cytokine toxicities of CAR T cells. Switchable CARs are specific to bispecific adaptors, such as folate-FITC, biotinylated antibody, PNE-Fab and Co-LOCKR96,97,101 and can direct a universal CAR T cell to target distinct antigens. Split, universal and programmable (SUPRA) CARs consist of a set of leucine-zipper universal CARs (zipCARs) and leucine-zipper scFv (zipFv) domains, which specifically bridge the zipCARs to various antigens. The SUPRA CAR system can fine-tune T cell activation and perform combinatorial logic operations (AND, NOT, OR, AND-NOT).84,85 Inhibitory CARs contain inhibitory domains derived from immune checkpoint proteins (PD-1 or CTLA-4), which are able to reduce off-tumor toxicities of CAR T cells by inhibiting T cell activation upon binding an antigen expressed on non-malignant cells.331 SynNotch CARs employ a co-expressed synNotch receptor to drive the expression of a CAR to achieve AND logic. The synNotch and CAR can target different antigens, resulting in improved specificity and sensitivity of CAR T cell therapy.40,190 deg degron, INH inhibitory domain, TF transcription factor

Design and engineering of the synthetic Notch (synNotch) receptor. a The architecture of synNotch receptors consists of an extracellular sensor domain, a transmembrane Notch core region and an intracellular actuator domain (transcription factors, TFs). In synNotch receptors, the extracellular and intracellular domains (ICDs) can be completely swapped with diverse recognition domains (scFv, nanobody, or peptide tags) and TFs (transcriptional activators or repressors). The core Notch regulatory region comprises the transmembrane domain (TMD) and multiple proteolytic cleavage sites of wild-type Notch. Ligand binding to synNotch leads to the intracellular proteolytic cleavage and release of the membrane-tethered TF to translocate into the nucleus and regulate gene expression.31 sbL surface-bound ligand, scFv single-chain fragment variant, JMD juxtamembrane domain. b Evolution of the development of synNotch receptors. (Right, Upper) The modular configuration of prototype synNotch. (Middle, Upper) Enhanced synNotch (esNotch) incorporates an intracellular hydrophobic sequence (QHGQLWF, name as RAM7) derived from native Notch which significantly decreases ligand-independent activation.192 (Left, Upper) Synthetic intramembrane proteolysis receptors (SNIPRs) are fully humanized transcriptional receptors through systematic modular engineering of the original synNotch.42 (Right, Lower) The diffusible synNotch system can detect diffusible ligands anchored by engineered anchor cells, which enables creating a synthetic morphogen signaling system.35 sL soluble ligand. (Middle, Lower) Orthogonal chemically activated cell-surface receptors (OCARs) are engineered by replacing the extracellular sensor domain of synNotch into a chemically induced dimerization (CID) domain, which can achieve small molecule-triggered activation in a cis fashion.193 (Left, Lower) In OCAR-synNotch system, one part of cis-acting OCAR is sequestered by synNotch through the incorporation of coiled-coil dimer-forming peptides into them, which prevents small molecule-induced activation of OCAR when synNotch is in an inactive state. Once synNotch is activated by surface-bound ligands, the sequestered OCAR part is liberated and OCARs can be activated by the addition of inducers, which subsequently enhance synNotch signaling193

Design and engineering of modular extracellular sensor architecture (MESA) and generalized extracellular molecule sensor (GEMS). a, b The architecture of MESA (a) and GEMS (b) comprises the extracellular sensor domain, the transmembrane domain (TMD) and the intracellular actuator domain. The extracellular sensor domain potentially includes single-chain variable fragments (scFvs), nanobodies (Nbs), chemically induced dimerization (CID) proteins, or ligand-binding domains from native receptors. scFv-/Nb-based extracellular sensor domains must bind to non-overlapping epitopes on a single-ligand molecule. a The MESA receptor contains two different transmembrane chains, target chain (TC) and protease chain (PC). The TC intracellular domain (ICD) contains an engineered transcription factor (TF) and a protease cleavage sequence between the TMD and the TF. The PC ICD consists of a cognate protease (e.g., tobacco etch virus protease (TEVp) shown here). Ligand binding induces the heterodimerization of the MESA receptor, causing the TEVp to cleave its cognate cleavage sequence on the TC and releasing the TF to translocate into the nucleus and modulate gene expression. Depending on the types of TFs, synthetic promoter-driven transgene can be induced (e.g., tTA) or endogenous gene expression can be regulated (e.g., dCas9-VP64 activator).47,48 sL soluble ligand. b The GEMS receptor also contains two transmembrane chains, of which the ICDs are derived from various receptor tyrosine kinases (RTKs) and cytokine receptors. Ligand binding induces the dimerization of the GEMS receptor, activating intracellular signaling cascade. By rewiring natural signaling cascades, transgene expression can also be induced.53 At the intracellular juxtamembrane region alanine residues are inserted to modulate the conformation of ICD,201 thus reducing ligand-independent signaling.53 EpoR erythropoietin receptor, D1 EpoR D1 domain, D2 EpoR D2 domain, F93A substitution of phenylalanine at position 93 with alanine, Ala alanine. c Evolution of the development of MESA receptors. (Right, Upper) The modular configuration of prototype MESA.47,48 (Left, Upper) Systematic evaluation of TMD reveals that the choice of TMD significantly affects MESA performance. The TMD-modified MESA utilizing two different TMDs in the TC and PC can achieve reduced background signals and/or increased ligand-induced signals.194 (Right, Lower) In split-TEVp MESA system, computationally optimized split TEVp can be reconstituted via ligand-induced dimerization and therefore restore TEVp function. The split-TEVp enables MESA to achieve low background and high fold induction.195 (Left, Lower) dCas9-synRTK (dCas9- and RTK-based chimeric receptor) as an example of dCas9-synRs (synthetic dCas9-based receptors), employs split-dCas9-VP64 and split-TEVp as the intracellular actuator domain, by fusing them to different RTKs. The difference between dCas9-synRTK and split-TEVp MESA is that dCas9-synRTK can only sense native ligands since the extracellular domain and TMD of a dCas9-synRTK are derived from an intact RTK.196 nTEVp N-terminal TEVp, cTEVp C-terminal TEVp, dCas9n N-terminal deactivated Cas9, dCas9c C-terminal deactivated Cas9, RTK receptor tyrosine kinase. d Engineering of chimeric cytokine receptors to mimic cytokine receptor signaling using scFv and EpoR scaffold. ScFv/c-Mpl (S-Mpl) chimera contains a scFv-based extracellular sensor domain, the extracellular EpoR D2 domain and transmembrane/cytoplasmic domains of cytokine receptors (e.g., c-Mpl).210,211,212 Chimeric cytokine receptor constructs with different combination of the domains containing the extracellular scFv, EpoR scaffold and intracellular domain of cytokine receptors (e.g., gp130). Compared to Sg, SD1D2g-1A additionally contains the extracellular D1D2 domain and one alanine residue at the intracellular juxtamembrane region. But the extracellular D1/D2 domain is dispensable for signaling.205 e Evolution of the development of GEMS receptors. GEMS should be considered as an evolutionary version of prototype SD1D2g-1A. Through modular engineering, the GEMS platform is able to specifically target a range of soluble ligands and robust transgene expression with high signal-to-noise ratios.53 Based on GEMS, generalized engineered activation regulators (GEARs) capitalize on MS2 bacteriophage coat protein (MCP)-nuclear factor fusion proteins and the dCas9/sgRNA-MS2 system to rewire induced receptor signaling to endogenous gene expression.62 Advanced modular bispecific extracellular receptors (AMBERs) combine the GEMS system and designed ankyrin repeat proteins (DARPins). The high-throughput binder-screening technology, DARPin, can generate various new binders and endow AMBER with desired sensitivity and specificity towards new inputs.213 In addition to customizing target gene expression, GMES and its derivatives inevitably perturb the endogenous gene regulatory network. dCas9, deactivated Cas9; sgRNA, single guide RNA; MS2, MS2 hairpin
Chimeric antigen receptors (CARs)
CARs are a best-known class of synthetic receptor systems, which have already been approved for CAR T cell therapies by the U.S. Food and Drug Administration (FDA)6,68 (Supplementary Note 1). They represent a success of advances in synthetic biology to pioneer a new generation of therapeutics, motivating the continuous optimization of CAR-design and the development of new synthetic receptor systems throughout the field.
Currently, CARs have been widely armed in immune cells like T cells, NK cells and macrophages (reviewed in refs. 5,69,70). Besides, innate T cells including invariant natural killer T (iNKT) cells, mucosal associated invariant T (MAIT) cells and gamma delta T (γδT) cells have been developed as promising immune effector cells, because they display intrinsic antitumor microenvironment (TME) capacities and minimal risks of graft versus host disease (GvHD) (reviewed in refs. 71,72,73). CAR mainly consists of the extracellular domain (ECD), the transmembrane domain (TMD) and the intracellular domain (ICD) (Fig. 3a). The modular characteristics of its structure confer it advantages to be highly amenable to modification and redesign.
The ECD can be segmented into the signal peptide (SP) and the ligand-binding domain (LBD). The SP prompts the transmembrane receptor protein to be translocated to the plasma membrane, which usually is cleaved from mature CAR protein co-translationally74,75 (Supplementary Note 2). The LBD, also called antigen-recognition domain, is typically a single-chain fragment variant (scFv).76,77 The scFv is a compact artificial construct that comprises the immunoglobulin light and heavy chain variable regions connected by a flexible linker78 (Supplementary Note 2). The scFv is widely adopted in CAR construction due to its compact size, high affinity and specificity for antigen recognition.77 By changing the scFv, CARs can specifically recognize different antigens on various cancer cells (e.g., cluster of differentiation 19 (CD19), mesothelin, CEA, B-cell maturation antigen (BCMA), CD38) by one-to-one authentication,39 and subsequently activate cancer-killing of CAR T cells. Besides simply replacing the scFv, CARs can be equipped with bispecific antibodies consisting of two different linked antigen-recognition domains (also referred to as ‘tandem CARs’)79 (Fig. 3c and Table 1). As a result, tandem CAR T cells can recognize different antigens expressed in a single cancer cell (e.g., human epidermal growth factor receptor 2 (Her2) and interleukin 13 receptor alpha 2 (IL13Rα2),80 CD19 and CD2081), and therefore reduce the possibility of tumor escape.82,83
Recently, Wong and colleagues invented an intriguing generalized CAR platform, the split, universal, and programmable (SUPRA) CAR system for T cell therapy84 (Fig. 3c and Table 1). In this system, the conventional CAR architecture is split into two elements: (1) a soluble zipFv by fusing a scFv to a leucine zipper and (2) a universal zipCAR containing the remnant transmembrane and intracellular domains attached to an extracellular cognate leucine zipper.84 By adding different zipFv proteins, a unique zipCAR-expressing T cell can retarget different tumor antigens.84 Moreover, SUPRA CARs can fine-tune T cell activation and program the Boolean logic operation, which enhances the safety and efficacy of T cell therapy.84,85 Alternatively, various switchable CAR systems with bispecific adapters are under way (refs. 86,87,88,89,90,91,92,93,94,95 and also reviewed in refs. 96,97) (Fig. 3c and Table 1). More information about UniCAR is shown in Supplementary Note 3.
As mentioned previously, conventional CARs can only target surface-bound ligands while being unable to sense soluble ligands. To redirect the response of CAR T cells to soluble cues, Chang et al. engineered a CAR that enabled T cells to respond robustly to diverse soluble ligands via dimerization-induced mechanotransduction mechanisms98 (Table 1).
The TMD is usually a hydrophobic α-helix that spans the plasma membrane and functions to anchor CAR proteins to the membrane.68,99 Besides controlling membrane integration, the TMD also regulates key interactions between CARs, such as assembly, activation and high-order clustering.68,99,100 Compared to ECD and ICD, TMD has received relatively less attention and research. Almost all the TMDs used in CARs are derived from natural T cell proteins, such as CD8, CD28, CD4 and CD3ζ.99,101 Nonetheless, studies have indicated that the TMDs certainly impact the stability and function of CAR.102,103 More recently, Elazar et al., demonstrated that the specific oligomeric state programmed by de novo designed TMDs can tune CAR function and CAR T cell activity, as well as decrease cytokine releasing relative to the commonly used CD28 TMD.100
By linking the extracellular antigen-binding and transmembrane domains, the hinge region functions as a linker, providing the flexibility to access sterically hindered epitopes99,101 (Supplementary Note 2). Studies have revealed much more crucial roles of the hinge region per se or it being coupled with the TMD than initially expected. These results demonstrate that the length and composition of the hinge can affect CAR functionalities, including fine-tuning CAR signaling activity, improving antitumor efficacy, and lowering cytokine release or neurotoxicity.102,103,104,105,106,107,108,109,110 Therefore, in CAR T cell engineering, it is essential for systematic evaluation and optimization of the hinge and the TMD to ensure optimal performance and reliability.68,77,99,111,112,113
The ICD transmits activation signals upon the antigen’s binding to the ECD. The ICDs of most well-studied CARs contain a CD3ζ-derived signaling moiety, which has three immunoreceptor tyrosine-based activation motifs (ITAMs)77,114 (Supplementary Note 4). From the first-generation (1 G) of CAR derived in the 1990s115 to now, the structure of CAR per se has been constantly evolving up to the fifth-generation (5 G) (Fig. 3b and Table 1), aiming to increase specificity and minimize off-target toxicity.114,116
The first-generation (1 G) CARs only contain ITAMs to provide activating signaling with none co-stimulatory domain115 (Fig. 3b and Table 1). Though 1 G CARs were proved to be able to activate T cells and control tumor in mice,117,118,119 they failed to achieve antitumor responses in subsequent clinical studies.120 The reason could be the CD3ζ alone is insufficient to activate resting T lymphocytes or trigger the production of optimal amounts of cytokines.121,122 To solve these problems, second-generation (2 G) CARs were created by incorporating a costimulatory domain (Supplementary Note 4) on the basis of 1 G CARs (Fig. 3b and Table 1), which enables the activation of a second signal when stimulated by a tumor antigen.123,124,125,126,127,128 This improvement has achieved the enhancement of cytokine production, CAR T cell persistence and antitumor efficacy.125
And the third-generation (3 G) CARs are designed to combine multiple costimulatory domains to further enhance CAR T cell potency129,130,131,132 (Fig. 3b). Although potential benefits including prolonged persistence and increased antitumor efficacy have been demonstrated both in vitro and in vivo,130,131,132,133,134,135,136,137 some clinical results did not prove a significant superiority of 3 G CAR T cells to the 2 G CAR T cells.138,139,140 As the available data was obtained from relatively small and heterogenic samples, it is still too early to draw a conclusion, and further large-scale studies are required to fully evaluate their feasibility.
The fourth-generation (4 G) CARs have been engineered from the 2 G CARs to constitutively or inducibly secrete cytokines (e.g., IL-12, IL-7, IL-15, IL-18, and IL-23), enhancing the immune modulating capacities141,142,143,144,145,146,147,148,149,150 (Fig. 3b and Table 1). Upon CAR-mediated T cell activation, cytokines can be ideally released within targeted tumor locally, alleviating systemic side effects and solving the problem of insufficient production. Since the 4 G CARs can not only improve CAR T cell activation but also hijack host immune cells to enhance tumor killing,151,152 the 4 G CAR T cells are also referred to as T cells redirected for universal cytokine killing (TRUCKs).114,116,151,152 Compared with conventional CAR T cells, TRUCKs show enhanced expansion and antitumor activity in preclinical studies, especially in animal models of solid tumors. However, in practice, systemic side effects of released cytokines may occur upon entry into circulation. For example, in a clinical study, Rosenberg and colleagues modified tumor-infiltrating lymphocytes (TILs) to express IL-12 under a NFAT-inducible promoter to treat metastatic melanoma. They observed severe toxicity induced in most patients, which is likely attributed to the secreted IL-12.153 Here, more data derived from clinical trials are necessary to assess their safety and efficacy.
Distinct from TRUCKs, the immunomodulatory factor expression module of the 5 G CARs is replaced with a novel costimulatory domain which can activate a specific signaling pathway inside equipped CAR T cells114,116 (Fig. 3b and Table 1). Based on this excellent principle, several approaches are emerging, of which the addition of IL-2 receptor β-chain (IL-2Rβ) into CARs is a notable example.154 Upon activation by the antigen, the extra IL-2Rβ domain allows the activation of the JAK kinase and the STAT3/5 signaling pathways, which can empower CAR T cells to achieve superior antitumor effects with minimal toxicity in mouse models due to a better persistence and expansion in vivo. However, it could potentially increase the risk of CRS, thus requiring to be cautiously addressed in translational studies.154
CAR T cell therapy has evolved and gradually matured during the past decades, showing great therapeutic potential in blood and bone marrow cancers. However, challenges remain in CAR-based solid tumor immunotherapy due to tumor heterogeneity, inefficient trafficking and tumor infiltration, and an immunosuppressive TME.83,155,156,157,158,159 To facilitate CAR T cell therapy for solid tumors, several strategies have been developed. For instance, dual CAR, tandem CAR and UniCAR systems can recognize more than one antigen, helping mitigate tumor antigen escape. CAR T cells armed with matched chemokine receptor expression can permit trafficking and infiltration, achieving enhanced killing of solid tumors.160,161,162,163 Also, engineering CAR T cells to express heparanase degrading extracellular matrix (ECM) can promote tumor T cell infiltration and antitumor activity.164 In addition, the aforementioned TRUCKs were developed to overcome the drawbacks of the TME on CAR T cell therapy to treat solid tumors by immunomodulatory factors.142,144
However, it remains challenging to minimize CAR-immune cells’ off-target and off-tumor toxicity. In this context, the development of next-generation CARs has already been underway96,99,101 (Fig. 3c and Table 1). For example, the combinatorial logic control by dual CAR165,166 or synNotch CAR40,167,168 can enhance tumor targeting specificity via the presence of two or more antigens. On the other hand, a safety control by ON- and OFF-switch CAR can finetune CAR activity169,170,171,172 and kill-switch CAR can manage the lifespan of CAR T cells.173,174 These are promising strategies to improve the safety of CAR T cell immunotherapy in the future.
Furthermore, most CAR T cells under investigation currently are engineered by inserting the CAR construct into autologous T cells without disrupting the endogenous T-cell receptor protein (TCR) gene. Under this condition, the risk of GvHD, which is triggered by human leukocyte antigen (HLA)-TCR mismatching,175 can be avoided. To facilitate allogeneic “off-the-shelf” CAR T cell transplantation, T cell receptor α chain (TRAC) deletion using endonucleases, thereby disrupting cell surface expression of the αβ T cell receptor (TCRαβ), can successfully prevent graft-versus-host responses.175,176,177,178,179,180 Recently, CRISPR/Cas-mediated knockin technology enables the precise integration of CAR-encoding gene into TRAC locus in human peripheral blood T cells,181 which not only facilitates the production of allogeneic CAR T cells,181,182,183 but also enhances T cell potency as the edited T cells outperformed conventionally engineered CAR T cells.181,184 However, more recent studies revealed that the endogenous TCR plays a critical role in promoting long-term in vivo persistence of CAR T cells in not only animal models but also patients.185,186 These results collectively indicate that it is crucial to balance the intricate effects of removing endogenous TCR from CAR T cells in tumor immunotherapy.
Synthetic Notch (synNotch) receptors
In-depth studies of Notch receptors provide critical insights into molecular mechanisms of Notch receptors.187 And the intrinsic features including modularity and mechanical forces-triggered signaling independent of native ligands make Notch receptors ideally suitable for modular chimeric receptor engineering.188 Taking advantage of it, Lim and colleagues created the innovative synNotch receptor system by utilizing the transmembrane core domain of native Notch receptors (Fig. 4a and Table 1), alongside the extracellular sensor domain and intracellular actuator domain.31,40,41
Three intriguing works reported the modular synNotch receptors functioning with customized sensing and responsive behaviors in mammalian cells, including T cells.31,40,41 Morsut et al. demonstrated that synNotch can function orthogonally to control cell differentiation, spatial patterning and Boolean decisions.31 Meanwhile, Roybal et al. reported that synNotch can sense tumor antigen and then drive the expression of CARs (synNotch CARs as mentioned above) (Fig. 3c and Table 1), which allows the engineering of AND-gate T cells activated only by dual antigen recognition.40 Roybal et al. also reported that synNotch enables CAR T cells to yield customized therapeutic responses like secreting cytokines and antibodies in a very precise and localized manner.41 Compared to conventional CARs whose activation drives T cells to produce a native cytokine profile, synNotch can control the expression of extra user-defined cytokines.41 A more powerful combination of synNotch receptors with CARs for precise and effective cancer-killing is on the rise, which endows synNotch the great potential of becoming another synthetic receptor system applied in cancer immunotherapy.
Due to its superior performance, the synNotch platform has been widely adopted for designer cell-engineering,25,33,34 particularly for the engineering of CAR T cells40,41,189,190 and the programming of self-organizing multicellular structures.31,32,35 Although there are still challenges and limitations when it comes to practical biomedical applications, researchers are trying hard to figure out possible solutions. First, native Notch receptors have an inherent feature that both trans- and cis-interaction modes co-exist,31 so activation cannot be achieved due to cis-inhibition when the cognate ligand presents on the same surface as the receptor does (cis).31,191 To avoid cis-inhibition, the ‘flippase-out’ strategy is employed to achieve the mutually exclusive expression of the synNotch and its ligand in flippase recombinase transgenic Drosophila.191 Second, synNotch activation requires mechanical forces triggered by surface-bound ligands, making synNotch receptors unable to sense soluble ligands.31 To address this, Toda et al. engineered anchor cells which can tether the soluble ligands (e.g., diffusible GFP), thus enabling synNotch receptors to respond to diffusible synthetic morphogens35 (Fig. 4b and Table 1). Third, synNotch receptors display a high level of ligand-independent activation,192 which is also quite common for other synthetic receptor systems.20 Excitingly, a recent work reported an improved version of synNotch, named enhanced synthetic Notch (esNotch) receptor. By adding a native Notch-derived intracellular hydrophobic sequence, an incredible reduction (14.6-fold) in the background activity level was achieved192 (Fig. 4b and Table 1). Impressively, Roybal and colleagues have achieved systematic and modular improvements of the synNotch architecture, including modifications of the extracellular sensor domain, TMD, intracellular juxtamembrane domain (JMD) and actuator domain.42 The evolved synNotch system is referred as synthetic intramembrane proteolysis receptors (SNIPRs) (Fig. 4b and Table 1), which realized background reductions and enhanced ligand-induced signals.42 Meanwhile, SNIPRs can be fully humanized with humanized modules, minimizing the risk of immune rejection.42 The use of humanized syn-TFs, including synZiFTRs, not only retains the orthogonality of SNIPRs, but also largely reduces the immunogenic potential for cell-based therapies.42
In addition, Fussenegger and colleagues derived an orthogonal chemically activated cell-surface receptor (OCAR) system on the basis of synNotch by substituting conventional protein-specific LBDs with chemically induced dimerization (CID) domains (Fig. 4b). Induced by small molecules, the engineered OCARs on one cell can form heterodimers and trigger signal activation by releasing synTFs.193 When the OCAR system is co-expressed with the conventional synNotch on the same cell, one part of OCAR will be sequestered by the synNotch receptor through coiled-coil interactions, making OCAR unable to be activated by the inducer. When synNotch receptor is activated by the presence of sender cells, the OCAR can restore its responsiveness to the inducer and thus further enhance synNotch signaling by adding inducers. Due to their mechanism of action, OCAR systems exhibit an intrinsic off-switch, which might be used as a safety switch in the case of toxicity or malfunction193 (Fig. 4b and Table 1).
Modular extracellular sensor architecture (MESA)
Like synNotch receptors, the modular extracellular sensor architecture (MESA) receptors also take advantage of the proteolytic release of synTFs, which translocate to the nucleus and initiate transcriptional activation to execute orthogonal signals without interrupting endogenous signaling pathways47,48 (Fig. 5a and Table 1). But different from synNotch receptors that require mechanical forces induced by surface-tethered ligands,31 MESA receptors signal via soluble ligand-triggered receptor heterodimerization to perform the subsequent proteolytic cleavage.47,48
Each MESA receptor comprises two different single-pass transmembrane proteins, as their intracellular domains are distinct (Fig. 5a). MESA receptors also comprise three modular domains to transduce extracellular ligand-binding inputs into intracellular transcriptional outputs through synTF-releasing.47,48
By engineering the extracellular sensor domain, MESA receptors can be redirected to new ligands.47,48 However, the prototype of the MESA architecture also suffers from high level of ligand-independent activation possibly due to transient receptor dimerization during trafficking or on the cell surface.48 To address this, Leonard and colleagues have been taking efforts on the improvement of the MESA system, especially to reduce undesired background signals. A systematical strategy was employed to elucidate and refine the MESA architecture, demonstrating that the TMD optimization can lower the background signaling and elevate the target signaling194 (Fig. 5c and Table 1). In another study, authors exploited a computational design strategy to use the split-TEVp system for MESA optimization (Fig. 5c and Table 1), achieving both a low background and a high fold induction.195 In parallel, a similar dCas9-based synthetic receptor system termed ‘dCas9-synR’ was reported (Fig. 5c and Table 1), which is capable of coupling native input signals with the direct activation of user-defined output response programs.196
As discussed above, MESA and dCas9-synR are conceived as cell-surface receptors based on previous studies,20,28,197 but a recent study challenged this conclusion.198 In this study, the authors showed that both receptor systems did not work with purified protein ligands, but were only activated when using co-transfected ligands or the cell permeable molecule rapamycin as inducers. Therefore, they suspected that these two receptor systems function within the endoplasmic reticulum (ER), and should not be classified into cell-surface receptors.198 By carefully checking previous publications, we find in three works relevant to MESA, the authors have shown that MESA receptors could be activated by purified vascular endothelial growth factor (VEGF) protein,48,62 and MESA proteins expressed on cell surface were detected by flow cytometry.48,194 And yet the inconsistent conclusion drawn by the recent study might attribute to the different components of MESA constructs (e.g., extracellular linker and TMD) used in research,198 which could change the expression level of MESA on cell surface as indicated by previous works.48,194 Though the early study done by Schwarz et al. showing that MESA could be activated by purified VEGF supported MESA as a cell-surface receptor,48 a more recent work from Krawczyk et al. failed to replicate the original data.62 Using purified VEGF, few changes (in the range of about 1.1 ~ 1.2 fold) were observed, which strongly indicated that MESA did not achieve cell-surface receptor performance.62 Thus, as the bias in the literature exists,48,62,196,198 to give a precise characterization of both MESA and dCas9-synR systems, more replication attempts are definitely required.
Generalized extracellular molecule sensor (GEMS)
A clever insight into the EpoR architecture and signaling mechanisms199,200,201,202 spurs the development of chimeric cytokine receptors based on EpoR scaffold203,204,205,206,207,208,209,210,211,212 (Fig. 5d and Table 1). These chimeric cytokine receptors (e.g., SD1D2g-1A) contain extracellular scFv, extracellular EpoR D1D2 as well as TMD domains, and the intracellular domains of cytokine receptors (e.g., glycoprotein 130 (gp130) and EpoR)205 (Fig. 5d and Table 1). These chimeric cytokine receptors can employ robust ligand-dependent ON/OFF regulation and mimic the function of the native cytokine receptor system.205,206
Based on this prototype, the GEMS system is rationally designed and engineered. GEMS receptors comprise a standard transmembrane scaffold derived from erythropoietin receptor (EpoR) with F93A mutation (Fig. 5b and Table 1), abolishing native ligand (erythropoietin) sensitivity.53 Extracellular LBDs can be modularly replaced to sense and respond to a wide range of extracellular soluble ligands. Significantly, the ICDs employ various signaling domains to reroute inputs into distinct endogenous pathways (e.g., JAK/STAT, PI3K/Akt, PLCG and MAPK/ERK).53 In recent reports, the GEMS platform has evolved into the superior generalized engineered activation regulators (GEARs)62 and advanced modular bispecific extracellular receptors (AMBERs)213 (Table 1). The GEAR system combines endogenous signaling-induced synTF nuclear translocation and dCas9-based gene expression via the DNA-binding module (Fig. 5e). Various pathways (e.g., NFAT, nuclear factor kappa B subunit (NFκB), mitogen activated protein kinase (MAPK) or SMAD) activated by diverse receptors including GEMS receptors can be incorporated into engineered GEARs.62 The AMBER system is, in short, a type of programmable DARPin- and GEMS-based receptors (Fig. 5e), which inherits the characteristics and advantages of both systems. Thus, AMBERs can be engineered and refined to bind to new soluble ligands in a modular and predictable manner.213 For proof of concept, the GEMS and its derivates have been applied to engineering designer cells in vitro and in vivo for disease treatment (Fig. 2d) (refs. 53,62,213,214 and reviewed in refs. 18,19). Though showing promise in preclinical studies, challenges remain to be addressed before the translation of the GEMS system into clinics. A major challenge is the safety concern relevant to the immunogenicity of designer cells. Recently, Weber and colleagues engineered a material-genetic interface as the safety switch for mammalian therapeutic cells, in which they achieved the control of cell survival by allowing the designer cells to sense whether they were embedded in the hydrogels using a GEMS-based receptor.215
As discussed above, synthetic receptors hijacking natural signaling pathways usually inevitably activate endogenous transcription networks to some extent. Using synthetic promotors, GEMS receptors can induce user-defined transgenic target expression.53 The advantage is that GEMS receptors can generate high signal-to-noise ratios.18 However, the accompanying disadvantage is the perturbation of the gene regulatory network might affect the proliferation and survival of the cells being engineered.
Synthetic receptor design
The prospect of programming sophisticated customized functions in mammalian cells has fostered the investigations on the design, engineering and iterative improvement of synthetic receptors, which is led by increasing knowledge of the intrinsic structure and molecular acting mechanisms of natural receptors. Currently, primary strategies for synthetic receptor engineering include but are not limited to chimeric/fusion protein, directed evolution, rational design and de novo design (reviewed in ref. 67).
Engineering chimeric/fusion proteins means genetically integrating different functional peptides together referring to the architecture of a natural archetype. It is widely used for synthetic receptor engineering. The above-mentioned CARs, synNotch, MESA, GEMS, and chimeric cytokine receptors as well as Tango were originally constructed using this method67).
Directed evolution is one of the most popular strategies for protein engineering, which mimics the natural evolution and harnesses the power of mutation and selection.216,217 Through iterative cycles of random mutagenesis, followed by selection, engineered DREADDs (designer receptors exclusively activated by designer drugs)218 and AMBERs213 enable targeting new ligands with high specificity and/or sensitivity (Table 1).
Compared with directed evolution, rational design essentially requires the in-depth understanding of protein structures. Mechanistic insights into receptors have facilitated the rational design and engineering of CARs, synNotch and GEMS receptors. Advanced computation-assisted approaches have aided the rational design of CARs219,220 and MESA195 as well as other modular sensor-actuator systems221 with improved specificity and sensitivity. In silico approaches enable scientists to construct numerous receptor variants and conduct thousands of simulated experiments by computer, which dramatically reduces the amount of candidates to be tested in laboratory experiments.222,223
De novo design is an intriguing strategy for protein design with predetermined structures and functions. Distinct from rational design, de novo design aims to generate new proteins from scratch.224 With the deepening understanding of the principles of protein biophysics and the development of computational devices and algorithms, scientists have achieved the de novo protein design of synthetic receptors. Currently, various functional proteins, such as protein-binding proteins and TMDs, are de novo designed.225,226,227,228,229,230,231 And they can be incorporated into synthetic receptor engineering by the modular approach. For example, de novo designed TMDs have been employed in CAR engineering and achieve the outperformance relative to the native CD28 TMD (as discussed above and also in ref. 100).
More prominently, Baker and colleagues designed a switchable protein platform from scratch, termed ‘latching orthogonal cage-key proteins’ (LOCKR)57 (Table 1). Though not being a cell-surface receptor, LOCKR represents a breakthrough in the de novo design of protein switch, indicating a branch of future direction in synthetic receptor design. In this system, intermolecular cage-key interactions can competitively inhibit intramolecular cage-latch interactions, and thus free the latch peptide from the cage to perform different functions (e.g., binding, degradation, and nuclear export).57 Capitalizing on LOCKR technology as the core, engineered modular and tunable protein switches achieve gene expression and even feedback control in cells.57,58 The advent of evolutionary technologies opens broad avenues to computationally design, engineer and improve arbitrary synthetic receptor systems.
Different strategies or approaches, including machine learning, could be combined to facilitate the engineering and improvement of synthetic receptors. The latest advances in CAR engineering could be a good example. As afore introduced, intracellular signaling domains of a CAR play vital roles in T cell activation and tumor killing, but only limited sets of signaling domains were explored. To expand the repertoire of CAR signaling domains, a range of studies have been focusing on their systematic optimization. Among them, Goodman et al.232 and Si et al.233 individually constructed a small library of CARs containing natural or rational recombinant costimulatory domains, and screened out novel costimulatory domains which were proved to achieve enhanced cytotoxicity and T cell persistence, thus improving antitumor efficacy. Meanwhile, high-throughput screening has also been applied to identify new combinations of signaling domains234 or new synthetic signaling domains via shuffling or recombination.235,236 Of note, Lim and colleagues incorporated machine learning in their study, and took advantage of trained neural networks to predict the cytotoxicity and stemness of CARs with synthetic signaling domains.236
Increasing approaches are boosting the development and improvement of various synthetic receptors (Table 1). For practical use, selecting and optimizing synthetic receptors for a specific application could be greatly facilitated by the generalized and systematic framework presented in Fig. 6 (also reviewed in detail in ref. 20). Defining the goal is the first and crucial step before entering into the DBTL cycle. It includes narrowing down options of synthetic receptors adapting to engineered cell or gene therapy by specifying the required characteristics and setting performance criteria for the expected synthetic receptors. Notably, performance metrics can be adopted to quantitively evaluate the performance of synthetic receptors in different application scenarios20 (Fig. 6a).

Metrics- and “design-build-test-learn” (DBTL) cycle-based framework for synthetic receptor engineering. a Performance metrics can be used for quantitative assessment of the performance of synthetic receptor systems. Signaling output can be quantified by reporter fluorescence measurement, luciferase assay or various other approaches. The performance of a synthetic receptor is determined not only by its background signal and signal-to-noise ratio, but also by the therapeutic thresholds in practical applications. An optimal receptor should exhibit a low background signal (OB) which is lower than the minimum therapeutic threshold (Tmin), and meanwhile exhibit a high induced output signal (Oi) (Right, Upper). A synthetic receptor exhibiting low OB and high Oi may still fail in practical applications, which might be due to OB > Tmax (maximum therapeutic threshold) (Right, Lower) or Oi < Tmin (Right, Lower). In another case when OB > Tmin (Left, Lower), leaky expression should be cautious of. b A modified DBTL-based framework can direct researchers to choose or engineer synthetic receptor systems for their application in cell therapy or gene therapy. A “goal” step is to define design objectives for engineered cell or gene therapy and the standards to quantify the performance of synthetic receptors using performance metrics. In the DBTL cycle, apart from conventional approaches, more advanced and powerful approaches like computation-guided design, high-throughput automation techniques, machine learning and computational modeling can further accelerate the engineering and improvement of synthetic receptors for better clinical applications. The figure is adapted from refs. 20,239
Driven by defined design objectives, researchers could follow the DBTL pipeline to design and engineer synthetic prototypes, test and optimize the performance in a befitting model (e.g., cell lines), and further validate the performance in a practical context (e.g., engineered cell therapy or gene therapy in animal models) (Fig. 6b). Of note, in the “design” step, choosing different module candidates is of vital importance, which could significantly impact the properties of synthetic receptors. For example, to force the expression of synthetic receptors on cell surface, both SP and TMD are essential (discussed above), which also influence the cell-surface expression levels of receptors. Moreover, data-driven computation-guided design can be incorporated to achieve a more efficient rational design of synthetic receptors. The “build” and “test” steps are traditionally laborious, involving series of construct engineering and assessment of performance metrics. The high-throughput automation techniques developing rapidly can not only increase productivity but also reduce human errors in this process.237,238 In the “learn” step, researchers can decipher the data generated and create blueprints of synthetic receptors based on it. Usually trial-and-error approaches are employed to determine the optimum design subsequently. Currently, advanced computational modeling and machine learning are starting to play a central role to process and learn from masses of biological data, achieving a new paradigm of predictive design. By bridging the gap between the “learn” and “design” stages, they can greatly accelerate the DBTL cycle.239
Conclusions and perspective
The design and engineering of synthetic receptors have achieved great advancements, and the constant evolution of synthetic receptor systems accelerates their biomedical applications and clinical translations. As the most successful case, CARs are at the forefront, with six CAR T cell therapies already approved by the FDA6,68 and hundreds more undergoing clinical studies (ClinicalTrials.gov). In addition, other CAR-engineered cells including CAR NK and CAR macrophage cells are also being widely investigated in translational researches.5,240,241,242
Looking forward, an important goal of the field is to uncover the principles of design to guide the rational design of synthetic receptors with desired properties and functions. A growing number of reports have taken advantage of computational designing strategies for engineering and improving synthetic receptor systems like CARs,219,220 MESA195 and de novo protein switches.57,58 Among them, de novo protein design is further ahead, as different kinds of non-natural functional proteins have been created from scratch.225,226,227,228,229,230,231 Knowledge-driven and data-driven computational approaches have established interpretable models for de novo protein design.243,244,245,246,247,248,249 As discussed above, de novo designed proteins have already been incorporated into synthetic receptor engineering.57,58,100 We anticipate that various valuable synthetic receptors can be designed by de novo design strategies224,250 or by generative language models in future.251
Importantly, selecting functional synthetic receptor systems and integrating them with ‘chassis cells’ can potentially push the boundaries of synthetic receptor applications and develop novel cell therapeutics (Fig. 7). For example, the application of CARs in engineered-T cells has enhanced the specific tumor killing ability68 (Fig. 7a). More importantly, a combination of different synthetic receptor systems can exhibit synergistic effects and further enhance their performance, as studies have shown that synNotch CAR T cells exhibit a significantly enhanced safety and antitumor efficacy by combinatorial antigen recognition40,167,168,189,252 or ultrasensitive antigen-density sensing.190 As engineered stem cell therapy emerges, synthetic receptors can also be applied to engineer pluripotent stem cell-derived cell products3,70,175 (Fig. 7b, c), potentially being used to enhance their survival and engraftment ability via programmed communications with the host microenvironment2,3,253,254 (Fig. 7b). However, since some precautionary data and critical attitudes about stem cell therapy exist, future trials are needed to further investigate the safety and efficacy of stem cell therapy per se.2,255 Meanwhile, concerns over the safety of both genetic materials (e.g., transgene immunogenicity) and genetic modifications introduced also require to be carefully addressed in preclinical and clinical studies.3

The expanding potential therapeutic applications of synthetic receptors. a A variety of immune cells, such as T cells, innate T cells, NK cells, macrophages, dendritic cells and myeloid cells, can be directly isolated from patients and genetically engineered with CARs to enhance their antitumor capacity.68,332 Alternatively, these immune cells can be differentiated from CAR-engineered pluripotent stem cells (PSCs) as ‘off-the-shelf’ products. After being infused back into patients, these engineered immune cells interact with antigens on the tumor cells, leading to the activation of CAR immune cells to achieve cancer-killing.70,333 b, c Synthetic receptors could be integrated into PSCs to enhance original or program novel functionalities of the differentiated derivates for developing next-generation cellular therapeutics. b PSCs can differentiate into various cell types, like hepatocytes, neurons, muscle cells, etc., which are suitable for transplantation. One could imagine after being transplanted, cells engineered with synthetic receptors could sense and respond to host microenvironmental cues to promote the survival, proliferation and enhance tissue repair or regeneration.30 c Differentiated derivates from synthetic receptor-engineered PSCs might also be encapsulated and implanted in patients to avoid immunogenicity. These implantable therapeutic cells can sense various serum biomarkers (e.g., glucose, uric acid or thyroid hormone) and then trigger the activation of the corresponding therapeutic functions.16 d Synthetic receptor systems can be further designed and engineered with compact size and lower immunogenicity easier for in vivo gene therapy.42,64 These suitable synthetic receptor constructs can be delivered by non-viral vectors (e.g., lipid nanoparticle (LNP))334,335 or viral vectors (e.g., AAV)27
Moreover, synthetic receptors can also program in vivo gene therapy with an improved safety and efficacy (Fig. 7d). One promising approach for in vivo gene therapy is to employ adeno-associated virus (AAV) systems to deliver therapeutic genetic materials into the human body.256,257 Although AAVs have been increasingly used in clinical trials as novel gene therapies,258 their limited packaging capacity (~4.7 kb) hinders the loading of most synthetic receptors described above. To meet the clinical demands, designing or refining synthetic receptors with a compact size is required, and already being investigated.42,64 In addition, humanizing synthetic receptors with minimal immunogenicity is also of vital importance in advancing gene therapy as well as engineered cell therapy.42,64
In conclusion, modular synthetic receptors with desired functions have been engineered and applied to therapeutic applications. Meanwhile, advances keep facilitating and accelerating the development and evolution of synthetic receptor systems, making the field to shift from the prior trial-and-error mode to more knowledge- and data-driven modes. Briefly, we expect that rapid progress in synthetic receptor biology will keep revealing exceptional new openings to program gene therapies and engineered-cell therapies that have been unreachable by established approaches.
References
Naldini, L. Gene therapy returns to centre stage. Nature 526, 351–360 (2015).
Yamanaka, S. Pluripotent stem cell-based cell therapy—promise and challenges. Cell Stem Cell 27, 523–531 (2020).
Kimbrel, E. A. & Lanza, R. Next-generation stem cells — ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov. 19, 463–479 (2020).
Johnson, L. A. & June, C. H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res 27, 38–58 (2017).
Finck, A. V., Blanchard, T., Roselle, C. P., Golinelli, G. & June, C. H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 28, 678–689 (2022).
Cappell, K. M. & Kochenderfer, J. N. Long-term outcomes following CAR T cell therapy: what we know so far. Nat. Rev. Clin. Oncol. 20, 359–371 (2023).
Cavazzana, M., Bushman, F. D., Miccio, A., Andre-Schmutz, I. & Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat. Rev. Drug Discov. 18, 447–462 (2019).
Ferrari, G., Thrasher, A. J. & Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 22, 216–234 (2021).
Dunbar, C. E. et al. Gene therapy comes of age. Science 359, eaan4672 (2018).
Groen, E. J. N., Talbot, K. & Gillingwater, T. H. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 14, 214–224 (2018).
Kulkarni, J. A. et al. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16, 630–643 (2021).
Cehajic-Kapetanovic, J., Singh, M. S., Zrenner, E. & MacLaren, R. E. Bioengineering strategies for restoring vision. Nat. Biomed. Eng. 7, 387–404 (2023).
Kitada, T., DiAndreth, B., Teague, B. & Weiss, R. Programming gene and engineered-cell therapies with synthetic biology. Science 359, eaad1067 (2018).
Shalabi, H. et al. Beyond the storm — subacute toxicities and late effects in children receiving CAR T cells. Nat. Rev. Clin. Oncol. 18, 363–378 (2021).
Morris, E. C., Neelapu, S. S., Giavridis, T. & Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 22, 85–96 (2022).
Cubillos-Ruiz, A. et al. Engineering living therapeutics with synthetic biology. Nat. Rev. Drug Discov. 20, 941–960 (2021).
Yan, X., Liu, X., Zhao, C. & Chen, G. Q. Applications of synthetic biology in medical and pharmaceutical fields. Signal Transduct. Target. Ther. 8, 199 (2023).
Teixeira, P. A. & Fussenegger, M. Engineering mammalian cells for disease diagnosis and treatment. Curr. Opin. Biotechnol. 55, 87–94 (2019).
Scheller, L. & Fussenegger, M. From synthetic biology to human therapy: engineered mammalian cells. Curr. Opin. Biotechnol. 58, 108–116 (2019).
Manhas, J., Edelstein, H. I., Leonard, J. N. & Morsut, L. The evolution of synthetic receptor systems. Nat. Chem. Biol. 18, 244–255 (2022).
Brenner, M., Cho, J. H. & Wong, W. W. Synthetic biology: sensing with modular receptors. Nat. Chem. Biol. 13, 131–132 (2017).
Tolle, F., Stucheli, P. & Fussenegger, M. Genetic circuitry for personalized human cell therapy. Curr. Opin. Biotechnol. 59, 31–38 (2019).
Mansouri, M. & Fussenegger, M. Therapeutic cell engineering: designing programmable synthetic genetic circuits in mammalian cells. Protein Cell 13, 476–489 (2022).
June, C. H. & Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 379, 64–73 (2018).
Hyrenius-Wittsten, A. & Roybal, K. T. Paving new roads for CARs. Trends Cancer 5, 583–592 (2019).
Allen, G. M. & Lim, W. A. Rethinking cancer targeting strategies in the era of smart cell therapeutics. Nat. Rev. Cancer 22, 693–702 (2022).
Chung, H. K. et al. A compact synthetic pathway rewires cancer signaling to therapeutic effector release. Science 364, eaat6982 (2019).
Chung, H. K. & Lin, M. Z. On the cutting edge: protease-based methods for sensing and controlling cell biology. Nat. Methods 17, 885–896 (2020).
Martinez-Ara, G., Stapornwongkul, K. S. & Ebisuya, M. Scaling up complexity in synthetic developmental biology. Science 378, 864–868 (2022).
Lim, W. A. The emerging era of cell engineering: harnessing the modularity of cells to program complex biological function. Science 378, 848–852 (2022).
Morsut, L. et al. Engineering customized cell sensing and response behaviors using synthetic Notch receptors. Cell 164, 780–791 (2016).
Toda, S., Blauch, L. R., Tang, S. K. Y., Morsut, L. & Lim, W. A. Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science 361, 156–162 (2018).
Toda, S., Frankel, N. W. & Lim, W. A. Engineering cell-cell communication networks: programming multicellular behaviors. Curr. Opin. Chem. Biol. 52, 31–38 (2019).
Trentesaux, C., Yamada, T., Klein, O. D. & Lim, W. A. Harnessing synthetic biology to engineer organoids and tissues. Cell Stem Cell 30, 10–19 (2023).
Toda, S. et al. Engineering synthetic morphogen systems that can program multicellular patterning. Science 370, 327–331 (2020).
Schwarz, K. A. & Leonard, J. N. Engineering cell-based therapies to interface robustly with host physiology. Adv. Drug Deliv. Rev. 105, 55–65 (2016).
Uings, I. J. & Farrow, S. N. Cell receptors and cell signalling. Mol. Pathol. 53, 295–299 (2000).
Heldin, C. H., Lu, B., Evans, R. & Gutkind, J. S. Signals and receptors. Cold Spring Harb. Perspect. Biol. 8, a005900 (2016).
Sadelain, M., Brentjens, R. & Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 3, 388–398 (2013).
Roybal, K. T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016).
Roybal, K. T. et al. Engineering T cells with customized therapeutic response programs using synthetic Notch receptors. Cell 167, 419–432.e16 (2016).
Zhu, I. et al. Modular design of synthetic receptors for programmed gene regulation in cell therapies. Cell 185, 1431–1443.e16 (2022).
Coward, P. et al. Controlling signaling with a specifically designed Gi-coupled receptor. Proc. Natl Acad. Sci. USA 95, 352–357 (1998).
Conklin, B. R. et al. Engineering GPCR signaling pathways with RASSLs. Nat. Methods 5, 673–678 (2008).
Barnea, G. et al. The genetic design of signaling cascades to record receptor activation. Proc. Natl Acad. Sci. USA 105, 64–69 (2008).
Lee, D. et al. Temporally precise labeling and control of neuromodulatory circuits in the mammalian brain. Nat. Methods 14, 495–503 (2017).
Daringer, N. M., Dudek, R. M., Schwarz, K. A. & Leonard, J. N. Modular extracellular sensor architecture for engineering mammalian cell-based devices. ACS Synth. Biol. 3, 892–902 (2014).
Schwarz, K. A., Daringer, N. M., Dolberg, T. B. & Leonard, J. N. Rewiring human cellular input-output using modular extracellular sensors. Nat. Chem. Biol. 13, 202–209 (2017).
Kipniss, N. H. et al. Engineering cell sensing and responses using a GPCR-coupled CRISPR-Cas system. Nat. Commun. 8, 2212 (2017).
Maze, A. & Benenson, Y. Artificial signaling in mammalian cells enabled by prokaryotic two-component system. Nat. Chem. Biol. 16, 179–187 (2020).
Engelowski, E. et al. Synthetic cytokine receptors transmit biological signals using artificial ligands. Nat. Commun. 9, 2034 (2018).
Lange, S. et al. A chimeric GM-CSF/IL18 receptor to sustain CAR T-cell function. Cancer Discov. 11, 1661–1671 (2021).
Scheller, L., Strittmatter, T., Fuchs, D., Bojar, D. & Fussenegger, M. Generalized extracellular molecule sensor platform for programming cellular behavior. Nat. Chem. Biol. 14, 723–729 (2018).
Lee, D., Hyun, J. H., Jung, K., Hannan, P. & Kwon, H. B. A calcium- and light-gated switch to induce gene expression in activated neurons. Nat. Biotechnol. 35, 858–863 (2017).
Gao, X. J., Chong, L. S., Kim, M. S. & Elowitz, M. B. Programmable protein circuits in living cells. Science 361, 1252–1258 (2018).
Siciliano, V. et al. Engineering modular intracellular protein sensor-actuator devices. Nat. Commun. 9, 1881 (2018).
Langan, R. A. et al. De novo design of bioactive protein switches. Nature 572, 205–210 (2019).
Ng, A. H. et al. Modular and tunable biological feedback control using a de novo protein switch. Nature 572, 265–269 (2019).
Donahue, P. S. et al. The COMET toolkit for composing customizable genetic programs in mammalian cells. Nat. Commun. 11, 779 (2020).
Scheller, L. et al. Phosphoregulated orthogonal signal transduction in mammalian cells. Nat. Commun. 11, 3085 (2020).
Stein, V. & Alexandrov, K. Synthetic protein switches: design principles and applications. Trends Biotechnol. 33, 101–110 (2015).
Krawczyk, K., Scheller, L., Kim, H. & Fussenegger, M. Rewiring of endogenous signaling pathways to genomic targets for therapeutic cell reprogramming. Nat. Commun. 11, 608 (2020).
van der Stegen, S. J., Hamieh, M. & Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 14, 499–509 (2015).
Li, H. S. et al. Multidimensional control of therapeutic human cell function with synthetic gene circuits. Science 378, 1227–1234 (2022).
Bramswig, K. H. et al. Soluble carcinoembryonic antigen activates endothelial cells and tumor angiogenesis. Cancer Res. 73, 6584–6596 (2013).
McLean, C. A. et al. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866 (1999).
Javdan, S. B. & Deans, T. L. Design and development of engineered receptors for cell and tissue engineering. Curr. Opin. Syst. Biol. 28, 100363 (2021).
Labanieh, L. & Mackall, C. L. CAR immune cells: design principles, resistance and the next generation. Nature 614, 635–648 (2023).
Weber, E. W., Maus, M. V. & Mackall, C. L. The emerging landscape of immune cell therapies. Cell 181, 46–62 (2020).
Li, Y. R. et al. Advancing cell-based cancer immunotherapy through stem cell engineering. Cell Stem Cell 30, 592–610 (2023).
Cortes-Selva, D., Dasgupta, B., Singh, S. & Grewal, I. S. Innate and innate-like cells: the future of chimeric antigen receptor (CAR) cell therapy. Trends Pharmacol. Sci. 42, 45–59 (2021).
Li, Y. R., Wilson, M. & Yang, L. Target tumor microenvironment by innate T cells. Front. Immunol. 13, 999549 (2022).
Courtney, A. N., Tian, G. & Metelitsa, L. S. Natural killer T cells and other innate-like T lymphocytes as emerging platforms for allogeneic cancer cell therapy. Blood 141, 869–876 (2023).
Martoglio, B. & Dobberstein, B. Signal sequences: more than just greasy peptides. Trends Cell Biol. 8, 410–415 (1998).
Hegde, R. S. & Bernstein, H. D. The surprising complexity of signal sequences. Trends Biochem. Sci. 31, 563–571 (2006).
Srivastava, S. & Riddell, S. R. Engineering CAR-T cells: design concepts. Trends Immunol. 36, 494–502 (2015).
Jayaraman, J. et al. CAR-T design: elements and their synergistic function. EBioMedicine 58, 102931 (2020).
Holliger, P. & Hudson, P. J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 23, 1126–1136 (2005).
Grada, Z. et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2, e105 (2013).
Hegde, M. et al. Tandem CAR T cells targeting HER2 and IL13α2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052 (2016).
Shah, N. N. et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat. Med. 26, 1569–1575 (2020).
Majzner, R. G. & Mackall, C. L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226 (2018).
Milone, M. C. et al. Engineering enhanced CAR T-cells for improved cancer therapy. Nat. Cancer 2, 780–793 (2021).
Cho, J. H., Collins, J. J. & Wong, W. W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426–1438.e11 (2018).
Cho, J. H. et al. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun. 12, 792 (2021).
Tamada, K. et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 18, 6436–6445 (2012).
Urbanska, K. et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 72, 1844–1852 (2012).
Kim, M. S. et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J. Am. Chem. Soc. 137, 2832–2835 (2015).
Ma, J. S. et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl Acad. Sci. USA 113, E450–E458 (2016).
Rodgers, D. T. et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl Acad. Sci. USA 113, E459–E468 (2016).
Herzig, E. et al. Attacking latent HIV with convertibleCAR-T cells, a highly adaptable killing platform. Cell 179, 880–894.e10 (2019).
Lee, Y. G. et al. Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. 79, 387–396 (2019).
Raj, D. et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 68, 1052–1064 (2019).
Lajoie, M. J. et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 369, 1637–1643 (2020).
Qi, J. et al. Chemically programmable and switchable CAR-T therapy. Angew. Chem. Int. Ed. Engl. 59, 12178–12185 (2020).
Hong, M., Clubb, J. D. & Chen, Y. Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 38, 473–488 (2020).
Lee, S., Khalil, A. S. & Wong, W. W. Recent progress of gene circuit designs in immune cell therapies. Cell Syst. 13, 864–873 (2022).
Chang, Z. L. et al. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 14, 317–324 (2018).
Rafiq, S., Hackett, C. S. & Brentjens, R. J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 17, 147–167 (2020).
Elazar, A. et al. De novo-designed transmembrane domains tune engineered receptor functions. Elife 11, e75660 (2022).
Labanieh, L., Majzner, R. G. & Mackall, C. L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2, 377–391 (2018).
Alabanza, L. et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 25, 2452–2465 (2017).
Ying, Z. et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 25, 947–953 (2019).
Hudecek, M. et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 3, 125–135 (2015).
Jonnalagadda, M. et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol. Ther. 23, 757–768 (2015).
Brudno, J. N. et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 26, 270–280 (2020).
Majzner, R. G. et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 10, 702–723 (2020).
Wang, D. et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 12, eaaw2672 (2020).
Bister, A. et al. A novel CD34-derived hinge for rapid and efficient detection and enrichment of CAR T cells. Mol. Ther. Oncolytics 23, 534–546 (2021).
Cordoba, S. et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat. Med. 27, 1797–1805 (2021).
Guedan, S., Calderon, H., Posey, A. D. Jr. & Maus, M. V. Engineering and design of chimeric antigen receptors. Mol. Ther. Methods Clin. Dev. 12, 145–156 (2019).
Ellis, G. I., Sheppard, N. C. & Riley, J. L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 22, 427–447 (2021).
Larson, R. C. & Maus, M. V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 21, 145–161 (2021).
Tokarew, N., Ogonek, J., Endres, S., von Bergwelt-Baildon, M. & Kobold, S. Teaching an old dog new tricks: next-generation CAR T cells. Br. J. Cancer 120, 26–37 (2019).
Eshhar, Z., Waks, T., Gross, G. & Schindler, D. G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc. Natl Acad. Sci. USA 90, 720–724 (1993).
Abreu, T. R., Fonseca, N. A., Goncalves, N. & Moreira, J. N. Current challenges and emerging opportunities of CAR-T cell therapies. J. Control. Release 319, 246–261 (2020).
Hwu, P. et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 55, 3369–3373 (1995).
Kershaw, M. H., Westwood, J. A. & Hwu, P. Dual-specific T cells combine proliferation and antitumor activity. Nat. Biotechnol. 20, 1221–1227 (2002).
Cooper, L. J. et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood 101, 1637–1644 (2003).
Kershaw, M. H. et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 12, 6106–6115 (2006).
Brocker, T. & Karjalainen, K. Signals through T cell receptor-ζ chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 181, 1653–1659 (1995).
Brocker, T. Chimeric Fv-ζ or Fv-ε receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 96, 1999–2001 (2000).
Geiger, T. L., Nguyen, P., Leitenberg, D. & Flavell, R. A. Integrated src kinase and costimulatory activity enhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood 98, 2364–2371 (2001).
Haynes, N. M. et al. Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 100, 3155–3163 (2002).
Maher, J., Brentjens, R. J., Gunset, G., Riviere, I. & Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 20, 70–75 (2002).
Brentjens, R. J. et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 9, 279–286 (2003).
Imai, C. et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18, 676–684 (2004).
Kowolik, C. M. et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 66, 10995–11004 (2006).
Pule, M. A. et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 12, 933–941 (2005).
Carpenito, C. et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl Acad. Sci. USA 106, 3360–3365 (2009).
Milone, M. C. et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17, 1453–1464 (2009).
Tammana, S. et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum. Gene Ther. 21, 75–86 (2010).
Zhong, X. S., Matsushita, M., Plotkin, J., Riviere, I. & Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 18, 413–420 (2010).
Guedan, S. et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 3, e96976 (2018).
Weng, J. et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J. Hematol. Oncol. 11, 25 (2018).
Ramos, C. A. et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol. Ther. 26, 2727–2737 (2018).
Schubert, M. L. et al. Treatment of adult ALL patients with third-generation CD19-directed CAR T cells: results of a pivotal trial. J. Hematol. Oncol. 16, 79 (2023).
Morgan, R. A. et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010).
Till, B. G. et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 119, 3940–3950 (2012).
Enblad, G. et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin. Cancer Res. 24, 6185–6194 (2018).
Kerkar, S. P. et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 70, 6725–6734 (2010).
Chmielewski, M., Kopecky, C., Hombach, A. A. & Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71, 5697–5706 (2011).
Pegram, H. J. et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141 (2012).
Chmielewski, M. & Abken, H. CAR T cells releasing IL-18 convert to T-bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 21, 3205–3219 (2017).
Hu, B. et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 20, 3025–3033 (2017).
Adachi, K. et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 36, 346–351 (2018).
Avanzi, M. P. et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 23, 2130–2141 (2018).
Golumba-Nagy, V., Kuehle, J., Hombach, A. A. & Abken, H. CD28-ζ CAR T cells resist TGF-β repression through IL-2 signaling, which can be mimicked by an engineered IL-7 autocrine loop. Mol. Ther. 26, 2218–2230 (2018).
Rafiq, S. et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 36, 847–856 (2018).
Gardner, T. J. et al. Engineering CAR-T cells to activate small-molecule drugs in situ. Nat. Chem. Biol. 18, 216–225 (2022).
Chmielewski, M., Hombach, A. A. & Abken, H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 257, 83–90 (2014).
Chmielewski, M. & Abken, H. TRUCKS, the fourth‐generation CAR T cells: current developments and clinical translation. Adv. Cell Gene Ther. 3, e84 (2020).
Zhang, L. et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 21, 2278–2288 (2015).
Kagoya, Y. et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 24, 352–359 (2018).
Newick, K., Moon, E. & Albelda, S. M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther. Oncolytics 3, 16006 (2016).
D’Aloia, M. M., Zizzari, I. G., Sacchetti, B., Pierelli, L. & Alimandi, M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 9, 282 (2018).
Marofi, F. et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell. Res. Ther. 12, 81 (2021).
Kirtane, K., Elmariah, H., Chung, C. H. & Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: review of the literature and challenges ahead. J. Immunother. Cancer 9, e002723 (2021).
Yan, T., Zhu, L. & Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 12, 14 (2023).
Di Stasi, A. et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113, 6392–6402 (2009).
Moon, E. K. et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 17, 4719–4730 (2011).
Cadilha, B. L. et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci. Adv. 7, eabi5781 (2021).
Lesch, S. et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 5, 1246–1260 (2021).
Caruana, I. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 21, 524–529 (2015).
Kloss, C. C., Condomines, M., Cartellieri, M., Bachmann, M. & Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 31, 71–75 (2013).
Hirabayashi, K. et al. Dual targeting CAR-T cells with optimal costimulation and metabolic fitness enhance antitumor activity and prevent escape in solid tumors. Nat. Cancer 2, 904–918 (2021).
Choe, J. H. et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 13, eabe7378 (2021).
Hyrenius-Wittsten, A. et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med. 13, eabd8836 (2021).
Wu, C. Y., Roybal, K. T., Puchner, E. M., Onuffer, J. & Lim, W. A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015).
Mestermann, K. et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 11, eaau5907 (2019).
Jan, M. et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 13, eabb6295 (2021).
Labanieh, L. et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 185, 1745–1763.e22 (2022).
Straathof, K. C. et al. An inducible caspase 9 safety switch for T-cell therapy. Blood 105, 4247–4254 (2005).
Diaconu, I. et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol. Ther. 25, 580–592 (2017).
Depil, S., Duchateau, P., Grupp, S. A., Mufti, G. & Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 19, 185–199 (2020).
Torikai, H. et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119, 5697–5705 (2012).
Valton, J. et al. A multidrug-resistant engineered CAR T cell for allogeneic combination immunotherapy. Mol. Ther. 23, 1507–1518 (2015).
Poirot, L. et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 75, 3853–3864 (2015).
Qasim, W. et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 9, eaaj2013 (2017).
Benjamin, R. et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet 396, 1885–1894 (2020).
Eyquem, J. et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017).
MacLeod, D. T. et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol. Ther. 25, 949–961 (2017).
Zhang, J. et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature 609, 369–374 (2022).
Maus, M. V. T-cell tweaks to target tumours. Nature 543, 48–49 (2017).
Stenger, D. et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 136, 1407–1418 (2020).
Wang, Z. et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 18, 2188–2198 (2021).
Kopan, R. & Ilagan, M. X. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 (2009).
Gordon, W. R. et al. Mechanical allostery: evidence for a force requirement in the proteolytic activation of Notch. Dev. Cell 33, 729–736 (2015).
Williams, J. Z. et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 370, 1099–1104 (2020).
Hernandez-Lopez, R. A. et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science 371, 1166–1171 (2021).
He, L., Huang, J. & Perrimon, N. Development of an optimized synthetic Notch receptor as an in vivo cell-cell contact sensor. Proc. Natl Acad. Sci. USA 114, 5467–5472 (2017).
Yang, Z. J., Yu, Z. Y., Cai, Y. M., Du, R. R. & Cai, L. Engineering of an enhanced synthetic Notch receptor by reducing ligand-independent activation. Commun. Biol. 3, 116 (2020).
Mahameed, M., Wang, P., Xue, S. & Fussenegger, M. Engineering receptors in the secretory pathway for orthogonal signalling control. Nat. Commun. 13, 7350 (2022).
Edelstein, H. I. et al. Elucidation and refinement of synthetic receptor mechanisms. Synth. Biol. 5, ysaa017 (2020).
Dolberg, T. B. et al. Computation-guided optimization of split protein systems. Nat. Chem. Biol. 17, 531–539 (2021).
Baeumler, T. A., Ahmed, A. A. & Fulga, T. A. Engineering synthetic signaling pathways with programmable dCas9-based chimeric receptors. Cell Rep. 20, 2639–2653 (2017).
Chen, Z. & Elowitz, M. B. Programmable protein circuit design. Cell 184, 2284–2301 (2021).
Zhou, J., Ge, Q., Wang, D., Guo, Q. & Tao, Y. Engineering a modular double-transmembrane synthetic receptor system for customizing cellular programs. Cell Rep. 42, 112385 (2023).
Livnah, O. et al. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283, 987–990 (1999).
Remy, I., Wilson, I. A. & Michnick, S. W. Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993 (1999).
Constantinescu, S. N., Huang, L. J., Nam, H. & Lodish, H. F. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol. Cell 7, 377–385 (2001).
Seubert, N. et al. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol. Cell 12, 1239–1250 (2003).
Kawahara, M., Kimura, H., Ueda, H. & Nagamune, T. Selection of genetically modified cell population using hapten-specific antibody/receptor chimera. Biochem. Biophys. Res. Commun. 315, 132–138 (2004).
Pihkala, P., Kawahara, M., Ueda, H. & Nagamune, T. An antigen-mediated selection system for mammalian cells that produce glycosylated single-chain Fv. Biochem. Biophys. Res. Commun. 324, 1165–1172 (2004).
Liu, W., Kawahara, M., Ueda, H. & Nagamune, T. Construction of a fluorescein-responsive chimeric receptor with strict ligand dependency. Biotechnol. Bioeng. 101, 975–984 (2008).
Liu, W., Kawahara, M., Ueda, H. & Nagamune, T. The influence of domain structures on the signal transduction of chimeric receptors derived from the erythropoietin receptor. J. Biochem. 145, 575–584 (2009).
Sogo, T. et al. T cell growth control using hapten-specific antibody/interleukin-2 receptor chimera. Cytokine 46, 127–136 (2009).
Tanaka, K., Kawahara, M., Ueda, H. & Nagamune, T. Selection and growth regulation of genetically modified cells with hapten-specific antibody/receptor tyrosine kinase chimera. Biotechnol. Prog. 25, 1138–1145 (2009).
Kawahara, M. et al. Growth promotion of genetically modified hematopoietic progenitors using an antibody/c-Mpl chimera. Cytokine 55, 402–408 (2011).
Saka, K., Kawahara, M., Ueda, H. & Nagamune, T. Activation of target signal transducers utilizing chimeric receptors with signaling-molecule binding motifs. Biotechnol. Bioeng. 109, 1528–1537 (2012).
Saka, K., Kawahara, M. & Nagamune, T. Reconstitution of a cytokine receptor scaffold utilizing multiple different tyrosine motifs. Biotechnol. Bioeng. 110, 3197–3204 (2013).
Saka, K., Kawahara, M. & Nagamune, T. Quantitative control of intracellular signaling activity through chimeric receptors incorporating multiple identical tyrosine motifs. Biotechnol. Bioeng. 111, 948–955 (2014).
Strittmatter, T. et al. Programmable DARPin-based receptors for the detection of thrombotic markers. Nat. Chem. Biol. 18, 1125–1134 (2022).
Bojar, D., Scheller, L., Hamri, G. C., Xie, M. & Fussenegger, M. Caffeine-inducible gene switches controlling experimental diabetes. Nat. Commun. 9, 2318 (2018).
Jerez-Longres, C. et al. Engineering a material-genetic interface as safety switch for embedded therapeutic cells. Biomater. Adv. 150, 213422 (2023).
Bloom, J. D. & Arnold, F. H. In the light of directed evolution: pathways of adaptive protein evolution. Proc. Natl Acad. Sci. USA 106, 9995–10000 (2009).
Lane, M. D. & Seelig, B. Advances in the directed evolution of proteins. Curr. Opin. Chem. Biol. 22, 129–136 (2014).
Dong, S., Rogan, S. C. & Roth, B. L. Directed molecular evolution of DREADDs: a generic approach to creating next-generation RASSLs. Nat. Protoc. 5, 561–573 (2010).
Krokhotin, A. et al. Computationally guided design of single-chain variable fragment improves specificity of chimeric antigen receptors. Mol. Ther. Oncolytics 15, 30–37 (2019).
Giordano-Attianese, G. et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat. Biotechnol. 38, 426–432 (2020).
Glasgow, A. A. et al. Computational design of a modular protein sense-response system. Science 366, 1024–1028 (2019).
Chu, H. Y. & Wong, A. S. L. Facilitating machine learning-guided protein engineering with smart library design and massively parallel assays. Adv. Genet. 2, 2100038 (2021).
Hie, B. L. & Yang, K. K. Adaptive machine learning for protein engineering. Curr. Opin. Struct. Biol. 72, 145–152 (2022).
Huang, P. S., Boyken, S. E. & Baker, D. The coming of age of de novo protein design. Nature 537, 320–327 (2016).
Tinberg, C. E. et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature 501, 212–216 (2013).
Lu, P. et al. Accurate computational design of multipass transmembrane proteins. Science 359, 1042–1046 (2018).
Chen, Z. et al. Programmable design of orthogonal protein heterodimers. Nature 565, 106–111 (2019).
Vorobieva, A. A. et al. De novo design of transmembrane β barrels. Science 371, eabc8182 (2021).
Cao, L. et al. Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022).
Wang, J. et al. Scaffolding protein functional sites using deep learning. Science 377, 387–394 (2022).
Wu, K. et al. De novo design of modular peptide-binding proteins by superhelical matching. Nature 616, 581–589 (2023).
Goodman, D. B. et al. Pooled screening of CAR T cells identifies diverse immune signaling domains for next-generation immunotherapies. Sci. Transl. Med. 14, eabm1463 (2022).
Si, W. et al. Design of diversified chimeric antigen receptors through rational module recombination. iScience 26, 106529 (2023).
Gordon, K. S. et al. Screening for CD19-specific chimaeric antigen receptors with enhanced signalling via a barcoded library of intracellular domains. Nat. Biomed. Eng. 6, 855–866 (2022).
Castellanos-Rueda, R. et al. speedingCARs: accelerating the engineering of CAR T cells by signaling domain shuffling and single-cell sequencing. Nat. Commun. 13, 6555 (2022).
Daniels, K. G. et al. Decoding CAR T cell phenotype using combinatorial signaling motif libraries and machine learning. Science 378, 1194–1200 (2022).
Hillson, N. et al. Building a global alliance of biofoundries. Nat. Commun. 10, 2040 (2019).
Liu, A. P. et al. The living interface between synthetic biology and biomaterial design. Nat. Mater. 21, 390–397 (2022).
Kitano, S., Lin, C., Foo, J. L. & Chang, M. W. Synthetic biology: learning the way toward high-precision biological design. PLoS Biol. 21, e3002116 (2023).
Daher, M. & Rezvani, K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 11, 45–58 (2021).
Myers, J. A. & Miller, J. S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 18, 85–100 (2021).
Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21, 799–820 (2022).
Koepnick, B. et al. De novo protein design by citizen scientists. Nature 570, 390–394 (2019).
Anishchenko, I. et al. De novo protein design by deep network hallucination. Nature 600, 547–552 (2021).
Dauparas, J. et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
Ferruz, N., Schmidt, S. & Hocker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Huang, B. et al. A backbone-centred energy function of neural networks for protein design. Nature 602, 523–528 (2022).
Lutz, I. D. et al. Top-down design of protein architectures with reinforcement learning. Science 380, 266–273 (2023).
Madani, A. et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 41, 1099–1106 (2023).
Leman, J. K. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Ferruz, N. & Höcker, B. Controllable protein design with language models. Nat. Mach. Intell. 4, 521–532 (2022).
Dannenfelser, R. et al. Discriminatory power of combinatorial antigen recognition in cancer T cell therapies. Cell Syst. 11, 215–228.e5 (2020).
Madl, C. M., Heilshorn, S. C. & Blau, H. M. Bioengineering strategies to accelerate stem cell therapeutics. Nature 557, 335–342 (2018).
Tewary, M., Shakiba, N. & Zandstra, P. W. Stem cell bioengineering: building from stem cell biology. Nat. Rev. Genet. 19, 595–614 (2018).
Hoang, D. M. et al. Stem cell-based therapy for human diseases. Signal Transduct. Target. Ther. 7, 272 (2022).
Wang, D., Tai, P. W. L. & Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378 (2019).
Li, C. & Samulski, R. J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 21, 255–272 (2020).
Kuzmin, D. A. et al. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 20, 173–174 (2021).
Hwu, P. et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor γ chain. J. Exp. Med. 178, 361–366 (1993).
Wang, J. et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum. Gene Ther. 18, 712–725 (2007).
Chockley, P. J., Ibanez-Vega, J., Krenciute, G., Talbot, L. J. & Gottschalk, S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat. Biotechnol. 41, 1434–1445 (2023).
Zah, E., Lin, M. Y., Silva-Benedict, A., Jensen, M. C. & Chen, Y. Y. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 4, 498–508 (2016).
Han, X., Wang, Y., Wei, J. & Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 12, 128 (2019).
Jia, H. et al. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 12, 57 (2019).
Schneider, D. et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J. Immunother. Cancer 5, 42 (2017).
Tong, C. et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood 136, 1632–1644 (2020).
Spiegel, J. Y. et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med. 27, 1419–1431 (2021).
Hegde, M. et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 21, 2087–2101 (2013).
Lanitis, E. et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 1, 43–53 (2013).
Ruella, M. et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 126, 3814–3826 (2016).
Schneider, D. et al. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med. 13, eabc6401 (2021).
Wilkie, S. et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 32, 1059–1070 (2012).
Brenner, M. J., Cho, J. H., Wong, N. M. L. & Wong, W. W. Synthetic biology: immunotherapy by design. Annu. Rev. Biomed. Eng. 20, 95–118 (2018).
Flugel, C. L. et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat. Rev. Clin. Oncol. 20, 49–62 (2023).
Huang, K. et al. Remote control of cellular immunotherapy. Nat. Rev. Bioeng. 1, 440–455 (2023).
Leung, W. H. et al. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight 5, e124430 (2019).
Carbonneau, S. et al. An IMiD-inducible degron provides reversible regulation for chimeric antigen receptor expression and activity. Cell Chem. Biol. 28, 802–812.e6 (2021).
Kosti, P. et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2, 100227 (2021).
Nguyen, N. T. et al. Nano-optogenetic engineering of CAR T cells for precision immunotherapy with enhanced safety. Nat. Nanotechnol. 16, 1424–1434 (2021).
Cao, W. et al. A reversible chemogenetic switch for chimeric antigen receptor T cells. Angew. Chem. Int. Ed. Engl. 61, e202109550 (2022).
Li, H. S. et al. High-performance multiplex drug-gated CAR circuits. Cancer Cell 40, 1294–1305.e4 (2022).
Choi, B. D. et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 37, 1049–1058 (2019).
Chen, Y. Y. Increasing T cell versatility with SUPRA CARs. Cell 173, 1316–1317 (2018).
Kawahara, M. & Nagamune, T. Engineering of mammalian cell membrane proteins. Curr. Opin. Chem. Eng. 1, 411–417 (2012).
Ogawa, K., Kawahara, M. & Nagamune, T. Construction of unnatural heterodimeric receptors based on IL-2 and IL-6 receptor subunits. Biotechnol. Prog. 29, 1512–1518 (2013).
Scheller, J., Engelowski, E., Moll, J. M. & Floss, D. M. Immunoreceptor engineering and synthetic cytokine signaling for therapeutics. Trends Immunol. 40, 258–272 (2019).
Sockolosky, J. T. et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 359, 1037–1042 (2018).
Hirai, T. et al. Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J. Clin. Invest. 131, e139991 (2021).
Zhang, Q. et al. A human orthogonal IL-2 and IL-2Rβ system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci. Transl. Med. 13, eabg6986 (2021).
Kalbasi, A. et al. Potentiating adoptive cell therapy using synthetic IL-9 receptors. Nature 607, 360–365 (2022).
Deckers, J. et al. Engineering cytokine therapeutics. Nat. Rev. Bioeng. 1, 286–303 (2023).
Park, J. S. et al. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. Proc. Natl Acad. Sci. USA 111, 5896–5901 (2014).
Armbruster, B. N., Li, X., Pausch, M. H., Herlitze, S. & Roth, B. L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl Acad. Sci. USA 104, 5163–5168 (2007).
Gomez, J. L. et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507 (2017).
Bonaventura, J. et al. High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nat. Commun. 10, 4627 (2019).
Zhang, S. et al. Molecular basis for selective activation of DREADD-based chemogenetics. Nature 612, 354–362 (2022).
Urban, D. J. & Roth, B. L. DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu. Rev. Pharmacol. Toxicol. 55, 399–417 (2015).
Arber, C., Young, M. & Barth, P. Reprogramming cellular functions with engineered membrane proteins. Curr. Opin. Biotechnol. 47, 92–101 (2017).
Wang, L. et al. Use of DREADD technology to identify novel targets for antidiabetic drugs. Annu. Rev. Pharmacol. Toxicol. 61, 421–440 (2021).
Allen, G. M. et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science 378, eaba1624 (2022).
Inagaki, H. K. et al. Visualizing neuromodulation in vivo: TANGO-mapping of dopamine signaling reveals appetite control of sugar sensing. Cell 148, 583–595 (2012).
Kroeze, W. K. et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015).
Wang, W. et al. A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat. Biotechnol. 35, 864–871 (2017).
Kim, M. W. et al. Time-gated detection of protein-protein interactions with transcriptional readout. Elife 6, e30233 (2017).
Fink, T. et al. Design of fast proteolysis-based signaling and logic circuits in mammalian cells. Nat. Chem. Biol. 15, 115–122 (2019).
Petschnigg, J. et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 11, 585–592 (2014).
Yao, Z. et al. A global analysis of the receptor tyrosine kinase-protein phosphatase interactome. Mol. Cell 65, 347–360 (2017).
Saraon, P. et al. A drug discovery platform to identify compounds that inhibit EGFR triple mutants. Nat. Chem. Biol. 16, 577–586 (2020).
Lim, S. H. et al. CFTR interactome mapping using the mammalian membrane two-hybrid high-throughput screening system. Mol. Syst. Biol. 18, e10629 (2022).
Quijano-Rubio, A. et al. De novo design of modular and tunable protein biosensors. Nature 591, 482–487 (2021).
Lee, S. & Wong, W. W. The most logical approach to improve CAR T cell therapy. Cell Syst. 11, 421–423 (2020).
Qudrat, A. & Truong, K. Engineering synthetic proteins to generate Ca(2+) signals in mammalian cells. ACS Synth. Biol. 6, 582–590 (2017).
Qudrat, A. & Truong, K. Antibody-based fusion proteins allow Ca(2+) rewiring to most extracellular ligands. ACS Synth. Biol. 7, 531–539 (2018).
Jing, J. et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca(2)(+) influx. Nat. Cell Biol. 17, 1339–1347 (2015).
He, L. et al. Optical control of membrane tethering and interorganellar communication at nanoscales. Chem. Sci. 8, 5275–5281 (2017).
Hooper, R. & Soboloff, J. STIMATE reveals a STIM1 transitional state. Nat. Cell Biol. 17, 1232–1234 (2015).
Ma, G. et al. Optogenetic toolkit for precise control of calcium signaling. Cell Calcium 64, 36–46 (2017).
Nguyen, N. T. et al. CRAC channel-based optogenetics. Cell Calcium 75, 79–88 (2018).
Tan, P., He, L., Huang, Y. & Zhou, Y. Optophysiology: illuminating cell physiology with optogenetics. Physiol. Rev. 102, 1263–1325 (2022).
Pham, E., Mills, E. & Truong, K. A synthetic photoactivated protein to generate local or global Ca(2+) signals. Chem. Biol. 18, 880–890 (2011).
He, L. et al. Near-infrared photoactivatable control of Ca(2+) signaling and optogenetic immunomodulation. Elife 4, e10024 (2015).
Ishii, T. et al. Light generation of intracellular Ca(2+) signals by a genetically encoded protein BACCS. Nat. Commun. 6, 8021 (2015).
Nguyen, N. T., He, L., Martinez-Moczygemba, M., Huang, Y. & Zhou, Y. Rewiring calcium signaling for precise transcriptional reprogramming. ACS Synth. Biol. 7, 814–821 (2018).
Ma, G. et al. Optogenetic engineering to probe the molecular choreography of STIM1-mediated cell signaling. Nat. Commun. 11, 1039 (2020).
Kyung, T. et al. Optogenetic control of endogenous Ca(2+) channels in vivo. Nat. Biotechnol. 33, 1092–1096 (2015).
Wang, T. et al. Caffeine-operated synthetic modules for chemogenetic control of protein activities by life style. Adv. Sci. (Weinh.) 8, 2002148 (2021).
Ma, G. et al. Optogenetic control of voltage-gated calcium channels. Angew. Chem. Int. Ed. Engl. 57, 7019–7022 (2018).
He, L. et al. Engineering of a bona fide light-operated calcium channel. Nat. Commun. 12, 164 (2021).
Sakemura, R. et al. A tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol. Res 4, 658–668 (2016).
Wang, Z., Guo, Y. & Han, W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 8, 896–925 (2017).
Fedorov, V. D., Themeli, M. & Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 5, 215ra172 (2013).
McNerney, M. P., Doiron, K. E., Ng, T. L., Chang, T. Z. & Silver, P. A. Theranostic cells: emerging clinical applications of synthetic biology. Nat. Rev. Genet. 22, 730–746 (2021).
Li, Y. R., Zhou, Y., Kramer, A. & Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer 7, 1059–1073 (2021).
Kang, M. et al. Nanocomplex-mediated in vivo programming to chimeric antigen receptor-M1 macrophages for cancer therapy. Adv. Mater. 33, e2103258 (2021).
Rurik, J. G. et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022).
Acknowledgements
We acknowledge funding received from the National Key Research and Development Program (2019YFA0903800 and 2019YFA0110800 to W.L.), the National Natural Science Foundation of China (32201193 to F.T.; 31621004 to Q.Z. and W.L.), and the China Postdoctoral Science Foundation (2022M713133 to T.C.).
Author information
Authors and Affiliations
Contributions
W.L., Q.Z., and F.T. conceived the idea and supervised the work. F.T., T.C., and W.L. drafted the initial manuscript with inputs and further editing from all other authors. F.T. and T.C. drew the figures. F.T., T.C., L.Z., and Q.G. collected related literature. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests. As an editorial board member of the journal, Q.Z. did not participate in any reviewing process of this review article.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Teng, F., Cui, T., Zhou, L. et al. Programmable synthetic receptors: the next-generation of cell and gene therapies. Sig Transduct Target Ther 9, 7 (2024). https://doi.org/10.1038/s41392-023-01680-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01680-5
