
Abstract
Small GTPases including Ras, Rho, Rab, Arf, and Ran are omnipresent molecular switches in regulating key cellular functions. Their dysregulation is a therapeutic target for tumors, neurodegeneration, cardiomyopathies, and infection. However, small GTPases have been historically recognized as “undruggable”. Targeting KRAS, one of the most frequently mutated oncogenes, has only come into reality in the last decade due to the development of breakthrough strategies such as fragment-based screening, covalent ligands, macromolecule inhibitors, and PROTACs. Two KRASG12C covalent inhibitors have obtained accelerated approval for treating KRASG12C mutant lung cancer, and allele-specific hotspot mutations on G12D/S/R have been demonstrated as viable targets. New methods of targeting KRAS are quickly evolving, including transcription, immunogenic neoepitopes, and combinatory targeting with immunotherapy. Nevertheless, the vast majority of small GTPases and hotspot mutations remain elusive, and clinical resistance to G12C inhibitors poses new challenges. In this article, we summarize diversified biological functions, shared structural properties, and complex regulatory mechanisms of small GTPases and their relationships with human diseases. Furthermore, we review the status of drug discovery for targeting small GTPases and the most recent strategic progress focused on targeting KRAS. The discovery of new regulatory mechanisms and development of targeting approaches will together promote drug discovery for small GTPases.
Similar content being viewed by others

Introduction
Small GTPases are a large family of low molecular weight enzymes that hydrolyze GTP. It contains more than 150 members, which can be divided into five families: Rat Sarcoma (Ras), Rhodopsin (Rho), Ras-related in brain (Rab), ADP-ribosylation factor (Arf), and Ras-related nuclear protein (Ran), on the basis of their sequence homology and physiological functions [Fig. 1a]. Small GTPases play essential roles in regulating a wide range of cellular activities including cell survival, cell cycle progression, proliferation, apoptosis, differentiation, adhesion and migration as well as subcellular events such as cytoskeletal dynamics and vesicle trafficking. They serve as key molecular switches that mediate cellular processes through recycling between GTP-bound state “active (on)” and GDP-bound state “inactive (off)” [Fig. 1c]. Generally, guanine nucleotide exchange factors (GEFs) bind to the GDP-bound small GTPases and facilitate the exchange from GDP to GTP,1 which induces conformational change that permits binding of effectors to produce distinct signal outputs. Conversely, GTPase-activating proteins (GAPs) terminate small GTPases signaling by stimulating GTP hydrolysis.2,3,4 For Rho and Rab family members, there is another class of proteins which regulate the activation of small GTPases: guanine nucleotide dissociation inhibitors (GDI).5 They bind to GDP-bound forms of GTPases, preventing GDP dissociation and maintaining their inactive state.6 The release of GDIs from these small GTPases prior to GDP-GTP exchange is tightly regulated by GDI displacement factors.7

Overview of small GTPases biology. Phylogenetic tree (a), and five conserved boxes (G1–G5) (b, d), of Ras, Rho, Rab, Arf and Ran families. c Molecular switch diagram: GEF-mediated GDP-GTP exchange for activation, GAP-mediated GTP hydrolysis for deactivation, GDI stabilizes the GDP-bound form of Rho and Rab proteins, and the GTP-bound small GTPases interact with effector proteins. e Structural difference of Switch regions between GDP-state (PDB: 4LPK, light purple) and GTP-bound state (PDB: 6GOD, pink). Conformational ensembles of Switch II (f), and Switch I (g), regions of HRAS are generated from NMR structures (PDB: 1CRP and 2LCF)
Ras family is the root of the small GTPase superfamily according to the phylogenetic reconstruction analysis, so the small GTPase family is also known as Ras superfamily.8 Ras family consists of 36 members falling into seven subfamilies: Ras (KRAS4A, KRAS4B, NRAS, and HRAS), Ral, Rheb, Rap, Rad, Rit and DIRAS, which serve as central nodes of a wide range of signaling pathways controlling cell survival, proliferation, differentiation, migration, and adhesion.9,10 As the first human oncogene to be identified, Ras has been well established as an oncogenic driver in multiple human cancers. Hotspot mutations in Ras drastically impair its GAP-mediated GTP hydrolysis activity, lead to constitutive activation of Ras and drive cell transformation and tumor initiation in different cancer models. Germline mutations of Ras result in aberrant activation of the downstream Mitogen-Activated Protein Kinase (MAPK) pathway and cause developmental disorders in patients, known as RASopathies. In addition, new members of Ras subfamily are being discovered through sequence homology screen and their functions are distinct from well-characterized Ras subfamily members like HRAS, KRAS, and NRAS.11 For example, DIRAS subgroup, consisting of DIRAS-1, DIRAS-2, and DIRAS-3 (Noey2), were found to be downregulated in cancer and act as tumor suppressors.12,13,14,15 Their biochemical properties and physiological functions remain to be further elucidated prior to being established as therapeutic targets of tumors. The Rho family comprises 22 members including Rho-related proteins (RhoA) subfamily, cell division control protein 42 homolog (Cdc42) subfamily, and Ras-related C3 botulinum toxin substrate (Rac) subfamily that are involved in cytoskeletal organization.16 Dysregulation of the key members within Rho family, such as RhoA and Cdc42, facilitates tumor progression via promoting epithelial to mesenchymal transition (EMT) and invasion of tumor cells.17,18 Mutations of Rho family members also have been linked to the development of neurologic and vascular diseases.19,20,21 Rab, the largest family of the small GTPases with more than 60 members, regulates membrane trafficking between different intracellular organelles and the plasma membrane.22,23 Aberrant regulation of Rab-mediated vesicle trafficking promotes tumor progression through enhancing cell migration/invasion.24,25,26,27,28,29 Rab mutations have also been implicated in neurologic diseases due to dysfunctional membrane trafficking.30,31 Arf is the fourth family of this superfamily, containing six members that are involved in several processes of membrane trafficking and tumor cell migration.32 Additionally, lack of ARF may cause brain diseases due to insufficient neuron migration.33 Finally, Ran family, the most abundant protein in cells with only one recognized member, is involved in nuclear transport.34 Ran GEFs interact with Ran in the nucleus, allowing GTP–bound Ran to bind to and transfer its client proteins from nucleus to cytoplasm.35 GTP-bound Ran is subsequently dissociated and inactivated by the engagement of Ran GAPs located in the cytoplasm. Ran-mediated nuclear-cytoplasmic transport accelerates the cell cycle and efficiency of DNA repair, facilitating proliferation in tumor cells. Additionally, defective Ran has been found in the development of Alzheimer’s disease and other brain diseases.36
Despite being famous therapeutic targets in human diseases, small GTPases have been termed as “undruggable” for decades, mainly owing to the absence of pharmacologically targetable pockets within wild types and mutant isoforms. Early efforts were devoted to developing indirect targeting strategies that interfere with GEFs mediated activation of small GTPases. As the most frequently mutated oncogene in human cancer, KRAS has been established as a paradigm for targeting Ras proteins and other small GTPases. Extensive studies have been focused on targeting KRAS post-translational modifications, membrane trafficking, upstream regulators (e.g., GEFs, GAPs), downstream effectors (e.g., MEK-ERK signaling, PI3K-AKT signaling), synthetic lethal partners and metabolic alterations. However, single agent treatment targeting the above targets have had insufficient efficiency in RAS-mutant cancer patients, and combinatorial targeting its downstream pathways also failed due to severe clinical toxicity. With emerging new targeting approaches and improvements in covalent drug design, the success of Sotorasib, an allele-specific inhibitor of KRASG12C, finally marked the dawn of a new era in targeting small GTPases directly. Inspired by the accelerated approval of Sotorasib for the treatment of KRASG12C mutant NSCLC, an upsurge in the development of KRAS inhibitors has spawned many new cutting-edge strategies, such as fragment-based screening, macromolecular inhibitors, PROTACs, genetic targeting and antibodies targeting covalent inhibitor based neoepitopes. Many more hotspot mutant alleles of KRAS including G12D/S/R have proven to be targetable in the past few years, providing valuable insights into the development of inhibitors of remaining KRAS mutant alleles and other Ras isoforms.37,38,39 Despite the rapid progress made in targeting KRAS during the past decade, hotspot mutations such as G12V, G13, and Q61 in KRAS, as well as Q61R/L/H in NRAS, still lack targeting methods. With the successful clinical application of KRASG12C inhibitors in NSCLC, the rapid onset of drug resistance driven by heterogenous mechanisms remains a major inevitable and urgent challenge that needs to be addressed to maximize clinical benefits. Moreover, development of selective inhibitors targeting other members of small GTPases is lagging, and the vast majority of small GTPases remain untargeted.
In this review, we provide an overview for structural properties, physiological functions, post-translational modifications, and regulation of small GTPases that underpin their fundamental roles as molecular switches in cells. Pathologic dysregulations of small GTPases in various human diseases including tumors, neurodegenerative diseases, cardiomyopathies, and infections are summarized. More importantly, we review the recent strategic developments in targeting small GTPases directly and indirectly, highlighting the breakthrough targeting strategies of KRAS and discussing crosstalk between Ras and Rho/Ran family proteins in tumorigenesis. We hope that this review will lay the groundwork for developing GTPases-based cooperative targeting. Next, we summarize mechanisms responsible for resistance of KRASG12C inhibitors in clinical trials and discuss potential combination strategies to prevent or delay resistance. In the last session, we discuss the future directions for targeting small GTPases based on the most recent outcomes in clinical trials and drug resistance studies, new regulatory mechanisms, and new targeting strategies.
Small GTPases biology
Structural basis for molecular switch
The primary structure of small GTPases is divided into two segments, G-domain (GTP-binding domain) for nucleotide-binding and C-terminal hypervariable region for membrane association. In 1988, the crystal structures of HRAS G-domain in complex with GDP/GTP were determined, which served as a prototype to understand structural properties shared among different small GTPases in Ras superfamily.40 Despite sequence divergences, small GTPases adopt canonical Rossmann fold composed of six β-sheets and five α-helices. There are five conserved motifs termed as G1–G5 representing five important loops for nucleotide binding and structural regulations41,42 [Fig. 1b, d]. G1 (residue 10–16 in Ras) is the first loop in the protein and participates in binding to nucleotide phosphates. Therefore, G1 is referred to as the P-loop, and it contains two oncogenic mutation hotspots, G12 and G13, in KRAS, as well as the G12V mutation in Cdc42.43,44 G2 (residue 32–38 in Ras) consists of the majority of Switch I, Thr35 (Thr35 in Ras, Rac1, and Cdc42 and Thr37 in RhoA) located in Switch I is a conserved residue for interacting with γ-phosphate and magnesium ion.45 As such, Switch I exhibits structural plasticity directly related to the nucleotide binding. G3 (residue 57–61 in Ras) spans across part of Switch II and helix 2. G3 harbors another oncogenic hotspot, Q61, which is frequently mutated in NRAS, HRAS, and Cdc42 because the sidechain of Q61 plays an important role in GTP-hydrolysis.43,44 G4 (residue 116–119 in Ras) and G5 (residue 145–147) are two loop regions for anchoring guanine bases and ribose and dictating the binding selectivity to guanosine over adenosine (ATP/ADP).46 In G4, K117 interacts with ribose, and its disease-related mutations (K117N and K117R) are characterized as “fast-cycling” because the weakened binding between protein and ribose of guanine nucleotide leads to higher exchange rates between bound and unbound nucleotides.47,48 Both K117 and K147 serve as hotspots for PTMs in the G-domain of Ras. Beyond the intramolecular interactions that involve the G1-G5 motifs, Switch I (residue 30–38) [Fig. 1e, g] and Switch II (residue 59–67) [Fig. 1e, f] are two structural regions that underpin the function of molecular switches for small GTPases. They possess significant structural plasticity by changing their conformations between GDP- and GTP-bound states.42 The different conformations of Switch regions dictate the specificity of small GTPases to interact with different regulatory proteins (GEFs, GAPs, GDIs) and downstream effectors. Moreover, the switch regions are highly dynamic and present as structural ensembles containing multiple coexisting and interconverting conformations. Both structure and dynamics of switch II are regulated by nucleotide binding, which have been exploited as a structural basis for developing inhibitors.49,50
Biochemistry of small GTPases: regulations and effector interactions
As the molecular switch, binary cycling between GDP’OFF’- and GTP’ON’-bound states is a signature process of small GTPases in regulating signal transduction [Fig. 1c]. The cycling is highly regulated by two major categories of regulatory proteins: GEFs and GAPs.3 To activate small GTPases, GEFs recognize the GDP-bound conformation of switch regions and facilitate the GDP-GTP exchange. In the GTP-bound state, small GTPases adopt distinct conformations in both Switch I and Switch II regions compared to the GDP-bound state [Fig. 1e–g]. Activated small GTPases interact with a variety of catalytically distinct downstream effectors such as RAF kinases and PI3K in Ras pathways. GAPs bind to the active small GTPases and catalyze the GTP hydrolysis through an arginine finger. Following this, the protein returns to the GDP-bound “OFF” state. Many oncogenic mutations impair GEF- and GAP-regulations; the GAP-deficiency due to disrupted GAP interactions with the mutant GTPases is characterized as a major consequence of the mutation to drive tumorigenesis. GEFs and GAPs can be shared within a small GTPases subfamily.8 Among different subfamilies, GEFs and GAPs are structurally distinct but mechanistically similar. In a recent study, a non-arginine finger GAP, RGS3, has been found to facilitate GTP hydrolysis of oncogenic KRASG12C mutant in tumor cell lines.51 Additionally, GDP-binding affinity is relatively weak for Rho and Rab families, and GDIs are required for maintaining their GDP-binding inactive state.5
Signaling pathways
Ras
Ras family proteins are coordinating or participating in multiple important signaling pathways in cells [Fig. 2a]. The intracellular signal transductions coordinated by RAS branch or RAS subfamily members (HRAS, NRAS, and KRAS) such as MAPK and PI3K-AKT, which are some of the most characterized pathways in research of cell biology and human diseases, especially in tumors. The MAPK pathway relaying cellular signal through RAS-RAF-MEK-ERK plays a critical role in cell cycle and proliferation. The MAPK cascade is initiated by activation of receptor tyrosine kinases (RTKs) upon extracellular ligand binding. The activated RTKs recruit growth factor receptor-bound protein 2 (GRB2) and RAS-GEF (SOS1). RAS-GEF then activates RAS by catalyzing GDP-GTP exchange, which switches RAS from its “OFF” to “ON” state. The activated RAS binds to RAF kinases and the signal is transduced downstream through RAF-MEK-ERK phosphorylation cascades and regulates a wide variety of cellular processes including cell proliferation, survival, cell cycle progression and differentiation. Ras proteins also regulate the PI3 Kinase (PI3K)-AKT-mTOR pathway, which plays an important role in regulating cell survival, proliferation, and motility.52,53 Both RAS and RTK can activate PI3K and recruit it to the plasma membrane; PI3K phosphorylates lipid molecule PIP2 to PIP3, and PIP3 triggers AKT phosphorylation by PDK154 and mTORC2.55,56 The signal amplitude of PI3K-AKT is modulated by lipid phosphatase PTEN, which converts PIP3 to PIP2. As such, PTEN serves as a suppressor to tumorigenesis.57,58 RalGEF (RalGDS) is another downstream effector of RAS proteins and catalyzes the nucleotide exchange and activates another RAS family member, RAL. RAL GTPases located at a downstream signaling node in the RAS pathway engage with several downstream factors involved in transcription, trafficking, tumor cell invasion and metastasis.59,60 Phospholipase C (PLCε) is positioned at the center of another RAS-mediated pathway in regulating cell differentiation, migration, and secretion.61,62 PLCε contains a catalytic domain, two RAS-associating (RA) domains, and a GEF domain.63 In one mechanism, PLCε is activated by RAS and then hydrolyzes PIP2 to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 then induces the release of Ca2+ from the endoplasmic reticulum while DAG activates protein kinase C (PKC) and protein kinase D.61,64,65 In a different mechanism, PLCε mediates nucleotide exchange of another RAS family member, RAP1, and the activated RAP1 binds to one RA domain of PLCε and activates PLCε activity as a positive feedback loop.63,66 PLCε can also be activated by G protein-coupled receptor (GPCR)-G protein-RHO pathway.67,68 RAS-association domain family (RASSF) proteins are another group of RAS effectors; there are ten members identified and they contain Ras-association (RA) domain. RASSFs connect Ras to Hippo pathway in apoptosis (RASSF1-6) and p53 regulation in tumor suppression (RASSF7-10). Among these, RASSF1 and RASSF5 (NORE1A) are the best characterized members in terms of their interactions with Hippo kinase ortholog MST1 (mammalian sterile twenty).69,70

Cellular signaling coordinated by Ras and Rho subfamilies. a Ras-mediated signaling pathways include RAS-RAF-MEK-ERK, RAS-PI3K-AKT, RAS-RALGDS-RAL, RAS-PLCε, RAS-RASSF, the crosstalk between Ras, and other small GTPases including CDC42, RAC1 and RAP1, RHEB, and RAL are highlighted by red color. b Cytoskeletal dynamics are regulated by Rho, Rac and CDC42 through their main downstream signaling nodes such as ROCK, mDIA1, LIMK, PAK, WRC, and N-WASP. This figure was created with BioRender.com
Rho
RHO GTPases play critical roles in modulating cytoskeletal dynamics [Fig. 2b]. RHO, RAC, and CDC42 are the three best characterized subfamilies. They modulate cell polarity, morphology, migration, and cell cycle progression.71,72 RND and RhoBTB are two other subfamilies. RND proteins include RNDs and RhoE and antagonize the RHO signaling in different tissues. However, the RhoBTB subfamily is still poorly characterized. RHO-associated coiled-coil containing kinase (ROCK), one of the downstream effectors of RHOA, phosphorylates MLC (myosin light chain) and leads to the activation of myosin, which is involved in cytoskeletal reorganization.73,74,75 ROCK also regulates actin assembly through ROCK-LIMK (LIM kinase)-COFILIN phosphorylation cascades. Interestingly, LIMK is also regulated by RAC1 and CDC42: the activated forms of both bind to PAK (p21-activated kinase) and enhance PAK/LIMK association, leading to increased phosphorylation of LIMK by PAK.16,76 RHOA-mDIA1-ACTIN is another RHOA pathway in which mDIA1, a formin family protein, (Diaphanous-related formin) mediates actin nucleation and elongation as well as stabilization of microtubules.
Ras-related C3 substrate (RAC) subfamily proteins mediate cell migration by affecting cytoskeletal dynamics. WASP family verprolin homologous protein (WAVE) and PAK are two well-characterized effectors of RAC1. RAC1 binds to WRC (WAVE regulatory complex) and subsequently actives ARP2/3 complex,77 which plays an important role in actin polymerization and formation of lamellipodia.78,79 PAK is a shared signaling node located downstream of both RAC1/CDC42 and RHOA. PAK-LIMK-COFILIN pathway is essential in stabilizing the structure of actin filaments, leading to the accumulation of these filaments at the leading edge of moving cells.80 Interestingly, CRAF, MEK1 and ERK3 are substrates of PAK, indicating PAK serves as a regulator of RAS-driven MAPK pathway.81 CDC42 regulates a wide spectrum of cellular processes including cell migration, polarity, transformation, division, and invasion. Besides PAK, Wiskott Aldrich syndrome protein (WASP) is another main CDC42 effector and the CDC42-bound N-WASP activates ARP2/3 complex in a similar manner as RAC1-bound WRC, meaning this pathway also regulates actin.82,83
There are several instances of crosstalk between RHO and RAS proteins.84,85 Activation of CDC42, RAC1, and RHOA is necessary for RAS-induced malignant transformation of fibroblasts.86,87,88,89 Loss of CDC42 in HRASG12V-transformed cells leads to significant changes in morphology and inhibition of the cell cycle and proliferation.85 Constitutive activation of CDC42 prevents EGFR degradation and leads to sustained MAPK signaling.90 CDC42 activates PAK4 at the Golgi apparatus, which is important in RAS-mediated transformation.91
Rab
Ras-like proteins in brain (RAB), ADP-ribosylation factor (ARF), and Ras-like nuclear (RAN) proteins are three families of small GTPases that are important in regulating vesicle trafficking (RAB and ARF) and nucleocytoplasmic transport (RAN). The RAB proteins comprise the largest family of RAS superfamily with more than 60 members and each has its own effector proteins. Rab effectors include a wide range of proteins such as tether proteins (p115, GM130, Giantin, Golgin-84, Rabenosyn-5 etc.), regulatory proteins (Rabphilin-3, Rabip4 etc.), adapter proteins (Rabaptins, Rabankyrin-5, Tip47, etc.), motors/motor adapters, scaffold proteins, SNARE proteins, kinases and phosphatases. And the list of Rab effectors is growing with new Rab downstream proteins being identified.23,92,93 Through interactions with specific effectors, RABs coordinate multiple steps of membrane trafficking between different organelles and the plasma membrane, which are related to endocytosis, recycling, secretion and degradation22,94,95 [Fig. 3a]. RAB proteins control membrane identity and spatiotemporal dynamics of vesicle traffic through their C-terminal prenylation and interactions with effectors.96 RAB1, RAB22, RAB6, RAB33, RAB40, and RAB2 coordinate a series of steps during trafficking between endoplasmic reticulum (ER) and Golgi. RAB5 and RAB21 participate in regulating endocytosis97,98 while RAB4 and RAB11 are responsible for recycling endosomes. RAB5, RAB14, and RAB22 together mediate early formation and early-to-late maturation of phagosome,99 after which the late phagosome is sorted to lysosome under the coordination of RAB7.100,101 As such, RAB7 has a special role in targeting of late endosome, late phagosome, and autophagosome to lysosome for degradation, in which RAB24 and RAB33 regulate the formation of autophagosome together.99,102 RAB8 coordinates biosynthetic trafficking from Golgi to the plasma membrane and mediates insulin-stimulated translocation and fusion of GLUT4 (glucose transporter)-coated vesicles targeting the plasma membrane together with RAB10, RAB13, and RAB14.103,104,105,106 RAB8 also participates in regulating ciliogenesis with RAB17 and RAB23. RAB3, RAB26, RAB27, RAB29, RAB34, and RAB37 direct secretory vesicles and granules to the plasma membrane.107,108,109,110 RAB32 and RAB38 are involved in the biogenesis of melanosomes and RAB32 also controls mitochondrial fission.111,112 RAB31 is reported to drive intraluminal vesicle formation and prevent multivesicular endosome degradation during the biogenesis of exosomes.113 RAB18 has a still debated role in maintaining lipid droplet biogenesis and homeostasis.114,115 More recently, RAB22A has been shown to mediate a new type of non-canonical autophagosome formation, namely Rafeesome (RAB22A mediated non-canonical autophagosome fused with early endosome), and transfer activated STING between cells to promote anti-tumor immunity.116

Cellular trafficking coordinated by Rab, Arf and Ran subfamilies. a Multiple vesicle trafficking steps are coordinated by Rab proteins: (1) ER to Golgi by RAB1, (2) Golgi to plasma membrane by RAB8, (3) Secretory vesicles and granules trafficking between Golgi and plasma membrane by RAB3, RAB26, RAB27 and RAB37, (4) Golgi to melanosome by RAB32 and RAB38 for biogenesis of melanosome, (5) Golgi to ER by RAB2, RAB6, RAB22, RAB33 and RAB40, (6) GLUT4 vesicle trafficking to plasma membrane by RAB8, RAB10 and RAB14, (7-9) Plasma membrane to early endosome (RAB5), then to late endosome (RAB7) and finally to lysosome (RAB7), (10-11) Early endosome to recycling endosome (RAB4), then to plasma membrane (RAB11), (12) Early phagosome to late phagosome by by RAB5, RAB14 and RAB22, (13) Late phagosome to lysosome by RAB7, (14-15) Autophagosome budding from plasma membrane by RAB24 and RAB33 and trafficking to lysosome by RAB7, (16) Lipid droplet biogenesis and homeostasis mediated by RAB18, (17) Mitochondrial fission mediated by RAB32, (18) Endocytosis by RAB5 and RAB21, (19) Ciliogenesis by RAB8, RAB17 and RAB23. b Vesicle trafficking and cytoskeletal dynamics are regulated by Arf proteins including ARF, ARL and SAR. c, Nucleocytoplasmic cargo transport through NPC is controlled by RAN. This figure was created with BioRender.com
Arf
Similar to RAB, Arf family proteins regulate multiple steps of vesicle transport117 and modulate dynamics of cytoskeleton proteins such as actin filaments and microtubules [Fig. 3b].118,119 Arf family is further divided into ARF, ARL (ARF-like), and SAR subfamilies. ARF1 is the best characterized ARF protein. During vesicle budding, GTP-bound ARF1 recruits coat protein-I (COPI) complex and induces positive membrane curvature for budding vesicle.120,121,122,123 Thus, ARF controls vesicle budding, transport between the Golgi and ER as well as transport of secretory vesicles and endosomes. Like ARF1, ARF3, ARF4, and ARF5 regulate formation of COPI-coated vesicles through ARF–COPI interactions. ARF1 also promotes the assembly of actin in the Golgi apparatus.124 ARF6 modulates actin polymerization and membrane traffic through colocalization with and activating PIP5-kinase, which is responsible for generation of PIP2 and directly interacts with PIP2 on the plasma membrane.125,126,127,128,129 ARLs constitute the largest subfamily of Arf family. There are 21 ARLs and they have a broad spectrum of functions, mainly involving endosome–Golgi trafficking, lysosome trafficking, cilia formation, actin remodeling and tubulin assembly. ARL2 cooperates with tubulin cofactors (TBCC, TBCD, and TBCE) in modulating αβ-tubulin assembly and microtubule dynamics.130,131 SAR1 has been well known for its cargo transportation from ER to Golgi.132 Activated SAR1 accumulates in ER, where it recruits SEC23/ SEC24 heterodimer to form a pre-budding complex. Then, the pre-budding complex binds to SEC13/31, leading to the assembly of coat protein- complex II (COPII)-coated vesicles, which are required for ER-to-Golgi cargo transport.133
As far as crosstalk with other small GTPases families is concerned, ARF6 binds to RhoGEF and Kalirin, recruiting them to the plasma membrane for Rac activation in the scenario of Rac-mediated cytoskeletal remodeling and membrane ruffling.134 TBC (Tre2–Bub2–Cdc16) domain is one key catalytic component in RabGAP proteins.135 Vacuole-living bacterial pathogens employ the effector proteins containing TBC-like motifs to catalyze RAB inactivation for counteracting host cell defenses, of which the binding of TBC to ARF proteins is a necessary step for implementing the GAP function.136
Ran
RAN represents a single member small GTPases family and is located close to RAB family in the phylogenetic tree. RAN plays an essential role in coordinating nucleocytoplasmic transport through the nuclear pore complex, which is involved in mitotic spindle formation137,138 and nuclear envelope assembly139,140 [Fig. 3c]. RanGTP enhances the binding of importin to the cargo and promotes the release of exportin. The asymmetric distribution of the RanGTP and RanGDP between the cytoplasm and nucleus, mediated by cytosol-localized RanGAP and nucleus-localized RanGEF respectively, generates a gradient to ensure proper transport direction of the cargo.141,142
Post-translational modifications (PTMs)
PTMs constitute an important category of mechanisms in regulating the function of small GTPases.143 C-terminal modifications in HVR are essential for membrane association of many small GTPases.144 In Ras family proteins, after synthesis is completed in ribosomes, the CAAX motif of cytosolic Ras proteins undergoes farnesylation, endoproteolysis and carboxylmethylation. The farnesylated C-terminal tail serves as the primary moiety responsible for binding to the plasma membrane.145 CAAX motif is located at the carboxyl-terminal, which can be farnesylated by cytosolic prenyltransferases, termed farnesyltransferase (FTase).146,147 FTase enhances the hydrophobicity of RAS via modifying the cysteine with 15-carbon farnesyl isoprenoid in CAAX motif covalently, and it is followed by the cleavage of AAX residues by RAS-converting enzyme (RCE1) in ER.148 RAS is then modified by isoprenylcysteine carboxylmethyltransferase (ICMT) and α‑carboxyl group of the farnesylated cysteine is carboxylmethylated by ICMT.149 A secondary modification through PTMs in the proceeding region of CaaX is needed to assist membrane attachment, which varies among different isoforms.150 Palmitolytion occurs in HVR of HRAS, NRAS and KRAS4A, KRAS4B, using its polybasic patch (lysine repeats) for attaching to the membrane together with the farnesylation.150 Farnesylation, prenylation, geranylgeranylation, and palmitolytion are four major C-terminal lipid modifications responsible for docking small GTPases to the membrane.
In G-domain of Ras proteins, PTMs have been continuously discovered within the last decade. Nitrosylation at C118 by reactive nitrogen species or oxidation by reactive oxygen species intermediates promote Ras activation via facilitating GDP-GTP exchange151 impeding GAP-mediated GTP hydrolysis152,153 while monoubiquitination at K117 in HRAS promotes its activation by rendering a fast-cycling state.154 K104 in KRAS is a hotspot for PTMs. However, the roles of K104 acetylation in regulating biochemical properties and oncogenicity in KRAS remain elusive.155,156,157 While the biochemical and structural properties of K104 monoubquitination in KRAS were characterized, its functional phenotype remains undetermined.158,159 Acetylation and methylation are two types of PTMs recently found in KRAS. Acetylation is found at multiple sites in the effector lobe and K5 methylation occurs in all different isoforms.144 The regulatory roles of the acetylation and methylation remain unclear and need further characterization. K147 in G5 motif is another hotspot for PTMs in KRAS, including monoubiquitination, methylation and acetylation, suggesting that potential interplay between different PTMs might provide an additional level of regulation. Phosphorylation at Y32 and Y64 by SRC reduces binding capacity of RAS proteins to downstream effectors and in particular decrease the oncogenicity of KRASG12 mutants.160,161,162 The inhibitory effects by tyrosine phosphorylation can be reversed by SHP2 phosphatase, thus SHP2 has been developed as an important regulatory target for KRAS-driven cancers. Sumoylation was found at K42 in HRAS, NRAS, and KRAS and linked to the activation of RAS, but the mechanism is still undetermined.163,164 In a recent study, an autophosphorylation occurs at A59T mutant of HRAS and KRAS by transferring a phosphate group from the bound GTP, and the phosphorylation at T59 subsequently inhibits GTP hydrolysis and decreases binding to RAF proteins, which reveals a new mechanism by promoting nucleotide exchange and weakening the effector binding.165
In Rho family, phosphorylation and AMPylation are two major modifications in Switch I and Switch II regions of RhoA, Cdc42, and Rac1.166 Y32 in Cdc42 and Rac1, and Y34 in RhoA in Switch I region are highly conserved residues in small GTPases families and their AMPylation is found in all three isoforms, which block interactions with downstream effectors.167 EGF-stimulated Y64 phosphorylation in Cdc42 increases its binding to Rho GDI and is implicated in regulating the cellular localization and transformation.168 S71, located at the end of Switch II, is recognized as a substrate site of AKT kinase. S71 phosphorylation inhibits activation of Rac1 and Cdc42.169,170 In a recent study, S71 phosphorylation mediates the interaction between Rac1 and 14-3-3, an important scaffold protein involved in multiple signaling pathways.171 Multiple lysines in RhoA and Rac1 are involved in ubiquitination-dependent proteasome degradation, which regulates the protein expression level and impact on cytoskeleton dynamics and cell migration.172,173,174,175 HACE1 E3 ubiquitin-ligase, a tumor-suppressor, catalyzes ubiquitination and recruits Ubiquitin-proteasome system preferentially to the activated Rac1. This serves as a protection mechanism to ameliorate activation caused by point mutation176 and has also been reported to reduce reactive oxygen species generated by Rac1-dependent NADPH oxidases.177
Rab proteins coordinate vesicle trafficking and secretion, and the cooperation of C-terminal prenylation and regulator proteins such as Rab GDI and Rab GEF are important in sorting Rab to different subcellular locations.178 Phosphorylation is a major PTM that has been functionally characterized in most Rab proteins.93 Many kinases such as CDK, PINK1, LRRKs, PKCs, and SRC have been identified to phosphorylate different Rab proteins. Phosphorylation of Rab1 and Rab4 modulate Rab-related mitosis179,180 and S72 phosphorylation by TBK1 enhances the binding of Rab7 to FLCN-FNIP1, which targets damaged mitochondria during process of mitophagy.181 S72 phosphorylation by leucine-rich repeat kinase 1 (LRRK1) and dephosphorylation by PTEN in Rab7a dynamically regulate EGFR trafficking and degradation.182,183 Many Rab proteins such as Rab3, Rab8, Rab10, Rab12, Rab29, Rab35, and Rab43 can be phosphorylated by the pathological LRRK2 variants at the Thr/Ser residues located in Switch II.184,185 The phosphorylation by LRRK2 leads to a decreased affinity to Rab GDIs and likely alters the distribution between cytosol and membranes. Meanwhile it is also implicated in ciliogenesis through modulating interactions of RAB8A, RAB10, and RAB12 with RILPL1/2.185 Upon activation by mitochondrial depolarization, PTEN-induced kinase 1 (PINK1) phosphorylate S111 in Rab8A, which blocks Rabin8 (Rab8 GEF)-mediated Rab8A activation.186 LRRK1 and PINK1 are two important kinases in development of Parkinson’s disease (PD). As such, phosphorylation of Rabs by LRRK1 and PINK are implicated in PD progression.187 Other PTMs such as AMPylation, phosphocholination, and adenylylation as consequences of bacterial infection interrupt GDP-GTP cycle of Rab proteins (mainly Rab1 and Rab35 by phosphocholination), and these PTMs mainly occur at Tyr and Ser residues in switch II.188,189,190,191,192 Palmitoylation at C83 and C84 in Rab7 promotes its interactions with retromer complex and mediates endosome to trans-Golgi network trafficking of the lysosomal sorting receptors.193,194 K38 in Rab7A is subjected to ubiquitination by PARKIN and deubiquitination by USP32, which alters interactions of Rab7A with its effector and regulates the Rab7A-dependent endosome pathway during PD progression.195,196 An interesting interplay among β2-adrenergic receptor (β2-AR), HACE1 ubiquitination ligase, and Rab11A has been observed. HACE1 mediates recycling of β2-AR through ubiquitinating RAB11A at K145 whereas the β2-AR/HACE1 interactions are required for activation of HACE1.197 During infection by pathogenic bacterial like Legionella pneumophila, one important mechanism used by the bacteria to compromise host immune defenses is to ubiquitinate multiple RABs by the bacterial effector proteins (equivalent to E3 ligase) in an uncommon E1/E2-independent manner.198,199
Arf proteins have an N-terminal amphipathic helix adjacent to G-domain and myristoylation at the beginning of N-terminal is conserved in Arf proteins. Upon GDP-GTP exchange, activated Arfs release the myristoylated N-terminal to attach to the membrane.200,201 Acetylation at Y2 in the amphipathic N-terminal helix of ARL3, Arl14 and Arfrp1 is critical for its subcellular localizations such as recruitment to Golgi membranes.202,203,204 Methionine acetylation at N-terminal of ARL8A/B is necessary for lysosome localization and regulates lysosomal transport in cells.205 Palmitoylation of C8/C9 in the N-terminal of ARL13B is found to regulate protein trafficking and stability during cilia formation.206 Additionally, C-terminal SUMOylation of ARL13 is found to regulate ciliary targeting of sensory receptors.207 In comparison with other small GTPases, phosphorylation, ubiquitination, and other PTMs have been scarcely investigated for Arf proteins.118
Unlike other small GTPases, Ran shuffles between cytoplasmic space and nucleus rather than binding to the cellular membrane through terminal modifications. Five lysine residues (K37, K60, K71, K99, K159) located in important functional regions such as P-loop, Switch regions and G5 motif in Ran are identified with acetylation by mass spectrometry.208 Using unnatural amino acid incorporation strategy, a dynamic acetylation network associated with these sites has been revealed to regulate its basic GTPase activities, formation of nuclear import/export complex, and subcellular location in a site-dependent manner.209,210 K134 acetylation promotes Ran activation through releasing Ran from Ran-Mog1 complex and enabling RCC1 (Ran-GEF) binding. K134 acetylation in Ran has been reported to regulate the subsequent chromosome segregation in mitosis.211 Besides acetylation, other PTMs in Ran have been rarely characterized.
Taken together, in addition to GAP/GEF/GDI-mediated regulations, PTMs add another layer of regulation to the activity of small GTPases. In particular, many modifications occur at the switch regions. Along with technical development of proteomics, more PTMs have been found, bringing more complexity and opportunities to discover novel mechanisms in regulation and targeting of small GTPases. Many of these newly identified PTMs have been poorly characterized in terms of their effects on internal structural dynamics, GTPase cycle, molecular interactions, and relevance in diseases. There are enzymes that are responsible for installing modifications, such as transferases, kinases, E3 ligases, as well as those with roles for removing modifications including phosphatases, deacetylases, deubiquitinases, demethylases. Many of these functions are still largely unknown for most of the newly-discovered PTMs found in small GTPases. As such, the discovery of PTMs and their mechanistic characterizations has created an exciting time in the research of small GTPases.
Small GTPases in human diseases
Ras
Ras family proteins are associated with a wide range of human diseases (Table 1). Somatic KRAS mutations are present in many different human cancers, including pancreatic adenocarcinoma, colorectal cancer, non-small cell lung cancer, cholangiocarcinoma, multiple myeloma, uterine cancer, endometrial cancer, gastric cancer, testicular cancer, cervical adenocarcinoma, diffuse large B-cell lymphoma, breast cancer, acute myeloid leukemia, chronic lymphocytic leukemia, bladder cancer, and cutaneous malignant melanoma212 [Fig. 4]. Mutations of other isotypes, NRAS and HRAS are found in many human cancers as well. For instance, NRAS Q61L is one of dominant drivers of melanoma. The most commonly mutated residues in Ras proteins include G12, G13, and Q61, as these mutants impede GTP hydrolysis and lead to constitutive activation of Ras.213 In addition to tumors, RASopathies are a group of genetic disorders caused by germline mutations in genes that encode proteins of RAS/MAPK pathway and their regulatory proteins.214,215,216 The developmental disorders characterized as RASopathies include neurofibromatosis, Noonan syndrome, and Costello syndrome.217 These conditions share common clinical features such as facial abnormalities, cognitive impairment, and congenital heart defects, due to aberrant activations of RAS/MAPK pathway.218 Furthermore, abnormalities in the RAS-MAPK pathway have been found in nearly 80% of therapy-relapsed neuroblastoma samples, which indicates that personalized therapy targeting specific RAS mutations as well as a sensitivity to MEK inhibition may be effective in the treatment of neuroblastoma.219,220 While neuroblastoma was initially believed to be largely attributed to other cellular mechanisms such as activating ALK or inactivating ATRX mutations, refractory neuroblastoma cases have been shown to express mutant Ras proteins, most commonly NRAS Q61K and HRAS Q61K.220 Furthermore, activation and increased expression of RALs are observed in many RAS-driven tumors.221 RALs are further shown to contribute to anchorage-independent cell growth and invasion of multiple cancers such as lung cancer, colorectal cancer, melanoma, pancreatic cancer and bladder cancer;59,221,222 RALA is required for anchorage-independent growth while RALB is necessary for the survival of tumor cells.223 Increased expression and activation of both RALs are associated with enhanced tumor growth. Owing to its broad impacts in many cellular processes, RASA1 associated disorders are characterized by capillary and arteriovenous malformations, which contribute to heart failure in affected patients.224

Small GTPases in human diseases. Distribution of mutations or aberrant expression of small GTPases across human diseases including cancers and neurologic diseases. This figure was created with BioRender.com
Rho
The Rho family affects a wide variety of cellular functions and thus contributes to many human pathologies [Fig. 4], (Table 1). Primarily, Rho, Cdc42, and Rac proteins are associated with tumorigenesis and cardiovascular disease, including lymphomas, Kaposi’s sarcoma, prostate cancer, gastric cell carcinoma, breast cancer, non-small cell lung cancer, squamous cell carcinoma, and glioblastoma. Movement of tumor cells is important in tumor progression and processes including formation and extension of pseudopods, establishment of new adhesion sites, contraction of cell bodies, and retraction of the tails are regulated by many different mechanisms.225 Rho subfamily is one of the key regulators in cell-matrix adhesion and cytoskeletal reorganization, thus regulating the invasion of tumor cells.225 Cdc42 communicates with the RAS subfamily to induce RAS-mediated transformation into carcinogenic cells through cell cycle progression.226 Additionally, Rac1 has been linked to the development of atherosclerosis and vascular disease. Rac1 is essential in the production of reactive oxygen species by NADPH oxidase as well as the migration of endothelial cells of blood vessels during shear stress, two processes that are vital contributors to atherosclerosis.21 The formation of reactive oxygen species following cardiac myocyte injury is one of the key processes in the development of cardiac hypertrophy. In one study, DL0805 has been shown to be a possible therapeutic agent in cardiovascular disease by acting as a vasorelaxant in rat thoracic aortas through inhibition of the Rho/ROCK signaling pathway.227 RhoA is required for the phosphorylation of myosin light chain, a key event in the regulation of vascular smooth muscle cell contraction. Rho-kinase and nitric oxide have been shown to have opposite effects on lipid metabolism. Nitric oxide activates hepatic sterol regulatory element-binding protein-2 which is a transcriptional factor for cholesterol metabolism and the expression of LDL receptors, decreasing the burden of cardiovascular disease. RhoA suppresses whole body energy consumption by inhibiting AMPK, thus contributing to dyslipidemia.228 As such, Rho plays a crucial rule in the pathogenesis of coronary artery vasospasm, which can cause angina and myocardial infarction. In the central nervous system, Rho acts by regulating axonogenesis, neuronal migration, and synaptic plasticity, and mutations in Rho proteins contribute to several neurologic disorders. Increased level of Rho was found in postmortem brains of patients with Huntington’s disease and Alzheimer’s disease.19,20 Cdc42 G12V and Q61L mutations, found in the same positions as RAS hotspot mutations, result in constitutively active proteins that exhibit oncogenic activities.229 Cdc42 is especially important in migration of tumor cells. In thyroid cancer, overexpression of Cdc42 increases production of lactic acid and polarization of M2 macrophages, which function to inhibit T cells and allow cancer cells to proliferate uninhibited.230 Cdc42 is also thought to have an essential role in the regulation of epileptic seizures, as pretreatment with ML141, a Cdc42 inhibitor, was found to reduce seizure severity.231 Finally, in the skin, constitutively active Rac1 (RACV12) in mice resulted in the development of lesions similar to human psoriasis and inhibition prevented psoriasis hyperplasia in xenografts. Rac1 modulation was shown to affect epidermis-immune reactions in inflammatory pathways including STAT3, NFKB, and zinc finger protein 750,232 contributing to the pathophysiology of melanoma, squamous cell carcinoma, and neutrophil immunodeficiency. Additionally, Rac1 is commonly hyper-expressed in some types of cancer, including urothelial carcinoma. Inhibition of Rac1 has been shown to prevent metastasis of bladder cancer.233 In non-cancerous pathology, Rac1 contributes to pulmonary hypertension via its role in NO-mediated smooth muscle relaxation.234 Disruption of Rac1 macrophage regulation has been shown to increase disease stability in atherosclerosis as well,235 highlighting its role in development of the disease. RhoA and Rac1 have been shown to be crucial in the development of some rheumatologic diseases as well, including rheumatoid arthritis236 and systemic sclerosis.237
Rab
Dysregulation in the Rab family is implicated in human diseases related to disrupted intracellular membrane trafficking and vesicle movement [Fig. 4], (Table 1). Rab is predominantly expressed in the brain and thus has a large effect on the development of neurologic diseases, such as neurodegenerations including PD and AD.238 PD is a disease largely caused by death of neuronal cells due to accumulation of misfolded proteins in the substantia nigra. Rab29a and Rab39 have been shown to play important roles in the development of PD, as inactivating mutations in these proteins prevent proper expression of surface receptors, such as AMPA, required for proper secretory trafficking.30 In AD, aberrant expression of Rab5 results in abnormally large early endosomes due to dysfunctional membrane trafficking. In mice, Rab5 overexpression was found to induce AD-like neurons and pathology that was previously attributed to AB/Beta amyloid, signifying that Rab5 may be a worthwhile therapeutic target for AD.31 Aberrant expression of Rab25 and Rab31 have been implicated in development of multiple types of cancer.239,240 Rab35 mutations can activate PI3K and AKT in tumor progression.241 Dysfunction in the Rab pathway has also been known to contribute to hearing loss and cone-rod dystrophy.242 Gain of function mutations in Rab7 have been linked to the development of Charcot-Marie-Tooth Type 2B disease.243 Furthermore, Rab mutations have been implicated in other immune disorders,244 such as Carpenter syndrome (RAB23),245 Griscelli syndrome (RAB27A),246 and Hermansky-Pudlak syndrome (RAB38).247,248
Ran
Ran is overexpressed in breast and lung cancers and knockdown of Ran leads to a reduction of Met receptor expression that contributes drug resistance of trastuzumab and gefitinib249 [Fig. 4], (Table 1). Ran expression is upregulated in metastatic colorectal cancers250 and downregulation of Ran increases senescence in normal cells. The activation of Ran is critical for the activity of nuclear-cytoplasmic transport. Increased level of Ran and thus nuclear-cytoplasmic transport allow the cell to evade DNA damage-induced cell cycle arrest and senescence, which is vital for cancer cells.251 In neurologic conditions, reduced expression of Ran was observed in AD36 [Fig. 3]. It also plays a crucial role in frontotemporal lobar degeneration by regulating TDP-43 induced retinal degeneration.36
Arf
The Arf family plays key roles in tumor progression and invasion, in particular tumor angiogenesis252 [Fig. 4], (Table 1). During neurological development, Arf6 is highly expressed in the human brain and Arf6 dysfunction leads to defects in the cellular migration that contribute to neuronal disease.253 Defective Arf-related processes are involved in autosomal recessive periventricular heterotopia, retinitis pigmentosa, and amyotrophic lateral sclerosis as a consequence of defective neuron migrations. ARL3 is implicated in pathogenesis of gliomas. Low expression of ARL3 in gliomas predicted poor prognosis, likely due to its essential role in angiogenesis and immune cell infiltration in the microenvironment of the cancer.254 Arf6 is involved in the accumulation of cholesterol in podocytes in diabetic kidney disease.255 Legionella pneumophila and Rickettsia prowazekii, the bacteria responsible for Legionnaire’s disease and epidemic typhus respectively, utilize a Type IV secretion system to infect target cells. One of these effector molecules is RalF, which contains a domain homologous with Arf GEFs.256 The bacteria utilize this homology to recruit Arf to bacteria-containing vacuoles and allow the bacteria to proliferate.257 Furthermore, ARL13B mutations have been shown to contribute to the pathophysiology of inherited disorders such as Meckel-Gruber syndrome, Joubert syndrome, and nephronophthisis. ARL13B is found to contribute to the structure of the ciliary membrane and its defects prevent cellular transport and diffusion across the membrane from occurring properly.258 ARL6 mutants with disrupted nucleotide binding are reported to cause ciliary transport defects in Bardet-Biedl syndrome.259,260 Finally, SAR1B is a member of the Arf family which plays a key role in secretions of chylomicrons into the small intestine, and mutations in this protein result in Chylomicron retention disease.261
Targeting small GTPases in human diseases
Due to their significant prevalence in human diseases, there has been a long journey for developing strategies to target small GTPases. Among them, Ras family proteins such as KRAS have been established as a paradigm for targeting Ras proteins and other small GTPases. Since the first KRASG12C inhibitor (Sotorasib or AMG510) has been approved by the FDA in November 2021,262,263 many new targeting strategies have been discovered during the last decade through concerted efforts of chemical biology, structural biology, and fragment-based drug discovery (FBDD). In this section, we will review the progress made in the field of targeting Ras and other small GTPases with an emphasis on emerging strategies. We will divide the topic into two major categories: direct targeting and indirect interventions. Because of the similarity in the structure and basic biological function shared between different small GTPases families, we expect this summary will booster drug discovery progress for other small GTPases through dissemination of the methods developed for KRAS.
Direct targeting
Assuming that GTP and ATP share similar chemical structures and most of the kinase inhibitors are ATP analogs, the early attempts to inhibit Ras were focused on developing molecules to compete with GTP. However, this has been in vain because the picomolar affinity between GTP/GDP and Ras proteins is untamable. Moreover, the lack of effective pockets at the protein surface poses another tremendous challenge for applying the traditional drug development methods to target Ras proteins. As such, the progress for targeting Ras proteins has been stalled for decades and Ras has been notoriously described as “undruggable”. More recently, the field has regained new momentum as the pockets associated with the switch regions have been successfully exploited for small molecule binding as well as the surfaces at the effector lobe for macromolecular inhibitor binding. Most of the inhibitors are designated to interrupt the GEF-mediated Ras activation process (GDP-GTP exchange) or interfere with effector binding. Until very recently, mutation-specific covalent binding strategies (PROTACs, CLAMPs, etc.) have been developed for targeting KRAS, which serve as proof of concepts and technical repertoire for targeting other Ras proteins and small GTPases. Therefore, the dawn of the “druggable” is rising.
Exploiting structural plasticity of Switch regions
Direct targeting relies on exploring structural motifs at protein surfaces for accommodating ligand binding with structural specificity. In the last decade, many milestones have been reached in targeting KRAS by diversified efforts of structure-based drug discovery. Most of the ligand binding pockets are associated to switch regions of KRAS due to their significant structural plasticity. Their conformations and dynamic properties are tightly modulated by GDP/GTP binding or interactions with regulatory or effector proteins [Fig. 1f, g]. As a proof of the conformational selection theory, a recent NMR study reveals that many reported ligand binding pockets co-exist as minor conformations in dynamic assembles mainly comprised of switch regions in the KRAS unbound to ligand,49 indicating that the structural plasticity and dynamics are the key factors to guide structure-based drug screen and design for KRAS and other small GTPases. According to the location, there are two major cavities for ligand binding, Switch I/II pocket (a pocket between Switch I and Switch II) and Switch II pocket.
Switch I/II pocket (SI/II-P)
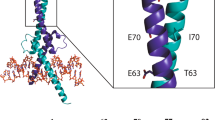
Switch I/II pocket is a shallow and small pocket located between Switch I and Switch II [Fig. 5b], previously tagged as “undruggable”. This stigma has been removed by advancements of FBDD efforts over last decade. A key characteristic of the FBDD approach is to pick up small molecules with low molecular weight (<300 kDa) and weak affinity but high potential for optimizations and expansion in chemical space. When applying FBDD to Ras proteins, two structure-based methods, nuclear magnetic resonance (NMR) and virtual screen, play important roles in finding fragment hits binding to SI/II-P. In 2012, DCAI compound (Kd: ~1.5 mM, Genentech) and compound 12 (Kd: 190 μM, Vanderbilt) were discovered by NMR-based FBDD. Both of these compounds bind to Switch I/II pocket and block Ras-SOS (RasGEF) interactions, which are required for Ras activation.264,265 A year later, Kobe2602, a compound derived from a virtual screening, was shown to bind to a similar pocket in HRAST35S mutant and reduce Ras-Raf binding and downstream signaling.266 Following this path, a fragment screening conducted on active KRAS (bound to GMPPCP) by NMR led to the identification of BI-2852, which binds to SI/II-P at nanomolar affinity (KD).267 BI-2852 binds to both inactive and active KRAS, diminishes protein–protein interactions (PPIs) between KRAS and GEF/GAP/effectors, and demonstrates unambiguous dose-dependent reduction in phosphorylation level of ERK and MEK kinase with antiproliferation effects in cell models.

Direct targeting of small GTPases by small molecules. a G12C-specific inhibitors bind to SII-P with the covalent warheads, other ligands specifically bind to SII-P of KRAS hotspot mutants (G12D/S/R). b Compounds bind to SI/II-P of WT KRAS and different mutants
NMR is evidently instrumental in FBDD efforts to target SI/II-P with respect to its advantages in screening, structure mapping, hit validation, and optimization. Two recent articles have reviewed the progress made on structural and dynamic characterizations and drug discovery for small GTPases by NMR.268,269 Other strategies have also been employed to develop SI/II-P binders. A comparative screening conducted between Ras complexed with an intracellular antibody fragment (i.e., anti-Ras VH chain) and Ras alone can elucidate the compound that binds to the VH epitope. The compound Abd-7 binds to SI/II-P, interrupts interactions of KRAS with its effector proteins, and reduces its downstream signaling.270 The Abd compounds were further optimized by fusing with the functional groups screened by the crystal soaking method and the fused compound, ch-3, exhibits enhanced potency.271 Besides this, a pan-Ras inhibitor (3144) from virtual screening has been found to interact with different sites located at SI/II-P and displays anti-tumor activity in a xenograft mouse model and the human breast cancer cell line (MDA-MB-231) that carry KRAS G13D mutation.272 Natural products constitute another important resource enriched with naturally occurring molecular architectures to be exploited for drug discovery. Tricyclic indolopyrrole alkaloid and indoloisoquinolinones (15R) were identified from a natural products screen and structurally verified to bind to SI/II-P of GMPPCP-bound KRASG12D.273
Switch II pocket (SII-P)
SII-P consists of Switch II, helix-3 (H3), and the end of β-sheet 1 (β1). It is a well-known pocket [Fig. 5a] for G12C inhibitors including Sotorasib. The earliest effort to screen ligand binding to this pocket by NMR was in 1997, which led to the identification of a sugar derivative ligand, SCH-54292.274,275 Even with limited affinity, the discovery of SCH-54292 provides an alternative possibility to target the allosteric site instead of directly outcompeting GDP or GTP. In 2013, a series of high-affinity SII-P ligands have been developed by taking advantage of endogenous reactivity from the cysteine sidechain of oncogenic KRASG12C mutant, an oncogenic mutation presenting in 13% NSCLC patients in the United States.276 A combinatory approach was applied by integrating two moieties containing a lead binding to SII-P and an electrophilic warhead covalently attaching to the thiol group of the cysteine.276 This serves as a milestone to target KRAS and opens a new avenue that drives the potent G12C inhibitors approved by FDA or under evaluations of clinical trials, such as Sotorasib from Amgen,262,263 Adagrasib (MRTX-849) from Mirati, and JNJ-74699157 (ARS-3248, an updated version of ARS 1620) from J&J.277,278,279,280,281 Medicinal chemistry plays a key role in evolving compounds with greater potency and efficacy by optimizing chemical spaces for both the SII-P binding moiety and warhead. It has been proven that binding of G12C SII-P covalent inhibitors reshapes conformation of switch regions to favor GDP-binding by abrogating the interactions between γ-phosphate and Switch II region.282 As such, the KRAS mutant is retained in the inactive “off” state and the KRASG12C-driven tumor growth is inhibited.
G12C only represents ~12% of KRAS mutations in human cancer according to the statistics from COSMIC database, but targeting other oncogenic KRAS mutations remains challenging. Inspired by the successes of the covalent G12C ligands, covalent ligands for G12S and G12R mutants have been recently developed. In the G12S ligand, an electrophilic group, β-lactone, specifically reacts with a serine residue and has been attached to the tetrahydropyridopyrimidine moiety of the G12C ligand Adagrasib.283 The G12S covalent ligands, G12Si-1/5 exhibit inhibitory effects to both KRAS GTP-loading and phosphorylation of downstream ERK in tumor cell lines containing KRAS G12S mutation. Similarly, a compound that contains an α, β-diketoamide warhead (compound-4) selectively binds to GDP-bound G12R mutant through covalent binding to the sidechain nitrogen of arginine 12. In contrary to the G12C mutant, the G12R mutation leads to severe loss of GTP hydrolysis activity and dominantly exists in the GTP-bound state, thus the current version of the G12R covalent ligand shows limited activity in cells and further optimization is needed.284
G12D is the most prevalent KRAS mutation in pancreatic cancer and colorectal cancer, however, the aspartic acid mutation does not render the same chemical reactivity as the cysteine mutation. Through chemical space optimization based on the G12C inhibitors, a noncovalent KRASG12D inhibitor, MRTX1133, has been recently reported with good selectivity and potency.39,285 Starting from the established SAR of the G12C inhibitor Adagrasib, a pyrido[4,3-d] pyrimidine scaffold was chosen and a substitution of the reactive warhead with [3.2.1] bicyclic diamino substituent at the C4 position was made for selectively interacting with the negatively charged sidechain and the neighboring residues in the P-loop. Then, further optimizations were made with a substituent 2-fluoro-pyrrolizidine at the C2 position and with a substituent 7-fluoro-8-ethynylnaphthyl at the C7 position in order to sterically fit better into SII-P of G12D mutant. The final product termed MRTX1133 displays a sub-picomolar binding affinity in a non-covalent manner. Furthermore, MRTX1133 induces tumor regression to most of the PDAC tumor models through effective inhibition of G12D signaling.38,286
Presently, there are inhibitors targeting different alleles of KRAS including G12C, G12D, G12S, and G12R under development and KRAS is evidently becoming druggable. One key question is whether SII-P is the only accessible pocket to the covalent ligands and the GDP-loaded protein. In a new study, the G12C covalent ligands Adagrasib, MRTX1257, and G12D ligand MRTX-EX185 have been found to reversibly interact with SII-P of GDP-bound WT KRAS, KRASG12D, and WT HRAS. MRTX-EX185 also binds to GTP-bound form of all three.287,288 More interestingly, all three MRTX compounds exhibit in-cell target engagement across WT KRAS and the hotspot mutants including G12C, G12D, G13D, Q61H, and Q61L. Furthermore, MRTX-EX185 engages G12V and Q61R mutants in cells. Unlike MRTX compounds, Sotorasib exclusively interacts with the G12C mutant. Both MRTXs and Sotorasib bind to SII-P but the residues involved in the interactions are partially different, which led to the emergence of different secondary KRAS mutations in relapsed tumors following treatment with different drugs.289 Taken together, SII-P widely exists across Ras isoforms and KRAS hotspot mutants as well as both GDP- and GTP-bound forms, confirming that the structure of SII-P and dynamic modulations of switch region conformations in different hotspot mutants is important for discovery of novel mutant-specific ligands to address the stringent needs of precision medicine. Furthermore, the strategies and experimental methods evolved during the last decade for targeting SII-P as well as SI/II-P of KRAS shed light on drug discovery for other Ras isoforms and small GTPases.
In addition to target the SII-P, the progress achieved with G12C covalent inhibitors also revived the concept of competing with GDP/GTP in binding to the guanine nucleotide-binding pocket. Compound SML-8-73-1 represents another type of G12C covalent inhibitor by binding to the nucleotide pocket and rendering KRAS locked in an inactive conformation with reduced cellular activity.290,291 Ras-like proteins (RALA and RALB) are located at Ras branch in the phylogenetic tree of small GTPases and share high structural similarity with Ras proteins. Interestingly, Rals are also located at a downstream signaling node of the Ras pathway (Ras-RalGDS-Ral). Activation and upregulation of Rals are observed in many Ras-driven tumors, indicating the potential of Rals as cooperative targets with Ras for cancer treatments.221 In a parallel study for Ral inhibitors, two compounds, RBC8 and BQU57 from virtual screen, were found to bind to a pocket mainly composed by residues in Switch II with ~100 μM affinity. They inhibit tumor growth in lung cancer xenograft models.292
Macromolecular inhibitors
Therapeutic macromolecules such as peptide ligands, proteins, and antibodies represent an alternative Ras targeting approach. Compared with small molecule ligands, macromolecular ligands exhibit improved selectivity and binding capacity to compete with endogenous PPI partners such as GEFs and effectors.293
Cyclic peptide ligands
A cyclic peptide screened from bacteriophage display binds to part of SII-P of KRASG12D mutant with 10 nm affinity (KD) and six-fold selectivity over WT KRAS [Fig. 6a]. It disrupts Ras activation and effector binding to SII-P in vitro and inhibits proliferation in G12D mutant cancer cell models and KRASG12D-driven tumor growth in animal models.294,295,296 In another study, a bicyclic peptidyl pan-Ras inhibitor, cyclorasin B4-27, binds to GTP-bound form of WT KRAS and G12V mutant with ~20 nm affinity (KD). NMR characterizations indicate the binding site of B4-27 is mainly overlapped with SI/II-P.297,298 With continuous improvement in chemical compositions, cellular permeability, and metabolic stability, B4-27 displays good potency in blocking Ras-effector interactions and induces apoptosis of multiple cancer cell lines containing KRAS or HRAS mutants. A 16-mer cyclic peptide, screened from a naïve library, binds to non-switch-region of Cdc42 at nanomolar affinity and inhibits RAS-driven tumor cell proliferation and invasion in a Cdc42-dependent manner.299

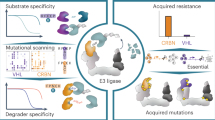
Direct targeting of small GTPases by macromolecules and emerging paradigm. a Cyclic peptides and staple peptides bind to KRAS and RALB. b Protein ligands interact with switch regions or allosteric lobe of KRAS. c Newly-developed targeting strategies and drug screening methods
Through an integrative approach of mRNA display, in vitro translation, and unnatural amino acid substitution, a cyclic peptide, KD2, was identified to preferably bind to GTP-bound KRASG12D mutant at a groove mainly involving SII-P region and block the interactions of KRAS with CRAF RBD.300 Its selectivity to the mutation allele and nucleotide-bound form presents a promising approach to target the challenging oncogenic mutant.
Staple peptides
Staple peptides [Fig. 6a] are capturing increased interests in developing peptide inhibitors for KRAS and other small GTPases. The design of staple peptides harnesses the potentials of binding affinity and selectivity inherited from the peptide motifs located at the PPI interface. In the early attempt, a helical peptide derived from a crucial helical Ras-binding motif (αH: F929-N944) at SOS surface was designed with hydrogen bond surrogate (HBS) strategy to stabilize the helical conformation. The HBS helical peptide binds to a surface spanning both Switch I and II at Ras with an affinity (KD) ~ 100 μM.301 By engineering the helical motif with hydrocarbon insertions, the helical peptide ligand binds to Ras with nanomolar affinity by fitting into SI/II-P and blocks guanine nucleotide binding.302 The hydrocarbon helical peptide demonstrates inhibitory effects in cancer cell models containing KRAS G12 or G13 mutations and Drosophila melanogaster model corresponding to G12V mutation. In a recent study, αH and another helical motif (αH: 960–975) from Ras-interaction interface of SOS are engineered together via a synthetic linker, termed as CHDSos-5.303 As an extended staple peptide or proteomimic, biophysical characterizations reveal CHDSos-5 can bind to both switch regions in nucleotide bound form in contrast to preferable binding of HBS to the nucleotide-free Ras. CHDSos-5 resists proteolysis, enters cells through micropinocytosis, and exerts inhibitory effects by suppressing downstream signaling in cancer cells with high levels of micropinocytosis, such as T24 (HRASG12V) bladder and H358 (KRASG12C) lung cancer cells. CHDSos-5 binds WT HRAS with six to eight folds higher affinity to G12V/D/C mutant. Moreover, it interacts with KRAS, RAN, and multiple Rabs according to chemoproteomic analysis, suggesting that potential off-target effects be taken into account. Screened from a naïve library, a “mini-protein” containing two helical components and a loop linker shows picomolar binding affinity to the switch regions of KRAS with an interesting observation that the interactions induce formation of a groove connecting SI/II-P and SII-P.304 Similar to Ras, staple peptides derived from RLIP76 RBD, a Ral effector, selectively binds to Rals with affinity at single digit micromolar (KD) and interrupts the PPIs.305
Peptide drugs including cycling peptides and staple peptides demonstrate many advantages like high affinity to compete with the endogenous PPIs of small GTPases. As such, the effective inhibition is achieved through interrupting interactions with GEFs or effectors, which are difficult for small molecules. A major limitation for therapeutic peptides is their short plasma circulation time because the natural peptide bonds are prone to enzymatic cleavages. Other limitations include low oral bioavailability and cell penetration. To address these issues, strategies such as chemical modifications, incorporation of unnatural amino acids, adjusting molecular size, conjugations to other functional molecules or peptides have been employed.306 For peptide ligands targeting small GTPases, further developments to improve selectivity towards different subfamily isoforms or disease mutants are also needed.
Protein ligands
Protein ligands [Fig. 6b] developed for targeting Ras proteins within the last decade mainly include antibody mimetics/fragments, monobody, and other interacting proteins. The prototypical immunoglobulin antibody contains multiple disulfide bonds that are volatile in the reducing environment of cytosol. A cysteine-free antibody mimetic with low molecular weight (<20 KDa), DARPin K27, was obtained from phage display, binds to a surface spanning both Switch regions of KRAS with 4 nm affinity (KD) and exhibits potent intracellular inhibitions to Ras downstream signaling.307 Other DARPins, K13 and K19, bind to helix α3-loop 7-α4 in the allosteric lobe of KRAS and inhibit downstream signaling through interruption of GEF-mediated activation, dimerization, and possible allosteric perturbations on conformation of switch regions.308 NS1 monobody, a synthetic binding protein, binds to α4-β6-α5 region in the allosteric lobe of KRAS and HRAS, but not NRAS. It inhibits RAS interaction with RAF kinase by disrupting Ras dimerization at the membrane surface.309 In another study, a monobody, 12VC1, exhibits up to 400-fold selectivity for binding to GTP-bound G12V and G12C mutants compared to WT KRAS. It suppresses ERK phosphorylation and KRAS-driven cancer cell proliferation in vitro and tumor growth in vivo.310 In contrary to NS1 monobody, 12VC1 binds to an interface composing of switch I and switch II. Besides this, intrabody, the cysteine-less antibody fragments in low molecular weight, have been developed to target Ras proteins.311,312,313,314 Switch pockets of KRAS are also major targets for protein-ligand binding: Affimer K3 binds to SII-P, Affimer K6 binds to SI/II-P,315 miniprotein 225–11 binds to SI/II-P with midpicomolar affinity304 and R11.1.6 binds to SI/II-P with nanomolar affinity.316 JAM20, another monobody, binds to SI/II-P of all Ras isoforms with nanomolar affinity at both GDP- and GTP-bound states.317 It competes with RAF-RBD, thereby effectively inhibiting the Ras downstream signaling and Ras-driven transformation in vitro and in vivo.
Applications of macromolecules to target intracellular proteins remain clinically limited due to the practical difficulty in entering cells. Nevertheless, the cases of DARPins, NS1, and 12VC1 point out that interactions occurring at regions in the allosteric lobe and switch I/II modulate Ras functions through distinct mechanisms. These binding proteins can also be used as probes to find small molecules that selectively bind to the allosteric sites. Furthermore, the effector lobe (a.a. 1−86) in Ras is strictly conserved, while the allosteric lobe (a.a. 87–166) determines the differences among different Ras isoforms. As such, targeting the allosteric lobe can be an alternative strategy to develop isoform-specific Ras inhibitors.
Emerging paradigms
Many cutting-edge targeting approaches [Fig. 6c] are evolving on the basis of chemical biology, medicinal chemistry, and structural biology, such as targeted degradations by PROTACs. Other attractive platforms have also been developed for targeting Ras, including molecular glues and CLAMPs. Regulation mechanisms such as membrane associations and genetic modulations, as well as targeting the allosteric lobe, offer potential new angles for RAS inhibition.
Targeted degradations
It is not surprising that KRAS is an attractive battlefield for method developments and applications of PROTACs. ARS-1620 and Adagrasib, established as G12C covalent inhibitors, were integrated with E3-ligase binders to develop PROTAC degraders for targeting KRASG12C mutant. The KRASG12C PROTAC degraders demonstrate potent inhibition of KRAS downstream signaling and antiproliferation effects.318,319 Following the path of protein ligands, DARPins, NS1, 12VC1, intrabodies and Raf-RBD were fused to functional domain of E3 ligase respectively for the targeted degradation of RAS.310,320,321 These engineered protein degraders not only exhibit specificity on target degradation and inhibition, also demonstrate their merit to investigate the degradation efficiency dependencies on nucleotide-bound forms and mutation types.
The targeted degradations pave a new avenue to eliminate the oncogenic mutations instead of direct inhibition. As a prerequisite for targeting, the degrader relies on high-affinity ligands that have been developed for binding KRAS. Besides, when applying PROTAC degraders containing a protein ligand, effective delivery into targeted tumor cells will be a rate-limiting step for their future applications in clinics.
New directions
Membrane association is important for activation of Ras proteins and most small GTPases. In the case of Ras proteins, the dimerization of Ras at the membrane surface is recognized as a regulating event to induce dimerization of Raf kinases for their activation.322,323 Therefore, targeting membrane-association surface of Ras might provide another opportunity to inhibit downstream signaling. Protein ligands, DARPin-K13/K19 and NS1 monobody, bind to the allosteric lobe and demonstrate the inhibitory effects on downstream signaling. NMR characterization of a chemical compound (cmpd2) reveals that the ligand binds to SI/II-P adjacent to Ras-membrane interface and shifts RAS spatial orientation equilibrium, presenting a unique manner to inhibit Ras-Raf signaling.324 In another study, compounds generated by the repurposing of fendiline block proliferation of KRAS-driven tumor cells through perturbing membrane localization of KRAS, yet the binding sites of these compounds were not able to be characterized.325
Exploring the dynamic properties of switch regions is a promising direction to be pursued. An ensemble-based virtual screen has been developed by using conformational ensembles of switch regions and conserved G-boxes derived from experimental restrains of NMR, which is helpful to find the lowly populated pockets suitable for ligand binding.49,326 In a recent work, a strategy using conformation-locking antibodies for molecular probes (CLAMPs) has been developed to stabilize rare conformation by antibodies, which provides an experimental approach for ligand screen and interaction studies based on these rare conformations.327
Similar to PROTAC, molecular glues represent a concept able to induce neo-protein-protein associations by small molecule ligands.328 Bifunctional ligands were designed by chemically linking two ligands together: one ligand contains a KRAS binding module while the second contains a cyclophilin A/ FKBP12 binding module.329 The bifunctional ligand successfully induced the non-native associations of KRAS to cyclophilin A/FKBP12 in cells and blocked KRAS-BRAF interactions in vitro.
At the genetic level, G-quadruplex (G4) sequences in the promoter region of KRAS have been shown to regulate transcription of KRAS. G4 structures are sensitive to changes of microenvironments in the nucleus of tumor cells. The unique structural configurations of G4 likely present a different opportunity to target KRAS.330,331,332 In a recent study, the structure of KRAS G4 in complex with the natural products berberine and coptisine has been determined by NMR, which delineate the structural basis for ligand binding specificity and downregulation of KRAS transcription caused by the ligands.333 The complex structures further prove the potential of G4 as a therapeutic target. However, targeting G4 does not render selectivity toward different KRAS oncogenic mutations. Further pharmacological evaluations are required, with G4 possibly acting as a cooperative target for lowering transcription of the oncogenic proteins.
Progress of targeting other major small GTPases: Rho, Arf, and Rab
Drug discovery efforts targeting other families of small GTPases are not as advanced as those targeting the Ras family and nearly none have advanced to clinical trials.
Rho proteins, Rac proteins, and CDC42 hold immense interest for developing molecular modulators (activators/inhibitors).334,335,336 Altered expression, rather than aberrant mutations, of Rho family proteins is a major driver of diseases. As such, Rho/GEF interactions are the primary mechanism being targeted. NSC23766 is an early example discovered from virtual screening; it inhibits Rac-GEF interactions through binding to a groove located in Rac/GEF interface and does not interfere with interactions of Rho and CDC42 with their specific GEFs.337 NSC23766 inhibits proliferation, anchorage-independent growth, and invasion of the prostate cancer PC-3 cells. EHT 1864, another Rac1 inhibitor, was reported to selectively bind to Rac1 and interfere with guanine nucleotide association. EHT 1864 potently inhibits Rac1 downstream signaling and blocks Rac1-driven transformation of NIH 3T3 cells.338,339 ZCL278 is another ligand obtained from virtual screening and specifically binds to CDC42/intersectin (CDC42 GEF) interface. It blocks microspike formation in 3T3 fibroblast cultures, neuronal branching, and actin-based motility and migration in PC-3 cells.340 Another ZCL compound, ZCL367, binds to CDC42 with improved selectivity and inhibits proliferation and migration of multiple tumor cell lines, including the lung cancer cells containing EGFR or KRAS G12S mutations.341 CASIN (CDC42 activity-specific inhibitor) is another ligand that interferes with GEF-mediated activation. It interrupts CDC42-mediated actin dynamics and induces mobilization of hematopoietic stem cells from bone marrow to peripheral blood in mouse models.342 ARN22089 compound is obtained through in-silico screening based on the CDC42/PAK6 complex structure and it recognizes the GTP-bound “on” state of CDC42, blocking the interaction between CDC42 and its downstream effectors.343 ARN22089 displays various inhibitory effects on tumor cell growth and angiogenesis in vitro and triggers pro-inflammatory and apoptotic signaling as well as growth of BRAF mutant mouse melanomas through inhibition of MAPK pathway. A series of compounds screened by time-resolved fluorescence methods can block proliferation and migration of breast carcinoma cells through interrupting interactions between CDC42 and the scaffold protein IQGAP1.344 In a recent study, a covalent ligand, DC-Rhoin04, has been discovered to bind to an allosteric pocket mainly composed by switch II next to residue C107 in RHOA and inhibits migration and invasion of tumor cells. This binding has been verified and characterized by crystallization.345 ML141 binds to CDC42 in a reversible non-competitive manner and inhibits CDC42 selectively. It is reported to suppress division and induce death of basal-like breast cancer cells and enhance their sensitivity to tamoxifen treatment in vivo.346
Arf family and their regulatory proteins are important therapeutic targets in cancer treatment.252 There are several natural products that have been discovered that bind to and inhibit Arf proteins. Brefeldin A (BFA) is a fungi metabolite identified to significantly alter protein transportations between Golgi and the ER.347,348 The structure of ternary complex formed by ARF1, Sec7 catalytic domain of GEF, and BFA corroborates that BFA binds to a hydrophobic pocket at ARF1/Sec7 interface and blocks GTP exchange for activation.349 BFA has been previously used as a molecular probe to interrogate functionality of ARF1, but a BFA derivative was recently reported to putatively bind to the ARF1/SEC interface and show potential anti-leukemia activity.350 LM11 is an inhibitor that is screened based on the complex structure of ARF1 with Sec7 domain of ARNO (Arf GEF). It binds to the ARF1/Sec7 interface similarly to BFA, inhibits activation of ARF1 rather than ARF6, and blocks both ARF1-mediated trafficking at Golgi and ARNO-dependent migration of Madin-Darby canine kidney cells.351 Chlortetracycline (CTC) is a tetracycline-class antibiotic obtained from a high throughput assay using active ARF6 attached on the membrane surface. It is a non-nucleotide-competitive ligand that likely inhibits GEF-mediated activation by binding to an allosteric pocket and blocks ARF6-mediated migration and invasion of breast tumor cell lines.352 Of note, membrane association is critical for Arf activation and thus important in evaluating drug actions.353
Dysregulations of Rab proteins are involved in many human pathologies, including neurodegenerative disease and bacterial infections. Although Rabs are established as an important category of therapeutic targets, their drug development progress is lagging compared to the Ras and Rho families. More complex regulatory mechanisms, structural and sequence similarities shared between Rab members and other small GTPases families, and the requirements for passing through the blood-brain barrier pose challenges to Rab inhibitor drug discovery.354 CID1067700 is a compound obtained from a high throughput screen and acts as a GTP-competitive inhibitor with selectivity towards RAB7 in biochemical assays;355 it displays inhibitory effects in disease models of ischemic stroke356 and lupus.356 NAB2 (N-arylbenzimidazole) was identified to alleviate α-Synuclein induced toxicity in PD cell models, and a following chemoproteomic study reveals that RAB1 is a target of NAB2 for its rescuing effects.357 Exploring the allosteric pockets on the Rab protein surfaces has also been pursued based on structure-based computational analysis.358,359 Relative to other families, drug discovery efforts toward Ran are limited, though there have been some early attempts to interfere with RAN-GEF interactions.360,361 Nevertheless, the altered expression of Ran is closely related to tumorigenesis in different cancer types, indicating the potential of Ran to be the therapeutic target for these tumors.362
Taken together, there are many compounds identified as inhibitors or modulators to Rho, Arf, and Rab. Although some of these compounds have been well characterized, most of the molecules were identified through virtual screening or in vitro high throughput assays before being tested with functional assays. Structural characterizations, such as protein-ligand complex structures determined by crystallization or NMR, have been recognized as an indispensable criterion for evaluating a new drug candidate,363 but the complex structures for inhibitors binding to Rho, Arf, or Rab are still missing. Considering the experience of targeting KRAS, these molecules can serve as the starting point for further in-depth characterizations and optimizations of small GTPases inhibitors. Future drug development would be accelerated by adopting the versatile strategies developed for targeting KRAS and characterizing the drug actions.
Indirect targeting
The early efforts to inhibit Ras proteins mainly involve targeting the regulatory mechanisms rather than direct targeting. Presently, indirect targeting represents another important branch to block the abnormal cell proliferation driven by the non-targetable hotspot mutations or serves as a combinatory approach together with direct targeting to enhance the inhibition of Ras downstream pathways and overcome the drug resistance of current KRAS inhibitors. Indirect targeting includes targeting post-translation modifications, membrane association, upstream regulators, downstream effectors, synthetic lethal partners, and metabolic rewiring [Fig. 7a].

Strategies for indirectly targeting RAS and emerging approaches for targeting oncogenic RAS with immunotherapy. a Targeting key post-transcriptional modifications of RAS protein as well as upstream and downstream factors of RAS signaling have been proved as promising strategies for interfering with RAS activity and exhibiting anti-tumor effects in RAS mutant-driven tumors. Inhibitors targeting the key factors controlling RAS post-translational modifications and membrane trafficking (FTase, PDE6δ), upstream mediators (RTK receptors, SHP2, SOS1/2), and downstream effectors (RAF, MEK, ERK, CDK4/6, PI3K, AKT, mTOR1, AURKA, PLK) are listed. FDA-approved drugs are highlighted in blue, others are still under preclinical or clinical study. b Combination of KRASG12C inhibitors with immune checkpoint blockade and development of bispecific antibodies with T cell engager targeting neoepitopes derived from KRASG12C inhibitor-mediated covalent modification of KRASG12C. This figure was created with BioRender.com
Targeting the post-translational modifications and membrane trafficking of small GTPases: FTase, PDEδ
Membrane targeting is important for the activation and function of most small GTPases. RAS undergoes a series of post-translational modifications (PTMs) to attach on cell membranes and to subsequently interact with downstream effectors and transduce signals. Therefore, interfering with key PTMs of RAS has been identified as one of the weaknesses of RAS, widening the chemical space in the discovery of RAS inhibitors. Since FTase is the first rate-limiting enzyme of the CAAX processing, it was hypothesized that inhibiting FTase would disrupt RAS cellular location and dampen downstream signaling. Thus, many efforts were made to block the activity of FTase. Unfortunately, although several FTase inhibitors (FTIs) showed great anti-tumor potency in preclinical studies, phase II and phase III clinical trials on solid tumors showed resistance to FTIs.364,365,366 One explanation provided for this disappointing failure is that KRAS and NRAS mutations are susceptible to alternative prenylation mediated by geranylgeranyltransferase-I (GGTase-I) which promotes RAS membrane localization and oncogenic activation when FTase is blocked.365,367,368
Recently, there has been a renewed hope in the use of FTIs in HRAS-mutant cancers. HRAS is prenylated exclusively by FTase and it is theoretically much more sensitive to FTIs.367 Indeed, a phase II clinical trial (NCT02383927) of FTI tipifarnib showed inspiring anti-tumor activity in patients bearing recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC) with HRAS mutations.369 This study also found that patients with high mutant HRAS variant allele frequency (VAF) have more clinical benefit after tipifarnib administration, which not only supports the breakthrough therapy designation approval of tipifarnib in HNSCC patients with VAF ≥ 20%, but also accelerates the repurposing of tipifarnib in tumor types harboring HRAS mutations and demonstrates that VAF of mutant HRAS might be a promising biomarker for tipifarnib efficacy.
After post-translational modifications, phosphodiesterase6δ (PDE6δ) binds and escorts farnesylated Ras to the plasma membrane and thereby enhances RAS signaling activity.370 This finding calls for the development of inhibitors disrupting the interaction between RAS and the farnesyl-binding pocket of PDE6δ. Deltarasin is the first proven benzimidazole inhibitor targeting the farnesyl-binding site of PDE6δ with KD = 7.6 ± 1.3 nM.371 In KRAS-driven human pancreatic ductal adenocarcinoma cells (hPDACs), Deltarasin induces decreased membrane localization of KRAS, inhibits downstream MAPK signaling transduction, and significantly inhibits tumor cell growth both in vitro and in vivo. However, Deltarasin was found to show a “switch-like” inhibition of proliferation, which could be explained by its general cytotoxicity or its off-target effect of binding different membrane proteins, such as GPCRs.372,373 Deltazinone 1 is another pyrazolopyridazinone inhibitor that has a similar binding method to PDE6δ as Deltarasin.372 Deltazinone 1 (KD = 8 ± 2 nM) exhibits higher specificity to PDE6δ and less general cytotoxicity than Deltarasin, however, it requires >20 μM to show inhibitory effects on RAS signaling and cell proliferation in cell-based assays. This limitation might be due to the fast release of inhibitors from PDE6δ by the allosteric engagement of release factor Arl2, a small GTPase belonging to ARF family. This suggests that inhibitors with slow off-rates or covalent binding modes could overcome this limitation.374 Deltasonamides were later developed as more potent PDE6δ inhibitors with much stronger non-covalent interactions, dramatically reducing release caused by Arl2.374 (E)-N′-((3-(tert-butyl)-2-hydroxy-6,7,8,9-tetrahydrodibenzo [b, dfuran-1-yl] methylene)-2,4-dihydroxybenzohydrazide (NHTD) was recently discovered as a new PDE6δ inhibitor via high throughput molecular docking-based screening. NHTD treatment causes KRAS delocalization and showed significant anti-tumor activity in KRAS-driven non-small cell lung cancer (NSCLC) xenografts with good tolerance.375
In vitro study identified benzyl indazole compound CHS-111 as a RHOA inhibitor which acts through reducing RHOA membrane association and inhibiting its interaction with GEF Vav.376 In addition, through phenotypic screening, Secramine A was reported to repress CDC42 activation and downstream signaling pathway via stabilizing its interaction with RhoGDI1, which prevents CDC42 membrane attachment and GDP-GTP exchange.377 Ras proteins are prenylated by FTase or GGTase-I as indicated previously. Rho family and Rab family are generally geranylgeranylated by GGTase-I and II respectively, which facilitates their membrane association and activation. Nitrogen-containing BPs (N-BPs), used to treat skeletal diseases, were found to have inhibitory effect on GGTase-I.378,379 Later, N-BP derivatives such as risedronate or zoledronic acid, were shown to induce apoptosis of myeloma or mesothelioma tumor cells in vitro through inhibiting prenylation of Rab proteins.380,381
Targeting RAS activation by upstream regulators: GEFs, SHP2, STK19
Since mutationally activated RAS was first discovered in human cancer in 1982,382 mutations of RAS have been well established as the drivers of diverse types of lethal cancers, such as colorectal cancer, pancreatic cancer, and lung cancer.383,384,385,386,387,388 Oncogenic mutations of RAS usually disrupt the GAP-mediated GTP hydrolysis and lead to constitutive activation. Thus, developing medications to interfere with the upstream events that are responsible to regulate GTPase activities of RAS constitutes an alternative approach to dampen RAS-driven tumor growth.
GEFs
Targeting GEFs or other related regulatory protein kinases that regulate RAS activity is an alternative anti-RAS strategy. Particularly, son of sevenless 1 (SOS1) has become a target that gained focus in RAS-driven tumor therapy. SOS1 is a major GEF for RAS389 and essential for RAS activation by facilitating GDP-GTP exchange. Thus, interfering with SOS1-mediated RAS activation is a strategy to inhibit RAS. The first SOS1 modulator was identified by serendipity and subsequent cocrystal analysis. It was shown to bind to a hydrophobic pocket in the CDC25 domain adjacent to the catalytic site of SOS1, which interacts with Switch II region of RAS.390 The early generation SOS1 modulators are interestingly activating RAS-mediated MAPK pathways instead of inhibiting it in cells,390,391,392,393,394,395 thus the development of RAS-inhibitors by targeting SOS1 has been rudimentary. Until recently, the discovery of quinazoline molecules, including BAY-293 from Bayer396 and BI-3406 from Boehringer Ingelheim397 shed light on the development of SOS1 inhibitors. Intriguingly, these two inhibitors both targeted the same hydrophobic pocket that previously reported to cause SOS1 mediated RAS activation. The mechanism of action (MOA) for their inhibition of SOS1 are identical, which could be explained by disrupting the RAS binding epitope on SOS1 interaction sterically.396,398 BAY-293 was reported to show anti-proliferative activity in both KRAS mutant and wild-type human cancer cell lines, and the efficacy of this could be enhanced by the combination of ARS-853, a covalent inhibitor of KRASG12C 396. BI-3406 dampens the key interaction between Tyr884SOS1-Arg73RAS for RAS activation, exhibits improved potency in KRAS-driven tumors both in vitro and in vivo, and synergizes with MEK inhibitor trametinib, resulting in remarkable tumor regressions with adequate tolerance.397,398 BI 1701963, the analog of BI-3406, is the only SOS1 inhibitor that has moved forward into clinical study. BI 1701963 alone or in combination with trametinib (NCT04111458), KRASG12C inhibitor Adagrasib (NCT04975256), and with KRASG12C inhibitor BI 1823911 as well as midazolam (NCT04973163) are now in phase I clinical studies. However, BI 1701963 alone or in combination with irinotecan in patients with advanced bowel cancer with KRAS mutations (NCT04627142) and BI 1701963 alone or in combination with MEK inhibitor BI 3011441 in patients with KRAS mutant non-small cell lung cancer and colorectal cancer (NCT04835714) were terminated recently due to the unexpected toxicity. Combinatorial treatment strategy is well characterized to prevent or delay resistance and improve antitumor efficacy. However, the termination of the indicated clinical trials warns us that recognizing and minimizing combination-induced toxicity is one of the main challenges for developing combinatory therapy in clinic.399,400 Designing trials that reflect toxicity and intermittent dosing in early clinical trials400 may provide great insight to strike a balance between efficacy and toxicity.
Considering advancements made on targeting Ras proteins, other small GTPases have attracted increased attention since new approaches to target small GTPases have been developed. Some of these methods include disrupting the interaction between small GTPases and GEFs, inhibiting nucleotide binding, impairing GEF-regulated guanine nucleotide exchange, and interfering with membrane binding. Most small molecule inhibitors in this field are based on the strategy that inhibits the binding of small GTPases to their GEFs. For example, Y16 was identified to specifically inhibit RHOA activation through disrupting Rho GEF LARG binding to RhoA.401 LM11, a noncompetitive ARF1 inhibitor, targets the binding of ARF1 to GEF ARNO at the Golgi and significantly decreased metastatic capability of breast cancer cells in a zebrafish model.351,402 Similar to quinazoline inhibitors binding to SOS1, Sec7 inhibitor H3 (SecinH3) can also disrupt the interaction of ARF1 and ARF6 with ARNO by inhibiting the Sec7 catalytic domain in ARNO. This was later found to show benefits in treating breast tumors,403 colorectal tumors404 and non-small-cell lung tumors405 both in vitro and in vivo. NSC23766 was characterized as a small molecule that binds to a putative binding pocket in Rac1 that interacts with Rac-GEFs Trio and Tiam-1 rather than Vav,337 and it can significantly impede BCR-ABL-induced myeloproliferative disease in xenograft model in vivo.406 Further studies showed that NSC23766 derivative EHop-016 potently inhibits Rac at low micromolar levels (IC50 = 1.1 μM) via binding to a deeper pocket in Rac and disrupting the key interactions between Rac1 and Rac-GEF Vav1.407 This inhibitor holds promise for treating metastatic cancers evidenced by its effective anti-tumor activity in multiple in vitro and in vivo studies.407,408,409,410 AZA1, which is also a derivative of NSC23766, inhibits the binding of Cdc42 and Rac1 with their GEFs.411 In a preclinical advanced colon cancer mouse model, AZA197, an AZA1 analog with improved selectivity on Cdc42, delayed tumor growth and significantly prolonged mouse survival via inhibiting Cdc42 and its downstream PAK1 signaling pathway.412
GAPs
Stimulating RAS hydrolysis through activating RAS-specific GAPs, such as p120GAP, neurofibromin (NF1), and RGS3, is an essential regulation to turn off the activated RAS.51,413,414,415 Most RAS mutant alleles show reduced intrinsic GTPase activity in comparison with WT RAS, except for KRASG12C allele.416 Moreover, RAS mutation impairs GAP-mediated GTP hydrolysis, leading to constitutive RAS activation. RGS3, a conventional GAP for heterotrimeric G protein α subunit (Gα), has been recently discovered with capacity in catalyzing GTP-hydrolysis for WT and mutated KRAS, including G12C, G12V, G12D, G13D, and G13V.51,417 Compared with canonical Ras-GAP, RGS3 catalyzes GTP hydrolysis of KRAS through a conserved asparagine residue instead of the arginine finger. Current G12C inhibitors recognize and interact exclusively with the GDP-bound protein, but the majority of the G12C mutant remains binding to GTP in tumor cells due to defective GAP mediated hydrolysis. Thus, RGS3 has been established as an alternative GAP to return the G12C mutant back to the GDP-bound “OFF” state for inhibitor actions. Despite a slower hydrolysis rate mediated by RGS3 compared to RAS-GAP, discovery of a regulatory role of a large G-protein GAP in GTP hydrolysis of KRAS presents a new angle to understand biochemical properties of the oncogenic mutants and might create a new opportunity for alleviating KRAS-driven tumorigenesis.
SHP2
Src homology-2-containing protein tyrosine phosphatase 2 (SHP2) serves as a vital regulatory node that activates RAS/MEK/ERK signaling and promotes proliferation of tumor cells by either recruiting GRB2-SOS1 complex to plasma membrane or directly dephosphorylating RAS at Y32.418 Of note, recent studies demonstrated the essential role of SHP2 in KRAS-driven tumor progression, showing that genetic ablation or pharmacological inhibition of SHP2 did dramatically delay tumor growth in KRAS amplified gastric carcinoma, KrasG12D driven pancreatic tumor, and NSCLC tumor models with KrasG12D or KrasG12V mutation, This demonstrates that targeting upstream SHP2 is a potential approach to inhibit RAS activation, including wild-type RAS amplification and RAS mutants.419,420,421,422 Furthermore, these three studies also noted that SHP2 is tightly associated with the resistance caused by MEK inhibitors and its inhibition in conjunction with MEK inhibitor treatment blocks the rebound of MAPK signaling and thus results in a more potent anti-tumor effect. Presently, there are fifteen SHP2 inhibitors that have moved into clinical trials and most of them are still in the early stages. According to the binding mode, SHP2 inhibitors can be classified into two categories: orthosteric inhibitors and allosteric inhibitors. Sodium stibogluconate (SSG) is the first orthosteric SHP2 inhibitor. Given that SSG can synergize with interferon-α2b (IFN-α2b), phase I clinical trials studying the combination of SSG and IFN-α2b in the treatment of advanced tumors (NCT00629200) and stage IV melanoma (NCT00498979) were launched and completed in 2018 and 2020, respectively. However, no objective regression of disease was observed in the trials423 and no follow-up research has been reported since then. TNO-155, a derivative of SHP099, is the first allosteric SHP2 inhibitor that entered the clinical trial stage. There have been eight clinical trials for TNO-155 mono- or combinational treatment for different types of tumors. A phase I study was launched in April 2017 that aimed to find the recommended dose for TNO-155 in combination with EGFR inhibitor nazartinib in patients with advanced solid tumors, including EGFR mutant NSCLC and KRASG12-mutant NSCLC (NCT03114319). Efficacy of TNO-155 in combination with PD-1 antibody spartalizumab or CDK4/6 inhibitor ribociclib in various types of advanced tumors is under evaluation in a phase I study (NCT04000529). In addition, a phase I study of TNO-155 in combination with KRASG12C inhibitor Adagrasib in patients suffering from advanced solid tumors that harbor KRASG12C mutations was initiated in April 2020 (NCT04330664). Additionally, a phase Ib/II study of TNO-155 with another KRASG12C inhibitor JDQ443 in KRASG12C mutant solid tumors is currently recruiting participants (NCT04699188). Concurrently, other SHP2 allosteric inhibitors, such as JAB-3068, HBI-2376, RMC-4630 etc. are in phase I trials. Of note, a phase I study on HBI -2376 in patients with advanced solid tumors harboring KRAS or EGFR mutations are now recruiting (NCT05163028) (Table 2) and RMC-4630 combined with LY3214996 (NCT04916236) and Sotorasib (NCT05054725) for the treatment of tumors harboring KRAS mutations are under phase I and phase II trials, respectively (Table 3).
STK19
Recently, a serine/threonine kinase, STK19, has been identified as a new NRAS activator by phosphorylating NRAS and subsequently enhancing the downstream NRAS-driven malignant transformation in melanoma cells. Knockdown STK19 by shRNAs has consistently inhibited NRAS activity, downstream signaling, and melanomagenesis in vivo.424,425 One of the STK19 inhibitors, ZT-12-037-01, was developed based on an initial hit from a chemical screen. ZT-12-037-01 is competitive inhibitor of ATP for STK19 with IC50 = 35 nM. The natural product chelidonine was also found to be a potent STK19 inhibitor with IC50 = 125.5 ± 19.3nM.425,426 Chelidonine not only suppresses NRAS-mediated signaling, but also significantly inhibits the growth of tumors with NRAS mutation. The discovery of ZT-12-037-01 and chelidonine opens the door of drug discovery utilizing STK19 for inhibition of NRAS mutant-driven tumors and provides valuable molecular probes for further investigation of the biological function of STK19. The progress on SHP2 and STK19 also proves that targeting well-characterized PTMs of Ras proteins and other small GTPases is a promising direction for indirect inhibition.
Targeting downstream effectors
For RAS, the best-characterized pathways include RAF-MEK-ERK and PI3K-AKT-mTOR, so the downstream effectors in these pathways are important drug targets in cancer treatments as well. Active RAS binds to RAF kinases, stimulating RAF dimerization and subsequent phosphorylation and activation, which leads to activation of MEK-ERK kinase cascade. This is an essential pathway regulating cell proliferation, survival, cell cycle progression, and differentiation. Multiple studies have validated RAF as the key mediator in mutant RAS-driven cancer growth and progression.427,428,429 However, current RAF inhibitors approved for treating BRAFV600E mutant melanoma are not effective in targeting RAS mutant cancer due to their unique pharmacologic features. They can specifically inhibit BRAFV600E monomers in BRAF mutant cancer cells, but in RAF WT cells, their binding to RAF induces RAF dimerization and allosteric transactivation of the second protomer, leading to paradoxical ERK signaling activation.430,431
Next-generation pan-RAF inhibitors, including LY3009120, belvarafenib (clinicaltrials.gov identifier NCT03284502), LXH-254 (NCT02607813, NCT04417621, NCT02974725, NCT03333343, and NCT04294160), lifirafenib (NCT03905148), BGB-3245 (NCT04249843), and TAK-580 (NCT03429803), can also induce WT RAF dimerization. However, they can bind both protomers of RAF dimer with equal potency and inhibit ERK signaling in RAS mutant cells.432,433,434,435,436,437 RAF dimer inhibitors have been reported to inhibit RAS mutant tumors in preclinical models, and several of them have been associated with partial responses in RAS-mutant cancer patients in phase I clinical trials. Two confirmed partial responses out of ten NRAS-mutant melanoma patients have been observed following belvarafenib treatment.432 Two patients (one endometrial cancer and one NSCLC) out of 66 with KRAS mutations had confirmed responses to lifirafenib treatment.438 Future large-scale clinical trials are warranted to validate their clinical efficacy. Outside of directly targeting RAF or RAF mutants, rigosertib was recently identified as an effective agent blocking the interaction between RAS-RAS-binding domain (RBD) and RAF family members.439 This finding opens a new door for inactivating RAS signaling.
MEK1/2 are the only well-characterized downstream substrates of RAF, making them attractive targets for treating RAS mutant cancer. Current allosteric MEK inhibitors are clinically approved for treatment of BRAFV600E mutant cancer either alone or in combination with RAF inhibitors.440 However, MEK inhibitors have only exhibited modest efficacy against RAS mutant tumors in clinical trials, with NRAS mutant melanoma being the only subtype showing responsiveness to MEK inhibition. Twenty percent of NRAS mutant-driven melanoma patients had partial response to MEK inhibitor binimetinib in a phase II clinical trial.441 Similarly, targeting ERK kinase, the downstream effector of MEK, failed to show clinical benefits in treating RAS mutant cancer. ERK inhibitor ulixertinib resulted in 18% partial response in NRAS mutant melanoma patients in a phase I clinical trial. Other ERK inhibitors have not been associated with objective tumor responses in RAS mutant tumors.442
There are two reasons that account for the limited efficacy of RAF dimer inhibitors, MEK inhibitors, and ERK inhibitors against RAS mutant tumors: upstream feedback release-induced MAPK pathway reactivation upon RAF dimer inhibitors/MEK inhibitors/ERK inhibitors treatment and limited on-target activity at maximum tolerated doses of the above inhibitors. MAPK pathway is tightly regulated through both transcriptional and post-translational mechanisms. Under normal physiological conditions, ERK activation causes feedback inhibition of RTK, RAS GEF SOS, RAF, and MEK through ERK-mediated phosphorylation. It also activates the transcription of negative regulators of the MAPK pathway including SPRY, SPRED, and DUSPs to ensure a controlled amplitude and duration of ERK signaling. Following treatment with RAF dimer inhibitors, MEK inhibitors, or ERK inhibitors, the MAPK pathway is inhibited, leading to the release of ERK-dependent negative feedback and reactivation of the pathway in the presence of the above inhibitors. On the other hand, studies from genetically engineered mouse models have shown that knock-out of either MEK1/2 or ERK1/2 led to embryonic lethality due to severe intestinal defects, underscoring that MAPK pathway is essential in normal tissue homeostasis. Inhibition of MAPK pathway by RAF dimer inhibitors/MEK inhibitors/ERK inhibitors will cause significant on-target toxicities to normal cells, which limits their efficacy in blocking the pathway in tumor cells.
Since single-agent RAF, MEK, or ERK inhibitors show limited efficacy in treating tumors with RAS mutations, combination strategies including vertical inhibition of multiple members (RAF, MEK, ERK, CDK4/6) in MAPK pathway, autophagy inhibitors hydroxychloroquine, chemotherapy, and immune checkpoint blockade, are being tested in the clinic. The PI3K-AKT-mTOR pathway is also a well-established downstream pathway of RAS. Binding of RAS to PI3K leads to activation of AKT and mTOR, which controls multiple cellular activities through regulation of protein translation and cell metabolism. Mutations in PI3K pathway including PI3K mutations, AKT mutations and amplification, or PTEN loss, are found to be concurrent with RAS mutations in human cancer, indicating that RAS activation may not be sufficient to fully activate PI3K signaling. Activation of PI3K-AKT-mTOR pathway has been reported to contribute to both intrinsic and acquired resistance to MAPK pathway inhibitors, which provides strong rationale for combinatory inhibition of MAPK and PI3K pathway in the treatment of RAS mutant cancer. Presently, alpelisib is the only PI3K inhibitor being utilized for the treatment of breast cancer, while leniolisib was recently assigned as new drug for activated PI3K Delta Syndrome (APDS). However, the safety concerns related to toxicity including hyperglycemia, rash, and diarrhea, caused by on-target effect and off-target effects of PI3K inhibitors are not negligible.443,444 Despite synergy observed in combination treatment in preclinical mouse models, clinical trials evaluating the efficacy of combined inhibition of MAPK and PI3K pathway failed due to toxicity in normal tissue. Isoform or mutant allele specific inhibitors with a wide therapeutic window will likely be better tolerated and more effective in targeting MAPK and PI3K signaling in RAS mutant cancer patients.
Targeting synthetic lethal partners in RAS mutant cancer
Synthetic lethal screens have been used to identify genes which are indispensable for the survival of RAS mutant cells but not RAS wild-type cells. Early RNAi screens using isogenic cell models or panels of KRAS mutant/WT cancer cell lines to identify RAS synthetic lethal genes yielded different targets among different studies. This high variability between independent screens could be attributed to the off-target effects in RNAi libraries. Later CRISPR-CAS9-based screens with lower off-target activities led to identification of regulators which are essential for RAS membrane localization including RCE1 and ICMT, RAS downstream effectors such as CRAF, and regulators of RAS-MAPK pathway such as SHOC2.428 Further studies employed in vivo CRISPR-CAS9 screen in either 3D organoid, tumor spheroid, or tumor xenograft models and found synthetic vulnerabilities including TGFBR2, TSC1, regulator of ferroptosis GPX4, and metabolic genes (NADK, KHK) in the context of mutant KRAS activation.445,446,447 However, none of the synthetic lethal targets have been able to transform into successful clinical approaches.
More recently, synthetic lethal screens have been employed to identify targets that function cooperatively with therapies targeting MAPK pathway including MEK inhibitors and KRASG12C inhibitors in RAS mutant cancer cells. Since the efficacy of MEK or KRASG12C inhibitors monotherapy in RAS mutant cancer is limited due to feedback reactivation of RAS downstream pathways, identification of potential combination strategies through synthetic lethal screens could be the key to achieve maximum and durable suppression of RAS signaling in tumors and improve patient response. Combinatorial CRISPR-CAS9 screen with MEK inhibitor treatments in RAS mutant cancer cells identified RTK signaling, anti-apoptotic proteins MCL1 and BCL2L1, and Centrosome-associated protein CENPE as synthetic targets which contribute to MEK inhibitor resistance.448,449,450 Interestingly, SHOC2 was identified as synthetic lethality again in KRAS mutant-driven lung and pancreatic cancer following MEK inhibitor treatment, which further validated its essential role in the context of RAS activation.448 The clinical success of KRASG12C inhibitors evoked a plethora of enthusiasm in finding synthetic lethal genes whose depletion will enhance sensitivity of KRASG12C mutant cancer cells to KRASG12C inhibitors. These efforts uncovered RTK such as IGF1R and EGFR, CPD, and Aurora kinase as potential therapeutic targets in combination with KRAS inhibitors.451,452
To date, synthetic lethal studies employing large panels of RAS mutant cell lines combined with 3D culture models or xenograft models to mimic in vivo tumor microenvironment have identified many RAS synthetic lethal targets. Increasing evidence indicates that the synthetic lethal vulnerabilities in RAS mutant tumors are unique to specific contexts of genetic background, cell lineage, different RAS mutant alleles, and tumor microenvironment.453 Thus, future synthetic lethal studies should consider the specific tumor context to better understand RAS dependencies during tumor initiation, progression, and drug resistance development.
Targeting autophagy
Research over the past decade has identified that RAS activation has a critical role in reprogramming metabolic process that promotes tumorigenesis and survival of tumor cells. Therefore, targeting metabolic alterations induced by mutant RAS is of great significance. In 2011, scientists found that introduction of HRasV12 or KrasV12 enhanced baseline autophagy in baby mouse kidney epithelial cells,454 and this finding was echoed by a later report that autophagic flux was elevated in KRAS-driven PDAC cell lines, which benefits the proliferation of KRAS-mutant tumor cells. Further pharmacological inhibition of autophagy by chloroquine has profoundly prolonged the survival of mice with KRAS mutant PDAC.455 The observed autophagy dependency of mutant RAS-driven cancer cells provides a strong rationale to inhibit autophagy in RAS-mutant tumors. In 2019, two publications further advanced this field. Interestingly, they both identified that inhibiting KRAS or its downstream signaling pathways increased the level of autophagy in PDACs, which serves as a protective mechanism for PDACs to support a high proliferative state.456,457 Combination of ERK inhibitor or MEK inhibitor with chloroquine synergistically inhibited tumor progression in murine tumor models. Noteworthy, a pancreatic tumor patient had a partial response with 50% reduction in tumor burden following treatment with trametinib plus hydroxychloroquine, while there was no obvious evidence of concurrent toxicity.457 As such, cooperatively targeting autophagy and MEK/ERK is a promising therapeutic strategy for treating KRAS mutant pancreatic tumors, as well as advanced gastrointestinal malignancies (NCT05221320), biliary cancer (NCT04566133), NSCLC (NCT04735068), and gastrointestinal cancer (NCT04214418) (Table 3).
Targeting mutant RAS-driven metabolic alteration in tumor cells
It has been known that tumor cells require a higher metabolic demand, including increased glucose to support their uncontrolled proliferation. Activating KRAS mutations were found to enhance glucose metabolism in different aspects, such as promoting the expression of glucose transporter GLUT1 to increase the uptake of glucose,458 inducing activity of key enzymes in glycolysis to increase the glycolytic flux, and supplying bioenergetic intermediates for other anabolic pathways.459,460,461,462,463 In addition, KRAS-driven PDAC was found to process glutamine in a non-canonical way mediated by glutaminase (GLS), aspartate transaminase 1/2, malate dehydrogenase 1 (MDH1), and malic enzyme 1 (ME1), which is essential for tumor cells to maintain redox state and proliferation. Glutamine is first catalyzed to glutamate by GLS in mitochondria, and glutamate-derived aspartate in mitochondria is then transported into the cytoplasm. Cytosolic aspartate is then transformed to oxaloacetate by GOT1, and oxaloacetate is further converted to malate and pyruvate via MDH1 and ME1, respectively. Ablation of any participating enzyme would induce dramatic suppression of tumor growth both in vitro and in vivo.464 Furthermore, lung adenocarcinoma cells bearing KRASG12V mutation displayed enhanced glutathione metabolism mainly triggered by the up-regulation of SLC7A11, which is responsible for importing cystine from the extracellular environment and promoting the synthesis of glutathione to sustain its specific metabolic fitness.465 Based on this finding, SLC7A11 inhibitor HG106 effectively inhibits the SLC7A11/glutathione axis and thus results in metabolic lethality in KRAS-mutant LUAD.465 In addition to altered glucose and glutamine metabolism, KRAS mutant NSCLC cancer cells aberrantly activate lipid biosynthesis and β-oxidation through Acyl-CoA synthetase long-chain family member 3 (ACSL3) mediated conversion of fatty acids into fatty Acyl-CoA esters. ACSL3 suppression reduces cellular ATP levels and impairs survival in KRAS mutant NSCLC cells.466 In addition to ACSL3, the expression of fatty acid synthase (FASN), the rate-limiting enzyme of fatty acid synthesis, was also identified to be upregulated in KRAS mutant lung cancer cells.467 In line with previous findings, FASN-mediated fatty acid synthesis and subsequently oxidized phospholipid remodeling through the Lands cycle were found to play essential roles in protecting mutant lung cancer cells from ferroptosis.468 FASN pharmacological inhibition using TVB-3664 exhibits anti-tumor activity in KRASG12D-driven lung cancer mouse model in vivo.468 Given that these distinct metabolic pathways are dispensable for normal cells, these interesting findings help define new approaches for exploiting the metabolic vulnerability of KRAS-driven cancer. Besides metabolic rewiring driven by oncogenic small GTPases such as KRAS, a recent study reported a reciprocal regulation of small GTPase RAC1 activity by a key metabolic enzyme LDHA through a catalytically independent mechanism.469,470 LDHA was found to bind to RAC1 and block RAC GAP access and maintain RAC1 in its active GTP bound state, leading to constitutive RAC1 activation and malignant progression and metastasis of breast cancer. This study links the noncanonical function of metabolic enzymes to small GTPases regulations, which opens a new avenue for studying the crosstalk between rewired metabolism and small GTPases signaling and foster identification of new therapeutic strategies targeting the interface between metabolic enzymes and small GTPases.
Resistance mechanisms and overcoming strategies
Resistance to KRASG12C inhibitor and combination Strategy
The distribution of KRAS mutations differs among various cancer types and notably, KRASG12C prevails in about 41% of lung adenocarcinomas,471,472 as well as in 3% of colorectal and 1% of pancreatic cancer.473,474,475 In the past two years, the FDA granted approval for two KRASG12C inhibitors, Sotorasib and Adagrasib, to NSCLC patients with KRASG12C mutation who have received at least one prior systemic therapy, demonstrating the dawn of the clinical application of KRASG12C inhibitor.278,476 Despite the impressive efficacy of KRASG12C inhibitors with around 30-45% response rate and nearly 7-month median progression-free survival in patients with KRASG12C mutant lung cancer, therapeutic responses in the clinic are usually short-lived, attributed to emerging resistance to monotherapy. The latest result of the first phase III trial of KRASG12C inhibitor Sotorasib in NSCLC patients with KRASG12C mutation (CodeBreak 200 study, NCT04303780) showed that 12-month treatment of Sotorasib pronouncedly improves the progression-free survival (PFS) with 24.8% and overall response rate (ORR) with 28.1% comparing to docetaxel, whose PFS is 10.1% and ORR is 13.2%. However, improvement of overall survival was not observed in this trial.477,478 Multiple studies revealed intrinsic, adaptive, and acquired resistance to KRASG12C inhibitors, and it is urgent to decipher resistance mechanisms and develop rational combinatorial treatment approaches to prevent the emergence of drug resistance to KRASG12C inhibitors.
The underlying mechanisms of intrinsic resistance to KRASG12C inhibitors [Fig. 8a] remain elusive. Genetic heterogeneity in different types of tumors and co-occurring mutations have been recently identified to confer intrinsic resistance toward KRASG12C inhibitors.479,480 Deep next-generation sequencing was launched in total of 330 NSCLC patients with KRAS mutations, and a panel of the most frequently concurrent mutations including TP53 (41%), STK11 (28%), KEAP1/NFE2L2 (27%), and KEAP1 (24%) were identified.481 Later, exploration of the effect of TP53, STK11 and KEAP1 co-mutation on the response of NSCLC patients with KRASG12C mutation to KRASG12C inhibitor was initiated (NCT03600883).482 The lowest response rate was observed in a group of wild-type STK11 and mutated KEAP1, whereas the highest response was found among patients with mutated STK11 and wild type KEAP1. However, the potential effects of STK11 and KEAP1 that modulate the clinical responses to KRASG12C inhibitor remain unclear since the number of patients included in co-occurring genomic alteration analysis was limited. Further analysis is ongoing in a phase Ib/II clinical trial of KRASG12C inhibitor, in KRASG12C mutant NSCLC harboring mutant STK11 and wild-type KEAP1 (NCT05276726). Upon KRAS activation, cyclin D/CDK4/6 complex get activated, and subsequently phosphorylates retinoblastoma (RB) and promotes tumor cells’ entry into S phase. KRASG12C inhibitor administration mainly arrests tumor cells in G0/G1 phase. Combined treatment with the CDK4/6 inhibitor further blocks escaping cells in the G1/S checkpoint, leading to enhanced cell cycle arrest and tumor regression.479 Clinical trials for combination of G12C inhibitors and CDK4/6 inhibitors are in progress (NCT04185883; NCT04165031). RGS3, a previously known GAP of G-protein signal transduction, was recently identified to mediate hydrolysis of GTP in KRAS mutants, which inactivates KRAS and provides the rationale for the antitumor efficacy of GDP bound KRASG12C inhibitor.51 Further validation in PDX models and patient samples demonstrated that RGS3 is negatively correlated with KRAS mutant signaling output, and patients who have lower RGS3 expression showed resistance to the therapy of G12C inhibitor. This exciting finding unveils a new mechanism for modulating the GTPases activity of KRAS mutants and suggests that RGS3 may serve as a possible biomarker to guide informed therapeutic strategy.

Intrinsic, adaptive, and acquired resistance to KRASG12C inhibitors. Resistance of KRASG12C inhibitors can be classified as (a) intrinsic, (b) adaptive, and (c) acquired resistance. a Lineage specific signaling between different tumor types, concurrent gene mutations with KRAS oncogenes, atypical GAP dependency/sensitivity have been reported to drive intrinsic resistance to KRASG12C inhibitors. b ERK signaling and PI3K/AKT signaling rebound driven by strong reactivation of different nodes such as RTK, SOS/SHP2, wild type RAS, or KRASG12C is the main cause of adaptive resistance to KRASG12C inhibitors. c Acquired genetic alterations in RTK, NRAS, HRAS, RAF, MEK, loss of function mutation of GAPs such as NF1, secondary mutations of KRASG12C including G13, Q61, R68, H95, Y96, and A146, as well as enhanced tumor plasticity including EMT and histologic transformation are associated with the acquired resistance to KRASG12C inhibitors. This figure was created with BioRender.com
Adaptive resistance limits the clinical benefit of almost all targeted therapies, and this has also limited the efficacy of G12C inhibitors51,289,452,475,483,484,485 [Fig. 8b]. A much lower response rate was observed in colorectal cancer than in NSCLC following treatment with KRASG12C inhibitor. Higher ERK signaling rebound driven by strong reactivation of EGFR limits the efficacy of KRASG12C inhibitors in KRASG12C mutant colorectal cancer. This finding motivates the development of combination therapy for colorectal cancer by co-targeting both KRASG12C and EGFR.486,487 Because EGFR/SHP2 signaling is a well-known upstream stimulus for activation of KRASG12C, co-targeting EGFR/SHP2 signaling and KRASG12C is an attractive strategy. A recent study reported a divergent response in KRASG12C lung cancer cells following KRASG12C inhibitor treatment through single-cell RNA sequencing.452 KRASG12C inhibition rendered some cells in a quiescent state whereas others rapidly escape inhibition. Drug-induced quiescent cells produce new KRASG12C, which can be activated by EGFR. Active AURK signaling further promotes KRAS-effector activation, which leads to adaptive cell cycle progression in the presence of the drug. Another study found activation of WT HRAS and NRAS by feedback relief through multiple RTKs as an alternative mechanism for the adaptive resistance to KRASG12C inhibitors in colorectal cancer cells,488 which is consistent with the previous findings that the two remaining WT RAS isoforms (NRAS and HRAS) also play crucial roles in promoting RTK/MAPK signaling activation and cell growth in KRAS mutant-driven tumor cells.489 This study also found combining KRASG12C inhibitors with an SHP2 inhibitor exhibited better anti-tumor efficacy in colorectal tumor PDX models than the KRASG12C inhibitor and EGFR inhibitor combination, largely because SHP2 inhibition can block RAS reactivation mediated by multiple RTKs upon KRASG12C inhibition.488 Therefore, to overcome adaptive resistance, combinatory therapies of G12C inhibitors with EGFR inhibitor (NCT04185883) and SHP2 inhibitor (NCT04185883) are currently under clinical evaluation.
Aside from its role in conferring adaptive resistance to KRASG12C inhibitors, RAS-MAPK signaling reactivation was also reported to contribute to acquired resistance to KRASG12C inhibitors in human lung cancer patients. In an old male patient with KRASG12C-mutant lung adenocarcinoma, the initial response was rapidly observed just a few days after taking Sotorasib, and some measurable tumors were noted to decrease in size. However, at week 13 post-treatment, some lesions began to grow, indicating the emergence of acquired resistance. At week 17, Sotorasib was stopped due to rapid tumor progression.490 Along with the rapid development of drug resistance, heterogeneous resistant mechanisms involving reactivation of MAPK and PI3K-AKT-mTOR signaling, epithelial-to-mesenchymal transition (EMT), immune evasion, metabolic rewiring, loss of KRASG12C allele and copy number were observed in multiple lesions derived from different sites in this patient.490 Meanwhile, in a 67-year-old patient with metastatic KRASG12C-driven NSCLC treated with Adagrasib,483 KRAS, NRAS, BRAF, and MAP2K1 mutations emerged in the cell-free DNA of this patient after disease progression and all led to the reactivation of RAS-MAPK signaling [Fig. 8c]. Of note, KRASG12C/Y96D secondary mutation was found to disrupt Adagrasib binding, which was sufficient to retain KRAS activation in the presence of Adagrasib.483 Other secondary mutations in SII-P of KRAS discovered in a clinical study cohort (NCT03785249), including R68S, H95D, H95Q, H95R, and Y96C, were reported to confer resistance to Adagrasib in vitro483 [Fig. 8c]. Interestingly, H95D, H95Q, and H95R mutations still showed marked response to Sotorasib treatment, implying that different drug-binding modes between Adagrasib and Sotorasib can potentially induce drug-specific resistance mutations.289 Hotspot mutations in Ras family or BRAF, such as KRASG12V, NRASQ61K, MRASQ71R, and BRAFG596R were illustrated to have a strong propensity to bypass KRASG12C inhibition and drive resistance via reactivating ERK signaling, which was found in 43 paired specimens obtained before and after the treatment of Sotorasib.484 Consistently, co-targeting ERK with G12C inhibitors led to an enhanced anti-tumor activity both in resistant cell models and PDX models.484 Additionally, distinct oncogenic fusions involving BRAF, ALK, RET and FGFR3 also emerged in progressive tumors from patients treated with Adagrasib.289 Thus, mutations in the MAPK pathway genes, as well as oncogenic mutations and fusions in RTK, constitute the acquired resistance to G12C inhibitors, and combinatorial inhibition of KRASG12C mutant and these cooperative targets such as RTKs (NCT04185883; NCT03785249; NCT04165031), MEK(NCT04185883), and RAF dimers (NCT05074810) has been proven to be an effective strategy to overcome resistance.
EMT has been found to trigger adaptive as well as intrinsic resistance to KRASG12C inhibitors in NSCLC cell lines485 [Fig. 8c]. EMT induces sustained activation of PI3K/AKT and reactivation of ERK in NSCLC tumor cells during the treatment of Sotorasib. Hence, the addition of PI3K inhibitor GDC-0941 into the cell lines can restore the sensitivity of Sotorasib. Triple combination of Sotorasib, GDC-0941 with SHP2 inhibitor SHP099 exhibits a more pronounced anti-tumor effect than Sotorasib dual combination with either GDC-0941 or SHP099.485 Moreover, a histologic transformation from adenocarcinoma to squamous cell carcinoma was discovered in two out of ten patients (nine NSCLC patients and one colorectal tumor patient) who developed acquired resistance after the treatment with KRASG12C inhibitor [Fig. 8c]. Intriguingly, no resistant mechanisms previously reported at the genomic level were found within these two patients (NCT03785249).289 Since KRAS mutations are rarely observed in squamous cell carcinoma.491 It is possible to speculate that this histological transformation may serve as an alternative way to escape the blockade of KRASG12C inhibitor.
Emerging targeting strategies
Since preclinical studies reported that KRASG12C inhibitor demonstrated greater efficacy in tumor growth inhibition in immuno-competent mice when compared with immune-deficient mice,492 it is vital to develop novel strategies to combine RAS inhibitors with immunotherapies to achieve greater clinical benefit for patients with RAS-mutants. It is well established that KRAS mutants contribute to the immunosuppressive tumor microenvironment through multiple mechanisms,493,494,495,496 including prompting M1 to M2 polarization of macrophages, inducing regulatory T cell differentiation by secreting TGF-β and IL-10, enhancing infiltration of myeloid-derived suppressor cells, and disrupting CD8+ cytotoxic T cells activation through modulation of immune checkpoint signaling. Therefore, combining targeted KRAS mutant inhibition with immune checkpoint blockade (ICB) is a straightforward strategy that has shown synergistic antitumor effects in the CT26 tumor xenograft model492 [Fig. 7b]. Meanwhile, adoptive cell therapies targeting KRAS mutants,497,498 tumor vaccines499 and antibodies targeting KRAS mutant peptide-MHC I complex500 are now in full swing [Fig. 7b]. Lately, a proof-of-concept study identified that neoantigen derived from ARS1620-mediated covalent modification of KRASG12C could be presented by MHC I in cancer cells. Antibodies recognizing these neoepitopes were developed and displayed immunotherapeutic efficacy in KRASG12C tumor cell lines.501 Remarkably, ARS1620-targeted antibody triggered cytotoxic T-cell response in tumor cell lines with intrinsic or acquired resistance to ARS1620 when co-cultured with peripheral blood mononuclear cells,501 demonstrating the potential to overcome drug resistance. Concurrently, a HapImmuneTM platform was established to discover antibodies targeting the Sotorasib-engaged KRASG12C peptide/MHC complexes.502 R023 scDb, which was developed by combining HapImmuneTM and T cell-engaging bispecific antibody platform, can selectively kill Sotorasib-pre-treated KRASG12C mutant lung cancer cells that show intrinsic resistance to Sotorasib.502 Lastly, Ras-driven cell transformation has also been reported to be dependent on the activity of Rho, Arf, and Ran proteins, which provides a strong rationale to target Ras, Rho, Arf, and Ran to synergistically suppress the proliferation of RAS-driven tumor cells and reverse RAS mediated malignant phenotype.503,504,505
Collectively, the approval of KRASG12C inhibitor ignited the enthusiasm of developing allele-specific inhibitors targeting RAS, and now the major direction of the pharmaceutical industry is developing inhibitors directly targeting different alleles of RAS. Nevertheless, exploring approaches that indirectly target RAS including its upstream regulators, downstream effectors, synthetic lethal partners, metabolic reprogramming, immune microenvironment, and signaling crosstalk is equally important since they are the most promising candidate targets to be applied in combinatorial therapies to enhance efficacy of direct RAS targeting inhibitors and combat drug resistance.
Concluding remarks
Since the establishment of association with human diseases, the journey of targeting small GTPases family has been laborious but also fulfilling. As a predominant oncogenic protein, KRAS has been established as a paradigm in the process of drug discovery towards small GTPases, with numerous targeting strategies developed during the recent decade. On the contrary, the efforts to target Rho, Arf, Rab, and Ran families are lagging and most of the identified compounds remain insufficiently characterized and lack follow-up optimizations. In 2021, Sotorasib received accelerated approval from the FDA to treat NSCLC harboring KRASG12C mutation during its phase II clinical trial. Now, this drug has been approved in 44 markets, including the United States, the European Union, the United Kingdom, and Japan. This has been reckoned as a milestone in proving the druggability of KRAS after its discovery 40 years ago. In 2022, AMGEN announced results from the global Phase 3 CodeBreaK 200 trial. 25% of patients showed PFS compared with 10% for chemotherapy reagent docetaxel, while OS between Sotorasib (10.6 months) and docetaxel (11.3 months) remaining unchanged. Treatment-related adverse events are 33% for Sotorasib relative to 40% for docetaxel. As such, the clinical performance and adverse effects are still under evaluation for further approval.38,506 Also in 2022, J&J announced they are terminating further clinical trials of another KRASG12C covalent inhibitor JNJ-74699157 (ARS-3248) due to “dose-limiting skeletal muscle toxicities and the lack of efficacy at the 100 mg dose”.280 This shows the adverse effects observed during clinical trials and its underlying mechanisms need to be further addressed. Also during 2022, the new ligands that selectively bind to KRAS G12D, G12S, and G12R mutants have been developed, renewing the frontier to target alleles-specific hotspot mutations of KRAS.506 The G12D inhibitor exhibits encouraging antitumor efficacy in PDAC tumor models38 and G12S/R inhibitors provide proof of concept approaches to inspire the future development of other KRAS mutants.
Despite the successful clinical application of KRASG12C inhibitors in NSCLC patients with KRASG12C mutations, resistance develops in almost all patients. Uncovering the sophisticated mechanisms of intrinsic, adaptive, and acquired drug resistance will be the key to developing novel combinatory strategies to target cancers driven by RAS mutations, which will facilitate the process of targeting other RAS isoforms and other small GTPases. KRASG12C inhibitors induced a quiescent state in a subpopulation of tumor cells and these quiescent cancer cells synthesized new KRASG12C mutant protein, escaping drug inhibition through EGFR and AURKA-mediated RAS signaling activation.452 This study explained why KRASG12C inhibitor treatment only had partial responses in most lung cancer patients. Further mechanistic studies into the divergent responses following KRASG12C inhibitor treatment and transcriptional modulations controlling new KRAS synthesis in drug-induced quiescent cells will uncover new therapeutic targets and modalities to overcome adaptive resistance to current KRASG12C inhibitors.452 KRAS inhibitors often trigger a cytostatic response in cancer cells and targeting a quiescent cell state will strengthen tumor targeting and likely lead to tumor cell death to achieve durable and complete responses in cancer patients. Acquired resistances of KRASG12C inhibitors are engendered by KRAS secondary mutations or fusions/mutations occurring at upstream RTKs or downstream RAF/MEK. 10 distinct alterations in KRAS, NRAS, BRAF, and MEK were identified in a KRASG12C NSCLC patient with acquired resistance to Adagrasib.483 In another clinical study, more than one potential resistance mechanism was identified in 41% (7/17) of patients with KRASG12C NSCLC or CRC.289 As such, the heterogeneity of resistance mechanisms poses challenges to both basic research and clinical treatments. Since it is difficult to target multiple heterogenous resistant mechanisms at the time of tumor relapse, we need to find a way to prevent the expansion of tumor heterogeneity along with KRASG12C inhibitor treatment. Mechanistic studies into the evolution of heterogenous resistant alterations under drug pressure through combining single-cell DNA sequencing and barcode tracing techniques will improve our understanding of the expansion of tumor heterogeneity and identify potential targets to delay the emergence of the acquired resistance.
Further exploration into uncovering new regulatory mechanisms underlying biological functions of small GTPases and developing mechanism-based targeting strategies are still needed for targeting different small GTPases and their hotspot mutations. Liquid-liquid phase separation (LLPS) offers a new angle to decipher the complex regulatory mechanisms in cells.507,508 The cytoplasmic tail of EGFR and GRB2 (Growth factor receptor-bound protein 2) form a binary condensate upon the phosphorylation of EGFR, and then LLPS recruits SOS at the membrane surface for Ras activation.509,510 RTK fusion proteins can condensate into de novo cytoplasmic membraneless granules, which further recruit and concentrate Grb2/SOS for initiating Ras-mediated MAPK signaling.511 A parallel study reveals that RTK forms condensate at the membrane surface and this process is critical in recruiting SHP2 and PLCγ1 and modulating their enzymatic activities.512 Other small GTPases such as Rac and Rab are also implicated in mediating LLPS.513,514 As introduced in the first session, PTMs play unique roles in modulating the structures and functions of small GTPases. Because they are highly diversified and heterogeneous, they remain largely undiscovered. Therefore, the continuous discovery of new PTMs and functional/structural characterizations will bring new insights into targeting regulation mechanisms. For instance, the finding of a dynamic process of NRAS phosphorylation mediated by STK19 led to the discovery of STK19 inhibitor to target NRAS mutant melanoma.424,426 Increasing efforts are also directed at defining new mediators of the oncogenic properties of Ras. A recent study identified EFR3A as a new interaction partner for oncogenic KRAS through mining interactomes of oncogenic Ras proteins. EFR3A binds specifically to active KRAS and further recruits phosphatidylinositol kinase PI4KA.515 This leads to the accumulation of PI4P and phosphatidylserine (PtdSer) at the plasma membrane, which promotes KRAS plasma membrane localization, nanoclustering, and activation of downstream signaling. Moreover, a selective PI4KA inhibitor showed synergistic anti-proliferative activity with KRASG12C inhibitors in KRASG12C mutant human cancer cell lines.515 Another study uncovered the essential role of PtdSer lipid transport proteins in maintaining PM PtdSer levels and KRAS PM localization, and further showed targeting PI4KIIIα to reduce PM PI4P and PtdSer levels inhibited KRAS-driven tumor growth in pancreatic cancer models.516 Recently, a new protein RASON encoded by long noncoding RNA (LINC00673) is reported to lock KRASG12D and KRASG12V in a GTP-bound state and thus lead to continuous KRAS activation.517 In the PDAC xenograft model, deletion of RASON sensitizes KRAS mutant PDAC to EGFR inhibitor and inhibits tumor growth. As such, identifying novel proteins that maintain/accelerate small GTPases activity might define more vulnerable targets to block small GTPases’ oncogenic signaling alternatively. The spatiotemporal dynamics of the subcellular distribution, transient dimerization, and switching between active/inactive conformations are not only important for the biological functions of GTPases but also useful for inferring mechanisms of drug actions. Although in-situ real-time visualization and quantification of these dynamic properties are still challenging, versatile engineered fluorescent biosensors and single molecular methods have been developed for measuring these properties of heterotrimeric G-protein518,519,520 and small GTPases521,522,523,524,525 in living cells.
Strategic innovations by adopting or developing new methods for targeting small GTPases are in high demand. Combinatory therapies of KRAS inhibitors with inhibitors of the upstream regulators or downstream effectors have been proven with enhanced anti-tumor activities. Targeting crosstalk between different small GTPases is another interesting avenue because accumulating evidence indicates that the transformation of RAS-driven tumor cells requires endogenous activities of Rho proteins or overexpression of Rho, Arf, and Ran.526 KRASG12C inhibitors have been reported to drive a pro-inflammatory tumor microenvironment and enhance the anti-tumor activity when combined with immune checkpoint blockade (ICB).492,527 However, a recent study indicates that G12C inhibitor selectively synergizes with ICB in inflamed tumors but not in non-inflamed tumors, which addresses a strong immunogenicity dependency of synergizing KRASG12C inhibitor with ICB and guides the selection of patients who might respond to this combination.528 Further mechanistic studies into the failure of KRASG12C inhibitors to synergize with ICB in non-inflamed tumors will be needed to provide novel strategies to improve the therapeutic effect of combined KRASG12C inhibition and ICB.
Instead of combining KRASG12C inhibitors with ICB, another attractive direction is developing bispecific T cell engagers targeting KRAS mutant peptide-MHC I complex on the surface of KRASG12C mutant cancer cells.501,502 Currently, these bispecific T cell engagers based on a KRASG12C inhibitor conjugated neoepitopes have been reported to cause cytotoxic T cells response in a co-culture system with T cells in vitro. Cancer cells need to be pretreated with a covalent KRASG12C inhibitor for a period to allow full engagement of the drug with KRASG12C mutant protein and its subsequent presentation by MHC. The dosing schedule of the covalent KRASG12C inhibitor and the antibody needs to be optimized to achieve maximum targeting efficiency. Further, in vivo bioavailability, safety profiles, and immunotherapeutic effects of these antibodies targeting KRASG12C inhibitors-based neoepitopes are waiting to be evaluated to move forward to its clinical application. Taken together, KRASG12C inhibitor-targeted immunotherapy or cotreatment of KRASG12C inhibitor with immunotherapy represents new directions in tackling drug resistance to KRASG12C inhibitors through remodeling the immunosuppressive tumor microenvironment driven by KRAS mutants.
Multi-omics have been undergoing fast evolution, which acts as a powerhouse for mapping gene regulatory networks at the cellular level and uncovering concerted targets at transcriptional and post-transcriptional levels. However, large-scale data outputs pose challenges in data mining and interpretation for the protein of interest, and more specific functional validations and characterizations are necessary. Protein structure is pivotal in drug design and complex structures of small GTPases with their regulatory proteins or downstream effectors are limited. This issue might be improved with the recent developments of cryoEM for structural determinations and deep learning methods for structural predictions such as AlphaFold Multimer529 and RoseTTA Fold.530 Understanding the structural plasticity of switch regions in small GTPases is a unique resource for developing inhibitors with desirable selectivity to the disease mutants. The exploitation of structural dynamics and lowly populated conformations for drug discovery has been promoted for years531,532 and has been attested by some of the recent studies on KRAS, such as CLAMPs,327 NMR-derived ensembles of the switch pockets49 and allosteric regulations.533 Now, it is time for bringing this concept from the bench to the bedside with more energetic efforts. New computational platforms using artificial intelligence for ultra-large-scale virtual screening are reshaping the workflow of drug discovery. They can unprecedentedly maximize the chemical space and elevate the time efficiency in drug screening.534,535,536,537 Recently, the Random nonstandard Peptide Integrated Discovery (RaPID) system has been successfully employed to screen macrocyclic peptide inhibitors for G-protein (Gαs).538 Two macrocyclic peptides are cell-permeable and nucleotide-state-selective; they can bind to the GDP- and GTP-bound Gαs by distinguishing the conformation of Switch II/helix-3 pocket. RaPID is established by combining mRNA display technique with in vitro translation system539,540 and the library diversity of macrocyclic peptides can reach a scale of 1000 molecules. Both Gα and small GTPases belong to the family of GTP-binding proteins and share a structural similarity, which foresees the potential applicability of RaPID to screen for macrocyclic peptide inhibitors of small GTPases. PROTACs are evolving on a fast track and will play a more important role in eliminating the disease mutants with the future development of more specific chemical compounds and protein ligands. Targeting Ras membrane association through PI4KA inhibitors represents a new indirect targeting strategy and holds promise for KRAS mutant cancer patients.515,516 Enhanced dependence on lipid biosynthesis and β-oxidation of KRAS mutant cancer cells also leads to new approaches to target the metabolic vulnerability of KRAS-driven cancers.466,467,468
In summary, many methodological innovations and monumental progresses have been made in targeting KRAS. However, the mission of drug discovery for small GTPases is far from being completed. For a protein family with over 150 members, there are only two drugs targeting KRAS G12C approved in the clinic. For KRASG12C inhibitors, further efforts are needed to improve its PFS rates, and combat drug resistance and adverse effects. Less progress has been made in targeting NRASQ61 mutation for treating melanoma and other hotspot mutations (G12V, G13, Q61H/L) have not yet been targeted. G12V mutation occurs in both KRAS and CDC42 and it is intractable by current targeting strategies because the sidechain of valine is less chemically amenable than others. Most of the KRAS inhibitors currently in clinical trials are targeting GDP-bound state by preventing KRAS activation. Future efforts are summoned to establish effective strategies to target the GTP-bound active conformation for directly turning off the oncogenic mutants. Progress of targeting Rho, Arf and Rab falls far behind Ras. Cross-activity is a practical concern raised during the development of isoform-specific ligands for Rho, Rab and Arf family proteins due to high sequence similarity and shared regulatory proteins within each subfamily. Discerning the subtle structural difference that potentially leads to uncovering different pocket properties and finding chemical reactive residues for covalent binding are keys to circumventig this obstacle, and a plethora of lessons can be learned from targeting KRAS. Therefore, drug discovery for Rho, Arf, Rab, and Ran should be illuminated rather than overshadowed by Ras, and the uncharted space for academic research and industry is enormous.
References
Rogge, R. D., Karlovich, C. A. & Banerjee, U. Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell 64, 39–48 (1991).
Tcherkezian, J. & Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 99, 67–86 (2007).
Bos, J. L., Rehmann, H. & Wittinghofer, A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 (2007).
Vigil, D., Cherfils, J., Rossman, K. L. & Der, C. J. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat. Rev. Cancer 10, 842–857 (2010).
Cherfils, J. & Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 (2013).
Dransart, E., Olofsson, B. & Cherfils, J. RhoGDIs revisited: novel roles in Rho regulation. Traffic 6, 957–966 (2005).
Sivars, U., Aivazian, D. & Pfeffer, S. R. Yip3 catalyses the dissociation of endosomal Rab-GDI complexes. Nature 425, 856–859 (2003).
Wennerberg, K., Rossman, K. L. & Der, C. J. The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 (2005).
Fernández-Medarde, A. & Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2, 344–358 (2011).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 (2011).
Bernal Astrain, G., Nikolova, M. & Smith, M. J. Functional diversity in the RAS subfamily of small GTPases. Biochem. Soc. Trans. 50, 921–933 (2022).
Kontani, K. et al. Di-Ras, a distinct subgroup of ras family GTPases with unique biochemical properties. J. Biol. Chem. 277, 41070–41078 (2002).
Lu, Z. et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 118, 3917–3929 (2008).
Sutton, M. N. et al. DIRAS3 (ARHI) blocks RAS/MAPK signaling by binding directly to RAS and disrupting RAS clusters. Cell Rep. 29, 3448–3459.e3446 (2019).
Sutton, M. N. et al. RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell death and are required for autophagy in murine ovarian cancer cells. Autophagy 14, 637–653 (2018).
Heasman, S. J. & Ridley, A. J. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701 (2008).
Sahai, E. & Marshall, C. J. RHO-GTPases and cancer. Nat. Rev. Cancer 2, 133–142 (2002).
Stone, L. Bladder cancer: Rho-sensitive pathway mediates metastasis. Nat. Rev. Urol. 13, 630 (2016).
Narayanan, K. L., Chopra, V., Rosas, H. D., Malarick, K. & Hersch, S. Rho kinase pathway alterations in the brain and leukocytes in Huntington’s disease. Mol. Neurobiol. 53, 2132–2140 (2016).
Aguilar, B. J., Zhu, Y. & Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res. Ther. 9, 97 (2017).
Bandaru, S. et al. Lack of RAC1 in macrophages protects against atherosclerosis. PLoS One 15, e0239284 (2020).
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 (2009).
Zhen, Y. & Stenmark, H. Cellular functions of Rab GTPases at a glance. J. Cell Sci. 128, 3171–3176 (2015).
Cheng, K. W. et al. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 10, 1251–1256 (2004).
Yoon, S. O., Shin, S. & Mercurio, A. M. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 65, 2761–2769 (2005).
Bravo-Cordero, J. J. et al. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. Embo J. 26, 1499–1510 (2007).
Park, M. H., Choi, K. Y. & Min do, S. The pleckstrin homology domain of phospholipase D1 accelerates EGFR endocytosis by increasing the expression of the Rab5 effector, rabaptin-5. Exp. Mol. Med. 47, e200 (2015).
Park, S. J. et al. HDAC6 sustains growth stimulation by prolonging the activation of EGF receptor through the inhibition of rabaptin-5-mediated early endosome fusion in gastric cancer. Cancer Lett. 354, 97–106 (2014).
Wheeler, D. B., Zoncu, R., Root, D. E., Sabatini, D. M. & Sawyers, C. L. Identification of an oncogenic RAB protein. Science 350, 211–217 (2015).
Lara Ordóñez, A. J., Fasiczka, R., Naaldijk, Y. & Hilfiker, S. Rab GTPases in Parkinson’s disease: a primer. Essays Biochem. 65, 961–974 (2021).
Pensalfini, A. et al. Endosomal dysfunction induced by directly overactivating Rab5 recapitulates prodromal and neurodegenerative features of Alzheimer’s disease. Cell Rep. 33, 108420 (2020).
Casalou, C., Faustino, A. & Barral, D. C. Arf proteins in cancer cell migration. Small GTPases 7, 270–282 (2016).
Donaldson, J. G. & Jackson, C. L. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 12, 362–375 (2011).
Rush, M. G., Drivas, G. & D’Eustachio, P. The small nuclear GTPase Ran: how much does it run? Bioessays 18, 103–112 (1996).
Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 8, 195–208 (2007).
Mastroeni, D. et al. Reduced RAN expression and disrupted transport between cytoplasm and nucleus; a key event in Alzheimer’s disease pathophysiology. PLoS One 8, e53349 (2013).
Hallin, J. et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 28, 2171–2182 (2022).
The KRAS(G12D) inhibitor MRTX1133 elucidates KRAS-mediated oncogenesis. Nat. Med. 28, 2017–2018 (2022).
Wang, X. et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS(G12D) inhibitor. J. Med. Chem. 65, 3123–3133 (2022).
de Vos, A. M. et al. Three-dimensional structure of an oncogene protein: catalytic domain of human c-H-ras p21. Science 239, 888–893 (1988).
Itzen, A. & Goody, R. S. GTPases involved in vesicular trafficking: structures and mechanisms. Semin. Cell Dev. Biol. 22, 48–56 (2011).
Wittinghofer, A. & Vetter, I. R. Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 80, 943–971 (2011).
Hobbs, G. A., Der, C. J. & Rossman, K. L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292 (2016).
Stengel, K. & Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell Signal 23, 1415–1423 (2011).
Spoerner, M., Herrmann, C., Vetter, I. R., Kalbitzer, H. R. & Wittinghofer, A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc. Natl Acad. Sci. USA 98, 4944–4949 (2001).
Rensland, H. et al. Substrate and product structural requirements for binding of nucleotides to H-ras p21: the mechanism of discrimination between guanosine and adenosine nucleotides. Biochemistry 34, 593–599 (1995).
Janakiraman, M. et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 70, 5901–5911 (2010).
Schubbert, S. et al. Biochemical and functional characterization of germ line KRAS mutations. Mol. Cell Biol. 27, 7765–7770 (2007).
Liu, D., Mao, Y., Gu, X., Zhou, Y. & Long, D. Unveiling the “invisible” druggable conformations of GDP-bound inactive Ras. Proc. Natl Acad. Sci. USA 118, e2024725118 (2021).
Lu, S. et al. Ras conformational ensembles, allostery, and signaling. Chem. Rev. 116, 6607–6665 (2016).
Li, C. et al. The G protein signaling regulator RGS3 enhances the GTPase activity of KRAS. Science 374, 197–201 (2021).
Sabatini, D. M. mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 6, 729–734 (2006).
Fruman, D. A. et al. The PI3K pathway in human disease. Cell 170, 605–635 (2017).
Balendran, A., Currie, R., Armstrong, C. G., Avruch, J. & Alessi, D. R. Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70 S6 kinase in vivo at Thr-412 as well as Thr-252. J. Biol. Chem. 274, 37400–37406 (1999).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
Smith, S. F., Collins, S. E. & Charest, P. G. Ras, PI3K and mTORC2—three’s a crowd? J. Cell Sci. 133, jcs234930 (2020).
Li, J. et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997).
Sansal, I. & Sellers, W. R. The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol. 22, 2954–2963 (2004).
Apken, L. H. & Oeckinghaus, A. The RAL signaling network: cancer and beyond. Int. Rev. Cell Mol. Biol. 361, 21–105 (2021).
Bodemann, B. O. & White, M. A. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer 8, 133–140 (2008).
Noh, D. Y., Shin, S. H. & Rhee, S. G. Phosphoinositide-specific phospholipase C and mitogenic signaling. Biochim. Biophys. Acta 1242, 99–113 (1995).
Zhang, R. Y., Du, W. Q., Zhang, Y. C., Zheng, J. N. & Pei, D. S. PLCε signaling in cancer. J. Cancer Res. Clin. Oncol. 142, 715–722 (2016).
Kelley, G. G., Reks, S. E., Ondrako, J. M. & Smrcka, A. V. Phospholipase C(epsilon): a novel Ras effector. EMBO J. 20, 743–754 (2001).
Berridge, M. J. & Inositol, R. F. I. phosphates and cell signalling. Nature 341, 197–205 (1989).
Dusaban, S. S. & Brown, J. H. PLCepsilon mediated sustained signaling pathways. Adv. Biol. Regul. 57, 17–23 (2015).
Shibatohge, M. et al. Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J. Biol. Chem. 273, 6218–6222 (1998).
Yu, O. M. & Brown, J. H. G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol. Pharmacol. 88, 171–180 (2015).
Kelley, G. G., Kaproth-Joslin, K. A., Reks, S. E., Smrcka, A. V. & Wojcikiewicz, R. J. H. G-protein-coupled Receptor Agonists Activate Endogenous Phospholipase Cϵ and Phospholipase Cβ3 in a Temporally Distinct Manner. J Biol. Chem. 281, 2639–2648 (2006).
Donninger, H., Schmidt, M. L., Mezzanotte, J., Barnoud, T. & Clark, G. J. Ras signaling through RASSF proteins. Semin. Cell Dev. Biol. 58, 86–95 (2016).
Dhanaraman, T. et al. RASSF effectors couple diverse RAS subfamily GTPases to the Hippo pathway. Sci. Signal. 13, eabb4778 (2020).
Sandrine Etienne-Manneville, A. H. Rho GTPases in cell biology. Nature 420, 629–635 (2002).
Lawson, C. D. & Ridley, A. J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 217, 447–457 (2018).
Amano, M. et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 (1996).
Kimura, K. et al. Regulation of myosin phosphatase by rho and rho-associated kinase (Rho-Kinase). Science 273, 245–248 (1996).
Clements, R. T., Minnear, F. L., Singer, H. A., Keller, R. S. & Vincent, P. A. RhoA and Rho-kinase dependent and independent signals mediate TGF-beta-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L294–L306 (2005).
Bishop, A. L. & Hall, A. Rho GTPases and their effector proteins. Biochem. J. 348, 241–255 (2000).
Eden, S., Rohatgi, R., Podtelejnikov, A. V., Mann, M. & Kirschner, M. W. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418, 790–793 (2002).
Takenawa, T. & Suetsugu, S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 8, 37–48 (2007).
Miki, H., Suetsugu, S. & Takenawa, T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. Embo J. 17, 6932–6941 (1998).
Rane, C.K. & Minden, A. P21 activated kinases: structure, regulation, and functions. Small GTPases 5, e28003 (2014).
Radu, M., Semenova, G., Kosoff, R. & Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 14, 13–25 (2014).
Rohatgi, R. et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97, 221–231 (1999).
Stradal, T. E. & Scita, G. Protein complexes regulating Arp2/3-mediated actin assembly. Curr. Opin. Cell Biol. 18, 4–10 (2006).
Baschieri, F. & Farhan, H. Crosstalk of small GTPases at the Golgi apparatus. Small GTPases 3, 80–90 (2012).
Stengel, K. R. & Zheng, Y. Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PLoS One 7, e37317 (2012).
Qiu, R. G., Chen, J., Kirn, D., McCormick, F. & Symons, M. An essential role for Rac in Ras transformation. Nature 374, 457–459 (1995).
Qiu, R. G., Chen, J., McCormick, F. & Symons, M. A role for Rho in Ras transformation. Proc. Natl Acad. Sci. USA 92, 11781–11785 (1995).
Qiu, R. G., Abo, A., McCormick, F. & Symons, M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol. Cell Biol. 17, 3449–3458 (1997).
Cheng, C. M. et al. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol. Cell Biol. 31, 983–997 (2011).
Wu, W. J., Tu, S. & Cerione, R. A. Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 114, 715–725 (2003).
Callow, M. G. et al. Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines. J. Biol. Chem. 277, 550–558 (2002).
Grosshans, B. L., Ortiz, D. & Novick, P. Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl Acad. Sci. USA 103, 11821–11827 (2006).
Homma, Y., Hiragi, S. & Fukuda, M. Rab family of small GTPases: an updated view on their regulation and functions. Febs j. 288, 36–55 (2021).
Mizuno-Yamasaki, E., Rivera-Molina, F. & Novick, P. GTPase networks in membrane traffic. Annu. Rev. Biochem. 81, 637–659 (2012).
Novick, P. Regulation of membrane traffic by Rab GEF and GAP cascades. Small GTPases 7, 252–256 (2016).
Pylypenko, O., Hammich, H., Yu, I. M. & Houdusse, A. Rab GTPases and their interacting protein partners: Structural insights into Rab functional diversity. Small GTPases 9, 22–48 (2018).
Chavrier, P., Parton, R. G., Hauri, H. P., Simons, K. & Zerial, M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 62, 317–329 (1990).
Simpson, J. C. et al. A role for the small GTPase Rab21 in the early endocytic pathway. J. Cell Sci. 117, 6297–6311 (2004).
Chua, C. E., Gan, B. Q. & Tang, B. L. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol. Life Sci. 68, 3349–3358 (2011).
Kinchen, J. M. & Ravichandran, K. S. Phagosome maturation: going through the acid test. Nat. Rev. Mol. Cell Biol. 9, 781–795 (2008).
Kitano, M., Nakaya, M., Nakamura, T., Nagata, S. & Matsuda, M. Imaging of Rab5 activity identifies essential regulators for phagosome maturation. Nature 453, 241–245 (2008).
Ortiz Sandoval, C. & Simmen, T. Rab proteins of the endoplasmic reticulum: functions and interactors. Biochem Soc. Trans. 40, 1426–1432 (2012).
Sun, Y., Bilan, P. J., Liu, Z. & Klip, A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc. Natl Acad. Sci. USA 107, 19909–19914 (2010).
Gulbranson, D. R. et al. RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis. Proc. Natl Acad. Sci. USA 114, E8224–e8233 (2017).
Roach, W. G., Chavez, J. A., Mîinea, C. P. & Lienhard, G. E. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem. J. 403, 353–358 (2007).
Sano, H., Roach, W. G., Peck, G. R., Fukuda, M. & Lienhard, G. E. Rab10 in insulin-stimulated GLUT4 translocation. Biochem. J. 411, 89–95 (2008).
Masuda, E. S. et al. Rab37 is a novel mast cell-specific GTPase localized to secretory granules. FEBS Lett. 470, 61–64 (2000).
Lara Ordónez, A. J. et al. RAB8, RAB10, and RILPL1 contribute to both LRRK2 kinase-mediated centrosomal cohesion and ciliogenesis deficits. Hum. Mol. Genet. 28, 3552–3568 (2019).
Oguchi, M. E., Okuyama, K., Homma, Y. & Fukuda, M. A comprehensive analysis of Rab GTPases reveals a role for Rab34 in serum starvation-induced primary ciliogenesis. J. Biol. Chem. 295, 12674–12685 (2020).
Onnis, A. et al. The small GTPase Rab29 is a common regulator of immune synapse assembly and ciliogenesis. Cell Death Differ. 22, 1687–1699 (2015).
Wasmeier, C. et al. Rab38 and Rab32 control post-Golgi trafficking of melanogenic enzymes. J. Cell Biol. 175, 271–281 (2006).
Alto, N. M., Soderling, J. & Scott, J. D. Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J. Cell Biol. 158, 659–668 (2002).
Wei, D. et al. RAB31 marks and controls an ESCRT-independent exosome pathway. Cell Res. 31, 157–177 (2021).
Li, C. et al. COPI-TRAPPII activates Rab18 and regulates its lipid droplet association. Embo J. 36, 441–457 (2017).
Jayson, C. B. K. et al. Rab18 is not necessary for lipid droplet biogenesis or turnover in human mammary carcinoma cells. Mol. Biol. Cell 29, 2045–2054 (2018).
Gao, Y. et al. Intercellular transfer of activated STING triggered by RAB22A-mediated non-canonical autophagy promotes antitumor immunity. Cell Res. 32, 1086−1104 (2022).
Jackson, C. L. & Bouvet, S. Arfs at a glance. J. Cell Sci. 127, 4103–4109 (2014).
Sztul, E. et al. ARF GTPases and their GEFs and GAPs: concepts and challenges. Mol. Biol. Cell 30, 1249–1271 (2019).
Cherfils, J. Arf GTPases and their effectors: assembling multivalent membrane-binding platforms. Curr. Opin. Struct. Biol. 29, 67–76 (2014).
Kozlov, M. M. & Taraska, J. W. Generation of nanoscopic membrane curvature for membrane trafficking. Nat. Rev. Mol. Cell Biol. 24, 63–78 (2023).
Faini, M., Beck, R., Wieland, F. T. & Briggs, J. A. Vesicle coats: structure, function, and general principles of assembly. Trends Cell Biol. 23, 279–288 (2013).
Beck, R. et al. Membrane curvature induced by Arf1-GTP is essential for vesicle formation. Proc. Natl Acad. Sci. USA 105, 11731–11736 (2008).
Springer, S., Spang, A. & Schekman, R. A primer on vesicle budding. Cell 97, 145–148 (1999).
Fucini, R. V. et al. Activated ADP-ribosylation factor assembles distinct pools of actin on Golgi membranes. J. Biol. Chem. 275, 18824–18829 (2000).
Donaldson, J. G. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J. Biol. Chem. 278, 41573–41576 (2003).
Honda, A. et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99, 521–532 (1999).
Ge, M., Cohen, J. S., Brown, H. A. & Freed, J. H. ADP ribosylation factor 6 binding to phosphatidylinositol 4,5-bisphosphate-containing vesicles creates defects in the bilayer structure: an electron spin resonance study. Biophys. J. 81, 994–1005 (2001).
Brown, F. D., Rozelle, A. L., Yin, H. L., Balla, T. & Donaldson, J. G. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 154, 1007–1017 (2001).
Langemeyer, L. & Ungermann, C. Vesicle transport: Exocyst follows PIP(2) to tether membranes. Curr. Biol. 32, R748–r750 (2022).
Nithianantham, S. et al. Tubulin cofactors and Arl2 are cage-like chaperones that regulate the soluble αβ-tubulin pool for microtubule dynamics. Elife 4, e08811 (2015).
Bhamidipati, A., Lewis, S. A. & Cowan, N. J. ADP ribosylation factor-like protein 2 (Arl2) regulates the interaction of tubulin-folding cofactor D with native tubulin. J. Cell Biol. 149, 1087–1096 (2000).
Saito, K., Maeda, M. & Katada, T. Regulation of the Sar1 GTPase cycle is necessary for large cargo secretion from the endoplasmic reticulum. Front. Cell Dev. Biol. 5, 75 (2017).
Bi, X., Corpina, R. A. & Goldberg, J. Structure of the Sec23/24–Sar1 pre-budding complex of the COPII vesicle coat. Nature 419, 271–277 (2002).
Koo, T. H., Eipper, B. A. & Donaldson, J. G. Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol. 8, 29 (2007).
Barr, F. & Lambright, D. G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 22, 461–470 (2010).
Dong, N. et al. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150, 1029–1041 (2012).
Kalab, P., Pu, R. T. & Dasso, M. The ran GTPase regulates mitotic spindle assembly. Curr. Biol. 9, 481–484 (1999).
Carazo-Salas, R. E., Gruss, O. J., Mattaj, I. W. & Karsenti, E. Ran-GTP coordinates regulation of microtubule nucleation and dynamics during mitotic-spindle assembly. Nat. Cell Biol. 3, 228–234 (2001).
Zhang, C. & Clarke, P. R. Chromatin-independent nuclear envelope assembly induced by Ran GTPase in Xenopus egg extracts. Science 288, 1429–1432 (2000).
Bamba, C., Bobinnec, Y., Fukuda, M. & Nishida, E. The GTPase Ran regulates chromosome positioning and nuclear envelope assembly in vivo. Curr. Biol. 12, 503–507 (2002).
Weis, K. Nucleocytoplasmic transport: cargo trafficking across the border. Curr. Opin. Cell Biol. 14, 328–335 (2002).
Mattaj, I. W. & Englmeier, L. Nucleocytoplasmic transport: the soluble phase. Annu. Rev. Biochem 67, 265–306 (1998).
Osaka, N. et al. Divergent mechanisms activating RAS and small GTPases Through Post-translational Modification. Front. Mol. Biosci. 8, 707439 (2021).
Campbell, S. L. & Philips, M. R. Post-translational modification of RAS proteins. Curr. Opin. Struct. Biol. 71, 180–192 (2021).
Ahearn, I., Zhou, M. & Philips, M. R. Posttranslational modifications of RAS proteins. Cold Spring Harb. Perspect. Med. 8, a031484 (2018).
Casey, P. J., Solski, P. A., Der, C. J. & Buss, J. E. p21ras is modified by a farnesyl isoprenoid. Proc. Natl Acad. Sci. USA 86, 8323–8327 (1989).
Seabra, M. C., Reiss, Y., Casey, P. J., Brown, M. S. & Goldstein, J. L. Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell 65, 429–434 (1991).
Choy, E. et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98, 69–80 (1999).
Dai, Q. et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J. Biol. Chem. 273, 15030–15034 (1998).
Cox, A. D., Der, C. J. & Philips, M. R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 21, 1819–1827 (2015).
Heo, J. & Campbell, S. L. Ras regulation by reactive oxygen and nitrogen species. Biochemistry 45, 2200–2210 (2006).
Baker, R. et al. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat. Struct. Mol. Biol. 20, 46–52 (2013).
Sasaki, A. T. et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal 4, ra13 (2011).
Baker, R. et al. Differences in the regulation of K-Ras and H-Ras isoforms by monoubiquitination. J. Biol. Chem. 288, 36856–36862 (2013).
Yang, M. H. et al. Regulation of RAS oncogenicity by acetylation. Proc. Natl Acad. Sci. USA 109, 10843–10848 (2012).
Yang, M. H. et al. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 11, 1072–1077 (2013).
Yin, G. et al. A KRAS GTPase K104Q mutant retains downstream signaling by offsetting defects in regulation. J. Biol. Chem. 292, 4446–4456 (2017).
Yin, G. et al. KRAS ubiquitination at lysine 104 retains exchange factor regulation by dynamically modulating the conformation of the interface. iScience 23, 101448 (2020).
Nair, V. V. et al. Monoubiquitination of KRAS at lysine104 and lysine147 modulates its dynamics and interaction with partner proteins. J. Phys. Chem. B 125, 4681–4691 (2021).
Bunda, S. et al. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc. Natl Acad. Sci. USA 111, E3785–E3794 (2014).
Bunda, S. et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 6, 8859 (2015).
Kano, Y. et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 10, 224 (2019).
Choi, B. H., Philips, M. R., Chen, Y., Lu, L. & Dai, W. K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J. Biol. Chem. 293, 17574–17581 (2018).
Choi, B. H., Chen, C., Philips, M. & Dai, W. RAS GTPases are modified by SUMOylation. Oncotarget 9, 4440–4450 (2018).
Johnson, C. W. et al. Regulation of GTPase function by autophosphorylation. Mol. Cell 82, 950–968.e914 (2022).
Olson, M. F. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases 9, 203–215 (2018).
Worby, C. A. et al. The fic domain: regulation of cell signaling by adenylylation. Mol. Cell 34, 93–103 (2009).
Tu, S., Wu, W. J., Wang, J. & Cerione, R. A. Epidermal growth factor-dependent regulation of Cdc42 is mediated by the Src tyrosine kinase. J. Biol. Chem. 278, 49293–49300 (2003).
Schoentaube, J., Olling, A., Tatge, H., Just, I. & Gerhard, R. Serine-71 phosphorylation of Rac1/Cdc42 diminishes the pathogenic effect of Clostridium difficile toxin A. Cell Microbiol 11, 1816–1826 (2009).
Kwon, T., Kwon, D. Y., Chun, J., Kim, J. H. & Kang, S. S. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J. Biol. Chem. 275, 423–428 (2000).
Abdrabou, A., Brandwein, D., Liu, C. & Wang, Z. Rac1 S71 Mediates the interaction between Rac1 and 14-3-3 proteins. Cells 8, 1006 (2019).
Wang, H. R. et al. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 302, 1775–1779 (2003).
Wei, J. et al. A new mechanism of RhoA ubiquitination and degradation: roles of SCF(FBXL19) E3 ligase and Erk2. Biochim. Biophys. Acta 1833, 2757–2764 (2013).
Chen, Y. et al. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 35, 841–855 (2009).
Oberoi, T. K. et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. Embo J. 31, 14–28 (2012).
Torrino, S. et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 21, 959–965 (2011).
Daugaard, M. et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat. Commun. 4, 2180 (2013).
Shinde, S. R. & Maddika, S. Post translational modifications of Rab GTPases. Small GTPases 9, 49–56 (2018).
Bailly, E. et al. Phosphorylation of two small GTP-binding proteins of the Rab family by p34cdc2. Nature 350, 715–718 (1991).
van der Sluijs, P. et al. Reversible phosphorylation-dephosphorylation determines the localization of rab4 during the cell cycle. Embo J. 11, 4379–4389 (1992).
Heo, J. M. et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 4, eaav0443 (2018).
Hanafusa, H. et al. LRRK1 phosphorylation of Rab7 at S72 links trafficking of EGFR-containing endosomes to its effector RILP. J. Cell Sci. 132, jcs228809 (2019).
Shinde, S. R. & Maddika, S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 7, 10689 (2016).
Steger, M. et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5, e12813 (2016).
Steger, M. et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6, e31012 (2017).
Lai, Y. C. et al. Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. Embo J. 34, 2840–2861 (2015).
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
Du, J. et al. Rab1-AMPylation by Legionella DrrA is allosterically activated by Rab1. Nat. Commun. 12, 460 (2021).
Müller, M. P. et al. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949 (2010).
Mukherjee, S. et al. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477, 103–106 (2011).
Oesterlin, L. K., Goody, R. S. & Itzen, A. Posttranslational modifications of Rab proteins cause effective displacement of GDP dissociation inhibitor. Proc. Natl Acad. Sci. USA 109, 5621–5626 (2012).
Goody, P. R. et al. Reversible phosphocholination of Rab proteins by Legionella pneumophila effector proteins. Embo J. 31, 1774–1784 (2012).
Modica, G. & Lefrancois, S. Post-translational modifications: how to modulate Rab7 functions. Small GTPases 11, 167–173 (2020).
Modica, G. et al. Rab7 palmitoylation is required for efficient endosome-to-TGN trafficking. J. Cell Sci. 130, 2579–2590 (2017).
Song, P., Trajkovic, K., Tsunemi, T. & Krainc, D. Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J. Neurosci. 36, 2425–2437 (2016).
Sapmaz, A. et al. USP32 regulates late endosomal transport and recycling through deubiquitylation of Rab7. Nat. Commun. 10, 1454 (2019).
Lachance, V. et al. Ubiquitylation and activation of a Rab GTPase is promoted by a β2AR-HACE1 complex. J. Cell Sci. 127, 111–123 (2014).
Qiu, J. et al. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533, 120–124 (2016).
Bhogaraju, S. et al. Phosphoribosylation oF Ubiquitin Promotes Serine Ubiquitination And Impairs Conventional Ubiquitination. Cell 167, 1636–1649.e1613 (2016).
Pucadyil, T. J. & Schmid, S. L. Conserved functions of membrane active GTPases in coated vesicle formation. Science 325, 1217–1220 (2009).
Liu, Y., Kahn, R. A. & Prestegard, J. H. Dynamic structure of membrane-anchored Arf*GTP. Nat. Struct. Mol. Biol. 17, 876–881 (2010).
Behnia, R., Panic, B., Whyte, J. R. & Munro, S. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 6, 405–413 (2004).
Setty, S. R., Strochlic, T. I., Tong, A. H., Boone, C. & Burd, C. G. Golgi targeting of ARF-like GTPase Arl3p requires its Nalpha-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 6, 414–419 (2004).
Yang, F., Li, T., Peng, Z., Liu, Y. & Guo, Y. The amphipathic helices of Arfrp1 and Arl14 are sufficient to determine subcellular localizations. J. Biol. Chem. 295, 16643–16654 (2020).
Hofmann, I. & Munro, S. An N-terminally acetylated Arf-like GTPase is localised to lysosomes and affects their motility. J. Cell Sci. 119, 1494–1503 (2006).
Roy, K. et al. Palmitoylation of the ciliary GTPase ARL13b is necessary for its stability and its role in cilia formation. J. Biol. Chem. 292, 17703–17717 (2017).
Li, Y. et al. SUMOylation of the small GTPase ARL-13 promotes ciliary targeting of sensory receptors. J. Cell Biol. 199, 589–598 (2012).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009).
de Boor, S. et al. Small GTP-binding protein Ran is regulated by posttranslational lysine acetylation. Proc. Natl Acad. Sci. USA 112, E3679–E3688 (2015).
Knyphausen, P., Kuhlmann, N., de Boor, S. & Lammers, M. Lysine-acetylation as a fundamental regulator of Ran function: Implications for signaling of proteins of the Ras-superfamily. Small GTPases 6, 189–195 (2015).
Bao, X. et al. Mitosis-specific acetylation tunes Ran effector binding for chromosome segregation. J. Mol. Cell Biol. 10, 18–32 (2018).
Chen, K., Zhang, Y., Qian, L. & Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 14, 116 (2021).
Prior, I. A., Lewis, P. D. & Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012).
Tajan, M., Paccoud, R., Branka, S., Edouard, T. & Yart, A. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr. Rev. 39, 676–700 (2018).
Hebron, K. E., Hernandez, E. R. & Yohe, M. E. The RASopathies: from pathogenetics to therapeutics. Dis. Model Mech. 15, dmm049107 (2022).
Yi, J. S., Perla, S. & Bennett, A. M. An assessment of the therapeutic landscape for the treatment of heart disease in the RASopathies. Cardiovasc. Drugs Ther. (2022). https://doi.org/10.1007/s10557-022-07324-0.
Jafry, M. & Sidbury, R. RASopathies. Clin. Dermatol 38, 455–461 (2020).
Kim, Y. E. & Baek, S. T. Neurodevelopmental aspects of RASopathies. Mol. Cells 42, 441–447 (2019).
Siskind, M. S., McCoy, C. E., Chobanian, A. & Schwartz, J. H. Regulation of intracellular calcium by cell pH in vascular smooth muscle cells. Am. J. Physiol. 256, C234–C240 (1989).
Eleveld, T. F. et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 47, 864–871 (2015).
Yan, C. & Theodorescu, D. RAL GTPases: biology and potential as therapeutic targets in cancer. Pharm. Rev. 70, 1–11 (2018).
Moghadam, A. R. et al. Ral signaling pathway in health and cancer. Cancer Med. 6, 2998–3013 (2017).
Oxford, G. et al. RalA and RalB: antagonistic relatives in cancer cell migration. Cancer Res. 65, 7111–7120 (2005).
Kitajima, S. & Barbie, D. A. RASA1/NF1-mutant lung cancer: racing to the clinic? Clin. Cancer Res. 24, 1243–1245 (2018).
Crosas-Molist, E. et al. Rho GTPase signaling in cancer progression and dissemination. Physiol. Rev. 102, 455–510 (2022).
Lamas, I., Merlini, L., Vještica, A., Vincenzetti, V. & Martin, S. G. Optogenetics reveals Cdc42 local activation by scaffold-mediated positive feedback and Ras GTPase. PLoS Biol. 18, e3000600 (2020).
Gong, L. et al. The vasorelaxant mechanisms of a Rho kinase inhibitor DL0805 in rat thoracic aorta. Molecules 17, 5935–5944 (2012).
Noda, K. et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS One 9, e110446 (2014).
Chen, S., Shu, L., Zhao, R. & Zhao, Y. Molecular dynamics simulations reveal the activation mechanism of mutations G12V and Q61L of Cdc42. Proteins 90, 1376–1389 (2022).
Huang, D. et al. Cdc42 promotes thyroid cancer cell proliferation and migration and tumor-associated macrophage polarization through the PTEN/AKT pathway. J. Biochem Mol. Toxicol. 36, e23115 (2022).
Zhang, Y., Liu, J., Luan, G. & Wang, X. Inhibition of the small GTPase Cdc42 in regulation of epileptic-seizure in rats. Neuroscience 289, 381–391 (2015).
Winge, M. C. et al. RAC1 activation drives pathologic interactions between the epidermis and immune cells. J. Clin. Investig. 126, 2661–2677 (2016).
Liu, L. et al. KDM6A-ARHGDIB axis blocks metastasis of bladder cancer by inhibiting Rac1. Mol. Cancer 20, 77 (2021).
Dilasser, F. et al. Smooth muscle Rac1 contributes to pulmonary hypertension. Br. J. Pharm. 179, 3418–3429 (2022).
Healy, A. et al. Statins disrupt macrophage Rac1 regulation leading to increased atherosclerotic plaque calcification. Arterioscler Thromb. Vasc. Biol. 40, 714–732 (2020).
Maruyama, K. et al. Bone-protective functions of Netrin 1 protein. J. Biol. Chem. 291, 23854–23868 (2016).
Margheri, F. et al. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, beta2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: failure of association in systemic sclerosis endothelial cells. Arthritis Rheum. 54, 3926–3938 (2006).
Kiral, F. R., Kohrs, F. E., Jin, E. J. & Hiesinger, P. R. Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol. 28, R471–r486 (2018).
Wang, S., Hu, C., Wu, F. & He, S. Rab25 GTPase: functional roles in cancer. Oncotarget 8, 64591–64599 (2017).
Chua, C. E. & Tang, B. L. The role of the small GTPase Rab31 in cancer. J. Cell Mol. Med. 19, 1–10 (2015).
Corallino, S. et al. A RAB35-p85/PI3K axis controls oscillatory apical protrusions required for efficient chemotactic migration. Nat. Commun. 9, 1475 (2018).
Jespersgaard, C. et al. A missense mutation in RAB28 in a family with cone-rod dystrophy and postaxial polydactyly prevents localization of RAB28 to the primary cilium. Investig. Ophthalmol. Vis. Sci. 61, 29 (2020).
Cioni, J. M. et al. Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72.e15 (2019).
Krzewski, K. & Cullinane, A. R. Evidence for defective Rab GTPase-dependent cargo traffic in immune disorders. Exp. Cell Res. 319, 2360–2367 (2013).
Jenkins, D. et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 80, 1162–1170 (2007).
Ménasché, G. et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 25, 173–176 (2000).
Osanai, K. et al. A mutation in Rab38 small GTPase causes abnormal lung surfactant homeostasis and aberrant alveolar structure in mice. Am. J. Pathol. 173, 1265–1274 (2008).
Osanai, K. et al. Altered lung surfactant system in a Rab38-deficient rat model of Hermansky-Pudlak syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L243–L251 (2010).
Yuen, H. F. et al. Ran GTPase promotes cancer progression via Met recepto-rmediated downstream signaling. Oncotarget 7, 75854–75864 (2016).
Wang, X. et al. Regulation of the small GTPase Ran by miR-802 modulates proliferation and metastasis in colorectal cancer cells. Br. J. Cancer 122, 1695–1706 (2020).
Cekan, P. et al. RCC1-dependent activation of Ran accelerates cell cycle and DNA repair, inhibiting DNA damage-induced cell senescence. Mol. Biol. Cell 27, 1346–1357 (2016).
Casalou, C., Ferreira, A. & Barral, D. C. The role of ARF family proteins and their regulators and effectors in cancer progression: a therapeutic perspective. Front. Cell Dev. Biol. 8, 217 (2020).
Falace, A. et al. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc. Natl Acad. Sci. USA 111, 2337–2342 (2014).
Wang, Y., Zhao, W., Liu, X., Guan, G. & Zhuang, M. ARL3 is downregulated and acts as a prognostic biomarker in glioma. J. Transl. Med. 17, 210 (2019).
Hu, J. et al. Small GTPase Arf6 regulates diabetes-induced cholesterol accumulation in podocytes. J. Cell Physiol. 234, 23559–23570 (2019).
Folly-Klan, M. et al. On the use of Legionella/Rickettsia chimeras to investigate the structure and regulation of Rickettsia effector RalF. J. Struct. Biol. 189, 98–104 (2015).
Rennoll-Bankert, K. E. et al. Which way in? The RalF Arf-GEF orchestrates rickettsia host cell invasion. PLoS Pathog. 11, e1005115 (2015).
Cevik, S. et al. Active transport and diffusion barriers restrict Joubert Syndrome-associated ARL13B/ARL-13 to an Inv-like ciliary membrane subdomain. PLoS Genet. 9, e1003977 (2013).
Fan, Y. et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat. Genet. 36, 989–993 (2004).
Wiens, C. J. et al. Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. J. Biol. Chem. 285, 16218–16230 (2010).
Sané, A. T. et al. Understanding chylomicron retention disease through Sar1b Gtpase gene disruption: insight from cell culture. Arterioscler. Thromb. Vasc. Biol. 37, 2243–2251 (2017).
Blair, H. A. Sotorasib: first approval. Drugs 81, 1573–1579 (2021).
Rosen, N. Finally, effective inhibitors of mutant KRAS. N. Engl. J. Med. 384, 2447–2449 (2021).
Maurer, T. et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl Acad. Sci. USA 109, 5299–5304 (2012).
Sun, Q. et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew. Chem. Int. Ed. Engl. 51, 6140–6143 (2012).
Shima, F. et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl Acad. Sci. USA 110, 8182–8187 (2013).
Kessler, D. et al. Drugging an undruggable pocket on KRAS. Proc. Natl Acad. Sci. USA 116, 15823–15829 (2019).
Marshall, C. B. et al. NMR in integrated biophysical drug discovery for RAS: past, present, and future. J. Biomol. NMR 74, 531–554 (2020).
Yin, G. et al. Early-stage structure-based drug discovery for small GTPases by NMR spectroscopy. Pharm. Ther. 236, 108110 (2022).
Quevedo, C. E. et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat. Commun. 9, 3169 (2018).
Cruz-Migoni, A. et al. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl Acad. Sci. USA 116, 2545–2550 (2019).
Welsch, M. E. et al. Multivalent small-molecule pan-RAS inhibitors. Cell 168, 878–889.e829 (2017).
Bergner, A. et al. KRAS binders hidden in nature. Chemistry 25, 12037–12041 (2019).
Taveras, A. G. et al. Ras oncoprotein inhibitors: the discovery of potent, ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorg. Med. Chem. 5, 125–133 (1997).
Ganguly, A. K. et al. Interaction of a novel GDP exchange inhibitor with the Ras protein. Biochemistry 37, 15631–15637 (1998).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Adagrasib response remains strong in NSCLC. Cancer Discov. 12, OF1 (2022).
Jänne, P. A. et al. Adagrasib in non-small-cell lung cancer harboring a KRAS(G12C) mutation. N. Engl. J. Med. 387, 120–131 (2022).
Janes, M. R. et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e517 (2018).
Wang, J. et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist 27, 536–e553 (2022).
Kwan, A. K., Piazza, G. A., Keeton, A. B. & Leite, C. A. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J. Exp. Clin. Cancer Res. 41, 27 (2022).
Ostrem, J. M. & Shokat, K. M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov. 15, 771–785 (2016).
Zhang, Z., Guiley, K. Z. & Shokat, K. M. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat. Chem. Biol. 18, 1177–1183 (2022).
Zhang, Z., Morstein, J., Ecker, A. K., Guiley, K. Z. & Shokat, K. M. Chemoselective covalent modification of K-Ras(G12R) with a small molecule electrophile. J. Am. Chem. Soc. 144, 15916–15921 (2022).
Zheng, Q., Peacock, D. M. & Shokat, K. M. Drugging the next undruggable KRAS Allele-Gly12Asp. J. Med. Chem. 65, 3119–3122 (2022).
Kemp, S. B. et al. Efficacy of a small molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov.13, 298−311 (2022).
Vasta, J. D. et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 18, 596–604 (2022).
Marshall, C. B. & Ikura, M. Hitting the hotspots. Nat. Chem. Biol. 18, 578–579 (2022).
Awad, M. M. et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 384, 2382–2393 (2021).
Hunter, J. C. et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl Acad. Sci. USA 111, 8895–8900 (2014).
Lim, S. M. et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int Ed. Engl. 53, 199–204 (2014).
Yan, C. et al. Discovery and characterization of small molecules that target the GTPase Ral. Nature 515, 443–447 (2014).
Pei, D., Chen, K. & Liao, H. Targeting Ras with macromolecules. Cold Spring Harb. Perspect. Med. 8, a031476 (2018).
Sakamoto, K. et al. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun. 484, 605–611 (2017).
Sogabe, S. et al. Crystal structure of a human K-Ras G12D mutant in complex with GDP and the cyclic inhibitory peptide KRpep-2d. ACS Med. Chem. Lett. 8, 732–736 (2017).
Sakamoto, K., Masutani, T. & Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 10, 21671 (2020).
Upadhyaya, P. et al. Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Angew. Chem. Int Ed. Engl. 54, 7602–7606 (2015).
Buyanova, M. et al. Discovery of a bicyclic peptidyl pan-Ras inhibitor. J. Med. Chem. 64, 13038–13053 (2021).
Tetley, G. J. N. et al. The discovery and maturation of peptide biologics targeting the small G-protein Cdc42: a bioblockade for Ras-driven signaling. J. Biol. Chem. 295, 2866–2884 (2020).
Zhang, Z. et al. GTP-state-selective cyclic peptide ligands of K-Ras(G12D) block its interaction with Raf. ACS Cent. Sci. 6, 1753–1761 (2020).
Patgiri, A., Yadav, K. K., Arora, P. S. & Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 7, 585–587 (2011).
Leshchiner, E. S. et al. Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl Acad. Sci. USA 112, 1761–1766 (2015).
Hong, S. H. et al. A Sos proteomimetic as a pan-Ras inhibitor. Proc. Natl Acad. Sci. USA 118, e2101027118 (2021).
McGee, J. H. et al. Exceptionally high-affinity Ras binders that remodel its effector domain. J. Biol. Chem. 293, 3265–3280 (2018).
Hurd, C. A. et al. Affinity maturation of the RLIP76 Ral binding domain to inform the design of stapled peptides targeting the Ral GTPases. J. Biol. Chem. 296, 100101 (2021).
Muttenthaler, M., King, G. F., Adams, D. J. & Alewood, P. F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 20, 309–325 (2021).
Guillard, S. et al. Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat. Commun. 8, 16111 (2017).
Bery, N. et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 10, 2607 (2019).
Spencer-Smith, R. et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 13, 62–68 (2017).
Teng, K. W. et al. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun. 12, 2656 (2021).
Tanaka, T. & Rabbitts, T. H. Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. Embo J. 22, 1025–1035 (2003).
Tanaka, T., Williams, R. L. & Rabbitts, T. H. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. Embo J. 26, 3250–3259 (2007).
Cetin, M. et al. RasIns: genetically encoded intrabodies of activated Ras proteins. J. Mol. Biol. 429, 562–573 (2017).
Yang, J. L. et al. A novel anti-p21Ras scFv antibody reacting specifically with human tumour cell lines and primary tumour tissues. BMC Cancer 16, 131 (2016).
Haza, K. Z. et al. RAS-inhibiting biologics identify and probe druggable pockets including an SII-α3 allosteric site. Nat. Commun. 12, 4045 (2021).
Kauke, M. J. et al. An engineered protein antagonist of K-Ras/B-Raf interaction. Sci. Rep. 7, 5831 (2017).
Wallon, L. et al. Inhibition of RAS-driven signaling and tumorigenesis with a pan-RAS monobody targeting the Switch I/II pocket. Proc. Natl Acad. Sci. USA 119, e2204481119 (2022).
Bond, M. J., Chu, L., Nalawansha, D. A., Li, K. & Crews, C. M. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent. Sci. 6, 1367–1375 (2020).
Zeng, M. et al. Exploring targeted degradation strategy for oncogenic KRAS(G12C). Cell Chem. Biol. 27, 19–31.e16 (2020).
Bery, N., Miller, A. & Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 11, 3233 (2020).
Lim, S. et al. Exquisitely specific anti-KRAS biodegraders inform on the cellular prevalence of nucleotide-loaded states. ACS Cent. Sci. 7, 274–291 (2021).
Zhou, Y., Prakash, P., Gorfe, A. A. & Hancock, J. F. Ras and the plasma membrane: a complicated relationship. Cold Spring Harb. Perspect. Med. 8, a031831 (2018).
Zhou, Y. et al. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 168, 239–251.e216 (2017).
Fang, Z. et al. Inhibition of K-RAS4B by a unique mechanism of action: stabilizing membrane-dependent occlusion of the effector-binding site. Cell Chem. Biol. 25, 1327–1336.e1324 (2018).
Wang, P. et al. Scaffold repurposing of fendiline: Identification of potent KRAS plasma membrane localization inhibitors. Eur. J. Med. Chem. 217, 113381 (2021).
Gupta, A. K. et al. Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem. Biol. Drug Des. 94, 1441–1456 (2019).
Davies, C. W. et al. Conformation-locking antibodies for the discovery and characterization of KRAS inhibitors. Nat. Biotechnol. 40, 769–778 (2022).
Schreiber, S. L. The rise of molecular glues. Cell 184, 3–9 (2021).
Zhang, Z. & Shokat, K. M. Bifunctional small-molecule ligands of K-Ras induce its association with immunophilin.Proteins Angew. Chem. Int. Ed. Engl. 58, 16314–16319 (2019).
Fleming, A. M., Zhou, J., Wallace, S. S. & Burrows, C. J. A role for the fifth G-track in G-quadruplex forming oncogene promoter sequences during oxidative stress: do these “spare tires” have an evolved function? ACS Cent. Sci. 1, 226–233 (2015).
Pramanik, S. et al. The human AP-endonuclease 1 (APE1) is a DNA G-quadruplex structure binding protein and regulates KRAS expression in pancreatic ductal adenocarcinoma cells. Nucleic Acids Res. 50, 3394–3412 (2022).
Teng, F. Y. et al. G-quadruplex DNA: a novel target for drug design. Cell Mol. Life Sci. 78, 6557–6583 (2021).
Wang, K. B. et al. Structural insight into the bulge-containing KRAS oncogene promoter G-quadruplex bound to berberine and coptisine. Nat. Commun. 13, 6016 (2022).
Lin, Y. & Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 10, 991–1010 (2015).
Murphy, N. P., Mott, H. R. & Owen, D. Progress in the therapeutic inhibition of Cdc42 signalling. Biochem. Soc. Trans. 49, 1443–1456 (2021).
Guiler, W., Koehler, A., Boykin, C. & Lu, Q. Pharmacological modulators of small GTPases of rho family in neurodegenerative diseases. Front. Cell Neurosci. 15, 661612 (2021).
Gao, Y., Dickerson, J. B., Guo, F., Zheng, J. & Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl Acad. Sci. USA 101, 7618–7623 (2004).
Shutes, A. et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 282, 35666–35678 (2007).
Onesto, C., Shutes, A., Picard, V., Schweighoffer, F. & Der, C. J. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 439, 111–129 (2008).
Friesland, A. et al. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl Acad. Sci. USA 110, 1261–1266 (2013).
Aguilar, B. J. et al. Inhibition of Cdc42-intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth. Cancer Biol. Ther. 20, 740–749 (2019).
Liu, W. et al. Rational identification of a Cdc42 inhibitor presents a new regimen for long-term hematopoietic stem cell mobilization. Leukemia 33, 749–761 (2019).
Jahid, S. et al. Structure-based design of CDC42 effector interaction inhibitors for the treatment of cancer. Cell Rep. 39, 110641 (2022).
Sayedyahossein, S. et al. Discovery of small molecule inhibitors that effectively disrupt IQGAP1-Cdc42 interaction in breast cancer cells. Sci. Rep. 12, 17372 (2022).
Sun, Z. et al. Covalent inhibitors allosterically block the activation of rho family proteins and suppress cancer cell invasion. Adv. Sci. 7, 2000098 (2020).
Chen, H. Y., Yang, Y. M., Stevens, B. M. & Noble, M. Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol. Med. 5, 723–736 (2013).
Lippincott-Schwartz, J. et al. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60, 821–836 (1990).
Chardin, P. & McCormick, F. Brefeldin A: the advantage of being uncompetitive. Cell 97, 153–155 (1999).
Mossessova, E., Corpina, R. A. & Goldberg, J. Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 12, 1403–1411 (2003).
Lu, X. X. et al. Semi-synthesis, cytotoxic evaluation, and structure-activity relationships of brefeldin a derivatives with antileukemia activity. Mar. Drugs 20, 26 (2021).
Viaud, J. et al. Structure-based discovery of an inhibitor of Arf activation by Sec7 domains through targeting of protein-protein complexes. Proc. Natl Acad. Sci. USA 104, 10370–10375 (2007).
Macia, E. et al. Chlortetracycline, a novel Arf inhibitor that decreases the Arf6-dependent invasive properties of breast cancer cells. Molecules 26, 969 (2021).
Benabdi, S. et al. Family-wide analysis of the inhibition of Arf guanine nucleotide exchange factors with small molecules: evidence of unique inhibitory profiles. Biochemistry 56, 5125–5133 (2017).
Jordan, K. L., Koss, D. J., Outeiro, T. F. & Giorgini, F. Therapeutic targeting of Rab GTPases: relevance for Alzheimer’s disease. Biomedicines 10, 1141 (2022).
Agola, J. O. et al. A competitive nucleotide binding inhibitor: in vitro characterization of Rab7 GTPase inhibition. ACS Chem. Biol. 7, 1095–1108 (2012).
Qin, Y. et al. CID1067700, a late endosome GTPase Rab7 receptor antagonist, attenuates brain atrophy, improves neurologic deficits and inhibits reactive astrogliosis in rat ischemic stroke. Acta Pharm. Sin. 40, 724–736 (2019).
Hatstat, A. K. et al. Chemoproteomic-enabled characterization of small GTPase Rab1a as a target of an N-arylbenzimidazole ligand’s rescue of Parkinson’s-associated cell toxicity. RSC Chem. Biol. 3, 96–111 (2022).
Kumar, A. P., Verma, C. S. & Lukman, S. Structural dynamics and allostery of Rab proteins: strategies for drug discovery and design. Brief Bioinform. 22, 270–287 (2021).
Kumar, A. P. & Lukman, S. Allosteric binding sites in Rab11 for potential drug candidates. PLoS One 13, e0198632 (2018).
Haggag, Y. A. et al. Nano-encapsulation of a novel anti-Ran-GTPase peptide for blockade of regulator of chromosome condensation 1 (RCC1) function in MDA-MB-231 breast cancer cells. Int. J. Pharm. 521, 40–53 (2017).
Haggag, Y. A. et al. Novel Ran-RCC1 inhibitory peptide-loaded nanoparticles have anti-cancer efficacy in vitro and in vivo. Cancers 11, 222 (2019).
Boudhraa, Z., Carmona, E., Provencher, D. & Mes-Masson, A. M. Ran GTPase: a key player in tumor progression and metastasis. Front. Cell Dev. Biol. 8, 345 (2020).
Dang, C. V., Reddy, E. P., Shokat, K. M. & Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508 (2017).
James, G. L., Brown, M. S., Cobb, M. H. & Goldstein, J. L. Benzodiazepine peptidomimetic BZA-5B interrupts the MAP kinase activation pathway in H-Ras-transformed Rat-1 cells, but not in untransformed cells. J. Biol. Chem. 269, 27705–27714 (1994).
James, G. L., Goldstein, J. L. & Brown, M. S. Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J. Biol. Chem. 270, 6221–6226 (1995).
Fiordalisi, J. J. et al. High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J. Biol. Chem. 278, 41718–41727 (2003).
Whyte, D. B. et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 272, 14459–14464 (1997).
Rowell, C. A., Kowalczyk, J. J., Lewis, M. D. & Garcia, A. M. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 272, 14093–14097 (1997).
Ho, A. L. et al. Tipifarnib in head and neck squamous cell carcinoma with HRAS mutations. J. Clin. Oncol. 39, 1856–1864 (2021).
Chandra, A. et al. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 14, 148–158 (2011).
Zimmermann, G. et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 497, 638–642 (2013).
Papke, B. et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat. Commun. 7, 11360 (2016).
Martín-Gago, P., Fansa, E. K., Wittinghofer, A. & Waldmann, H. Structure-based development of PDEδ inhibitors. Biol. Chem. 398, 535–545 (2017).
Martín-Gago, P. et al. A PDE6δ-KRas inhibitor chemotype with up to seven H-bonds and picomolar affinity that prevents efficient inhibitor release by Arl2. Angew. Chem. Int. Ed. Engl. 56, 2423–2428 (2017).
Leung, E. L. et al. Identification of a new inhibitor of KRAS-PDEδ interaction targeting KRAS mutant nonsmall cell lung cancer. Int. J. Cancer 145, 1334–1345 (2019).
Chang, L. C. et al. Signaling mechanisms of inhibition of phospholipase D activation by CHS-111 in formyl peptide-stimulated neutrophils. Biochem. Pharm. 81, 269–278 (2011).
Pelish, H. E. et al. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat. Chem. Biol. 2, 39–46 (2006).
van Beek, E., Pieterman, E., Cohen, L., Löwik, C. & Papapoulos, S. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem. Biophys. Res. Commun. 264, 108–111 (1999).
Bergstrom, J. D., Bostedor, R. G., Masarachia, P. J., Reszka, A. A. & Rodan, G. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch. Biochem. Biophys. 373, 231–241 (2000).
Roelofs, A. J. et al. Selective inhibition of Rab prenylation by a phosphonocarboxylate analogue of risedronate induces apoptosis, but not S-phase arrest, in human myeloma cells. Int. J. Cancer 119, 1254–1261 (2006).
Okamoto, S. et al. Zoledronic acid induces apoptosis and S-phase arrest in mesothelioma through inhibiting Rab family proteins and topoisomerase II actions. Cell Death Dis. 5, e1517 (2014).
Cox, A. D. & Der, C. J. Ras history: the saga continues. Small GTPases 1, 2–27 (2010).
Haigis, K. M. et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 40, 600–608 (2008).
Humpton, T. J. et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 9, 1268–1287 (2019).
Kortlever, R. M. et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315.e1314 (2017).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337.e310 (2018).
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J. & Der, C. J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014).
Simanshu, D. K., Nissley, D. V. & McCormick, F. RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017).
Chardin, P. et al. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338–1343 (1993).
Burns, M. C. et al. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl Acad. Sci. USA 111, 3401–3406 (2014).
Abbott, J. R. et al. Discovery of aminopiperidine indoles that activate the guanine nucleotide exchange factor SOS1 and modulate RAS signaling. J. Med. Chem. 61, 6002–6017 (2018).
Hodges, T. R. et al. Discovery and structure-based optimization of benzimidazole-derived activators of SOS1-mediated nucleotide exchange on RAS. J. Med. Chem. 61, 8875–8894 (2018).
Burns, M. C. et al. High-throughput screening identifies small molecules that bind to the RAS:SOS:RAS complex and perturb RAS signaling. Anal. Biochem. 548, 44–52 (2018).
Abbott, J. R. et al. Discovery of quinazolines that activate SOS1-mediated nucleotide exchange on RAS. ACS Med. Chem. Lett. 9, 941–946 (2018).
Sarkar, D. et al. Discovery of sulfonamide-derived agonists of SOS1-mediated nucleotide exchange on RAS using fragment-based methods. J. Med. Chem. 63, 8325–8337 (2020).
Hillig, R. C. et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc. Natl Acad. Sci. USA 116, 2551–2560 (2019).
Hofmann, M. H. et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov. 11, 142–157 (2021).
Ramharter, J. et al. One atom makes all the difference: getting a foot in the door between SOS1 and KRAS. J. Med. Chem. 64, 6569–6580 (2021).
Jaaks, P. et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 603, 166–173 (2022).
Lopez, J. S. & Banerji, U. Combine and conquer: challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 14, 57–66 (2017).
Shang, X. et al. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl Acad. Sci. USA 110, 3155–3160 (2013).
Xie, X. et al. Suppression of breast cancer metastasis through the inactivation of ADP-ribosylation factor 1. Oncotarget 7, 58111–58120 (2016).
Zhao, H. et al. Endothelial Robo4 suppresses breast cancer growth and metastasis through regulation of tumor angiogenesis. Mol. Oncol. 10, 272–281 (2016).
Pan, T. et al. Cytohesins/ARNO: the function in colorectal cancer cells. PLoS One 9, e90997 (2014).
Bill, A. et al. Anti-proliferative effect of cytohesin inhibition in gefitinib-resistant lung cancer cells. PLoS One 7, e41179 (2012).
Thomas, E. K. et al. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell 12, 467–478 (2007).
Montalvo-Ortiz, B. L. et al. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 287, 13228–13238 (2012).
Castillo-Pichardo, L. et al. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 7, 546–555 (2014).
Martin, H. et al. Pak and Rac GTPases promote oncogenic KIT-induced neoplasms. J. Clin. Investig. 123, 4449–4463 (2013).
Okada, T. et al. Integrin-α10 dependency Identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discov. 6, 1148–1165 (2016).
Zins, K., Lucas, T., Reichl, P., Abraham, D. & Aharinejad, S. A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS One 8, e74924 (2013).
Zins, K., Gunawardhana, S., Lucas, T., Abraham, D. & Aharinejad, S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of PAK1 activity. J. Transl. Med. 11, 295 (2013).
Xu, G. F. et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62, 599–608 (1990).
Trahey, M. & McCormick, F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 238, 542–545 (1987).
Moore, A. R., Rosenberg, S. C., McCormick, F. & Malek, S. RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 19, 533–552 (2020).
Hunter, J. C. et al. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 13, 1325–1335 (2015).
Cox, A. D. & Der, C. J. Filling in the GAPs in understanding RAS. Science 374, 152–153 (2021).
Glembotski, C. C. The role of the unfolded protein response in the heart. J. Mol. Cell Cardiol. 44, 453–459 (2008).
Wong, G. S. et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 24, 968–977 (2018).
Ruess, D. A. et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 24, 954–960 (2018).
Mainardi, S. et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 24, 961–967 (2018).
Mai, T. T. & Lito, P. A treatment strategy for KRAS-driven tumors. Nat. Med. 24, 902–904 (2018).
Yi, T. et al. Phosphatase inhibitor, sodium stibogluconate, in combination with interferon (IFN) alpha 2b: phase I trials to identify pharmacodynamic and clinical effects. Oncotarget 2, 1155–1164 (2011).
Yin, C. et al. Pharmacological targeting of STK19 inhibits oncogenic NRAS-driven melanomagenesis. Cell 176, 1113–1127.e1116 (2019).
Asquith, C. R. M. & Temme, L. STK19: a new target for NRAS-driven cancer. Nat. Rev. Drug Discov. 19, 579 (2020).
Qian, L. et al. Targeting NRAS-mutant cancers with the selective STK19 kinase inhibitor chelidonine. Clin. Cancer Res. 26, 3408–3419 (2020).
Sanclemente, M. et al. c-RAF ablation induces regression of advanced Kras/Trp53 mutant lung adenocarcinomas by a mechanism independent of MAPK signaling. Cancer Cell 33, 217–228.e214 (2018).
Wang, T. et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168, 890–903.e815 (2017).
Blasco, M. T. et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and C-RAF. Cancer Cell 35, 573–587.e576 (2019).
Hatzivassiliou, G. et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 (2010).
Poulikakos, P. I., Zhang, C., Bollag, G., Shokat, K. M. & Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010).
Yen, I. et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature 594, 418–423 (2021).
Yao, Z. et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 28, 370–383 (2015).
Peng, S. B. et al. Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28, 384–398 (2015).
Monaco, K. A. et al. LXH254, a potent and selective ARAF-sparing inhibitor of BRAF and CRAF for the treatment of MAPK-driven tumors. Clin. Cancer Res. 27, 2061–2073 (2021).
Sun, Y. et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 19, 774–785 (2017).
Karoulia, Z. et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell 30, 485–498 (2016).
Desai, J. et al. Phase I, open-label, dose-escalation/dose-expansion study of lifirafenib (BGB-283), an RAF family kinase inhibitor, in patients with solid tumors. J. Clin. Oncol. 38, 2140–2150 (2020).
Athuluri-Divakar, S. K. et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell 165, 643–655 (2016).
Yaeger, R. & Corcoran, R. B. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 9, 329–341 (2019).
Ascierto, P. A. et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 14, 249–256 (2013).
Sullivan, R. J. et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a phase I dose-escalation and expansion study. Cancer Disco. 8, 184–195 (2018).
Chia, S. et al. Novel agents and associated toxicities of inhibitors of the pi3k/Akt/mtor pathway for the treatment of breast cancer. Curr. Oncol. 22, 33–48 (2015).
Esposito, A., Viale, G. & Curigliano, G. Safety, tolerability, and management of toxic effects of phosphatidylinositol 3-kinase inhibitor treatment in patients with cancer: a review. JAMA Oncol. 5, 1347–1354 (2019).
Michels, B. E. et al. Pooled in vitro and in vivo CRISPR-Cas9 screening identifies tumor suppressors in human colon organoids. Cell Stem Cell 26, 782–792.e787 (2020).
Takahashi, N. et al. 3D culture models with CRISPR screens reveal hyperactive NRF2 as a prerequisite for spheroid formation via regulation of proliferation and ferroptosis. Mol. Cell 80, 828–844.e826 (2020).
Yau, E. H. et al. Genome-wide CRISPR screen for essential cell growth mediators in mutant KRAS colorectal cancers. Cancer Res. 77, 6330–6339 (2017).
Sulahian, R. et al. Synthetic lethal interaction of SHOC2 depletion with MEK Inhibition in RAS-driven cancers. Cell Rep. 29, 118–134.e118 (2019).
Szlachta, K. et al. CRISPR knockout screening identifies combinatorial drug targets in pancreatic cancer and models cellular drug response. Nat. Commun. 9, 4275 (2018).
Yu, C. et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 41, 191–203 (2022).
Han, K. et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141 (2020).
Xue, J. Y. et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425 (2020).
Ku, A. A. et al. Integration of multiple biological contexts reveals principles of synthetic lethality that affect reproducibility. Nat. Commun. 11, 2375 (2020).
Guo, J. Y. et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 (2011).
Yang, S. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 (2011).
Bryant, K. L. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 25, 628–640 (2019).
Kinsey, C. G. et al. Protective autophagy elicited by RAF → MEK → ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 25, 620–627 (2019).
Yun, J. et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 (2009).
Racker, E., Resnick, R. J. & Feldman, R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc. Natl Acad. Sci. USA 82, 3535–3538 (1985).
Kole, H. K., Resnick, R. J., Van Doren, M. & Racker, E. Regulation of 6-phosphofructo-1-kinase activity in ras-transformed rat-1 fibroblasts. Arch. Biochem. Biophys. 286, 586–590 (1991).
Amendola, C. R. et al. KRAS4A directly regulates hexokinase 1. Nature 576, 482–486 (2019).
Ying, H. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012).
Stincone, A. et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 90, 927–963 (2015).
Son, J. et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013).
Hu, K. et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 130, 1752–1766 (2020).
Padanad, M. S. et al. Fatty ACID OXIDATION MEDIATED by Acyl-CoA synthetase long chain 3 is required for mutant KRAS lung tumorigenesis. Cell Rep. 16, 1614–1628 (2016).
Gouw, A. M. et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl Acad. Sci. USA 114, 4300–4305 (2017).
Bartolacci, C. et al. Targeting de novo lipogenesis and the lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 13, 4327 (2022).
Liu, J. et al. Metabolic enzyme LDHA activates Rac1 GTPase as a noncanonical mechanism to promote cancer. Nat. Metab. 4,1830–1846 (2022).
Osaka, N. & Sasaki, A.T. Beyond Warburg: LDHA activates RAC for tumour growth. Nat. Metab. 4,1623−1625 (2022).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016).
Jordan, E. J. et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 7, 596–609 (2017).
Giannakis, M. et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 15, 857–865 (2016).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
Kim, D., Xue, J. Y. & Lito, P. Targeting KRAS(G12C): from inhibitory mechanism to modulation of antitumor effects in patients. Cell 183, 850–859 (2020).
Nakajima, E. C. et al. FDA approval summary: sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin. Cancer Res. 28, 1482–1486 (2022).
Amgen® in LUMAKRAS®/LUMYKRAS® (Sotorasib) demonstrates superior progression-free survival over decetaxel in first positive phase 3 trial of a KRas G12C inhibitor in non-small cell lung cancer. (2022).
ClinicalTrails.gov in study to compare AMG 510 “Proposed INN Sotorasib” With Docetaxel in Non Small Cell Lung Cancer (NSCLC) (CodeBreak 200) (2022).
Skoulidis, F. et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 5, 860–877 (2015).
Skoulidis, F. & Heymach, J. V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19, 495–509 (2019).
Arbour, K. C. et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin. Cancer Res. 24, 334–340 (2018).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Tanaka, N. et al. Clinical acquired resistance to KRAS(G12C) Inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Disco. 11, 1913–1922 (2021).
Zhao, Y. et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 599, 679–683 (2021).
Adachi, Y. et al. Epithelial-to-mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C-mutant non-small cell lung cancer. Clin. Cancer Res. 26, 5962–5973 (2020).
Amodio, V. et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 10, 1129–1139 (2020).
Koleilat, M. K. & Kwong, L. N. Same name, different game: EGFR drives intrinsic KRAS(G12C) inhibitor resistance in colorectal cancer. Cancer Discov. 10, 1094–1096 (2020).
Ryan, M. B. et al. KRAS(G12C)-independent feedback activation of wild-type RAS constrains KRAS(G12C) inhibitor efficacy. Cell Rep. 39, 110993 (2022).
Young, A., Lou, D. & McCormick, F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 3, 112–123 (2013).
Tsai, Y. S. et al. Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J. Clin. Investig. 132, e155523 (2022).
Acker, F. et al. KRAS mutations in squamous cell carcinomas of the lung. Front Oncol. 11, 788084 (2021).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Hamarsheh, S., Groß, O., Brummer, T. & Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 11, 5439 (2020).
Dias Carvalho, P. et al. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res. 78, 7–14 (2018).
Ischenko, I. et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat. Commun. 12, 1482 (2021).
Punekar, S. R., Velcheti, V., Neel, B. G. & Wong, K. K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol, 37,1–19 (2022).
Bear, A. S. et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat. Commun. 12, 4365 (2021).
Tran, E. et al. T-Cell Transfer Therapy Targeting Mutant KRAS in cancer. N. Engl. J. Med. 375, 2255–2262 (2016).
KRAS vaccine (mRNA-5671) https://www.modernatx.com/research/product-pipeline (2021).
Douglass, J. et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 6, eabd5515 (2021).
Zhang, Z. et al. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell 40, 1060–1069.e1067 (2022).
Hattori, T. et al. Creating MHC-restricted neoantigens with covalent inhibitors that can be targeted by immune therapy. Cancer Discov.13 132−145 (2022).
Sahai, E., Olson, M. F. & Marshall, C. J. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. Embo J. 20, 755–766 (2001).
Olson, M. F., Paterson, H. F. & Marshall, C. J. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 394, 295–299 (1998).
Danen, E. H., Sonneveld, P., Sonnenberg, A. & Yamada, K. M. Dual stimulation of Ras/mitogen-activated protein kinase and RhoA by cell adhesion to fibronectin supports growth factor-stimulated cell cycle progression. J. Cell Biol. 151, 1413–1422 (2000).
Ledford, H. Cancer drugs are closing in on some of the deadliest mutations. Nature 610, 620–622 (2022).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Lin, C. W. et al. A two-component protein condensate of the EGFR cytoplasmic tail and Grb2 regulates Ras activation by SOS at the membrane. Proc. Natl Acad. Sci. USA 119, e2122531119 (2022).
Huang, W. Y. C. et al. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science 363, 1098–1103 (2019).
Tulpule, A. et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell 184, 2649–2664.e2618 (2021).
Lin, C. C. et al. Receptor tyrosine kinases regulate signal transduction through a liquid-liquid phase separated state. Mol. Cell 82, 1089–1106.e1012 (2022).
Zhao, Y. G. & Zhang, H. Phase separation in membrane biology: the interplay between membrane-bound organelles and membraneless condensates. Dev. Cell 55, 30–44 (2020).
Floris, E. et al. Physics of compartmentalization: how phase separation and signaling shape membrane and organelle identity. Comput Struct. Biotechnol. J. 19, 3225–3233 (2021).
Adhikari, H. et al. Oncogenic KRAS is dependent upon an EFR3A-PI4KA signaling axis for potent tumorigenic activity. Nat. Commun. 12, 5248 (2021).
Kattan, W.E. et al. Components of the phosphatidylserine endoplasmic reticulum to plasma membrane transport mechanism as targets for KRAS inhibition in pancreatic cancer. Proc. Natl Acad. Sci. USA 118, e2114126118 (2021).
Cheng, R. et al. A novel protein RASON encoded by a lncRNA controls oncogenic RAS signaling in KRAS mutant cancers. Cell Res. 33, 30–45 (2022).
Maziarz, M. et al. Revealing the activity of trimeric G-proteins in live with a versatile biosensor design. Cell 182, 770–785.e716 (2020).
Asher, W. B. et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nat. Methods 18, 397–405 (2021).
Asher, W. B. et al. GPCR-mediated β-arrestin activation deconvoluted with single-molecule precision. Cell 185, 1661–1675.e1616 (2022).
Huang, W. Y. C., Alvarez, S., Kondo, Y., Kuriyan, J. & Groves, J. T. Relating cellular signaling timescales to single-molecule kinetics: a first-passage time analysis of Ras activation by SOS. Proc. Natl Acad. Sci. USA 118, e2103598118 (2021).
Weeks, R., Zhou, X., Yuan, T. L. & Zhang, J. Fluorescent biosensor for measuring ras activity in living cells. J. Am. Chem. Soc. 144, 17432–17440 (2022).
Wu, Y. W. Spatiotemporal imaging of small GTPase activity using conformational sensors for GTPase activity (COSGA). Methods Mol. Biol. 2262, 259–267 (2021).
Goswami, D. et al. Membrane interactions of the globular domain and the hypervariable region of KRAS4b define its unique diffusion behavior. Elife 9, e47654 (2020).
Murakoshi, H. et al. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl Acad. Sci. USA 101, 7317–7322 (2004).
Schaefer, A. & Der, C. J. RHOA takes the RHOad less traveled to cancer. Trends Cancer 8, 655–669 (2022).
Briere, D. M. et al. The KRAS(G12C) inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol. Cancer Ther. 20, 975–985 (2021).
Mugarza, E. et al. Therapeutic KRAS(G12C) inhibition drives effective interferon-mediated antitumor immunity in immunogenic lung cancers. Sci. Adv. 8, eabm8780 (2022).
Yin, R., Feng, B. Y., Varshney, A. & Pierce, B. G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 31, e4379 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Boehr, D. D., Dyson, H. J. & Wright, P. E. An NMR perspective on enzyme dynamics. Chem. Rev. 106, 3055–3079 (2006).
Alderson, T. R. & Kay, L. E. NMR spectroscopy captures the essential role of dynamics in regulating biomolecular function. Cell 184, 577–595 (2021).
Chao, F. A. et al. Insights into the cross talk between effector and allosteric lobes of KRAS from methyl conformational dynamics. J. Am. Chem. Soc. 144, 4196–4205 (2022).
Vamathevan, J. et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18, 463–477 (2019).
Sadybekov, A. A. et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 601, 452–459 (2022).
Crunkhorn, S. Screening ultra-large virtual libraries. Nat. Rev. Drug Discov. 21, 95 (2022).
Gorgulla, C. et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 580, 663–668 (2020).
Provenzi, L. et al. Pain-related increase in serotonin transporter gene methylation associates with emotional regulation in 4.5-year-old preterm-born children. Acta Paediatr. 109, 1166–1174 (2020).
Passioura, T. & Suga, H. A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets. Chem. Commun. 53, 1931–1940 (2017).
Yamagishi, Y. et al. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 18, 1562–1570 (2011).
Sideris, M. et al. The role of KRAS in endometrial cancer: a mini-review. Anticancer Res. 39, 533–539 (2019).
Maru, Y. et al. Kras activation in endometrial organoids drives cellular transformation and epithelial-mesenchymal transition. Oncogenesis 10, 46 (2021).
Fu, Y. et al. Ductal activation of oncogenic KRAS alone induces sarcomatoid phenotype. Sci. Rep. 5, 13347 (2015).
O’Hayre, M. et al. Inactivating mutations in GNA13 and RHOA in Burkitt’s lymphoma and diffuse large B-cell lymphoma: a tumor suppressor function for the Gα13/RhoA axis in B cells. Oncogene 35, 3771–3780 (2016).
Palomero, T. et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 46, 166–170 (2014).
Sakata-Yanagimoto, M. et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 46, 171–175 (2014).
Yoo, H. Y. et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 46, 371–375 (2014).
Gill, M. B., Turner, R., Stevenson, P. G. & Way, M. KSHV-TK is a tyrosine kinase that disrupts focal adhesions and induces Rho-mediated cell contraction. Embo J. 34, 448–465 (2015).
Chen, X. et al. CSTF2-induced shortening of the RAC1 3’UTR promotes the pathogenesis of urothelial carcinoma of the bladder. Cancer Res. 78, 5848–5862 (2018).
Li, J. et al. Increased expression of Rac1 in epilepsy patients and animal models. Neurochem Ress 41, 836–843 (2016).
Zhang, Y. et al. Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett. 293, 189–197 (2010).
Haga, R. B. & Ridley, A. J. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases 7, 207–221 (2016).
Kakiuchi, M. et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 46, 583–587 (2014).
Radisky, D. C. et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436, 123–127 (2005).
Li, Q. et al. Rac1 activates non-oxidative pentose phosphate pathway to induce chemoresistance of breast cancer. Nat. Commun. 11, 1456 (2020).
Stallings-Mann, M. L. et al. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci. Transl. Med. 4, 142ra195 (2012).
Liu, Y. et al. Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung Cancer 63, 375–382 (2009).
Lai, S. Y. et al. Activated Vav2 modulates cellular invasion through Rac1 and Cdc42 in oral squamous cell carcinoma. Oral. Oncol. 44, 683–688 (2008).
Zhong, H. et al. Overexpression of microRNA-19a-3p promotes lymph node metastasis of esophageal squamous cell carcinoma via the RAC1/CDC42-PAK1 pathway. Transl. Cancer Res. 10, 2694–2706 (2021).
Nihei, T. et al. Prognostic impacts of Rho-kinase activity in circulating leucocytes in patients with vasospastic angina. Eur. Heart J. 39, 952–959 (2018).
Gong, L. L. et al. Coptisine exert cardioprotective effect through anti-oxidative and inhibition of RhoA/Rho kinase pathway on isoproterenol-induced myocardial infarction in rats. Atherosclerosis 222, 50–58 (2012).
Ma, D. et al. Inhibition of KLF5-Myo9b-RhoA pathway-mediated podosome formation in macrophages ameliorates abdominal aortic aneurysm. Circ. Res 120, 799–815 (2017).
Oesterle, A., Laufs, U. & Liao, J. K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 120, 229–243 (2017).
Anaruma, C. P. et al. Rock protein as cardiac hypertrophy modulator in obesity and physical exercise. Life Sci. 254, 116955 (2020).
Wang, H. et al. Tax-interacting protein 1 coordinates the spatiotemporal activation of Rho GTPases and regulates the infiltrative growth of human glioblastoma. Oncogene 33, 1558–1569 (2014).
Fortin, S. P. et al. Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol. Cancer Res. 10, 958–968 (2012).
Fortin Ensign, S. P., Mathews, I. T., Symons, M. H., Berens, M. E. & Tran, N. L. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front. Oncol. 3, 241 (2013).
Lionarons, D. A. et al. RAC1(P29S) induces a mesenchymal phenotypic switch via serum response factor to promote melanoma development and therapy resistance. Cancer Cell 36, 68–83.e69 (2019).
Davis, M. J. et al. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl Acad. Sci. USA 110, 912–917 (2013).
Maldonado, M. D. M. & Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in cancer. Cancer Res. 78, 3101–3111 (2018).
Ambruso, D. R. et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl Acad. Sci. USA 97, 4654–4659 (2000).
Saveri, P. et al. Charcot-Marie-Tooth Type 2B: a new phenotype associated with a novel RAB7A mutation and inhibited EGFR degradation. Cells 9, 1028 (2020).
Handley, M. T. et al. Warburg Micro syndrome is caused by RAB18 deficiency or dysregulation. Open Biol. 5, 150047 (2015).
Wilson, G. R. et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am. J. Hum. Genet. 95, 729–735 (2014).
Cunha, D. L. et al. REP1 deficiency causes systemic dysfunction of lipid metabolism and oxidative stress in choroideremia. JCI Insight 6, e146934 (2021).
Yin, Y. X. et al. Increased expression of Rab25 in breast cancer correlates with lymphatic metastasis. Tumour Biol. 33, 1581–1587 (2012).
Liu, L. & Ding, G. Rab25 expression predicts poor prognosis in clear cell renal cell carcinoma. Exp. Ther. Med. 8, 1055–1058 (2014).
Li, Y., Jia, Q., Zhang, Q. & Wan, Y. Rab25 upregulation correlates with the proliferation, migration, and invasion of renal cell carcinoma. Biochem. Biophys. Res. Commun. 458, 745–750 (2015).
Cao, C. et al. Expression of Rab25 correlates with the invasion and metastasis of gastric cancer. Chin. J. Cancer Res. 25, 192–199 (2013).
He, H. et al. Identification and characterization of nine novel human small GTPases showing variable expressions in liver cancer tissues. Gene Expr. 10, 231–242 (2002).
Ma, Y. F. et al. Expression of Ras-related protein 25 predicts chemotherapy resistance and prognosis in advanced non-small cell lung cancer. Genet Mol. Res. 14, 13998–14008 (2015).
Zhang, J. et al. Overexpression of Rab25 contributes to metastasis of bladder cancer through induction of epithelial-mesenchymal transition and activation of Akt/GSK-3β/Snail signaling. Carcinogenesis 34, 2401–2408 (2013).
Ding, B. et al. Knockdown of Ras-related protein 25 (Rab25) inhibits the in vitro cytotoxicity and in vivo antitumor activity of human glioblastoma multiforme cells. Oncol. Res. 25, 331–340 (2017).
Hu, C., Chen, B., Zhou, Y. & Shan, Y. High expression of Rab25 contributes to malignant phenotypes and biochemical recurrence in patients with prostate cancer after radical prostatectomy. Cancer Cell Int. 17, 45 (2017).
Serão, N. V., Delfino, K. R., Southey, B. R., Beever, J. E. & Rodriguez-Zas, S. L. Cell cycle and aging, morphogenesis, and response to stimuli genes are individualized biomarkers of glioblastoma progression and survival. BMC Med Genom. 4, 49 (2011).
Li, H. et al. SRPX2 and RAB31 are effective prognostic biomarkers in pancreatic cancer. J. Cancer 10, 2670–2678 (2019).
Scelo, G. et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat. Commun. 5, 5135 (2014).
Li, X. et al. High expression of Rab31 confers a poor prognosis and enhances cell proliferation and invasion in oral squamous cell carcinoma. Oncol. Rep. 45, 1182–1192 (2021).
Soelch, S. et al. Rab31-dependent regulation of transforming growth factor ß expression in breast cancer cells. Mol. Med. 27, 158 (2021).
Chen, K. et al. Rab31 promotes metastasis and cisplatin resistance in stomach adenocarcinoma through Twist1-mediated EMT. Cell Death Dis. 14, 115 (2023).
Zu, T. et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl Acad. Sci. USA 110, E4968–E4977 (2013).
Oldrini, B. et al. EGFR feedback-inhibition by Ran-binding protein 6 is disrupted in cancer. Nat. Commun. 8, 2035 (2017).
Sheng, K. L., Pridham, K. J., Sheng, Z., Lamouille, S. & Varghese, R. T. Functional blockade of small GTPase RAN inhibits glioblastoma cell viability. Front. Oncol. 8, 662 (2018).
Sheng, C. et al. Knockdown of Ran GTPase expression inhibits the proliferation and migration of breast cancer cells. Mol. Med. Rep. 18, 157–168 (2018).
Kurisetty, V. V. et al. RAN GTPase is an effector of the invasive/metastatic phenotype induced by osteopontin. Oncogene 27, 7139–7149 (2008).
Ning, J., Liu, W., Zhang, J., Lang, Y. & Xu, S. Ran GTPase induces EMT and enhances invasion in non-small cell lung cancer cells through activation of PI3K-AKT pathway. Oncol. Res. 21, 67–72 (2013).
Zafar, S. et al. Creutzfeldt-Jakob disease subtype-specific regional and temporal regulation of adp ribosylation factor-1-dependent Rho/MLC pathway at pre-clinical stage. J. Mol. Neurosci. 56, 329–348 (2015).
Sheen, V. L. Periventricular heterotopia: shuttling of proteins through vesicles and actin in cortical development and disease. Science 2012, 480129 (2012).
Sivadasan, R. et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19, 1610–1618 (2016).
Szymanska, K., Hartill, V. L. & Johnson, C. A. Unraveling the genetics of Joubert and Meckel-Gruber syndromes. J. Pediatr. Genet. 3, 65–78 (2014).
Wolf, M. T. & Hildebrandt, F. Nephronophthisis. Pediatr. Nephrol. 26, 181–194 (2011).
Acknowledgements
The authors are grateful to Prof. Xiaofeng Zhu, Prof. Liwu Fu, Prof. Xiaoshi Zhang, Prof. Peirong Ding, Prof. Haoxian Yang, Prof. Dandan Li, Prof. Xue Hou, Prof. Jianwei Chen, Prof. Qing Lv, Prof. Zheng Yang, Prof. Hongman Xue, and Prof. Chenju Yi from Sun Yat-sen University for helpful discussion, Dr. Zhixiong Sun from Columbia University and Dr. Yun Huang from Howard Hughes Medical Institute/Weill Cornell Medicine for editing. This work was supported by grants from National Key R&D Program of China (2022YFC2305400, 2022YFC3400600), Shenzhen Fundamental Research Program (JCYJ20220530145011025), National Natural Science Foundation of China (82282717, 82073311), Natural Science Foundation of Sichuan Province (2022JDTD0025).
Author information
Authors and Affiliations
Contributions
Conceptualization, writing and editing, G.Y. and Y.G.; Writing and editing, J.H., J.P., B. L., J.S.; Graphic preparations and editing, G. Y., J. H., H.J., Y.Z., S.G., J.W. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yin, G., Huang, J., Petela, J. et al. Targeting small GTPases: emerging grasps on previously untamable targets, pioneered by KRAS. Sig Transduct Target Ther 8, 212 (2023). https://doi.org/10.1038/s41392-023-01441-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01441-4
