
Abstract
Gastric cancer (GC) ranks fifth in global cancer diagnosis and fourth in cancer-related death. Despite tremendous progress in diagnosis and therapeutic strategies and significant improvements in patient survival, the low malignancy stage is relatively asymptomatic and many GC cases are diagnosed at advanced stages, which leads to unsatisfactory prognosis and high recurrence rates. With the recent advances in genome analysis, biomarkers have been identified that have clinical importance for GC diagnosis, treatment, and prognosis. Modern molecular classifications have uncovered the vital roles that signaling pathways, including EGFR/HER2, p53, PI3K, immune checkpoint pathways, and cell adhesion signaling molecules, play in GC tumorigenesis, progression, metastasis, and therapeutic responsiveness. These biomarkers and molecular classifications open the way for more precise diagnoses and treatments for GC patients. Nevertheless, the relative significance, temporal activation, interaction with GC risk factors, and crosstalk between these signaling pathways in GC are not well understood. Here, we review the regulatory roles of signaling pathways in GC potential biomarkers, and therapeutic targets with an emphasis on recent discoveries. Current therapies, including signaling-based and immunotherapies exploited in the past decade, and the development of treatment for GC, particularly the challenges in developing precision medications, are discussed. These advances provide a direction for the integration of clinical, molecular, and genomic profiles to improve GC diagnosis and treatments.
Similar content being viewed by others

Introduction
Gastric cancer (GC) remains one of the most common cancer types worldwide. According to the GLOBOCAN 2020 report, the global morbidity and mortality of GC rank fifth and fourth, respectively, with more than one million newly diagnosed cases and approximately one fatal case in every 13 cancer-related deaths.1 More than 95% of GC cases are adenocarcinomas.2 Men are twice as likely as women to suffer and die from GC.3 Despite a decline in the global prevalence and death rate of GC, rates remain high in Eastern Asian countries, which account for more than 70% of newly diagnosed and death cases of GC in the world.1,4 Notably, in both low-risk and high-risk regions, the incidence of GC is elevated in populations younger than 50 years, which may be linked to increased obesity and gastric microbiome dysbiosis associated with modern lifestyle.5 Thus, many challenges remain in controlling GC.
GC is generally categorized as cardia and non-cardia subtypes, which arise from the upper stomach and the mid-distal stomach, respectively. Each subtype has distinct epidemiological characteristics and risk factors.6 Non-cardia GC is more prevalent in Eastern Asian populations, while cardia GC is more common in Western countries.7 Chronic infection by Helicobacter pylori (H. pylori) is the dominant risk factor for the development of non-cardia GC.8 H. pylori infection, however, is generally not associated with cardia GC and may even reduce the risk of cardia GC in some populations.9 The molecular mechanism of H. pylori infection-mediated GC has not been completely elucidated. Prolonged H. pylori infection is thought to lead to chronic gastritis, where gastric acid secretion is inhibited by inflammatory mediators such as tumor necrosis factor-α (TNF-α) and interleukins. The loss of gastric acidity further exacerbates H. pylori infection and inflammation, causing parietal damage, ulcers, and atrophy of the stomach.10,11 Other contributors to non-cardia GC development include smoking tobacco, drinking alcohol, and consuming salt-preserved food or red/processed meat, which can cause destruction of stomach mucosa and enhance persistency of H. pylori infection.12,13,14 These factors are also associated with cardia GC,15 whereas obesity and gastroesophageal reflux disease are recognized as risk factors specifically linked to cardia but not non-cardia GC.16 In addition, infection with Epstein–Barr virus (EBV) is an important etiological agent responsible for ~10% of GC, frequently in male patients and the cardia subtype.17 EBV infection can promote the hypermethylation of tumor suppressor genes, inflammation of gastric mucosa, and immune evasion of the host, resulting in gastric carcinogenesis.18 As sustained infection with H. pylori and EBV can cause chronic inflammatory stress in the stomach, there is emerging attention to GC risk and co-infection by both pathogens, since H. pylori co-infection with EBV increases the occurrence of GC19,20 and may stimulate aggressiveness of GC.21
In addition to environmental and lifestyle factors, genetic aberrations (including gene mutations, chromosomal alterations, transcriptional dysregulations, and epigenetic modifications) are indispensable co-contributors in GC carcinogenesis.22 Approximately 10% of GC cases have a familial aggregation profile, and 1–3% have a confirmed hereditary mutation.23 The major type of hereditary GC is the autosomal dominant hereditary diffuse gastric cancer (HDGC) characterized by diffuse histopathological features. HDGC is frequently associated with a loss-of-function mutation in the Cadherin-1 (CDH1) gene encoding E-cadherin, which is essential for cell–cell adhesion and maintenance of the epithelial cell phenotype. E-cadherin also plays vital roles in signaling pathways that regulate cell survival, proliferation, migration, and invasion.24,25 The link between the CDH1 gene mutation and the diffuse type of GC was first identified in a large Aboriginal family in New Zealand in 1998 by Guilford and colleagues.26 Molecular genetic testing for the CDH1 gene mutation is a recommended approach for confirming the diagnosis and family studies of HDGC.27
The treatment and prognosis for GC largely depend on cancer staging, which is usually evaluated using the American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) system. This system describes the extent of tumor invasion into the gastric wall layers (T category), the spread of the tumor to nearby lymph nodes (N category), and the migration of cancer cells to other organs (M category).28 The overall staging of GC is assigned from large staging groups after the combination of the TNM information, ranging from earliest stage 0 (carcinoma in situ) to stages I through IV; the larger number, the more advanced the cancer is with the larger extent of spread.29 Surgery is the primary approach for treating GC in all stages, especially for those in the early stage.30 Chemotherapy or chemoradiation is the main therapeutic intervention applied either before surgery to shrink the tumor or after surgery to kill any remaining cancer cells.31 For advanced GC patients with unresectable local cancer, recurrence, or metastasis, chemotherapy is usually the first-line treatment to control cancer progression for as long as possible, and a combination of chemotherapy with targeted therapy, immunotherapy, or radiation therapy may be adopted.2
Because GC is morphologically heterogeneous, decisions about therapy and predictions for patient survival rely on histopathological classifications. The traditional Lauren classification has been widely used in clinical practices since it was introduced in 1965. This classification divides GC into intestinal type with glandular growth pattern, diffuse type with poorly cohesive cells, and mixed type.32 The intestinal-type GC occurs more commonly in men and the elderly and is associated with H. pylori-related chronic gastritis as well as gastroesophageal reflux disease. The diffuse-type GC, usually with poorer clinical outcomes, is more prevalent in women and the younger populations and is more relevant to dysfunction in cell adhesion, as found in CDH1-mutated hereditary cases.33 The other broadly used histology classification is the World Health Organization (WHO) guidelines issued in 2010 and updated most recently in 2019, which characterizes GC as papillary, tubular, mucinous, and poorly cohesive types followed by several subdivisions under each category.34 Japanese pathologists also use the Nakamura classification or the Japanese Gastric Cancer Association (JGCA) classification, which can distinguish differentiated tumors from undifferentiated tumors.35,36 Although the histopathological classifications provide recommendations for surgery and chemotherapy selections, they are insufficient to guide personalized treatments for GC patients.
With the recent advances in genome analysis, biomarkers have been identified with clinical importance for GC diagnosis, treatment, and prognosis. These include molecules in growth factor pathways (e.g., the human epidermal growth factor receptor 2 (HER2)), regulators of the cell cycle and apoptosis (e.g., the tumor protein p53 (encoded by TP53 gene)), cell adhesion factors (such as E-cadherin), immune checkpoint control modulators programmed death 1 and programmed death-ligand 1 (PD-1/PD-L1), and other molecules relevant to DNA, RNA, exosome, or epigenetic modifications.37,38 HER2 is the first clinically used molecular biomarker for GC patients. Approximately one-fifth of GC cases are HER2-positive, and determination of HER2 expression using immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) is mandatory for patients diagnosed with advanced GC.39 In 2010, the international Trastuzumab for Gastric Cancer (ToGA) phase III clinical study showed that the HER2 monoclonal antibody trastuzumab co-administered with cisplatin plus capecitabine or fluorouracil (5-FU) had better therapeutic outcomes compared to chemotherapy alone.40 Later in the same year, trastuzumab was approved by the United States Food and Drug Administration (FDA) as the first targeted drug used in combination with chemotherapeutic drugs for first-line treatment of HER2-positive metastatic GC.
To facilitate further development of personalized therapies for GC, molecular classifications have been introduced. Two large-scale, comprehensive genome-wide and molecular analyses on gastric tumors resulted in two major molecular classifications that partially overlap and complement. One proposed by The Cancer Genome Atlas (TCGA) research network in 2014 classified GC into four subtypes: EBV-positive (EBV+), microsatellite instable (MSI), genomically stable (GS), and chromosomal unstable (CIN).41 The Asian Cancer Research Group (ACRG) in 2015 classified GC into MSI, microsatellite stable or epithelial-mesenchymal transition (MSS/EMT), MSS positive for TP53 (MSS/TP53+), and MSS with loss of TP53 (MSS/TP53−) subtypes.42 Comprehensive molecular characterization of these GC subtypes shows clinical implications for GC treatment and prognosis (Table 1).43,44 With the development of immunotherapy in cancer management, the molecular classifications of GC have helped predict patients’ responsiveness to immunotherapy. Subgroups of GC patients with EBV+, high degree of MSI, or high burden of mutation are more likely to have a survival benefit from anti-PD-1 drugs like nivolumab and pembrolizumab.43
The identification of biomarkers and molecular classification have also provided important clues to improve early diagnosis and therapeutic interventions for rare GC types with unique histopathological characteristics, such as gastric signet-ring cell carcinoma (GSRCC). GSRCC is classified into diffuse, undifferentiated, and poorly cohesive types, noted for their poorly cohesive single cells and absence of gland formation.45 There are many clinical challenges in the diagnosis and treatment of GSRCC. GSRCC exhibits distinct epidemiology, oncogenesis processes, and therapeutic sensitivity profiles compared to other subtypes of diffuse GC.46,47 Moreover, GSRCC cases are frequently diagnosed at an advanced stage, in part because of the impracticality of using endoscopy and the lack of pathological tests for early stage screening.48 The regimen for treating GSRCC is still controversial, and overtreatment with chemotherapy may occur with detrimental results because of this lack of adequate predictive biomarkers.49 Since mutations in the CDH1 gene50 and high CLDN18-ARHGAP 26/6 fusion51 have been reported in GSRCC patients, GSRCC is considered a GS subtype of TCGA molecular classification,49 and the high CLDN18.2 expression found among advanced GSRCC patients has provided a novel therapeutic option of CLDN18.2-targeted therapy.52 In addition, high MSI was found in 3.5% of GSRCC, and this specific group of GSRCC patients may benefit from immunotherapy using PD-1 inhibitors.53,54
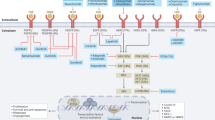
Since the first successful gastric resection in the 1880s, there has been tremendous progress in diagnosis and therapeutic strategies (Fig. 1) and significant improvements in patient survival in the long combat against GC. However, because GC is often asymptomatic until it progresses to higher malignancy levels, cases are often diagnosed at advanced stages, leading to unsatisfactory prognosis and high recurrence rates. The 5-year survival rates are as high as 68–80% for stage I GC, and then decrease sharply as the diagnosed staging becomes advanced, to 46–60% for stage II, 8–30% for stage III, and only 5% for stage IV.55 Resistance to chemotherapy and targeted drugs contributes to poor survival in GC.56,57 Therefore, identifying new biomarkers for early diagnosis and therapeutic selectivity and sensitivity is the main challenge in GC management. The modern molecular classifications support the important roles of signaling pathways like EGFR/HER2, p53, PI3K, immune checkpoint pathways, and cell adhesion signaling molecules in GC tumorigenesis, progression, metastasis, and therapeutic responsiveness. Four targeted drugs and two immune checkpoint inhibitors have already been approved by the FDA for GC treatment. Still, the relative significance of these signaling pathways in GC, their temporal activation and interaction with GC risk factors, and crosstalk among them is not well understood. There has been increasing attention to signaling pathways and the identification of novel therapeutic targets in GC research. In this article, the regulatory roles of signaling pathways in GC and potential biomarkers or therapeutic targets are reviewed. Furthermore, the current GC treatment and the development of signaling pathway-based targeted or immunotherapies will be discussed.

Timeline of selected key findings and significant therapy developments in gastric cancer. The major milestones for risk factor identification, classification and staging, and therapy developments for GC are listed. Chemotherapy regimens: FAM: fluorouracil (5-FU) + mitomycin C + doxorubicin; FAMTX: methotrexate + 5-FU + doxorubicin; ECF: epirubicin + cisplatin + 5-FU; TPF: docetaxel + cisplatin + 5-FU; FLOFOX: oxaliplatin + 5-FU + leucovorin; XELOX: capecitabine (Xeloda) + oxaliplatin; S-1: tegafur (5-FU prodrug) + 5-chloro-2,4-dihydroxypyridine (CDHP) + oteracil potassium (Oxo), in a molar ratio of 1:0.4:1. EBV Epstein–Barr virus, TCGA The Cancer Genome Atlas, ACRG Asian Cancer Research Group. This figure was created with Biorender.com
Signaling pathways in gastric cancer and therapeutic implications
MAPK signaling pathway
The mitogen-activated protein kinase (MAPK) signaling pathway is one of the most complicated cellular pathways involved in GC progression, including proliferation, migration, invasion, and metastasis.58 MAPKs are a large family of serine/threonine protein kinases that are responsible for cellular response to multiple extracellular stimuli.59 Each canonical single MAPK cascade pathway consists of at least three core kinases: MAPKKKs, MAPKKs, and MAPKs.60 The MAPK signaling pathway is shared by five cascades, which are accordingly named after the components of each MAPK tier: the extracellular signal-related kinases ERK (ERK1/2), Jun amino-terminal kinases (SAPK/JNK1,2,3), p38-MAPK (p38α, p38β, p38γ, and p38δ), ERK5, and ERK3/4.61
The MAPK/ERK signaling cascade is triggered by binding of extracellular factors to receptors including tyrosine kinases (RTKs), EGFR, and G protein-coupled receptors (GPCRs), and is sometimes triggered by vascular endothelial growth factor and its receptor (VEGF/VEGFR). Under physiological conditions, MAPK signaling is triggered through the activation of RAS proteins (KRAS, HRAS, and NRAS), a family of small guanine triphosphatases (GTPases) that integrate signals from a collection of upstream factors.62 RTK-RAS signaling pathway alterations are reported in about 37% of GC.63 In its GTP-bound activated condition, RAS undergoes a conformational shift in the switch I and II regions, which facilitates interactions with a variety of downstream effectors, including the RAF family of kinases (ARAF, BRAF, and CRAF).64,65 BRAF mutation occurs in all types of cancers and up to 11% in GC.66 Once activated, RAF kinases phosphorylate and activate MEK1/2 kinases, which in turn activate ERK1/2 kinases.67 ERK1/2 are vital sensors of proliferation, differentiation, and survival signals.68 Elevated p-ERK1/2 is an independent prognostic factor of poor survival in GC cases.69 The activated ERK1/2 kinases then phosphorylate a series of substrates that conduct critical biological processes.68,70 In GC, the MAPK/ERK pathways are involved in the regulation of cell motility by coordinating the activity of MMPs, cell adhesion, and EGFR-induced disassembly of focal adhesions, thus governing cell migration and invasion.59,71 Generally, the ERK3/4 MAPKs are considered atypical because of the absence of a tyrosine residue and the presence of the Ser-Glu-Gly motif in their activation loop.72 ERK5 can be activated by growth factors and oxidative stress and is essential for cell survival, normal development of the early embryo, and the vascular system.73
The JNK subgroup of MAPKs is encoded by three distinct genes: MAPK8 (which encodes JNK1), MAPK9 (which encodes JNK2), and MAPK10 (which encodes JNK3).74 The JNK1/2 subtypes are ubiquitously expressed, whereas JNK3 is expressed primarily in the heart, brain, and testis.75,76 JNKs are activated by stress signals and proinflammatory stimuli such as heat shock and oxidative stress. MKK4 and MKK7 kinases are the upstream regulators of JNKs. Activated JNKs subsequently phosphorylate downstream c-Jun and JunD and activate transcription factors.77 An important JNK target is the transcription factor activating protein-1 (AP-1).78 Activation of JNKs leads to cell proliferation, apoptosis, or transformation.79 Interactions can occur between JNKs and the other MAPK pathways; JNK subtypes can activate p38-MAPK, while several upstream regulators in the p38-MAPK module are shared by the JNK isoforms. Studies have shown that JNK1/2 is involved in the sensitization of p38-MAPK inhibition to cisplatin-induced cell death, and the elevated level of reactive oxygen species (ROS) mediates the activation of JNK1/2 by P38-MAPK inhibition.80 Compared to wild-type controls, JNK1 knockout mice showed a significant decrease in gastric carcinogenesis mediated by N-methyl-N-nitrosourea.81 Consequently, JNK1 is involved in tumor initiation as well as progression and is a promising target for the prevention of GC.
The p38-MAPK is selectively activated by upstream MAPK kinase (MKK) 3 and MKK6 kinases.82 The major downstream targets of p38-MAPK are protein kinases and transcription factors such as MAPK-activated protein kinase 2 (MK2), mitogen- and stress-activated protein kinase 1 (MSK1), p53, transcription factor ELK1, and activating transcription factor 2 (ATF2).83 The p38-MAPK pathway features a complicated regulation in cancers. Several studies showed that p38 acts as an oncogenic factor and plays a key role in pathological events related to tumor progression, such as inflammation, invasion, and angiogenesis84,85 (Fig. 2). Activation of the p38-MAPK/AP-1 pathway is positively related to chemotherapy resistance in human GC cells.86 On the other hand, a wealth of evidence supports the role of p38-MAPK as a tumor suppressor, inducing cell apoptosis by way of the activation of p53.87,88 Cell cycle arrest is another possible consequence of tumor suppression by p38, carried out by downregulating ERK and JNK signaling pathways, thus restricting RAS transformation.89

Main signaling pathways and fundamental factors in gastric cancer. The major signaling and crosstalk of MAPK, HER2, PI3K/AKT/mTOR, HGF/c-Met, p53, Wnt/β-catenin, and NF-κB pathways, as well as their regulatory roles in cellular processes, are illustrated. GPCRs G-protein-coupled receptors, HGF hepatocyte growth factor, c-MET c-mesenchymal-epithelial transition factor, EGFR epidermal growth factor receptor, HER2/3/4 human epidermal growth factor receptor 2/3/4, MAPKKKs mitogen-activated protein kinase kinase kinases, RTKs receptor tyrosine kinases, RAS rat sarcoma, RAF rapidly accelerated fibrosarcoma, MKK mitogen-activated protein kinase kinase, SAPK/JNK jun amino-terminal kinase, p38-MAPKs p38 group of mitogen-activated protein kinases, MEK mitogen-activated protein kinase kinase, ERK1/2 extracellular signal-related kinase 1/2, PI3K phosphoinositide 3-kinase, AKT protein kinase B, mTORC1/2 mammalian target of rapamycin complex 1/2, PTEN phosphatase and tensin homolog, PDK1 phosphoinositide-dependent protein kinase 1, TSC1/2 tuberous sclerosis complex 1/2, p70S6K1 phosphorylation of ribosomal p70S6 kinase 1, 4E-BP1 eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1, NF-κB nuclear factor kappa-B, GSK3 glycogen synthase kinase 3, BAD Bcl-xl/Bcl-2-asociated death promoter, Casp9 cysteinyl aspartate specific proteinase 9, MDM2 murine double minute 2, p53 tumor protein 53, EMT epithelial-mesenchymal transition, LRP5/6 low-density lipoprotein receptor-related protein 5/6, CKIα casein kinase Iα, APC adenomatous polyposis coli, TCF/LEF T-cell factor/lymphoid enhancer factor, TNFR tumor necrosis factor receptor, TLR toll-like receptors, IKK IκB kinase. This figure was created with Biorender.com
RAS/RAF/MAPK and PI3K/AKT/mTOR signal transduction pathways are the most dysfunctional pathways in multiple cancer types including GC.90,91 RTKs alterations in tumors lead to activation of both MAPK and PI3K pathways, and targeting the PI3K pathway was confirmed to promote cancer progression through MAPK signals and vice versa92 (Fig. 2). RAS mutations are the most common MAPK alterations observed in human cancer.93 The mutation frequency of KRAS in GC is 6.5%, and PIK3CA is 25%.94,95 Generally, the KRAS mutation is found in intestinal-type tumors whereas the NRAS mutation is reported to appear in diffuse and metastatic GC.96 Using pathway-based gene set enrichment analysis, MAPK/ERK gene features were found elevated in the intestinal subtype of GC. Genes involved in the RAS/ERK signaling cascade, including KRAS, EGFR, HER2, and MET, have been found amplified in a mutually exclusive manner in about two out of five GC patients.97
Migration and invasion of GC cells mediated by the MAPK/ERK signaling pathway involves various other factors.98,99,100 For example, Spondin 2 (SPON2) promotes the EMT of GC cells by activation of the MAPK/ERK1/2 pathway and consequently accelerates the metastasis of GC. Chemerin may act as a pro-invasive factor via induction of VEGF, IL-6, and matrix metalloproteinase-7 (MMP-7) in GC, and the process relies on the phosphorylation of ERK1/2.101 ERK also mediates GC migration and invasion by regulating the activity of downstream proteins like MMPs.71 Other studies have demonstrated that RAS/MAPK signal transduction is involved in the proliferation of GC cells.
Recent studies have shown that epigenetic regulation can affect GC cell growth and metastasis through MAPK/ERK pathways.102 Micro RNAs (miRNAs) are multipotent in the regulation of various cellular pathways and play a fundamental role in tumor biology. In particular, they have been found to regulate MAPKs like ERK1/2 and JNK and to modulate proliferation, survival, and metastasis of GC cells.103 miR-592 overexpression has been identified to promote proliferation, migration, and invasion of GC by targeting Sprouty 2 (SPRY-2) through the MAPK/ERK and PI3K/AKT signaling pathways.104 In addition to miRNAs, some long non-coding RNAs (lncRNAs) are involved in tumorigenesis and the progression of GC mediated by the MAPK/ERK signaling pathway.105 For example, lncRNA CASC2 suppresses the proliferation of GC cells by regulating the ERK1/2 and JNK/MAPK signaling pathways.106
HER2 signaling pathway
The frequency of HER2-positive tumors ranges from 4.4% to 53.4% in gastric/gastroesophageal cancer,107,108 and HER2-positive tumors are generally associated with more aggressive cancer and tumor recurrence.109,110 HER2 amplification/overexpression has been confirmed to play a critical role in GC tumorigenesis and development,111 and is a therapeutic target and biomarker for GC patients.112 The HER2 gene, also known as receptor tyrosine-protein kinase erbB-2, p185, or neu, is located on the human chromosome 17 (17q12),113 and is a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. The EGFR family consists of four members, HER1 (ERBB1, EGFR), HER2 (ERBB2), HER3 (ERBB3), and HER4 (ERBB4),114 all of which are identified to participate in regulating tumor cell growth, proliferation, and migration. Although the four human HER genes are located on different chromosomes, all of them are composed of an intracellular domain with tyrosine kinase properties, a lipophilic transmembrane domain, and a cysteine-rich extracellular domain containing the ligand-binding pocket.115
EGFR family members exist as monomers on the cell surface, but dimerize once the ligand binds to the extracellular domain, followed by the transphosphorylation of intracellular domains.116 The binding of ligands to the extracellular domain of HER1, HER3, and HER4 leads to the formation of kinase-active hetero-oligomers.117 Specific ligands for HER2 have not been identified, though it becomes constitutively activated following its heterodimerization with other family members (HER1 and/or HER3),118 thereby triggering different and complicated signal transduction cascades. Moreover, spontaneous formation of various heterodimers increases with amplification of the HER2 gene.119 Heterodimers containing HER2 provide a stronger signal and have significantly higher ligand-binding affinity than homodimers or heterodimers with other family members. For instance, in several HER2-induced cancers, the HER2/HER3 dimer, the most potent EGFR family heterodimer, is indispensable for tumorigenesis and tumor maintenance.120 Therefore, restricting the dimerization of HER2 with other EGFR family members, particularly HER3, might provide an efficient treatment strategy for HER2-positive tumors.
HER1 and HER2 are overexpressed in a heterogeneous manner in GC. HER3 and HER4 have also been detected in 20.7% and 13.3% of GC, respectively.121 Several studies proved the negative correlation between high HER3 expression levels and survival of GC patients.122 HER2 overexpression was also found to be a poor prognostic indicator in GC.109,123 HER2 overexpression drives tumorigenesis through the formation of spontaneous receptor homodimers, or heterodimers with other EGFR family members, resulting in activated downstream signaling cascades, such as PI3K/AKT/mTOR and MAPK/ERK1/2.124,125 This promotes tumor cell proliferation, differentiation, survival, angiogenesis, and metastasis125,126,127 (Fig. 2). For example, the HER2/HER3 heterodimers transduce PI3K signaling through direct binding of HER3 and the p85 subunit of PI3K.128
Trastuzumab (Herceptin), the first anti-HER2 monoclonal antibody targeting the extracellular domain of the HER2 protein, has been an acknowledged treatment for both early stage and metastatic HER2-positive breast cancer for decades.129 Trastuzumab interferes with HER2 signaling in tumors via various mechanisms: inhibition of dimerization, antibody-dependent cellular cytotoxicity, receptor internalization and/or degradation, and suppression of the PI3K/AKT/mTOR signaling cascades. Trastuzumab was also the first targeted agent approved as standard treatment for HER2-positive advanced GC based on the results of the ToGA trial.40 In the ToGA trial, it was found that there existed primary and secondary resistance to HER2 blockage in GC patients. Several potential mechanisms may explain this: alteration in HER2 dimers; activation of downstream signaling pathways such as PI3K/AKT, mTOR, and MAPK/ERK; and absence of downstream regulators or alternative transduction pathway from the insulin-like growth factor receptor (IGFR).130 In 2017, Deguchi et al.131 investigated HER2 expression and the occurrence of phosphatase and tensin homolog (PTEN) loss or PI3K mutation in 264 GC cases and reported the absence of PTEN in 34.5% of HER2-positive patients. No response was observed in patients with PTEN deficiency who received trastuzumab. PTEN deficiency and/or PI3KCA mutation leads to abnormal activation of the downstream AKT/mTOR signaling cascade, leading to ineffective inhibition of HER2.132 A peptidomimetic that binds extracellular subdomain IV and a nucleic-acid aptamer that binds the extracellular domain of HER2 have been found to downregulate the HER2-dependent signaling pathways, providing a promising novel treatment of HER2-positive GC and other tumors.133,134 In brief, a comprehensive understanding of the complicated interplay between the EGFR family and downstream signaling pathway cascades would assist in identifying patients who might benefit from EGFR family targeted therapies.
PI3K/AKT/mTOR signaling pathway
The phosphoinositide 3-kinase (PI3K) pathway plays a key role in the proliferation and survival of various cancer cells including GC.135,136,137 The PI3K/AKT/mTOR signaling pathway promotes tumor progression in GC through several mechanisms, including the inhibition of apoptosis, induction of drug resistance, metastasis, and angiogenesis138 (Fig. 2). PI3K/AKT/mTOR pathway alteration plays a vital part in resistance to HER2-targeted therapy and chemoresistance in GC and several other solid tumors.127,139,140
PI3K is a broad family of lipid kinases consisting of three different classes (I, II, and III) that stand at the top of the PI3K/AKT/mTOR cascade.141 Class I PI3K is categorized into class IA and IB and is more tightly related to tumor progression.142 Classes II and III PI3Ks have been identified to contribute to the regulation of mTOR activation and autophagy.143 The activation of PI3Ks is triggered by the binding of a variety of ligands to the oncogenic receptor tyrosine kinases including EGFR, IGFR, PDGFR (platelet-derived growth factors receptor), and other growth factors.135,136,144 Activated PI3K catalyzes the phosphorylation of phosphatidylinositol diphosphate (PIP2) to phosphatidylinositol 3-phosphate (PIP3), which subsequently interacts with homology domain-containing proteins on the inner surface of the plasma membrane, resulting in conformational changes of downstream proteins.
AKT, also known as protein kinase B (PKB), normally exists in the cytoplasm.145 Upon activation of PI3K and PIP2, downstream AKT kinase translocates to the cell membrane, resulting in its conformational activation.146 AKT contains a central kinase domain with a threonine residue responsible for binding to the phosphoinositide-dependent protein kinase 1 (PDK1) and a C-terminal tail domain responsible for binding to the mammalian target of rapamycin complex 2 (mTORC2).147 While phosphorylation by PDK1 at Thr308 is fundamental, the activation of AKT also relies on phosphorylation by mTORC2 on Ser473.148,149 Phosphorylated AKT (p-AKT) plays an important part in the regulation of intracellular biological processes such as cell growth, survival, proliferation, apoptosis, EMT, metastasis, and angiogenesis.147 The lipid phosphatase and tensin homolog (PTEN), a well-known tumor suppressor gene that encodes a lipid phosphatase, is a negative regulator of PI3K signal conduction by converting PIP3 back to PIP2.150 PTEN dysfunction leads to constitutive activation of PI3K/AKT and downstream signaling, thereby stimulating cell proliferation and survival.151,152
mTOR is a highly conserved serine/threonine kinase that participates as an effector in the PI3K/AKT pathway.153 mTOR consists of two distinct functional complexes known as mTORC1 (mTOR, Raptor, and mLST8) and mTORC2 (mTOR, Rictor, mLST8, and mSIN1).154 Activation of both mTOR complexes is a vital consequence of RTK-based signaling transduction in tumors.155 The mTORC1 complex controls protein synthesis and cell growth by triggering the phosphorylation of ribosomal p70S6 kinase 1 (S6K1) at Thr229 and Thr389 and inactivating 4E-BP1 through direct phosphorylation.156,157 Activated S6K1 acts as a negative regulator and downregulates the PI3K pathway, subsequently suppressing adapter molecule insulin receptor substrate 1 (IRS-1), which obstructs the signaling between insulin growth factor 1 (IGF1) and PI3K.158 The inactivation of 4E-BP1 leads to a release of EIF4e from the dimer that triggers transcription of multiple genes.159 Activated AKT can interrupt the stable heterodimer tuberous sclerosis complex (TSC1/TSC2) by phosphorylating TSC2, thereby promoting the activity of mTORC1.158 In the progression of cancer, the activity of the PI3K/AKT pathway is elevated, and TSC1/TSC2 heterodimer is restrained by activated AKT, leading to mTORC1 activation and subsequent activation of the downstream factors (P70S6K1 and EIF4e).160,161 Another important substrate of AKT is GSK3, which promotes cell proliferation by regulating the production of cell cycle proteins like cyclin D1.162 AKT deactivates GSK3 by phosphorylation as well. GSK3 collaborates with mTORC1 by phosphorylating p70S6K1 at Ser371, which enhances mTORC1-mediated p70S6K1 phosphorylation on Thr389.163 Rictor is a critical component of mTORC2 and can function as a downstream substrate of GSK3.164 Alteration of mTORC2/Rictor influences the structure of actin and promotes cell proliferation by phosphorylating the downstream molecules165,166 (Fig. 2).
The PI3K/AKT/mTOR pathway is frequently altered in GC.108,167 From the TCGA molecular subtypes, most of the GC cases studied had different degrees of mutations in the PIK3CA gene and amplification of RTK genes such as EGFR and HER2.41,168,169 Mutations of the PIK3CA gene are likely to be late and isolated events in GC.95,170 The relationship between PIK3CA mutation and the prognosis of GC patients is controversial. Some reports identified that PIK3CA mutation promotes the risk of tumor aggressiveness, and the mutation in the exon 9 of PIK3CA has been identified as a helpful indicator for predicting prognosis in EBV-positive GC.171,172,173 Other studies declared no effective association between PIK3CA mutations and clinical outcome.174,175
Genomic amplification plays an important part in neoplastic progression. Amplification in PIK3CA is tightly associated with tumor progression, prognosis, and the emergence of drug resistance in GC.176 The amplification of PIK3CA leads to the elevation of AKT and p-AKT, thereby promoting migration, invasion, and lymph node metastasis in GC.176 LY294002, one specific inhibitor of PI3K, has been found to inhibit the activity of the ATP binding site of PI3K and lead to the reduction of p-AKT, which was closely associated with the proliferation and apoptosis of GC cells.177 Recently, APY0202, a small-molecule inhibitor of PIKfyve, has been found to be involved in inducing repression of autophagy and cell cycle arrest in an in vitro GC cell model, GC organoid model, and in vivo xenograft GC model.178
AKT acts as a central character in the activation of the PI3K axis.179,180 Elevated AKT and p-AKT expression was observed in over 74% of GC.181 The abnormal expression of p-AKT was closely related to PI3K and HER2 overexpression, and the high p-AKT level was identified as a hallmark of tumor progression, metastasis, and poor prognosis in GC.182,183 Lymphangiogenesis plays a crucial role in metastasis, recurrence, and prognosis in early GC.184 A previous study confirmed that p-AKT plays a significant role in the angiogenesis of GC via VEGF-A activation.185 Subsequently, several studies proved that inhibition of p-AKT/p-mTOR in vitro leads to a remarkable decrease of VEGF-C and VEGF-D in gastric tumor cells, and the authors proposed that lymphangiogenesis of GC might be efficiently regulated by the AKT/mTOR/VEGF-C/VEGF-D signaling pathway.186 mTOR can be activated via multiple upstream factors and acts as a bridge in a variety of downstream signaling pathways. mTOR stands at the terminus of the PI3K/AKT/mTOR signaling cascade and is one of the most independent elements of the PI3K axis.187 The mutations in upstream regulators from the different axes, such as EGFR, PI3K, and PTEN, can lead to over-activation of mTOR.188,189,190 Aberrant activation of mTOR has been detected in over 60% of GC cases.191 The dysregulation of mTOR activity participates in the regulation of GC cell growth and differentiation.167 In addition, some previous studies have identified that the expression of mTOR was much higher in GC tissues than in normal gastric tissues.192 Additionally, a positive link between elevated mTOR levels and pathological parameters like invasive depth and lymph node metastasis was found in GC.193 Therefore, mTOR expression can serve as a biomarker of not only the diagnosis of GC but also the invasiveness and metastasis of the tumor, and its prognostic role has been proven by the negative correlation with five-year survival rates of GC patients in cohort studies.193,194
The significant contribution of the PI3K/AKT/mTOR signaling pathway in the progression of GC suggests that this signal axis is a promising target for cancer therapy. From the results of existing clinical investigations in GC, the efficacy of PI3K inhibitors, AKT inhibitors, mTOR inhibitors, and other monotherapy were not as effective as dual PI3K/mTOR inhibitors or several combination therapies,195 suggesting that the restriction on the therapeutic effect by the heterogeneity of GC should be emphasized in designing new targeted medication regimens.
P53 signaling pathway
The main role of p53 lies in its involvement in the regulation of DNA repair as well as in the control of the cell cycle, apoptosis, and differentiation, which is mainly through DNA-protein and protein-protein interactions.196 It can induce aging or promote cell apoptosis and DNA repair,197 providing a mechanism to prevent the accumulation of potentially malignant or defective cells.198 In vertebrates, p53 can temporarily block the cell cycle by regulating checkpoints in G1/S and G2/M phases199 and these regulatory processes are closely related to the transcriptional activation of related genes by the p53 protein. Cyclins and cyclin-dependent kinases (CDKs) are the two major proteins involved in cell cycle progression.200 Functional analysis revealed that Reprimo (RPRM) is transcriptionally regulated by p53 and serves to arrest the cell cycle at the G2/M checkpoint, by inhibiting nuclear translocation of the Cdc2/cyclin B1 complex.201 Significant downregulation of RPRM has been described in GC cells expressing wild-type p53.202 With DNA damage, the cell cycle is arrested in the G2/M phase as monitored by p53-mediated downregulation of p21, which prevents the transmission of mutagenic damage.200
p53 is affected by many non-coding RNAs. For example, miR-181a can elevate the expression and activity of p53203 by targeting the tumor suppressor ataxia-telangiectasia mutated (ATM) gene.204 miR-650 enhances the function of p53 in gene transcription and promotes cell growth by the upregulating expression of the inhibitor growth family member 4 (ING4).205 TP53-inducible nuclear protein 1 (TP53INP1) is a key element in p53-mediated cell death and cell cycle arrest. The upregulation of both miR-17-5p and miR-20a in GC can promote cell growth by deregulating TP53INP1 and p21.206 In contrast, miR-499 can indirectly upregulate p53 and its downstream target p21, activating caspase-apoptosis pathways.207 Therefore, downregulation of miR-449 observed in GC cells is associated with cell survival advantages.207 Mutations in some key sites of the p53 gene can directly lead to abnormal cell proliferation, while polymorphisms at non-important functional regions of TP53 may also affect GC tumorigenesis.208 Studies have reported elevated expression levels of p53 in more than 75% of GC patients, and the mutation rate of the TP53 gene in all GC patients is ~30%, but it may vary in patients with different GC subtypes and etiologies.209,210 The polymorphism of codon 72 of the TP53 gene is closely associated with gastric carcinogenesis in the US population.211 TP53 gene mutation is the main reason for the loss of normal function of p53 protein,210,212 which is an important initiating factor for the occurrence and development of GC. Cell cycle regulators, especially p16INK4A (cyclin-dependent kinase inhibitor 2A, CDKN2A), are upregulated by p53 inactivation in precancerous GC and act as a barrier to disease progression.213 Co-deletion of CDKN2A and TP53 in dysplastic gastric organoids promotes the cancer phenotype and also induces replication stress, thereby exposing susceptibility to inhibitors of the DNA damage response.213 In humans, folic acid (vitamin B9) supplementation may play a vital role in the chemoprevention of GC since it can significantly increase the expression of p53 and decreases the expression of the Bcl-2 oncogene protein in the gastric mucosa.214,215
H. pylori infection can promote the accumulation of mutations in the TP53 gene, which has been reported to occur in 50% of gastric tumors.216 The proteasomal degradation of p53 may also be induced indirectly by H. pylori infection.217,218 In response to genotoxic stress, p53 triggers signaling pathways that lead to temporary cell cycle arrest, activating the repair process of DNA.219 Inactivation of p53 promotes genomic instability, which is a hallmark of cancer.220 Thus, inhibition of p53 can be a strategy for modulating host cell function in response to H. pylori.221 From the aspect of molecular mechanism, H. pylori can induce aberrant DNA methylation and downregulate the expression of genes involved in signal transduction pathways and tumor suppression.222 Previous studies have found that H. pylori infection induces DNA hypermethylation in the promoter regions of upstream-stimulated transcription factor genes USF1 and USF2, and inhibits their expression, which accompanies the development of gastric precancer.223 These transcriptional factors may act as tumor suppressors by regulating genes involved in stress and immune responses, inflammation, cell cycle control, and genome stability.224 USF1 also binds to p53 as UV-induced DNA damage occurs and prevents the interaction between p53 and the E3-ubiquitin ligase HDM2. This results in p53 stabilization and transient cell cycle arrest.225,226 In about half of GC patients, USF1 expression is lower in tumor tissue than non-tumor tissue, and 88% of patients with low USF1 expression have H. pylori infection.227 Low expression of p53 closely correlates to low expression of USF1, and low expression of both is associated with poor prognosis.227
HGF/c-MET signaling pathway
The mesenchymal epidermal transition factor (c-MET), which is encoded by the proto-oncogene MET, is a transmembrane receptor expressed on the surface of epithelial and endothelial cells.228 c-MET belongs to the receptor tyrosine kinase (RTK) family, and hepatocyte growth factor (HGF) is the specific ligand for c-Met.229 The canonical pathway is activated when HGF binds to c-MET, followed by the homodimerization of c-MET and trans-phosphorylation of its intracellular kinase domains.229 These changes form a docking site on c-MET that recruits effector molecules, thus triggering the signals that regulate cell survival, proliferation, migration, and morphogenesis.230The major downstream signaling pathways include Ras/MAPK, PI3K/AKT (Fig. 2), Wnt/β-catenin, and signal transducer and activator of transcription 3 (STAT3).230,231 There are also many distinct mechanisms of HGF-independent activation of c-MET (non-canonical activation), such as the phosphorylation of c-MET mediated by direct binding of des-gamma-carboxyl prothrombin at the intracellular kinase domain232 and crosstalk with other signaling pathways.233 While the HGF/c-MET pathway has important physiological functions in normal cellular processes, aberrant activation of this pathway is closely associated with tumor invasion and metastasis in many types of epithelial cancers, such as lung, breast, kidney, liver, ovarian, thyroid, and gastrointestinal tract cancers.234 Multiple mechanisms, which can be related to canonical or non-canonical activation or both, may be involved, including gene amplification, activating mutations, transcriptional modification, overexpression, enhanced stimulation by autocrine or paracrine HGF, interactions with other active cell surface receptors, and dysregulations under certain environmental conditions such as hypoxia and inflammation.235,236
MET gene amplification, high c-MET expression, and co-expression of HGF and c-MET have been found to be significant predictive factors for worse prognosis in GC.237,238,239 Although MET gene amplification is relatively rare (4–10%) in GC patients,240 c-MET protein overexpression has been detected in up to 82% of cases.241 This discrepancy may result from detection methods, whether c-MET protein detection based on both membranous and cytoplasmic staining had a more significant correlation with MET gene amplification, compared to that only on membranous IHC.242 Another important mechanism is the deletion mutation of the MET gene at exon 14 (METex14del mutation), which leads to delayed ubiquitination and degradation of c-MET protein.243 In a study of 230 patient specimens, including 42 GC, 13 tumor samples were found to contain the METex14del mutation, among which all had MET overexpression but only one had MET gene amplified.243 Notably, MET inhibitors inhibit the growth of patient tumor-derived cell lines from GC and colon cancer containing the METex14del mutation, suggesting that METex14del can be a potential biomarker for gastrointestinal malignancies.243
As an important regulator of many signaling pathways, the HGF/c-Met axis is closely associated with GC development and progression, tumor metastasis, and therapeutic response. Overexpression of c-MET is frequently observed in GC cases with an increased risk of distant metastasis to the liver244 or peritoneum.245 Recent studies have discovered that the c-MET signaling may be involved in H. pylori infection-related GC tumorigenesis and metastasis. Ito et al.246 found that both canonical and non-canonical activation of c-MET signaling in GC cells could be promoted by H. pylori infection through its virulence factor CagA protein. Furthermore, the phosphorylated active form of c-MET can be secreted in exosomes by H. pylori-infected GC cells and transferred to macrophages, which may consequently induce the pro-tumorigenic phenotype conversion of macrophages promoting tumor progression.247 Additionally, H. pylori infection could increase the intracellular level of heparinase (HPA), an endoglucuronidase found to be carcinomatosis-relevant, leading to the activation of multiple signaling pathways in human GC cells.248 Hao and colleagues observed that overexpression of HGF and HPA had a positive correlation with TNM stage, depth of invasion, and poor prognosis in GC patients.249 Their further mechanistic study suggested that HGF/c-MET can regulate HPA expression by activating PI3K/AKT and downstream nuclear factor kappa B (NF-κB) signaling. HPA can also mediate the shedding of heparin-binding HGF to enhance HGF liberation, which can jointly induce tumor metastasis.249 Therefore, the HGF/c-MET axis and HPA may be effective therapeutic targets for treating H. pylori-related GC.
c-MET has been a well-studied target for cancer treatment and numerous targeted inhibitors have been developed. Blocking HGF in cancer-associated mesenchymal stem cells, where HGF is hyper-produced, may also be a potential GC treatment strategy based on a recent in vivo study.250 Currently, the precise regulatory cascades of HGF/c-MET in GC cells have not been fully elucidated. Utilizing complimentary deoxyribonucleic acid microarray technology, Koh et al.251 identified several downstream molecules of HGF/c-MET signaling, including E-cadherin, urokinase plasminogen activator, and Kisspeptin, which are cell invasion and migration regulators. Moreover, two cell apoptosis modulators, Jun-B and lipocalin-2, are also recognized as interacting with the HGF/c-MET pathway.251 Another study demonstrated that the phosphorylation of RhoA, which is a biomarker highly mutated in diffuse GC patients, may be dependent on c-MET activity.252 Notably, a c-MET inhibitor prevented GC cell growth only in GC cells transfected with wild-type RhoA but not Y42 mutant RhoA in vivo and in vitro. Thus, the combined levels of c-MET and phosphorylated-RhoA should be used as predictors for prognosis and patient stratification to optimize targeted c-MET therapy.252
In addition to downstream effectors, upstream regulators of HGF/c-MET are also important biomarkers and potential targets in GC. The C-X-C motif chemokine ligand 12 (CXCL12) was found to induce interaction of c-MET with caveolin 1 in lipid rafts. This interaction can lead to activation of c-MET, thereby inducing EMT in GC cells and promoting cell migration. Further analysis in clinical samples also revealed a positive correlation between the CXCL12 receptor CXCR4 and c-MET phosphorylation as well as poor patient prognosis, indicating the clinical importance of the crosstalk between c-MET and CXCL12 in GC treatment.253 Several miRNAs have been reported to be involved in GC proliferation and metastasis by their regulation of HGF/c-MET expression. It has been reported that miR-1/34a/144/206 directly target the mRNA of c-MET.254,255,256,257 In contrast, miR-15a/16/195 are found to directly target HGF mRNA.258 These are negative regulators of HGF/c-MET expression, which are found down-regulated in GC tumors, implying their potential therapeutic applications to repress HGF/c-MET-mediated cell proliferation and migration in GC. Other in vitro studies have indicated that ETS homologous factor (EHF) may be critical to GC cell proliferation, apoptosis, cell cycle, EMT, and invasion via the activated c-Met pathway,258 whereas IL-10 secreted by cancer-associated macrophages (CAMs) may be involved in GC carcinogenesis.259 Nevertheless, the clinical significance of miRNAs, EHF, and IL-10 in GC diagnosis and treatment must be further verified.
The HGF/c-MET axis may also be involved in the therapeutic response of GC. In GC cells with HGF/c-MET activation, excessive transphosphorylated c-MET molecules are likely to interact with other receptor tyrosine kinases such as EGFR and HER2 forming heterodimers, which may allow bypass signaling to provoke resistance to corresponding targeted therapies.260,261,262 This provided a clue that co-inhibition of bypassing pathways may be a potential therapeutic application in treating GC. MET gene mutations can change the sensitivity of GC cells to targeted drugs by affecting the activation of downstream signaling pathways. Shen et al.263 recognized that GC patients carrying MET G1163R or D1228Y/N mutations are likely to show resistance to the TKI drug crizotinib, whereas patients with MET V1092L, D1228G, or Y1230H mutations could benefit from this targeted therapy. This indicates that MET mutation analysis may be useful for designing precision medication for GC.
Wnt/β-catenin signaling pathway
The Wnt/β-catenin signaling pathway is involved in cell proliferation, migration, and death, and is important for the development and homeostasis of some tissues.264,265,266 The β-catenin protein is a transcriptional coactivator in Wnt pathway, which has been found to be involved in a number of biological processes of tumor cells, including proliferation,267,268 anti-apoptosis,269 and infiltration transfer.270 The Wnt/β-catenin pathway is activated when the Wnt ligands bind to the seven-transmembrane receptor Frizzled (FZD) and the low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6).271 The Wnt-FZD-LRP5/6 trimer complex recruits disheveled (DVL) and axin through the intracellular domains of FZD and LRP5/6, thereby inhibiting β-catenin phosphorylation and ensuring β-catenin stability. β-catenin then detaches from degradation complexes and accumulates in the cytoplasm, enabling the Wnt pathway to promote cancer progression during the cell cycle.272,273,274 Elevated cytoplasmic and nuclear levels of β-catenin promote the cooperation of β-catenin with T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to activate the expression of Wnt-responsive genes275 (Fig. 2). Several mutant component molecules of typical Wnt signaling lead to aberrant activation of the Wnt/β-catenin pathway,276,277 which further contributes to the malignant transformation and invasion of GC.278,279
Upregulation of Wnt-1 ligands has been shown to promote advanced GC development.280 In contrast, Wnt-2 enhancement is closely associated with gastric tumor formation, invasion, or dissemination.281 Studies have found that Wnt-5a can stimulate the migration and invasion of GC cells, mainly through the activation of focal adhesion kinase (FAK) and the small GTP-binding protein Rac.282 Overall, dysregulation of Wnt/β-catenin signaling is observed in more than half of the patients and is considered a primary mechanism of GC development.276,283 Although persistent activation of Wnt/β-catenin signaling is shown to be related to chemoresistance,284,285 the mechanism remains largely unexplored. Several researchers found that activation of Wnt/β-catenin signaling can inhibit ferroptosis in GC cells by attenuating the production of intracellular lipid ROS or inducing glutathione peroxidase 4 (Gpx4) expression by the direct binding of β-catenin/transcription factor 7 like 2 (TCF7L2, also known as T cell factor 4, TCF4) transcriptional complex to the promoter region of Gpx4.286,287,288 The latter mechanism was verified by two studies demonstrating that deficiency in TCF4 promoted cisplatin-induced ferroptosis both in vivo and in vitro.286,289 Modulating ferroptosis through regulating Wnt/β-catenin signaling may be a potential therapeutic strategy for improving chemosensitivity in advanced GC.286 Finally, targeting Wnt/β-catenin signaling may also improve the therapeutic outcomes of radiotherapy and immunotherapy due to the involvement of ferroptosis.286,290 A recent study demonstrated that the Wnt/β-catenin signaling pathway is inversely correlated with the infiltration of T cells in the tumor microenvironment (TME), and, as a result, affects the therapeutic efficacy of PD-1 antibodies.289,291,292,293 It has been found that the disruption of the Wnt/β-catenin pathway in GC cells inhibited their migration and invasion.294 Meanwhile, down-regulation of Wnt/β-catenin may enhance the sensitivity of GC cells to PD-1 antibody.295,296 This result further suggests that jointly targeting to inhibit β-catenin and PD-1 jointly may be a potential and effective treatment for GC patients.
Different mechanisms can facilitate tumor cell survival and proliferation mediated by activated Wnt/β-catenin signaling in GC. β-catenin-activated CCL28, which is a mucosae-associated epithelial chemokine, can regulate T cells in vitro.297 In a clinically relevant mouse GC model established by Helicobacter felis (H. felis) infection and the carcinogen N-methyl-N-nitrosourea (MNU), using a Wnt signaling pathway inhibitor iCRT14 to inhibit β-catenin/TCF activity resulted in decreased CCL28 expression and Treg expression in the stomach cell infiltration.297 Furthermore, the anti-CCL28 antibody significantly attenuated Treg cell infiltration and tumor progression in the H. felis/MNU mouse model.297 This study extended the previous understanding of the oncogenic role of the Wnt/β-catenin pathway mainly through its control of cell proliferation, survival, and differentiation in GC, and confirmed that the immunoregulatory function of the β-catenin signaling pathway also plays an important role in tumor progression.297 More importantly, CCL28 blockade exhibits a surprising antitumor effect by inhibiting Treg cell infiltration, providing a new idea for the immunotherapy of GC.297,298 E-cadherin, a component of the β-catenin degradation complex, also plays a crucial role in negatively regulating Wnt signaling.299 β-catenin is in direct contact between cadherin and α-catenin, the latter interacting with the actin cytoskeleton to form tight cell-cell junctions.299,300 As cadherin may maintain the activity and function of β-catenin on the membrane during EMT by competing with its degradation mechanism, the ability of β-catenin to bind to cadherin is essential when the transcription proceeded because cadherin may stabilize β-catenin on the membrane by competing with its degradation mechanism during EMT.301,302 In brief, the connection between cadherin and β-catenin may be one of the mechanisms of the EMT process in GC,303 and may provide new options for GC diagnosis or therapeutic interventions in the future.304
NF-κB signaling pathway
The NF-κB family of transcription factors consists of several members—RelA, RelB, c-Rel, NF-κB1(p50), and NF-κB2(p52)—which form dimers (homo- and hetero-) and modulate the expression of a variety of genes.305 The typical dimer refers to the heterodimer of RelA and p50 subunits.306 The canonical or classical NF-κB pathway is activated by different receptors, including tumor necrosis factor receptors (TNFRs), Toll-like receptors (TLRs), and interleukin-1 (IL-1R). NF-κB is kept inactive in the cytoplasm bound to members of the IκB family (IκBα, IκBβ, and IκBγ).307 Upon stimulation, the IκB kinase (IKK) complex is activated, leading to phosphorylation of IκBα at Ser32 and Ser36 by IκBβ,308 followed by poly-ubiquitination and subsequent degradation of IκBα by the 26S proteasome (Fig. 2). Degradation of IκBα sets NF-κB free, and it translocates to the nucleus where it binds to the promoters of downstream target genes, thus promoting GC progression.309,310,311
The NF-κB signaling pathway is one of the most critical cellular signaling pathways and has an important role in apoptosis and cell survival.312,313 One of the main functions of NF-κB is regulation of transcription of inflammatory molecules. NF-κB can regulate the expression of many inflammatory mediator genes related to inflammation and immune response, including bcl-2, bcl-xl, cIAP, BIRC5, TRAF, COX-2, MMP-9, iNOS, and various cell cycle regulators.314,315 The NF-κB pathway also plays a key role in EMT and cancer stem cell activities316 and has an important role in tumor formation and tumor development through its anti-apoptotic effect. Inhibition of NF-κB signaling can induce apoptosis and cell cycle arrest in GC cells.317,318 In tumorigenesis and development, NF-κB is more likely to play a key linking role in signaling pathways. Proto-oncogene mutation affects upstream factors of the NF-κB signaling pathway, and these factors activate the NF-κB signaling pathway and downstream effectors and initiate gastric carcinogenesis.319 Uncontrolled NF-κB signals lead to the occurrence of many tumors, and the abnormal activation of NF-κB in tumors may be one of the main anti-apoptotic factors in GC cells.319,320 When activated, it can generate strong anti-apoptotic signals and accelerate tumor development.
At the same time, NF-κB can promote tumor formation by a non-apoptotic mechanism, by directly stimulating cell proliferation through the activation of the proto-oncogenes c-myc321 and CCND1 (encoding cyclin D1).322 As a target gene of NF-κB, CCND1 transcription initiated by NF-κB promotes the cell cycle transition from G1/G0 phase to the S phase, leading to cell proliferation and transformation into malignant and cancerous cells.323,324 NF-κB can also upregulate hypoxia-inducible factor 1 (HIF-1), which initiates gastric carcinogenesis by promoting tumor angiogenesis.325,326 Studies have shown that connective tissue growth factor (CTGF) is upregulated in clinical tissue specimens of GC.327 In vitro experiments have shown that high expression of CTGF in advanced GC cells significantly increases tumor metastasis, while RNA interference-mediated knockout of CTGF significantly inhibits cell metastasis.328 This process demonstrates the promotive effect of CTGF on GC invasion and metastasis via the downregulation of E-cadherin and activation of NF-κB (Fig. 2). Similar studies also found that the expression of proteinase-activated receptor-1 (PAR-1) stimulates NF-κB activation, thereby initiating the invasion and metastasis of GC.329 Additionally, it has been found that NF-κB activation is associated with the heparanase gene expression in GC and is significantly correlated with GC invasion-related features such as lymph node invasion, pathological stage, and depth of invasion.330,331 Therefore, NF-κB may become a potential therapeutic target for inhibiting GC invasion and metastasis.324
The upregulation of the NF-κB signaling pathway is involved not only in the occurrence of tumors but is also associated with chemoresistance and radioresistance.332,333 NF-κB inhibitors may enhance the efficacy of antitumor drugs or increase sensitivity. With the improvement of the rapid detection technology of NF-κB activity and the understanding of the mechanism of NF-κB activation, many drugs that inhibit the activation of NF-κB have been developed. Natural drugs targeting NF-κB have exhibited potential as chemotherapy for GC.334,335,336,337 For example, Ji and colleagues have reported that tetramethylpyraz, a natural alkaloid, induces GC cell apoptosis by downregulating NF-κB and cyclin D1.338 Therefore, screening chemotherapeutic drugs with NF-κB-targeting effects may be a potential strategy for improving chemotherapy.
TGF-β signaling pathway
Transforming growth factor-β (TGF-β) is a family of active polypeptides that are physiologically involved in embryonic growth and development, stem cell differentiation, wound healing, and inflammation regulation.339 The secretion disorder of the TGF-β family is closely associated with the development of tumors.340 The TGF-β family consists of three forms with similar biological functions: TGF-β1, TGF-β2, and TGF-β3.340 Among them, TGF-β1 has the highest expression level.341,342 TGF-β1 is a multifunctional cell growth factor and a multi-type cell proliferation inhibitor.343 TGF-β1 can inhibit the proliferation and differentiation of various cells by binding to its receptors, such as TGF-β R1.344 It is widely involved in cell morphological changes, adhesion, metastasis, and apoptosis.345,346 The expression of TGF-β1 and TGF-βR1 is closely related to the biological behavior and prognosis of malignant tumors.347 TGF-β1 is the signaling protein of the DPC4 (SMAD4) gene, a tumor suppressor gene. The Smad4 proteins, which have an important impact on the occurrence, development, and metastasis of malignant tumors,348 are vital downstream effectors of the TGF-β signaling pathway.349 TGF-β ligands bind to membrane receptors to form two types of receptor heterodimers, type I and II, which can activate downstream Smad2 and Smad3 proteins and then combine with Smad4 to form a transcription complex in the nucleus, thereby regulating the transcription of target genes and exerting inhibitory effects on cell growth.340,350
TGF-β1 is generally considered a negative cell growth regulator and is strongly correlated with the occurrence and progression of GC and its clinicopathological features.340 TGF-β1 in normal gastric mucosa is expressed mainly in the cytoplasm of epithelial cells and some mucous cells and in the cytoplasm of cancer cells in GC tissue.351 A retrospective study of 50 patients with GC after surgery found that the 5-year survival rate of patients with high TGF-β1 expression was significantly lower than that of patients with low TGF-β1 expression, indicating that the expression of TGF-β1 is closely related to the prognosis of GC patients.352 However, depending on the cell type and physiological environment, TGF-β1 can exhibit opposite effects. TGF-β1 has a significant growth inhibitory effect on cells of epithelial origin by preventing cells from the G1-S phase in vitro,353,354 and TGF-β1 expression is often reduced or absent in malignant tumors.355 TGF-β1 can also inhibit the proliferation and induce apoptosis of GC cell lines HSC-39 and HSC-43 in vitro.356,357 However, the results of another study showed that TGF-β1 protein was highly expressed in GC and increased as the differentiation degree decreased, indicating that TGF-β1 may play a role in the malignant transformation and proliferation of tumors.358 The high expression of TGF-β1 in GC cells may also be due to the blockade between TGF-β1 and receptors, resulting in an accumulation of TGF-β1;359,360 the elevated TGF-β1 level may promote tumor growth rather than inhibit it, but it does not lose its inhibitory effect on immune cells such as NK and LAK, leading to immune escape of cancer cells.361,362 Both TGF-β and its receptors are highly expressed in early penetrating GC tissues, which is related to the strong growth and infiltration ability of this type of GC.363,364
Moreover, the TGF-β signaling is one of the main inducers of EMT, which may be related to its crosstalk with the AMPK pathway.350 AMPK activation not only inhibits the EMT process of GC cells regulated by TGF-β, but also inhibits the production of TGF-β.365,366 Smad3 was found to play a key role in these two processes as well. AMPK can inhibit the phosphorylation and the nuclear translocation of Smad3 protein, thus inhibiting the transcriptional regulatory functions of TGF-β.366,367 Therefore, inhibiting the phosphorylation of Smad3 may serve as a new therapeutic target for GC.
Immune checkpoint signaling pathways
The growth and progression of cancer are directly related to the suppression of the immune system, where inhibitory immune checkpoints play a vital role. Immune checkpoints are modulators of the immune system that either promote (co-stimulatory molecules) or stop signaling (co-inhibitory molecules) in immune cells and control their activity, thus, playing a crucial role in maintaining immune homeostasis in immune cells.368,369 The first immune checkpoint molecule, cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), was discovered by Brunet et al. in 1987.370 its function was unclear until 1995, when Allison et al. revealed CTLA-4 to be an important immune checkpoint molecule with great potential as a target for cancer therapy.371 Immunosuppressive checkpoint molecules, such as PD-1, CTLA-4, T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), Lymphocyte-activation gene 3 (LAG-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT), are usually expressed on T cells and bind to their ligands on other cells, thereby triggering negative regulations on immune signaling pathways and preventing immune damage.369,372,373,374,375 In tumor cells, upregulation of ligands of these inhibitory immune checkpoints during tumor progression helps suppress antitumor immune responses and induce tumor immune escape.369,376 Therefore, targeting immune checkpoints is a vital approach of immunotherapy in cancer treatment.
Different immune checkpoint molecules and their ligand-receptor signaling are summarized in Fig. 3a. PD-L1 and PD-L2 are transmembrane proteins, which are considered co-suppressors of the immune response. Upon the binding of PD-L1/PD-L2 to PD-1, the proliferation and cytokine secretion of PD-1-positive T cells are reduced, while apoptosis is activated. For cancer cells with PD-L1/PD-L2 expression, attenuating host anti-tumor immune response provides survival advantages for the cancer cells.377,378 In the CD28/CTLA-4/B7 co-stimulatory pathway, CD28 is one of the proteins expressed on T cells that produce co-stimulatory signals required for the activation of T cells; CTLA-4 proteins located on T cells function to help keep the body’s immune responses in check; and B7-1/2 are checkpoint proteins on the membrane of activated antigen-presenting cells (APC).379 T cells can be activated when the T cell receptor (TCR) binds to the antigen and major histocompatibility complex (MHC) proteins on the APC, accompanied by CD28 binding to B7-1 (CD80) or B7-2 (CD86) on the APC.380 However, when B7-1/B7-2 binds to CTLA-4, the T cells are inactivated and unable to kill tumor cells in the body.381 Using an immune checkpoint inhibitor (an anti-CTLA-4 antibody) to block the binding of B7-1/B7-2 to CTLA-4 allows the T cells to be activated and kill tumor cells.382 The TIM-3/galactin-9 and LAG-3/galactin-3 pathways are similar to the PD-1/PD-L1 pathway. The binding of TIM-3 present on activated T cells to the ligand galactin-9 on tumor cells blocks the response of interferon-γ (IFN-γ) -producing CD4+ T helper 1 (Th1) cells and induces apoptosis of CD4+ and CD8+ T cells, resulting in immune tolerance.383 TIM-3 may also be co-expressed with PD-1 in tumor-infiltrating immune cells and act synergistically to mediate effector T cell depletion and dysfunction.384 LAG-3 on activated T cells is associated with reduced anti-cancer immune response by inhibiting CD8+ T cells upon binding to galactin-3 in tumor cells.373 TIGIT is a co-inhibitory receptor that is highly expressed in the tumor-infiltrating lymphocytes in various malignant cancers.385 TIGIT can downregulate the immune response either by competing for CD155 ligand binding with CD226 thereby reducing the CD266/CD155-dependent co-stimulation of T cells,386,387,388 or by directly transmitting inhibitory signals to effector cells.389 Among these pathways of immune checkpoints, the PD-1/PD-L1 signaling is the most widely studied as a diagnostic/prognostic biomarker as well as a therapeutic target of GC.

The immune checkpoint signaling pathways in gastric cancer and regulations on PD-L1 by H. pylori and EBV. a The immune checkpoint proteins PD-1 on the surface of T cells interact with the ligands PD-L1/PD-L2 on GC cells, or the aberrant CTLA-4 proteins on GC patient T cells interact with B7 on antigen-presenting cells, resulting in an immunosuppressive microenvironment, providing cancer cells with a survival advantage. TIGIT on the T cells membrane competes with the activation of CD226 binding to CD155 from the GC cells. Other immune checkpoint proteins, TIM-3 or LAG-3, interact with galectin-9 or galectin-3 released from GC cells, inhibiting the activation of T cells. b Chronic H. pylori or EBV infection, which are risk factors of GC, can induce upregulation of PD-L1 in GC cells via various signaling pathways and microRNAs, promoting immune escape. EBV Epstein–Barr virus, PD-1 programmed death 1, PD-L1/2 programmed death ligand 1/2, CTLA-4 cytotoxic T-lymphocyte-associated protein 4, TCR T-cell receptor, MHC major histocompatibility complex, TIGIT T cell immunoreceptor with Ig and ITIM domains, TIM-3 T cell immunoglobulin and mucin-domain containing-3, LAG-3 lymphocyte-activation gene 3, IFN-γ interferon gamma, JAK2 Janus kinase 2, STAT1 signal transducer and activator of transcription 1, IRF1 interferon regulatory factor 1, EBNA1 Epstein–Barr nuclear antigen 1, MAPK mitogen-activated protein kinase, NOD1 nucleotide-binding oligomerization domain-containing protein 1, SHH Sonic hedgehog protein, CagA cytotoxin-associated gene A, T4SS type IV secretion system. This figure was created with Biorender.com
Transcriptome analysis of the TCGA subtypes in GC has revealed that immune cell signaling is significantly upregulated in EBV+ or MSI subtypes compared to the other two subtypes.390 The different levels of immunomodulation shown by the four TCGA subtypes have opened a stratifying strategy for GC patients to maximize immunotherapy efficacy, while immune cell signaling has gained extensive attention in GC research. High content of immune cells, downregulation of genes involved in cytokine/chemokine pathways, and upregulation in PD-L1 and/or PD-L2 expressions are frequently found in EBV+ GC cases.391,392 In contrast, the MSI subtype is characterized by increased mutation rates and DNA hypermethylation profiles for DNA mismatch repair genes like MSH1, MSH2, MSH3, and MLH1, which results in alterations in length with short, repeated DNA sequences (microsatellites) and enhanced expression of neoantigens.41,393 Because of the increased neoantigen recognition and the corresponding expression of immune checkpoints in the tumor microenvironment, GC of MSI subtype exhibits high CD8+ T cell infiltration and is more sensitive to immune checkpoint inhibitors.394,395
Elevated mRNA levels of PD-1, PD-L1, and PD-L2 have been observed in GC patients.396 Yun et al.397 found that HER2, PD-L1, and PD-1 gene expressions in GC are related to staging and lymph node metastasis. The elevated PD-L1 expression is correlated with certain GC molecular subtypes. Liu et al.398 observed that PD-L1 was expressed in 59.3% of GC patients and correlated with MSI and EBV+ subtypes. H. pylori-positive gastric tumors have also been found to have higher PD-L1 expression and T cell hypo-responsiveness, which is considered one of the carcinogenesis mechanisms by H. pylori infection.399 During GC initiation and progression, chronic EBV or H. pylori infection induces immunomodulation from a pro-inflammatory state recruiting immune cell infiltrations to an immunosuppressive microenvironment where PD-L1 is upregulated in GC cells.400
However, different mechanisms are involved in EBV- and H. pylori-induced PD-L1 upregulation. In EBV-associated GC, the PD-L1 expression on tumor cells is triggered by interferon-γ (IFN-γ) via the JAK2/STAT1/interferon regulatory factor-1(IRF1) signaling pathway.401 The EBV nuclear antigen 1 (EBNA1), which is a transcription factor that maintains EBV genome copy number during cell division, may also be a regulator of IFN-γ-induced PD-L1 expression.401 Compared to other GC subtypes, EBV-associated GC displays low expression levels of the PD-L1-targeting miR-200 family, which may also contribute to the high expression of PD-L1.402
Upregulation of PD-L1 by H. pylori in gastric epithelial cells primarily involves the activation of upstream signaling pathways that promote PD-L1 expression. The two major pathways are the nucleotide-binding oligomerization domain-containing protein 1 (NOD1)-dependent activation of p38-MAPK pathway promoted by the H. pylori type 4 secretion system (T4SS) components including the effector protein CagA and peptidoglycan fragments,403 and the CagA-dependent activation of sonic hedgehog signaling pathway.404 Infection by H. pylori also negatively affects the expression of PD-L1 suppressor miRNAs, such asmiR-132 and miR-200b, which partially contribute to the elevated PD-L1 expression in H. pylori-positive GC405 (Fig. 3b). The overexpression of PD-L1 on GC cells inhibits T cell proliferation via the PD-1/PD-L1 inhibitory signaling and induces Treg differentiation from naive T cells, leading to immune escape. Paradoxically, several studies have reported that in advanced GC patients who underwent surgical resection or resection plus adjuvant chemotherapy, the H. pylori-positive patients have an improved survival compared to H. pylori-negative patients.406,407,408,409,410,411 In a retrospective study involving 49 advanced GC patients, Koizumi et al. observed that the H. pylori-positive patients had a significantly better prognosis than H. pylori-negative patients in the population of PD-L1-negative, while the prognostic difference was statistically insignificant between H. pylori-positive and H. pylori-negative patients in the PD-L1-expressing population. The H. pylori-positive/PD-L1-negative group showed a potential survival benefit even when the dose of adjuvant S-1 chemotherapy was reduced.411 Since the other immune-related parameters, including CD4, CD8, TLC, MMR proteins, and MSI status, did not exhibit a significant correlation with PD-L1 levels or H. pylori infection, the immune escape induced by H. pylori-dependent PD-L1 upregulation is likely the dominant mechanism of tumor cell survival and poor prognosis.411 Therefore, the PD-L1 expression should be taken into consideration when H. pylori infection is used as a prognostic factor in GC.
Although PD-L1 overexpression is more likely to be detected in GC with deeper tumor infiltration and lymph node metastasis,412,413 PD-L1 can be a positive prognostic biomarker. Detection of PD-L1 or detection of both HER2 and PD-1/PD-L1 in GC may provide a vital reference for stratifying patients who can benefit from checkpoint inhibitor immunotherapy or targeted therapy. As a result, regulatory factors that induce PD-L1 expression have gained attention in developing strategies to increase immunotherapy efficacy. IFN-γ signaling has been shown to be involved in regulating not only the expression level of PD-L1414 but also the binding affinity of PD-L2 to PD-1.415 Moreover, PD-L1 expression can be stimulated by inhibition of autophagy via the IFN-γ signaling pathway,414,416 implying that pharmacological modulation of autophagy may be a novel strategy for improving the efficacy of PD-L1 blockade. On the other hand, miR-105-5p was found as a negative regulator of PD-L1 expression, highlighting it as a potential biomarker for PD-1/PD-L1 immunotherapy and a target for combinational regimen.417 However, it should be noted that taking the timing and site of PD-L1 expression into consideration is necessary. Kim and colleagues reported that in the mouse GC model, 5-FU and oxaliplatin reduced the numbers of myeloid-derived suppressor cells to increase the anti-GC efficacy of the PD-1 inhibitor and promote tumor infiltration by CD8+ T cells.418 However, these chemotherapeutic agents might also mediate induction of PD-L1 expression in tumor cells leading to tumorigenesis of gastric epithelial cells and tumor progression.418
Genetic alteration of CTLA-4 in humans has been associated with GC development;419 however, CTLA-4 may not be a good target in treating cancer according to the current knowledge. Liu et al.420 reported that the association of CTLA-4 single nucleotide polymorphism with noncardiac GC is not significant in a Chinese population. A recent case report showed hyperprogression of the lymph nodes and liver lesions compressing the gastric stump from a 68-year-old patient with stage IV MSI subtype GC after receiving immunotherapy of durvalumab (PD-1 inhibitor) and tremelimumab (CTLA-4 inhibitor).421 More study is still needed to evaluate the therapeutic significance of CTLA-4 in GC.
TIM-3 is an independent indicator of poor prognosis in GC patients and may play an essential role in the progression, invasion, and metastasis of GC.383,422 TIM-3 expression is induced on NK cells and tumor-infiltrating T cells during the development of GC, making it a potential indicator for evaluating the tumor progression.375,423 Elevated expression of the TIM-3 ligand galectin-9 on cancer cells has been associated with blood vessel invasion and TNM stage in GC.374 However, the prognostic value of galectin-9 remains controversial. Long et al.424 and Jiang et al.425 reported that low expression of galectin-9 in GC patients was associated with poor survival, whereas the study from Wang et al.374 reported that galectin-9 expression negatively correlated with poor prognosis in GC patients.374 This discrepancy may occur because of differing functions of galectin-9 in different immune states of the patients. As the galectin-9 function remains poorly understood, further research is needed to clarify whether it has a possible tumorigenic role or tumor-suppressing activity. Therefore, TIM-3 is thought to be a relatively promising biomarker and therapeutic target for GC compared to its ligand. In preclinical studies, TIM-3 inhibitors showed similar effects to PD-1 inhibitors, and a combination of PD-1 and TIM-3 inhibitors enhances T cell responsiveness to tumor antigens with synergistic effects, suggesting that TIM-3 may be a useful target in treating GC resistant to anti-PD-1 immunotherapy.426,427 The expression of TIM-3 inhibitory ligands on GC cells might also be potential biomarkers for predicting the treatment response of PD-1 mAb.428 Targeting PD-1 and TIM-3 combination immunotherapy may have more therapeutic benefit than mono-immunotherapy for GC patients.
LAG-3 expression has a remarkable synergistic effect with PD-1 on promoting the immune escape of GC cells, which suggests it might be a biomarker of poor prognosis.369 Galectin-3, the ligand of the LAG-3 inhibitory pathway, was also found to be a potential indicator for poor prognosis in the diffuse type of GC. However, its utility as a prognostic marker may be population-dependent, since overexpression of galectin-3 was highly significant in the North American cohort but not in the Asian cohort.429 Targeting both LAG-3 and PD-1 has become an important cancer immunotherapy strategy.372,430 However, the understanding of LAG-3’s mechanism in GC is still minimal, and many fundamental questions remain unanswered. Elucidating the mechanism of LAG-3 in more detail should permit a more rational design for LAG-3-dependent immunotherapy.
TIGIT overexpression in the tumor microenvironment has been observed in GC patients, accompanied by upregulation of its ligands, CD155 and CD112, and is associated with immune escape led by CD8+ T cell suppression.431 In a co-culture system of T and GC cells, the TIGIT expressing peripheral blood CD8+ T cells from GC patients exhibited decreased cellular metabolism and impaired cell functions, which were mediated by TIGIT/CD155 signaling and could be reversed by blockade of CD155.386 This suggests that the TIGIT/CD155 pathway can be a GC prognostic indicator and a novel immunotherapy target for treating GC. Bioinformatic analysis revealed that epigenetic regulation (majorly methylation) of TIGIT can affect the prognosis and immunotherapeutic responsiveness of GC.432 High TIGIT expression can be utilized to identify patients who are likely to be sensitive immunotherapy thereby improving prognosis. On the other hand, TIGIT may be a potential target for designing epigenetic drugs.433 Since TIGIT and PD-1 can be highly co-expressed in CD8+ T cells,431 TIGIT is expected to be a target for potentiating the benefits of anti-PD-1 therapy.
Other signaling pathways involved in gastric cancer
Many other signaling pathways have been identified to be involved in GC. Briefly reviewed here are recent discoveries of the signaling pathways relevant to fibroblast growth factors and corresponding receptors (FGF and FGFR), signal transducer and activator of transcription 3 (STAT3), hypoxia-inducible factor-1 α (HIF-1α), Hedgehog, and Notch. Alterations of signaling molecules relevant to cell adhesion and cell junction in diffuse-type GC are also discussed here as distinct molecular characterizations from other histological subtypes.
The fibroblast growth factor receptors (FGFR) are transmembrane proteins expressed widely by different cell types. The FGFR family has 4 members, namely FGFR1, FGFR2, FGFR3, and FGFR4. FGFR1 mutations, FGFR2 amplification, and FGFR3 rearrangements are the most common FGFR alterations found in GC.434 When bound with fibroblast growth factors (FGF), FGFRs are activated through phosphorylation of the intracellular tyrosine kinase domain, which then activates several important cellular pathways, including the RAS/MAPK, the PIK3CA/AKT/mTOR, and the Janus kinase (JAK) pathways.435 Activation of these signaling pathways can affect angiogenesis, cell mitosis, differentiation, proliferation, and invasive processes.435 Dysregulation of the FGF-FGFR axis has been thought to contribute to GC carcinogenesis. Overproduction of FGF presumably promotes communication between epithelial and stromal cells in the tumor microenvironment, which is critical for tumorigenesis.434 Alterations of the FGFR gene are commonly observed in GC patients, which can be a diagnostic biomarker for GC.436 In a large cohort of Chinese GC samples, the prevalence of overall FGFR aberrations was 7%.437 In another cohort of GC samples, FGFR2 amplification was found in 4.1% of samples.432 A small Hong Kong GC cohort study reported that FGF18–FGFR2 signaling could upregulate yes-associated protein 1 (YAP1) oncogene expression by activating the MAPK pathway effector c-Jun.438 Cancers that are co-positive for FGFR2, c-Jun, and YAP1 alterations are associated with worse clinical outcomes, indicating the translational potential of FGFR2–c-Jun–YAP1 as a prognostic predictor and therapeutic target for GC.438 FGF18 has also been identified as a potential GC prognostic biomarker and therapeutic target, which can be negatively regulated by miR-590-5p to inhibit gastric tumorigenesis.439 In addition to tumorigenesis, the FGF-FGFR axis can affect GC invasion and metastasis. Huang et al. reported that upregulation of FGF7/FGFR2 signals can increase the expression of thrombospondin-1, an extracellular glycoprotein responsible for cell–matrix and cell–cell interactions, possibly by activating the PI3K/AKT/mTOR pathway, and finally lead to enhanced GC cell invasion and migration.440
STAT3 is known to be an oncogene that is hyperactivated in many types of cancer, including GC.441 The STAT3 pathway is activated by the binding of an extracellular cytokine such as IL-6 or an EGF family member such as HGF to the transmembrane cytokine receptor. Binding triggers the dimerization and transphosphorylation of JAKs, which provide docking sites for STAT3 molecules. The JAK dimers mediate phosphorylation of tyrosine 705 of STAT3, and the activated STAT3 is released from the kinase complex and subsequently translocates into the nucleus.441 As a transcriptional factor, nuclear STAT3 regulates the gene expression of a wide range of genes that are involved in promoting cancer cell growth, tumor invasion, and chemoresistance.442,443 The STAT3 pathway is significantly involved in the tumor progression and metastasis of GC. STAT3 signaling was reported to drive EZH2 epigenetic modification, which is associated with advanced TNM stage and poor prognosis.444 Analysis of patient samples revealed that increased survivin and STAT3 expression significantly correlated with concurrent H. pylori infection; moreover, their subcellular localizations are key factors influencing GC progression.445 Therefore, STAT3 and survivin expressions can be collectively used as potential prognostic biomarkers and therapeutic targets for GC. Additionally, JAK2/STAT3 signaling may play a key role in GC EMT and metastasis induced by IL-6446 or mesothelial-mesenchymal transition of GC.447 Recent studies on STAT3-related mechanisms in GC have focused on the regulation by miRNA and long non-coding RNA (lncRNA). miRNAs and lncRNAs are potential upstream regulators of STAT that may fulfill their functions as oncogenes or tumor suppressors by influencing STAT3 expression levels in GC cells.448,449,450,451 Notably, circular RNAs (circRNAs), a non-coding RNA subclass that serves as competitive endogenous sponges for miRNAs, thereby negatively regulating miRNAs,452 have been recognized as potential regulators in GC chemoresistance.453,454 Deng et al.448 recently reported that elevated circVAPA expression was observed in GC tissues compared to normal tissues; moreover, circVAPA may promote cisplatin resistance and tumor progression in GC by modulating miR-125b-5p/STAT3 axis, making it a potential target for GC treatment.
HIF-1α is the pivotal molecule responsible for cell adaptation to hypoxia.455 Under hypoxic conditions, the expression of HIF-1α is upregulated and the inhibition on HIF-1α by hydroxylases is relieved due to lack of oxygen. The activated HIF-1α translocates to the nucleus where it acts as a transcription factor exerting stimulatory or inhibitory regulation on the transcription of target genes responsible for metabolism, inflammation, vascular homeostasis, and tumorigenesis.456 The HIF-1α signaling pathway has been thought to promote GC progression by mediating tumor cell proliferation, angiogenesis, EMT, therapeutic resistance, and inhibition of cell apoptosis.457 HIF-1α expression may be a predictor of poor overall survival for GC patients.458,459 The HIF-1α/microRNAs and HIF-1α/lncRNAs axes have been confirmed to play critical roles in GC progression, metastasis, and chemoresistance. Lin et al.460 showed that hypoxia-induced HIF-1α/lncRNA-PMAN inhibits ferroptosis of GC cells in peritoneal metastatic GC. Zhao et al. found that HIF-1α/miR-17-5p axis may contribute to the tumor growth and metastasis of GC by negatively regulating programmed cell death 4 (PDCD4).461 On the other hand, dysregulated miR-27a,462 miR-421,463 and lncRNA-PVT1464 may be associated with HIF-1α-mediated cisplatin resistance in GC. Other newly identified HIF-1α-regulating downstream molecules that are closely related to GC EMT and metastasis include N-myc downstream-regulated gene 2 (NDRG2),465 CXCR4,466 liver X receptor α (LXRα),467 and RhoE.468 The underlying mechanism of HIF-1α-induced angiogenesis in GC may be relevant to the crosstalk between the HIF-1α pathway and the STAT3 pathway or β-catenin/VEGF signaling.469,470 HIF-1α has been proven to be a druggable target, and pharmacologic manipulation of HIF-1α is under investigation as a novel therapeutic approach to GC.
The Hedgehog signaling pathway not only plays an essential role in the growth and development of various tissues during embryonic development but is also an important signaling pathway necessary for maintaining the homeostasis of recognized tissues.471 The Hedgehog pathway interconnects with Wnt and FGF signaling, which is important during embryogenesis and tissue regeneration.472,473 Through aberrant activation of the Hedgehog signaling pathway, the upregulation of sonic hedgehog (SHH) can lead to pathological consequences of multiple types of cancers, such as gastric, esophageal, pancreatic, and prostate cancers.474 SHH is expressed in the fundic glands of the human stomach, and is strongly expressed in embryos.475 The activation of SHH signaling affects the transcription of cell cycle regulators such as PTCH1, FOXM1, and CCND2, ultimately modulating cell proliferation.476,477 PTCH1, an SHH receptor as well as SHH signaling target, is expressed in parietal and mesenchymal cells. High expression levels of SHH and PTCH1 are significantly associated with poor prognosis in GC, and a high expression level of PTCH1 may be associated with GC progression.478,479 Another SHH signaling target, FOXL1, is also expressed in mesenchymal cells and may contribute to the functional maturation of the parietal cell lineage.477 SHH regulates growth and differentiation within the gastric mucosa through an autocrine loop and FOXL1-mediated epithelial-mesenchymal interaction.480 In GC, the upregulation of SHH can indicate an involvement of autocrine signaling loops and epithelial-mesenchymal interactions in the regulation of parietal cell lineage differentiation or maturation.
The Notch signaling pathway is a highly conserved system that regulates the function of multiple cell types and plays a crucial role in cell differentiation, survival, and proliferation. Activation of the Notch signaling pathway has been observed in tumors. Its abnormal activation is involved in direct intercellular communication and plays an essential role in the formation, development, survival, proliferation, invasion, and metastasis of tumors.481,482 Notch signaling activation is associated with various cancers and was recently established as a critical pathway regulating gastric stem cell proliferation and differentiation.483 Notch induces excessive cell proliferation by upregulating the expression of nuclear transcription factor NF-κB.484 It also promotes epithelial cell proliferation and participates in gastric mucosal carcinogenesis. The reduction of Notch1 gene expression can inhibit the proliferation of GC cells and reduce the ability of tumor migration and invasion.485 Therefore, it is closely related to the occurrence, development, and metastasis of GC.485 Notch2 can upregulate PI3K/AKT signaling pathwayto enhance the invasive ability of GC cells.486 In addition to regulating proliferation, the Notch pathway regulates the differentiation of gastric antral epithelial cells, acting in a global manner.481 Therefore, the critical molecular differences in somatic versus sinus stem cell differentiation regulated by Notch signaling will be an important area of future research.482,487
Cell junction and cell adhesion proteins play key roles in the tumorigenesis of diffused GC. E-cadherin (encoded by CDH1 gene) is an adhesive junction protein. Germline CDH1 gene mutation leads to HDGC, while somatic mutation of CDH1 is also common in sporadic diffused GC.41 These findings highlight the key roles of CDH1 in the formation of diffused GC. CLDN18-ARHGAP fusions are also common in a subset of diffuse type GC, including GSRCC.51,488 CLDN18 gene encodes Claudin18 protein, a key component of tight junction, which functions to lock adjacent cells together to form a barrier between the external and internal environment.489 There are two Claudin18 isoforms, Claudin18.1 and Claudin18.2, which differ in the first exon of the CLDN18 gene.490 Claudin18.2 is mainly expressed by differentiated cells rather than stem cells of the gastric mucosa.490 The expression of Claudin18.2 is maintained in a large fraction of GCs. A meta-analysis by Ungureanu et al.491 demonstrated that Claudin 18.2 expression was observed in 34.2% of a combined total of 2055 patients in six studies. Xu et al.52 reported a high expression rate of Claudin 18.2 in advanced GSRCC patients. In addition, the disruption of cell polarity in GC exposes the Claudin 18.2 epitope on the surface of tumor cells, which makes it an ideal target for therapy to have strong specificity and low toxicity. On the other hand, the ARHGAP family, represented by ARHGAP26, mediates the hydrolysis of GTP in RhoA, leading to RhoA inactivation.492 The fusion of CLDN18 to ARHGAP causes ARHGAP over-expression and over-activation and RhoA inactivation. A highly prevalent RHOA gene mutation was also found in recent years by large-scale NGS studies of GC.493 RhoA is a small GTPase-like RAS and plays a key role in regulating the dynamics of the actin cytoskeleton and cell movement. However, the role of RhoA in regulating carcinogenesis is controversial since it is unclear whether RhoA mutation is loss-of -function or gain-of-function.494 The aberrations of CDH1, RHOA, and CLDN18-ARHGAP26 are enriched in the GS subset of GC according to TCGA.41 Understanding the crosstalk of these three gene aberrations will be key to revealing the mechanisms leading to tumorigenesis in diffused GC.
Another molecule related to cell adhesion is the trophoblast cell surface antigen 2 (Trop2) encoded by the TACSTD2 (tumor-associated calcium signal transducer 2) gene, which is a transmembrane glycoprotein and calcium signal transducer.495 It is structurally related to the epithelial cell adhesion molecule (EpCAM).495 Trop2 was initially discovered in trophoblast cells and is expressed in many normal human tissues.496 It is involved in embryonic development and implicated in several oncogenic signaling pathways, such as ERK/MAPK and NF-κB pathways.497,498 Trop2 has been found to be overexpressed in about half of GC (47–66% according to two studies).499,500 Trop2 may induce EMT and metastasis of GC by directly binding to and activating β-catenin, resulting in the accumulation of β-catenin in the nucleus to facilitate GC cell migration and invasion.501
The discussed signaling pathways in GC and the identified biomarkers or potential therapeutic targets are summarized in Table 2. Studies on molecular mechanisms have led to a better understanding of how different signaling pathways affect GC tumorigenesis, progression, metastasis, and resistance to therapeutic drugs. These observations will greatly help to identify new targets for anticancer drugs and novel biomarkers of diagnosis, prognosis, as well as personalized treatments for GC patients.
Crosstalk between different signaling pathways in gastric cancer
Studies in the emerging field of systems biology have emphasized the complexity of signaling webs during tumor progression. p38-MAPKs activation orchestrates cellular responses by regulating various downstream targets, such as protein kinases and transcription factors, including p53. The functional interaction between p38-MAPKs and p53 appears to occur at multiple levels. The p53 status can directly affect the outcome of p38-MAPKs signaling by negative feedback loops in cells with wild-type p53, altering the biological response of p38-MAPKs activation. Contradictory effects have been reported on the modulation of the p38-MAPKs pathway in cancer. In accordance with its role in p53 activation, it has been proposed that p38-MAPKs activation could act as an onco-suppressive pathway; however, there is also evidence suggesting that p38-MAPK signaling is highly active in various cancer types and promotes tumor growth.502,503 The mutant p53 gain-of-function transcriptional target and p38-MAPKs upstream MKK3 and MAP2K have been reported as targets for tumor therapy.504,505 In 2021, a study investigating the distinct molecular landscapes of gastroesophageal adenocarcinoma (GEAs) patients with different PD-L1 expression levels identified that tumors with mutations in p53, KRAS, and MAPK pathways were associated with higher PD-L1 combined positive scores (CPSs) in the mismatch repair proficiency and microsatellite stability (pMMR&MSS) subgroup. The data provide potential novel insights for patient selection according to the status of RAS/MAPK pathway alterations and p53 mutations and for the development of rational combination immunotherapies in GEAs.506
Hedgehog signaling is important in the regulation of proliferation, survival, and growth of various tissues, including the gastrointestinal tract. Seto et al.507 assessed crosstalk between MAPK and hedgehog signaling in the control of cell proliferation in GC. The immunohistochemistry (IHC) results of 35 GC samples suggested that PTCH expression was significantly associated with ERK1/2 phosphorylation as well as SHH expression. The RAS/MEK/ERK signaling cascade positively regulates the transcriptional activity of glioma-associated oncogene homolog 1 (GLI1), a nuclear mediator of the Hedgehog pathway, thereby inducing the expression of hedgehog target genes in GC cells.508 Jayati et al. found that hedgehog signaling contributes to inducing PD-L1 expression in GC, and PD-1/PD-L1 inhibition reverses GLI2-induced tolerance, such that combined inhibition of hedgehog signaling and immune checkpoints may be suitable for selected patients.509
PD-1/PD-L1 signaling is regulated by various pathways. In gastrointestinal stromal tumors (GIST), knockdown of PD-L1 inhibited the expression level of PI3K, p-PI3K, and p-AKT, whereas the alteration of PI3K/AKT/mTOR pathway blocked PD-1/PD-L1 and attenuated apoptosis of CD8+ T cells.510 Activation of the PI3K/AKT pathway mediates PD-L1-induced P-gp upregulation in GC drug resistance.511 Wang et al.416 reported that autophagy inhibition increased PD-L1 expression by increasing the p62/SQSTM1 level and activating nuclear NF-κB in GC, which can be abolished by p62/SQSTM1 inhibition or NF-κB knock down.
The extensive crosstalk between TGF-β signaling and other pathways is a perennial theme of TGF-β research. Several studies have shown that HER2 signaling interplays intimately with TGF-β/Smad in regulating mammary epithelial cell biology and breast cancer progression.512,513 The synergy between the TGF-β and HER2/RAS/MAPK signaling can induce the secretion of additional growth factors and cytokines, including TGF-β itself, which in turn induce EMT and tumor invasion.514,515 Wnt signaling benefits from extensive crosstalk with other signaling pathways, particularly TGF-β/bone morphogenic protein (BMP) signaling. Wnt and TGF-β signaling often interact to ensure normal tissue homeostasis by modulating the expression of main target genes, and aberrant signaling conduction in either pathway usually results in tumorigenesis. Lei et al.516 found that Wnt and TGF-β synergized in the transcriptional activation of the Wnt target gene encoding gastrin, a promoter of GC, indicating that Wnt and TGF-β signaling can cooperate to induce tumorigenesis. Furthermore, the level of Wnt pathway activation inversely associates with the level of Hedgehog pathway activation in gastric tissues. Yanai et al.517 demonstrated that the overexpression of glioma-associated oncogene homolog 1 (GLI1), the nuclear mediator of Hedgehog signaling, could restrain Wnt transcriptional activity, nuclear β-catenin accumulation, and proliferation of human GC cells. Referencing this crosstalk between Wnt and Hedgehog pathways may be valuable in developing targeted therapy for GC.
The crosstalk of the STAT3 pathway with other tumorigenic pathways also plays an important role in GC development. In MET-unamplified GC, HGF derived from cancer-associated fibroblasts (CAFs) promoted tumor proliferation, migration, and invasion via the activation of the HGF/STAT3/twist1 pathway. CAFs-derived HGF can activate IL-6/STAT3/twist1 pathway by upregulating the expression of the IL-6 receptor.518 Additionally, in vivo experiments revealed that HGF from CAFs promoted tumorigenesis and metastasis of MET-unamplified GC.518 STAT3/c‐Myc and mTOR/pyruvate kinase isozyme 2 (PKM2) signaling pathways were upregulated in human GC. Knockdown of c‐Myc in GC cells downregulated cell proliferation, and knockdown of both PKM2 and c‐Myc were more inhibitory in GC cells than knockout of c‐Myc or PKM2 alone. These observations indicate that co-inhibiting PKM2 and c‐Myc might better antagonize the malignant behavior of GC and c‐Myc might be considered a potential therapeutic target for GC.519
Studies have also investigated the crosstalk between downstream pathways of integrin and EGFR. By blocking the synthesis of FAK they detected the effect of crosstalk between EGFR and integrin signal pathways on the proliferation and invasion in a GC cell line, SGC7901, and proved FAK to be a key cross point of two signaling pathways, which makes it a more effective molecular target for GC therapy.520
Epigenetic modifications involved in different signaling pathways of GC
Epigenetic alterations refer to the mechanisms of heritable and reversible regulations on gene expression without changing genomic DNA sequence. Epigenetic modifications include DNA methylation, histone post-translational modification, chromatin remodeling, and change in non-coding RNAs expression. In the past two decades, many studies have highlighted the active roles of epigenetic dysregulations in GC initiation and development. Targeting epigenetic regulators, including the non-coding RNAs, regulatory genes, and the enzymes involved in DNA methylation and histone modification--DNA methyltransferases (DNMTs) and histone deacetylases (HDACs), could be a potential therapeutic approach.521
DNA methylation is the transfer of a methyl group from the cofactor S-adenosylmethionine to the C5 position of a cytosine within CpG islands, which are regions with repeated CG dinucleotide sequences located at the promotors of most genes. DNA methylation results in inhibition of gene expression.522 Under the TCGA classification, EBV-positive and MSI subtypes of GC tumors generally exhibit a CpG island methylator phenotype (CIMP) characterized by high DNA methylation levels at multiple loci, particularly the tumor suppressor genes.41 The CIMP may also be associated with H. pylori infection.523 In contrast, other GC subtypes may exhibit global hypomethylation associated with proto-oncogene activation and genomic instability.524 Alteration of DNA methylation is considered to be an early event of GC tumorigenesis, which mostly occurs in genes that regulate cell cycle (such as CDKN2A, CDKN1B, TP53, SMAD2), DNA repair (such as MLH1, MSH2), cell adherence (such as CDH1), and cell death (such as HRAS).524,525 Hypermethylation of CDH1 promotor plays a vital role in HDGC and is frequently found to accompany CDH1 mutations or loss of heterozygosity as a second hit to inactivate CDH1.526 Aberrant methylation also affects genes involved in cancer-related pathways. For instance, hypermethylation of the DKK3 gene, which is an inhibitory regulator of β-catenin, is commonly found in GC patients inducing activation of Wnt/β-catenin and poor survival.527 Hypermethylation of the tumor suppressor gene ADAMTS9 in GC associates with abnormal activation of the AKT/mTOR pathway and cancer progression.528
The post-translational modifications of histone, such as acetylation, methylation, ubiquitination, phosphorylation, and SUMOylation, are important epigenetic mechanisms for regulating chromatin structure and gene expression.529 Histone modification plays an important role in GC development relevant to overexpression of oncogenes or downregulation of tumor suppressor genes. Elevated expression of histone deacetylating enzymes HDAC1 and HDAC2 has been observed in human GC tissue samples, and correlates with TNM staging and chemoresistance.530 Aberrant upregulation of HDACs is associated with hypoacetylation of histone, which can lead to downregulation of tumor suppressor genes. Reduced acetylation levels of histone H3 and H4 have been suggested to be associated with p21 downregulation and GC progression.531,532 Additionally, dysregulation of histone methylation and acetylation is involved in the progression and EMT of GC by cooperative regulation with PI3K/AKT and Wnt signaling pathways.533,534
Chromatin remodeling is induced by histone modification and influences the interaction between chromatin-modifying proteins and DNA.535 Recent studies have shown that members of the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex family can function as tumor suppressor genes. A well-studied example is the ARID1A gene. Mutations or deletions of the ARID1A gene have been detected in 8-25% of GC and are associated with concurrent gain-of-function mutations of PIK3CA and microsatellite instability.536,537 Another study by Zhang and colleagues revealed that ARID1A may function as a suppressor of GC cell proliferation by modulating PI3K/AKT pathway via targeting PIK3CA and PDK1. This provides a novel strategy of using PI3K and AKT inhibitors to treat GC with PI3K and AKT overexpression due to loss or deficiency of ARID1A.538
Noncoding RNAs (ncRNAs) include lncRNAs, miRNAs, siRNAs, and PIWI-interacting RNAs (piRNAs). The regulatory, potential diagnostic, and therapeutic values of certain lncRNAs, miRNAs, and siRNAs have been discussed in the previous sections or specific signaling pathways. piRNAs are a class of ncRNAs that form complexes with PIWI nuclear proteins to cause histone modifications. Research on the role of piRNAs in GC is still limited. Several studies have shown differential piRNA expression profiles in tumors compared to non-tumor tissues, suggesting that piRNAs can be novel cancer biomarkers. Cheng et al.539 reported that piR-651 was overexpressed in human GC cells compared to normal gastric epithelial cells, and individuals at advanced GC stages had higher expression than those at earlier stages. Furthermore, restrained growth of two GC cell lines was observed after inhibition of piR-651, suggesting a potential therapeutic value for targeting piR-651. In contrast, piR-823 expression was found to negatively correlate with GC progression, indicating its tumor-suppressing function.540 There have been reports that the piRNA/PIWI complex regulates STAT and AKT pathways in colorectal cancer and liver cancer;541 however, these interactions have not yet been reported in GC.
Interplay among different epigenetic mechanisms should be considered in GC. DNA methylation and miRNAs are involved in regulatory feedback loops, while siRNAs and piRNAs can regulate both DNA methylation and histone modification. LncRNAs are regulated by DNA methylation yet can regulate DNA methylation. During this process, some lncRNAs interact with miRNAs,542 and the lncRNA-miRNA-mRNA pathway undergoes another epigenetic regulatory step before altering target genes in GC tissues.543 A deeper understanding is needed to establish the foundation for designing dual or multiple epigenetic-targeting strategies for GC treatment.
Progress in therapies for gastric cancer
Current therapies for gastric cancer
Even as chemotherapy, radiation therapy, targeted therapy, immunotherapy, and other treatment modalities continue to advance, surgery remains the only radical treatment for GC. The goal of the procedure is to accomplish radical resection, which means that the relevant local lymph nodes are eliminated, and the cutting edge is tumor-free. The two most common surgical procedures are distal gastrectomy and anastomosis of the esophagus with the small intestine after total gastrectomy.544,545 The type of procedure for patients who are surgical candidates depends on the various clinical TNM (cTNM) stages of the tumor28 (Fig. 4). According to the patient’s physical state, individualized care is required for patients who are unable to undergo surgery.

Current therapies for gastric cancer based on staging. Therapeutic interventions for GC at different stages are illustrated by icons. The majorly used drugs or regimens of chemotherapy, targeted therapy, and immunotherapy are listed. EMR endoscopic mucosal resection, ESD endoscopic submucosal dissection. S-1 is an oral agent that is converted to 5-FU in the body, which contains a 5-FU prodrug called tegafur and the two enzyme inhibitors 5-chloro-2,4-dihydroxypyridine (CDHP) and oteracil potassium (Oxo), in a molar ratio of 1:0.4:1. This figure was adapted and modified from “Gastric Cancer Staging” by Biorender.com (2022). Retrieved from https://app.biorender.com/biorender-templates. Icons were adapted from Adobe Express
However, two studies have shown that perioperative treatment, which contains preoperative neoadjuvant therapy and postoperative adjuvant chemotherapy, can effectively improve the 5-year survival rate of GC patients.546,547 Preoperative neoadjuvant therapy not only has good safety, but also significantly improves the tumor remission rate, R0 resection rate, and 5-year survival rate without raising the risk of postoperative complications or mortality, according to the results of the RESOLVE and PRODIGY clinical trials.548,549 Additionally, the outcomes of two clinical trials, JACCROGC07 and ARTIST-II, demonstrate that postoperative adjuvant chemotherapy can induce positive tumor responses, lower the rate of tumor recurrence and metastasis, and improve the disease-free survival rate (DFS).548,550
For stage I GC, endoscopic resection, which comprises endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD), has demonstrated success for treating early GC and is thus the primary option unless there is a significant risk factor, such as lymph node metastasis.551 The criteria for EMR and ESD have been expanded to include macroscopically intramucosal (cT1a) differentiated carcinomas >2 cm without ulcer and ≤3 cm with ulcer, and there is no appreciable difference in long-term survival, according to the findings of a multicenter, prospective single-arm research (JCOG0607) in Japan.552 EMR and ESD are indicated for intramucosal carcinoma with a diameter of <2 cm, differentiated type, and no ulcer. ESD is indicated for either intramucosal differentiated carcinoma with a diameter >2 cm and no ulcer, or intramucosal differentiated carcinoma with a diameter <3 cm and with ulcer.552
For patients who do not meet the criteria for either EMR or ESD, gastrectomy combined with regional lymph node dissection D1 or D2 can be performed by laparotomy or laparoscopy.553 All perigastric lymph nodes and left gastric artery lymph nodes, which have the highest risk of metastatic GC, are included in the scope of lymph node dissection D1.554 Lymph nodes along the common and proper hepatic arteries, the splenic hilum, and the splenic artery are all included in the scope of the lymph node dissection D2.554 According to a Taiwanese randomized clinical study, patients who underwent gastrectomy combined with lymph node dissection D2 had a greater chance of survival than those who underwent gastrectomy combined with lymph node dissection D1.555 To increase the precision of staging and prognosis, lymph node dissection requires at least 16 lymph nodes.556
Stage II GC is often treated with laparoscopic gastrectomy combined with lymph node dissection D2.557 Laparoscopic surgery has emerged as a superior option to the traditional laparotomy method. Laparoscopic surgery has been shown to be safe compared to traditional laparotomy, making it suitable for use as a standard surgical practice, according to the findings of the large-scale prospective investigations JCOG0912 and KLASS01 from Japan and Korea.558,559 To improve the tumor remission rate, adjuvant chemotherapy with XELOX (oxaliplatin plus capecitabine) or S-1 monotherapy regimens are needed postoperatively.557 Because multiple randomized controlled clinical trials have demonstrated that increasing radiation therapy does not increase overall survival (OS) rates following gastrectomy, postoperative radiation therapy is not advised.560
For Stage III advanced GC, the results of two phase III prospective randomized controlled clinical trials, CLASS01 and KLASS02, show that laparoscopic distal gastrectomy combined with D2 lymph node dissection is safer than traditional laparotomy, and reduces intraoperative blood loss, speeds up the recovery of gastrointestinal function, and reduces patient hospitalization time, with no appreciable difference in long-term survival.561,562 Preoperative neoadjuvant chemotherapy or chemoradiotherapy, and postoperative adjuvant chemotherapy are important for patients with advanced GC.557 Preoperative neoadjuvant chemotherapy can be administered using a number of regimens, including the SOX regimen (oxaliplatin plus S-1),563 XELOX (oxaliplatin plus capecitabine), FOLFOX (leucovorin plus fluorouracil plus oxaliplatin), and FLOT (fluorouracil plus leucovorin, oxaliplatin and docetaxel) regimens.564,565,566 DT45~50.4Gy coupled with platinum or paclitaxel is used in preoperative neoadjuvant chemoradiotherapy.567 In most cases, XELOX (oxaliplatin plus capecitabine) or SOX (oxaliplatin plus S-1) are used for postoperative adjuvant chemotherapy.563,564
Comprehensive therapy is required depending on the patient’s condition for locally advanced, unresectable GC.568 Concurrent chemoradiotherapy has been shown in several trials to be more successful than conventional chemotherapy or radiotherapy in reducing the tumor resection rate and increasing the remission rate when the patient is normally in excellent health, and can prolong the survival time of patients.569 There are three types of concurrent chemoradiotherapy: (1) DT45~50.4 Gy coupled with carboplatin and paclitaxel; (2) DT45~50.4 Gy coupled with cisplatin or oxaliplatin and 5-FU or capecitabine; and (3) DT45~50.4 Gy coupled with paclitaxel and 5-FU or capecitabine.567,570,571 However, chemotherapy or radiotherapy alone can be used if the tumor has spread to numerous lymph nodes and the patient might not tolerate concurrent chemoradiotherapy.572 Patients’ clinical symptoms, such as pain relief and bleeding reduction, as well as their quality of life, can be improved by radiotherapy.573 Chemotherapy alone can increase the survival rate of patients with poor overall health condition.574 Currently, 5-FU, cisplatin, oxaliplatin, paclitaxel, and irinotecan are the most widely utilized chemotherapy medicines. A phase III clinical trial revealed that the combination drug’s effective rate and median OS were dramatically increased.575
For Stage IV GC, only systemic antineoplastic medications can be utilized to extend patients’ lives at this point, because surgery is no longer an option due to the organ metastases of cancer cells.576 Chemotherapeutic medicines, molecular-targeted therapies, and immune checkpoint inhibitors are now the most widely utilized systemic antineoplastic medications. Trastuzumab,40 an anti-HER2 medicine, and ramucirumab, an anti-angiogenesis pathway drug, are the two regularly used molecular-targeted medications. The results of two clinical studies, REGARD and RAINBOW, demonstrated that patients receiving ramucirumab had a longer median survival time and OS rate.577,578 In addition, an immune checkpoint inhibitor PD-1 monoclonal antibody, such as nivolumab, can be used in the treatment of refractory cancer.46 In comparison to patients who merely received a placebo and supportive therapy, participants treated with nivolumab had a better OS rate, according to a Phase III randomized study ATTRACTION-2.579
Additionally, supportive care is crucial in the treatment of advanced GC since it can considerably increase patients’ nutritional and psychological status as well as their survival time.580
Advances in targeted therapy and immunotherapy for gastric cancer
Currently, the development of new drugs for GC focuses on targeted therapy and immunotherapy. Although molecular and cellular evidence suggests many different genes and signaling pathways play key roles in the initiation and progression of gastric cancer, only a fraction is druggable. The current druggable targets reflect the importance of the EGFR/HER2 and c-MET pathways associated with cell growth, the immune checkpoint pathways associated with immune escape, and the cell adhesion and cell junction signaling associated with invasion and metastasis. The most successful target in GC is HER2, which transduces growth signaling and induces proliferation, motility, and invasion of cells. The introduction of immune checkpoint inhibitors, mainly PD-1 antibodies also changed the scheme of GC treatment significantly. Other druggable targets in GC are growth factor receptors, such as EGFR, VEGFR, c-MET, and FGFR2, and enzymes involved in epigenetic regulations like DNMT and HDAC. In addition, a few membrane proteins that are overexpressed in GC cells, including Claudin18.2, Trop2, and Mucin 17 (MUC17), are also targeted by strategies such as antibodies, ADC, bi-specific antibodies, or CAR-T. These drugs are under fast clinical development, which may change the picture of GC treatment in the next few years.
HER2-targeted therapies
Drugs targeting HER2, including antibodies, antibody-drug conjugates (ADC), and small-molecule tyrosine kinase inhibitors, are being developed for cancer treatment. The monoclonal antibody trastuzumab was the first agent developed for HER2 targeting and can improve outcomes among women with HER2-positive breast cancer.581 In GC, the addition of trastuzumab to standard chemotherapy of HER2-positive GC may increase the survival of the patients.40
Although widely used, treatment with the HER2 antibody failed to maintain the control of the tumor, and drug resistance eventually developed. HER2 ADC was developed to further enhance the cytotoxicity of HER2 antibodies. Trastuzumab deruxtecan (DS-8201) is an ADC consisting of an anti-HER2 antibody with the same amino acid sequence as trastuzumab, a cleavable tetrapeptide-based linker, and a cytotoxic topoisomerase I inhibitor exatecan. In a phase II trial, treatment with DS-8201 led to significantly improved response and OS, in comparison to standard chemotherapy, among patients with HER2-positive pretreated GC.582 Disitamab vedotin (RC48) is another anti-HER2 ADC containing hertuzumab coupling monomethyl auristatin E (MMAE) by a cleavable linker. In phase II single-arm trial, disitamab vedotin showed promising activity with manageable safety in patients with advanced gastric or gastroesophageal junction cancer overexpressing HER2.583,584
Zanidatamab (ZW25) is a bi-specific antibody directed against the two HER2 domains targeted by trastuzumab and pertuzumab, respectively. Zanidatamab was evaluated in phase I study (NCT02892123) in heavily pretreated gastroesophageal adenocarcinoma patients (including prior HER2-targeted therapy). Zanidatamab is well tolerated with promising and durable anti-tumor activity, both as a single agent and in combination with chemotherapy, which may be a good candidate drug for trastuzumab-resistant GC.585
Small-molecule tyrosine kinase inhibitors targeting HER2 are also under development for GC treatment. Lapatinib, the first dual inhibitor of EGFR and HER2, was approved by the US FDA in 2007. It is suggested for use in combination with chemotherapy for the treatment of HER2 overexpressing breast cancer.586 In the phase III TRIO-013/LOGiC trial, lapatinib was tested in combination with chemotherapy in HER2-positive gastric and esophageal cancer. Unfortunately, the addition of lapatinib to chemotherapy did not increase OS.587 In another study, the combination of lapatinib with perioperative chemotherapy for resectable HER2-positive gastroesophageal adenocarcinoma did not improve response.588
EGFR-targeted therapies
Like HER2, EGFR also plays a key role in various cancer types. Unlike HER2, EGFR is mainly activated through mutations rather than gene amplification. EGFR gene mutations, including point mutations and exon 20 insertions, are driver mutations in non-small cell lung cancer (NSCLC). However, EGFR mutations in other tumor types including GC are much rarer, and their clinical significance is unclear. Cetuximab, a monoclonal antibody targeting EGFR, is effective in treating colorectal cancer. However, the addition of cetuximab to standard chemotherapy failed to show any improvement in the survival of GC patients in the phase III EXPAND trial.589 This study was performed in GC patients not selected by EGFR status, which may be the reason for its failure. Another EGFR antibody, panitumumab, also failed in the phase III trial in unselected GC patients.590 Learning from these results, researchers tested the anti-EGFR treatment in EGFR-amplified GC patients. In an early study, researchers identified 19 gastroesophageal cancers with EGFR amplification out of 363 screened patients (5%). The addition of cetuximab to chemotherapy in this small group of patients resulted in high tumor response rates.591 Thus, anti-EGFR may be effective in meticulously selected GC patients. More clinical trials are needed to prove this preliminary result.
VEGFR-targeted therapies
Blocking angiogenesis has been attempted in GC treatment with varied results. Angiogenesis is predominately regulated by VEGF/VEGFR signaling.592 Strategies for blocking angiogenesis signaling include neutralizing VEGF with antibodies, blocking VEGF receptors with antibodies, and inhibiting VEGF intracellular activities with small-molecule tyrosine kinase inhibitors. Unfortunately, targeting VEGF in GC has been unsuccessful. In the phase III AVAGAST study, bevacizumab, a monoclonal antibody against VEGF, was tested as first-line therapy in advanced GC. The combination of bevacizumab with chemotherapy failed to improve the OS of the patients; however, bevacizumab treatment was associated with increases in progression-free survival and overall response rate.593
Targeting VEGFR has achieved positive results in GC. In the phase III REGARD trial, the VEGFR2 antibody ramucirumab was tested in advanced gastric or gastroesophageal junction cancer. Ramucirumab monotherapy showed survival benefits in patients.577 Apatinib is a selective VEGFR2 small molecule tyrosine kinase inhibitor approved in China.594 Phase III clinical trial showed that apatinib monotherapy can increase the OS of repeatedly treated GC patients.595 Lenvatinib and regorafenib are multikinase inhibitors with anti-VEGFR activity. These drugs are currently being tested in combination with immune checkpoint inhibitors to treat GC in early clinical trials. Some positive initial results have been observed and the final efficacy needs to be confirmed in larger clinical trials.596,597
c-MET-targeted therapies
Rilotumumab is a monoclonal antibody targeting c-MET. In the phase II trial, rilotumumab showed some anti-tumor efficacy in gastric and gastroesophageal cancer.598 Unfortunately, in the pivotal phase III RILOMET-1 trial, the addition of rilotumumab to chemotherapy failed to improve the outcome of gastric and gastroesophageal cancer.599 Currently, research on c-MET inhibitor drugs mainly focuses on tyrosine kinase inhibitors. Savolitinib is a selective c-MET tyrosine kinase inhibitor that was granted approval in China for the treatment of metastatic NSCLC with MET exon 14-skipping alterations.600 In the VIKTORY umbrella trial, patients with metastatic GC were assigned to eight different biomarker groups to receive corresponding targeted drugs as second-line treatment.601 Savolitinib was assigned to treat patients with MET amplification. The overall response rate was 50% (10/20). The biomarker-assigned treatment cohort had encouraging response and survival rates when compared to conventional second-line chemotherapy.601
FGFR2-targeted therapies
There are two main strategies to target FGFRs: using TKIs or antibodies. AZD4547 (ABSK091) is an FGFR1/2/3 inhibitor. The phase II SHINE trial compared AZD4547 with paclitaxel as second-line treatment for FGFR2 amplified metastatic GC. Unfortunately, the trial failed to show improved outcome for those patients.602
Bemarituzumab is a first-in-class monoclonal antibody that selectively binds to FGFR2b, blocking ligand binding and induces antibody-dependent cell-mediated cytotoxicity (ADCC). The phase II FIGHT trial investigated the efficacy of bemarituzumab in the first-line treatment for metastatic gastric and gastroesophageal cancer patients. The addition of bemarituzumab to chemotherapy led to a 2-month improvement in progression-free survival (PFS) but failed to extend the OS. The duration of response was longer in patients with higher FGFR2b expression.603 This study indicates that bemarituzumab may be used for the first-line treatment of GC.
Claudin18.2-targeted therapies
Currently, different strategies are used to target Claudin18.2, including monoclonal antibodies, bispecific antibodies, CAR-T, and ADCs. Zolbetuximab (IMAB362) is a Claudin18.2 targeted antibody. The FAST study enrolled advanced gastric, gastroesophageal junction, and esophageal adenocarcinoma patients.604 The addition of zolbetuximab to chemotherapy can improve both PFS and OS. In addition, the side effects were manageable. The combinination of zolbetuximab and chemotherapy was generally tolerated. Zolbetuximab is currently being evaluated in phase III trials (NCT03653507, NCT03504397).
This initial success has attracted more attention to strategies that target Claudin18.2, especially CAR-T. CT041 is a Claudin18.2 targeted CAR-T drug. In phase I of a clinical trial in patients with previously treated digestive system cancers, CT041 showed an acceptable safety profile and encouraging overall response rate (ORR), as well as a 6-month overall survival rate. These initial results suggest that CT041 has promising efficacy in treating GC.605
Trop2-targeted therapies
Sacituzumab govitecan, the first-in-class anti-Trop2 antibody-drug conjugate (ADC), was approved by the US FDA in 2020 for the third-line treatment of metastatic triple-negative breast cancer (TNBC).606 Clinical trials are underway to expand the use of sacituzumab govitecan in multiple solid tumors, including GC. In the phase I/II IMMU-132-01 basket trial, sacituzumab govitecan was tested in refractory metastatic epithelial cancers.607 Efficacy was seen in several cancer cohorts, which suggests Trop-2 might be a broad target in solid tumors. Unfortunately, only five GC patients were included in this study and efficacy could not be determined. More studies are warranted to validate the efficacy of sacituzumab govitecan in GC.
Immune checkpoint-targeted therapies and other immunotherapies
Immunotherapy is a breakthrough in cancer treatment in the last decade. Immunotherapy in GC has also been progressing very rapidly. Cancer immunotherapy mainly comprises checkpoint inhibitors, adoptive immune cell therapy, and cancer vaccine. Checkpoint inhibitors have been approved to treat various types of solid tumors. Other adaptive immune cell therapies and cancer vaccines are still under clinical investigation in solid tumors.
GCs of MSI or EBV+ subtype according to TCGA classification are highly immunogenic with high expression of immune checkpoints, which makes them good candidates for cancer immunotherapy.608 Currently, PD-1 inhibitors have been successfully applied in GC treatment. The phase III ATTRACTION-2 study evaluated PD-1 inhibitor nivolumab for repeatedly treated advanced-stage gastric and gastroesophageal junction (G/GEJ) cancer.609 According to 2-year follow-up results, OS was significantly longer in the nivolumab group regardless of tumor PD-L1 expression.609 In the phase III KEYNOTE-062 trial, the PD-1 inhibitor pembrolizumab, alone or in combination with chemotherapy, was tested as first-line therapy in advanced GC. This trial found that pembrolizumab was not inferior to chemotherapy, and fewer adverse events were observed.610 Similarly, nivolumab was also tested as a first-line treatment of advanced gastric, gastro-esophageal junction, and esophageal adenocarcinoma in the phase III CheckMate 649 trial.611 Nivolumab with chemotherapy, compared to chemotherapy alone, resulted in significant improvements in OS in patients with a PD-L1 CPS of five or more.611 The PD-1 inhibitor might also benefit HER2-positive GC. In the phase III KEYNOTE-811 study, pembrolizumab was added to the standard trastuzumab plus chemotherapy for HER2-positive gastric or gastroesophageal junction cancer. According to interim analysis, the addition of pembrolizumab markedly reduces tumor size and significantly improves objective response rate.612
CTLA-4 is another important checkpoint. The CTLA-4 inhibitor ipilimumab has been approved in melanoma treatment.613 Unfortunately, targeting CTLA-4 in GC has been unsuccessful. In a phase II trial in pretreated late-stage GC, ipilimumab was not superior to supportive care.614 New strategies to combine inhibitors of PD-1 and CTLA-4 have also been tried. Cadonilimab (AK104) is a first-in-class PD-1/CTLA-4 bi-specific antibody developed by a Chinese biotech company. It received marketing approval from the National Medical Products Administration (NMPA) of China in 2022 for cervical cancer.615 In a phase Ib/II study, AK104 was evaluated in combination with chemotherapy for the first-line treatment of G/GEJ cancer (NCT03852251). AK104 showed promising activity and manageable safety.616 A phase III study of AK104 combined with chemotherapy as first-line therapy for G/GEJ cancer is underway (NCT05008783).
LAG-3 is another inhibitory checkpoint, which can be blocked by the antibody relatlimab. The combination of relatlimab and PD-1 antibody nivolumab has been shown to be safe and effective in melanoma.617 Relatlimab in combination with nivolumab is currently being tested in a phase II clinical trial for the first-line treatment in patients with G/GEJ cancer (NCT03662659). In another phase Ib study, relatlimab in combination with nivolumab was tested as an induction treatment prior to concurrent chemoradiation in patients with operable E/GEJ cancer (NCT03044613).
Monoclonal antibodies targeting TIGIT can effectively restore T cell function, exerting an anti-cancer effect.618 Tiragolumab is a potent TIGIT inhibitor that has entered clinical trials. Study showed that tiragolumab can enhance the effect of the PD-L1 antibody atezolizumab in non-small-cell lung cancer.619 Tiragolumab is also being tested in combination with atezolizumab and chemotherapy in a phase II, single-arm study for the first-line treatment of HER2-negative, unresectable, recurrent, or metastatic G/GEJ cancer (NCT04933227).
Adoptive immune cell therapy is another area of immunotherapy undergoing rapid development. CAR-T therapy lies at the center of adoptive immune cell therapy. CAR-T therapy is highly effective in treating hematopoietic tumors, sometimes leading to the complete remission of tumors. Several CAR-T therapies have been approved worldwide so far.620 However, CAR-T therapies have been less impressive in treating solid tumors, and no CAR-T therapy has been approved for solid tumors. As discussed earlier, Claudin18.2 targeted CAR-T is in rapid drug development for GC. Tumor vaccines are still in early clinical development, and their potential in cancer therapy needs to be tested vigorously.
Development of targeted therapies under preclinical/early clinical investigations
Several other targets are under preclinical or early clinical investigation that hold the potential to change the treatment of GC in the future. For instance, inhibitors for FAK, a non-receptor tyrosine kinase that regulates cell adhesion and cell survival,621 are currently under early clinical investigation. Many FAK inhibitors have been tested in various cancer types with disappointing results.621 IN10018 is a FAK inhibitor that showed robust efficacy in patients with platinum-resistant recurrent ovarian cancer.622 IN10018 is under evaluation in a phase I trial in previously treated locally advanced or metastatic G/GEJ adenocarcinoma (NCT05327231). Interestingly, a recent in vivo study showed that diffuse gastric cancer with RHO-A mutations was specifically sensitive to FAK inhibitor.494
Tyrosine receptor kinase (TRK) receptors, encoded by neurotrophic receptor tyrosine kinase (NTRK) genes, are predominantly expressed in neuronal tissue. Fusion of NTRK genes is a driver mutation;623 however, this kind of mutation is rare (<0.4%) in GC. The TRK inhibitor entrectinib is approved in the US and Europe for the treatment of patients with certain types of solid tumors expressing an NTRK gene fusion.624 GC patients with NTRK fusions can also be candidates for NTRK inhibitor therapy,625 but the efficacy of TRK inhibitors in treating GC requires further validation.
DKN-01 is a humanized monoclonal antibody that targets the DKK1 protein, which modulates Wnt/β-catenin signaling and is a crucial prognostic factor predicting tumor recurrence and survival in advanced GC patients.626 The FDA granted an Orphan Drug Designation to DKN-01 for the treatment of patients with G/GEJ cancer.627 DKN-01 is also an immunomodulatory combination partner for the treatment of cancer. In a phase III study, DKN-01 is under evaluation in combination with PD-1 antibody tislelizumab for the treatment of patients with locally advanced or metastatic G/GEJ cancer (the DisTinGuish study; NCT04363801).
AMG 199 is bi-specific antibody targeting CD3 and MUC17 that was designed to engage CD3+ T cells to MUC17-positive G/GEJ cancer cells, mediate redirected tumor cell lysis, and induce T cell activation as well as proliferation.628 A phase I clinical trial is being conducted to test AMG 199 in patients with MUC17-positive G/GEJ cancer (NCT04117958).
Strategies targeting DNA methylation and histone modification to treat GC majorly focus on inhibiting DNMTs and HDACs. Both DNMT inhibitors (such as 5-azacitidine and decitabine) and HDAC inhibitors (such as trichostatin A and valproic acid) can re-establish the expression of the tumor suppressor genes, particularly those involved in programmed cell death and therapeutic resistance. This gives them great potential for overcoming resistance by combination with chemotherapy and radiotherapy in GC treatments.524 In a phase I trial, the DNMT inhibitor 5-azacitidine was added to the neoadjuvant chemotherapy for GC. The treatment was well-tolerated with significant clinical and epigenetic responses.629 5-azacitidine may be worth further investigation in more clinical trials. In a phase 2 trial, the HDAC inhibitor vorinostat was added to the standard capecitabine-cisplatin chemotherapy for first-line treatment of GC. The objective response rate was 42%, which is acceptable; however, more adverse events were observed in comparison with the historical data of fluoropyrimidine-platinium doublet regimens.630 Due to the lack of selectivity and the incomplete understanding of the pharmacology of these HDAC inhibitors, side effects are the main considerations. Comprehensive testing in preclinical models is needed before HDAC inhibitors can proceed to clinical trials.
As summarized in Table 3 and Fig. 5, the development of growth factor or growth factor receptor antibodies, small molecule tyrosine kinase inhibitors, check point inhibitors, and adoptive immune cell therapies revolutionized treatment of GC. More novel therapies developed based on molecular biomarkers and signaling pathways are expected to improve precision medicine for GC.

Overview of targeted therapy and immunotherapy in gastric cancer. The representative therapeutic targets in GC and the corresponding targeted or immunotherapeutic agents that have entered clinical investigations are depicted. EGFR epidermal growth factor receptor, MAPK mitogen-activated protein kinase, HER2 human epidermal growth factor receptor 2, PI3K phosphoinositide 3-kinases, FGFR2 fibroblast growth factor receptor 2, VEGFR2 vascular endothelial growth factor receptor 2, FAK focal adhesion kinase, RhoA Ras homolog family member A, PD-1 programmed death 1, PD-L1/2 programmed death ligand 1/2, ADC antibody-drug conjugate, LRP5/6 low-density lipoprotein receptor-related protein 5/6, DKK Dickkopf, CTLA-4 cytotoxic T-lymphocyte-associated protein 4, CD3 cluster of differentiation 3, TIGIT T cell immunoreceptor with Ig and ITIM domains, LAG-3 lymphocyte-activation gene 3, DNMT DNA methyltransferase, HDAC Histone deacetylases. This figure was created with Biorender.com
Summary and perspectives
Compared to chemotherapy, targeted therapy for GC is safer and more effective. Some molecular-targeted drugs such as trastuzumab and apatinib have also been approved for the treatment of GC. The development of more effective drugs and the search for biomarkers with stronger sensitivity and specificity are still major challenges in the targeted treatment of GC. Owing to the interpatient and intratumor heterogeneity of GC, developing personalized therapy for GC patients has been the main demand in contemporary combat against GC. With the advent of technologies for genome-wide analysis and the establishment of novel preclinical models, treatment of GC has been moving toward precision medicine. The molecular classifications of GC enable more personalized targeted therapies and immunotherapies for GC patients and greater understanding of the molecular mechanisms underlying GC development, progression, metastasis, and therapeutic resistance. This has shed light on novel diagnosis/prognosis biomarkers and potential therapeutic targets. Principal signaling pathways mentioned here include MAPK, HER2, PI3K/AKT/mTOR, p53, Wnt/β-catenin, NF-κB, TGF-β, HGF/c-MET signaling pathways, and those involved in immunomodulation. Other signaling pathways with relatively limited research, such as FGF-FGFR, STAT3, HIF-1α, Hedgehog, and Notch signaling pathways, and the cell adhesion/junction-related signaling molecules, have also been discussed for molecular mechanisms and potential therapeutic targets. Among the identified targets from the molecular discoveries, several have at least entered phase II clinical investigations. These include HER2, EGFR, VEGFR, FGFR2, Claudin18.2, Trop2, c-MET, and the immune checkpoint molecule PD-1. However, the molecular mechanisms are generally not associated with a unique signaling pathway but with crosstalk or feedback loops. Bypass pathways are critical contributors to therapeutic resistance when mono-targeted therapy is used. Therefore, the development and verification of novel combination regimens are in urgent demand.
The immune checkpoint inhibitor PD-1 monoclonal antibody has been approved for the first-line treatment of GC. Recently, 18 patients with rectal cancer received nine doses of dostarlimab (a PD-1 blocker) intravenously for immunotherapy. After 6 months of treatment, all 18 patients achieved complete clinical remissions.631 This study strongly demonstrates that immunotherapy is the future trend to treat gastrointestinal tumors. Immunotherapy has good safety and a durable immune response. With the rapid development of the high-throughput and whole-exome sequencing for immunologic screening of mutant genes, more neoantigen-reactive tumor-infiltrating lymphocytes (TIL) will be identified in GCs, which means more specific immunogenic gene products can be developed. Therefore, traditional therapy combined with immunotherapy is the trend in GC treatment. The timing of immunotherapy, the selection of drug combinations and combined therapy dose, the management of treating-related adverse events, and the selection of biomarkers for predicting clinical efficacy all need further research, but it shows a good prospect in the treatment of GC.
Although the systematic treatment of GC has evolved rapidly in recent years, there are still limited drugs available in the clinic. Innovation is needed to speed up drug development for GC. We expect breakthroughs to be made in GC therapy by looking deep into the tumor microenvironment specific to GC, stratifying patients more precisely using next-generation sequencing (NGS), and individualizing treatment through organoid-based functional drug predictions. NGS, like whole-exome sequencing (WES), and novel technologies, like single cell sequencing for profiling genetic changes, are the basis for biomarker identification and precision medicine. However, the complexity of NGS data analysis and its high cost hinder its application in the clinic. It is important to lower the cost of clinical NGS sequencing and expand its use to cover most of the GC patients. This will help the discovery of low-frequency genetic aberrations and the development of novel therapies. The complexity of cancer genomics requires fine stratification of patients to receive corresponding drugs. This means there are few patients to receive each drug treatment, which hinders the evaluation of the treatment effect. The umbrella trial and the basket trial were designed to deal with this issue.632 In an umbrella trial, patients with the same type of cancer are stratified into different subgroups based on their molecular profiles, and patients in each subgroup are treated accordingly. In a basket trial, patients with different types of tumors but the same targets are grouped. The drug of interest is tested in this phenotype-heterogeneous but genotype-homogeneous group of patients. These two novel designs for clinical trials have been used for new drug discovery and personalized cancer treatment (Fig. 6). For example, the VIKTORY trial was the largest umbrella trial in GC, where GC patients were assigned to eight different biomarker groups based on NGS.601 This study demonstrated the efficacy of targeted therapy in certain molecular subgroups.

Essential technologies and processes for elevating biomarker-guided precision medicine. The next-generation sequencing and novel technologies like single cell sequencing for profiling genetic changes enable biomarker identification with higher precision. Biomarkers are the basis for molecular classification and patient stratifying. Meanwhile, biomarker-based novel therapy is developed as the target is selected. New therapeutic agents are developed with lead compound or biologics identified, followed by formulation optimization and possible combination designs. The patient-derived xenograft or organoid research models are useful tools for drug screening and molecular mechanism verifications. Finally, novel clinical trial designs like umbrella trials and basket trials enable precise evaluation of treatment effects under a fine stratification of patients. This figure was drafted with Biorender.com and modified using Adobe Photoshop
However, biomarker-based precision medicine is restricted by the low sensitivity and specificity of biomarkers in predicting sensitivity or resistance to drugs. In addition, most of the patients lack actionable targets even after extensive biomarker profiling. As an alternative to biomarker-based drug prediction, functional drug screening may be a new strategy for cancer precision medicine. The organoid technology combines stem cell niche and 3D extracellular matrix (ECM) for in-vitro cell culture. Stem cells can form organized cell structures resembling their tissue-of-origin at both cellular and structural levels. When cancer cells derived from human tumors are cultured under organoid conditions, they can be expanded and stably passaged like cancer cell lines. The tumor organoid can faithfully maintain the genotypes and phenotypes of the original tumor tissue. Most importantly, the tumor organoid also maintains the sensitivity of the original tumor to drugs. These features make the organoid an ideal tool for in-vitro functional drug screening. Observational studies have confirmed the consistency of organoid-based drug sensitivity to the clinic response of the patients receiving the same regime.633,634,635 Researchers around the world, including us, have been trying to establish patient-derived organoids to guide GC treatment.636,637,638 It is hoped that organoid-based drug screening will go from bench to bedside to benefit cancer patients.
Recognition of novel molecular targets has also paved the way for developing gene therapy as a promising molecular alternative in GC treatment, including gene silencing approaches to inactivate oncogenes, replacing defective tumor suppressor genes, introducing suicide genes, genetic immunotherapy, and so forth. The therapeutic potential of genetic approaches has been demonstrated in certain in vitro studies, such as a nanoparticle-delivered siRNA to suppress oncogene CFL1639 and a CRISPR/Cas9 system-delivered LncRNA PANDAR (promoter of CDKN1A antisense DNA damage activated RNA) to interact with p53 and competitively regulate CDKN1A transcription in GC cell lines.640 Like drug-based therapies, the major challenge of gene therapy lies in finding a way to circumvent non-responsiveness, which is caused by immunogenic effects after the delivery of genetic material. A newly published study reported that combining p53 mRNA nanotherapy with anti-PD-L1 therapy can reprogram the immune microenvironment for improved anti-cancer effects compared to monotherapy.641 This implies that proper formulation and combination design with an optimized delivery system will be the key to developing novel targeted therapy, immunotherapy as well as gene therapy that can circumvent therapeutic tolerance or resistance.
Beyond any doubt, early diagnosis and effective prevention strategies are indispensable to reducing the morbidity and mortality of GC. Lifestyle control and endoscopic screening have been useful prevention approaches. As H. pylori infection is the dominant risk factor for GC development, testing for H. pylori and chemo-eradication have been the primary prevention strategy for GC.642 Additionally, vaccines aimed at eradicating H. pylori are under development.643 For early medication managements, the identification of novel molecular markers driven by the NGS technologies could improve precision in both diagnosis and therapeutic interventions.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Ajani, J. A. et al. Gastric cancer, version 2. 2022, NCCN clinical practice guidelines in oncology. J. Natl Compr. Canc. Netw. 20, 167–192 (2022).
Ferlay, J. et al. Cancer statistics for the year 2020: an overview. Int. J. Cancer 149, 778–789 (2021).
Ilic, M. & Ilic, I. Epidemiology of stomach cancer. World J. Gastroenterol. 28, 1187–1203 (2022).
Arnold, M. et al. Is gastric cancer becoming a rare disease? A global assessment of predicted incidence trends to 2035. Gut 69, 823–829 (2020).
Wang, Z. et al. Identification of new susceptibility loci for gastric non-cardia adenocarcinoma: pooled results from two Chinese genome-wide association studies. Gut 66, 581–587 (2017).
Inoue, M. Public health interventions for gastric cancer control. Gastrointest. Endosc. Clin. N. Am. 31, 441–449 (2021).
Plummer, M., Franceschi, S., Vignat, J., Forman, D. & de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 136, 487–490 (2015).
Valenzuela, M. A., Canales, J., Corvalan, A. H. & Quest, A. F. G. Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J. Gastroenterol. 21, 12742–12756 (2015).
Mukaisho, K.-I., Nakayama, T., Hagiwara, T., Hattori, T. & Sugihara, H. Two distinct etiologies of gastric cardia adenocarcinoma: interactions among pH, Helicobacter pylori, and bile acids. Front. Microbiol. 6, 412 (2015).
Bagheri, V. et al. Cytokine networks and their association with Helicobacter pylori infection in gastric carcinoma. J. Cell Physiol. 233, 2791–2803 (2018).
Fock, K. M. Review article: the epidemiology and prevention of gastric cancer. Aliment Pharmacol. Ther. 40, 250–260 (2014).
Raei, N., Behrouz, B., Zahri, S. & Latifi-Navid, S. Helicobacter pylori infection and dietary factors act synergistically to promote gastric cancer. Asian Pac. J. Cancer Prev. 17, 917–921 (2016).
De Manzoni, G. & Roviello, F. Gastric Cancer: the 25-year R-Evolution (Springer, 2021).
Thrift, A. P. & El-Serag, H. B. Burden of gastric cancer. Clin. Gastroenterol. Hepatol. 18, 534–542 (2020).
Karimi, P., Islami, F., Anandasabapathy, S., Freedman, N. D. & Kamangar, F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 23, 700–713 (2014).
Rawla, P. & Barsouk, A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz. Gastroenterol. 14, 26–38 (2019).
Naseem, M. et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat. Rev. 66, 15–22 (2018).
Rihane, F. E. et al. Helicobacter pylori co-infection with Epstein-Barr virus and the risk of developing gastric adenocarcinoma at an early age: Observational study infectious agents and cancer. Ann. Med. Surg. 68, 102651 (2021).
Gareayaghi, N. et al. Epstein-Barr Virus and Helicobacter pylori co-infection in patients with gastric cancer and duodenale ulcer. New Microbiol. 44, 217–226 (2021).
Kashyap, D., Baral, B., Jakhmola, S., Singh, A. K. & Jha, H. C. Helicobacter pylori and epstein-barr virus coinfection stimulates aggressiveness in gastric cancer through the regulation of gankyrin. Msphere 6, e00751–00721 (2021).
Chia, N.-Y. & Tan, P. Molecular classification of gastric cancer. Ann. Oncol. 27, 763–769 (2016).
Taja-Chayeb, L. et al. Hereditary diffuse gastric cancer (HDGC). An overview. Clin. Res. Hepatol. Gastroenterol. 46, 101820 (2022).
McLean, M. H. & El-Omar, E. M. Genetics of gastric cancer. Nat. Rev. Gastroenterol. Hepatol. 11, 664–674 (2014).
Liu, X. & Chu, K.-M. E-cadherin and gastric cancer: cause, consequence, and applications. Biomed. Res. Int. 2014, 637308 (2014).
Guilford, P. et al. E-cadherin germline mutations in familial gastric cancer. Nature 392, 402–405 (1998).
Blair, V. R. et al. Hereditary diffuse gastric cancer: updated clinical practice guidelines. Lancet Oncol. 21, e386–e397 (2020).
Amin, M. B. et al. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 67, 93–99 (2017).
Venerito, M., Link, A., Rokkas, T. & Malfertheiner, P. Review: gastric cancer-clinical aspects. Helicobacter 24, e12643 (2019).
Coburn, N. et al. Staging and surgical approaches in gastric cancer: a systematic review. Cancer Treat. Rev. 63, 104–115 (2018).
Allen, C. J. et al. Chemotherapy versus chemotherapy plus chemoradiation as neoadjuvant therapy for resectable gastric adenocarcinoma: a multi-institutional analysis. Ann. Surg. 274, 544–548 (2021).
Lauren, P. The two histological main types of gastric carcinoma: diffuse and so‐called intestinal‐type carcinoma: an attempt at a histo‐clinical classification. Acta Pathol. Microbiol. Scand. 64, 31–49 (1965).
Petrelli, F. et al. Prognostic value of diffuse versus intestinal histotype in patients with gastric cancer: a systematic review and meta-analysis. J. Gastrointest. Oncol. 8, 148–163 (2017).
World Health Organization. WHO Classification Of Tumours: Digestive System Tumours. Report No. 9283244990 (World Health Organization (WHO), 2019).
Nakamura, K., Sugano, H. & Takagi, K. Carcinoma of the stomach in incipient phase: its histogenesis and histological appearances. Gan 59, 251–258 (1968).
Japanese Gastric Cancer Association. Japanese gastric cancer treatment guidelines 2018. Gastric Cancer 24, 1–21 (2020).
Matsuoka, T. & Yashiro, M. Biomarkers of gastric cancer: current topics and future perspective. World J. Gastroenterol. 24, 2818–2832 (2018).
Abbas, M. et al. Current and future biomarkers in gastric cancer. Biomed. Pharmacother. 103, 1688–1700 (2018).
Patel, T. H. & Cecchini, M. Targeted therapies in advanced gastric cancer. Curr. Treat. Options Oncol. 21, 70 (2020).
Bang, Y.-J. et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376, 687–697 (2010).
Cancer, G. A. R. N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014).
Cristescu, R. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 21, 449–456 (2015).
Tirino, G. et al. What’s new in gastric cancer: the therapeutic implications of molecular classifications and future perspectives. Int. J. Mol. Sci. 19, 2659 (2018).
Chivu-Economescu, M. et al. New therapeutic options opened by the molecular classification of gastric cancer. World J. Gastroenterol. 24, 1942–1961 (2018).
Machlowska, J. et al. State of the art for gastric signet ring cell carcinoma: from classification, prognosis, and genomic characteristics to specified treatments. Cancer Manag. Res. 11, 2151–2161 (2019).
Pernot, S. et al. Signet-ring cell carcinoma of the stomach: impact on prognosis and specific therapeutic challenge. World J. Gastroenterol. 21, 11428–11438 (2015).
Bamboat, Z. M. et al. Stage-stratified prognosis of signet ring cell histology in patients undergoing curative resection for gastric adenocarcinoma. Ann. Surg. Oncol. 21, 1678–1685 (2014).
Voron, T. et al. Is signet-ring cell carcinoma a specific entity among gastric cancers? Gastric Cancer 19, 1027–1040 (2016).
Li, Y., Zhu, Z., Ma, F., Xue, L. & Tian, Y. Gastric signet ring cell carcinoma: current management and future challenges. Cancer Manag. Res. 12, 7973–7981 (2020).
Humar, B. et al. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 69, 2050–2056 (2009).
Shu, Y. et al. Prognostic significance of frequent CLDN18-ARHGAP26/6 fusion in gastric signet-ring cell cancer. Nat. Commun. 9, 2447 (2018).
Xu, B. et al. Highly expressed Claudin18.2 as a potential therapeutic target in advanced gastric signet-ring cell carcinoma (SRCC). J. Gastrointest. Oncol. 11, 1431–1439 (2020).
Hirotsu, Y. et al. Deficiency of mismatch repair genes is less frequently observed in signet ring cell compared with non-signet ring cell gastric cancer. Med. Oncol. 36, 1–7 (2019).
Puccini, A. et al. Molecular profiling of signet-ring-cell carcinoma (SRCC) from the stomach and colon reveals potential new therapeutic targets. Oncogene 41, 3455–3460 (2022).
In, H. et al. Validation of the 8th Edition of the AJCC TNM Staging System for Gastric Cancer using the National Cancer Database. Ann. Surg. Oncol. 24, 3683–3691 (2017).
Janjigian, Y. Y. et al. Genetic predictors of response to systemic therapy in esophagogastric cancer genomic biomarkers in esophagogastric adenocarcinoma. Cancer Discov. 8, 49–58 (2018).
Zhang, S.-X. et al. Current advances and outlook in gastric cancer chemoresistance: a review. Recent Pat. Anticancer Drug Discov. 17, 26–41 (2022).
Magnelli, L., Schiavone, N., Staderini, F., Biagioni, A. & Papucci, L. MAP kinases pathways in gastric cancer. Int. J. Mol. Sci. 21, 2893 (2020).
Yang, M. & Huang, C.-Z. Mitogen-activated protein kinase signaling pathway and invasion and metastasis of gastric cancer. World J. Gastroenterol. 21, 11673–11679 (2015).
Plotnikov, A., Zehorai, E., Procaccia, S. & Seger, R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 1813, 1619–1633 (2011).
Lei, Y.-Y., Wang, W.-J., Mei, J.-H. & Wang, C.-L. Mitogen-activated protein kinase signal transduction in solid tumors. Asian Pac. J. Cancer Prev. 15, 8539–8548 (2014).
Simanshu, D. K., Nissley, D. V. & McCormick, F. RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017).
Deng, N. et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 61, 673–684 (2012).
Pai, E. F. et al. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 341, 209–214 (1989).
Milburn, M. V. et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 247, 939–945 (1990).
Pratilas, C. A., Xing, F. & Solit, D. B. Targeting oncogenic BRAF in human cancer. Curr. Top. Microbiol. Immunol. 355, 83–98 (2012).
Bonni, A. et al. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362 (1999).
Roskoski, R. Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66, 105–143 (2012).
Fujimori, Y. et al. Prognostic value of RKIP and p-ERK in gastric cancer. J. Exp. Clin. Cancer Res. 31, 30 (2012).
Balmanno, K. & Cook, S. J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 16, 368–377 (2009).
Akter, H. et al. Activation of matrix metalloproteinase-9 (MMP-9) by neurotensin promotes cell invasion and migration through ERK pathway in gastric cancer. Tumour Biol. 36, 6053–6062 (2015).
Long, W. et al. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Invest. 122, 1869–1880 (2012).
Deleris, P. et al. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J. Biol. Chem. 286, 6470–6478 (2011).
Gupta, S. et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760–2770 (1996).
Dérijard, B. et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76, 1025–1037 (1994).
Mohit, A. A., Martin, J. H. & Miller, C. A. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 14, 67–78 (1995).
Kallunki, T., Deng, T., Hibi, M. & Karin, M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87, 929–939 (1996).
Eferl, R. & Wagner, E. F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 (2003).
Weston, C. R. & Davis, R. J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 (2007).
Pereira, L., Igea, A., Canovas, B., Dolado, I. & Nebreda, A. R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 5, 1759–1774 (2013).
Shibata, W. et al. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 68, 5031–5039 (2008).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 (2011).
Cuenda, A. & Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 1773, 1358–1375 (2007).
Souma, Y. et al. Antiproliferative effect of SOCS‐1 through the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int. J. Cancer 131, 1287–1296 (2012).
Yan, X., Rui, X. & Zhang, K. Baicalein inhibits the invasion of gastric cancer cells by suppressing the activity of the p38 signaling pathway. Oncol. Rep. 33, 737–743 (2015).
Guo, X. et al. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 8, 1–9 (2008).
She, Q.-B., Bode, A. M., Ma, W.-Y., Chen, N.-Y. & Dong, Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 61, 1604–1610 (2001).
Bacus, S. S. et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 20, 147–155 (2001).
Hui, L. et al. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet. 39, 741–749 (2007).
Qu, J.-L. et al. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 41, 875–880 (2009).
Shen, B., Li, M., Wang, H., Xin, L. & Xie, J. Expression and clinical significance of the RAS/RAF/MAPK cell signaling pathway in gastric cancer. Int. J. Clin. Exp. Med. 11, 11682–11689 (2018).
Liu, P., Cheng, H., Roberts, T. M. & Zhao, J. J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644 (2009).
Ryan, M. B. & Corcoran, R. B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 15, 709–720 (2018).
van Grieken, N. C. T. et al. KRAS and BRAF mutations are rare and related to DNA mismatch repair deficiency in gastric cancer from the East and the West: results from a large international multicentre study. Br. J. Cancer 108, 1495–1501 (2013).
Karakas, B., Bachman, K. E. & Park, B. H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 94, 455–459 (2006).
Takahashi, N. et al. Clinicopathological features and prognostic roles of KRAS, BRAF, PIK3CA and NRAS mutations in advanced gastric cancer. BMC Res. Notes 7, 271 (2014).
Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 15, 306–316 (2014).
Zhong, J. et al. ZIC1 modulates cell-cycle distributions and cell migration through regulation of sonic hedgehog, PI3K and MAPK signaling pathways in gastric cancer. BMC Cancer 12, 1–10 (2012).
Fukui, H. et al. IL-22 produced by cancer-associated fibroblasts promotes gastric cancer cell invasion via STAT3 and ERK signaling. Br. J. Cancer 111, 763–771 (2014).
Yang, M. et al. NAIF1 inhibits gastric cancer cells migration and invasion via the MAPK pathways. J. Cancer Res. Clin. Oncol. 141, 1037–1047 (2015).
Wang, C. et al. Increased serum chemerin level promotes cellular invasiveness in gastric cancer: a clinical and experimental study. Peptides 51, 131–138 (2014).
Dong, C., Sun, J., Ma, S. & Zhang, G. K-ras-ERK1/2 down-regulates H2A. XY142ph through WSTF to promote the progress of gastric cancer. BMC Cancer 19, 1–11 (2019).
Wu, W. K. K. et al. MicroRNA dysregulation in gastric cancer: a new player enters the game. Oncogene 29, 5761–5771 (2010).
He, Y. et al. MiR-592 promotes gastric cancer proliferation, migration, and invasion through the PI3K/AKT and MAPK/ERK signaling pathways by targeting Spry2. Cell. Physiol. Biochem. 47, 1465–1481 (2018).
Yang, F. et al. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 279, 3159–3165 (2012).
Li, P., Xue, W.-J., Feng, Y. & Mao, Q.-S. Long non-coding RNA CASC2 suppresses the proliferation of gastric cancer cells by regulating the MAPK signaling pathway. Am. J. Transl. Res. 8, 3522–3529 (2016).
Stahl, P. et al. Heterogeneity of amplification of HER2, EGFR, CCND1 and MYC in gastric cancer. BMC Gastroenterol. 15, 7 (2015).
Sukawa, Y. et al. HER2 expression and PI3K-Akt pathway alterations in gastric cancer. Digestion 89, 12–17 (2014).
Jorgensen, J. T. & Hersom, M. HER2 as a prognostic marker in gastric cancer - a systematic analysis of data from the literature. J. Cancer 3, 137–144 (2012).
Kim, K. C. et al. Evaluation of HER2 protein expression in gastric carcinomas: comparative analysis of 1,414 cases of whole-tissue sections and 595 cases of tissue microarrays. Ann. Surg. Oncol. 18, 2833–2840 (2011).
Ruschoff, J. et al. HER2 testing in gastric cancer: a practical approach. Mod. Pathol. 25, 637–650 (2012).
Gravalos, C. & Jimeno, A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann. Oncol. 19, 1523–1529 (2008).
Schechter, A. L. et al. The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature 312, 513–516 (1984).
Hyman, D. M. et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 554, 189–194 (2018).
Fornaro, L. et al. Anti-HER agents in gastric cancer: from bench to bedside. Nat. Rev. Gastroenterol. Hepatol. 8, 369–383 (2011).
Iqbal, N. & Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol. Biol. Int. 2014, 852748 (2014).
Yarden, Y. & Sliwkowski, M. X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 (2001).
Cho, H.-S. et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 (2003).
Pahuja, K. B. et al. Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell 34, 792–806 (2018). e795.
Vaught, D. B. et al. HER3 is required for HER2-induced preneoplastic changes to the breast epithelium and tumor formation. Cancer Res. 72, 2672–2682 (2012).
He, X. X. et al. Protein expression of HER2, 3, 4 in gastric cancer: correlation with clinical features and survival. J. Clin. Pathol. 68, 374–380 (2015).
Abrahao-Machado, L. F. & Scapulatempo-Neto, C. HER2 testing in gastric cancer: an update. World J. Gastroenterol. 22, 4619–4625 (2016).
Tanner, M. et al. Amplification of HER-2 in gastric carcinoma: association with Topoisomerase IIalpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann. Oncol. 16, 273–278 (2005).
Di Fiore, P. P. et al. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell 51, 1063–1070 (1987).
Choi, B. et al. Single-molecule functional anatomy of endogenous HER2-HER3 heterodimers. Elife 9, e53934 (2020).
Rohlenova, K., Neuzil, J. & Rohlena, J. The role of Her2 and other oncogenes of the PI3K/AKT pathway in mitochondria. Biol. Chem. 397, 607–615 (2016).
Berns, K. et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12, 395–402 (2007).
Wang, Q. et al. PI3K-p110alpha mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene 35, 3607–3612 (2016).
Slamon, D. et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 365, 1273–1283 (2011).
Roukos, D. H. Targeting gastric cancer with trastuzumab: new clinical practice and innovative developments to overcome resistance. Ann. Surg. Oncol. 17, 14–17 (2010).
Deguchi, Y. et al. PTEN loss is associated with a poor response to trastuzumab in HER2-overexpressing gastroesophageal adenocarcinoma. Gastric Cancer 20, 416–427 (2017).
Huang, L.-T. et al. Durable clinical response to pyrotinib after resistance to prior anti-HER2 therapy for HER2-positive advanced gastric cancer: a case report. Front. Oncol. 9, 1453 (2019).
Mahlknecht, G. et al. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc. Natl Acad. Sci. USA 110, 8170–8175 (2013).
Kanthala, S. et al. Novel peptidomimetics for inhibition of HER 2: HER 3 heterodimerization in HER 2‐positive breast cancer. Chem. Biol. Drug Des. 85, 702–714 (2015).
Willems, L. et al. PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr. Oncol. Rep. 14, 129–138 (2012).
Cantley, L. C. The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 (2002).
Li, H., Prever, L., Hirsch, E. & Gulluni, F. Targeting PI3K/AKT/mTOR signaling pathway in breast cancer. Cancers 13, 3517 (2021).
Vara, J. Á. F. et al. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 30, 193–204 (2004).
Baselga, J. et al. Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of Trastuzumab Emtansine in HER2-positive metastatic breast cancer. Clin. Cancer Res. 22, 3755–3763 (2016).
Pungsrinont, T., Kallenbach, J. & Baniahmad, A. Role of PI3K-AKT-mTOR pathway as a pro-survival signaling and resistance-mediating mechanism to therapy of prostate cancer. Int. J. Mol. Sci. 22, 11088 (2021).
Bilanges, B., Posor, Y. & Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 20, 515–534 (2019).
Denley, A., Kang, S., Karst, U. & Vogt, P. K. Oncogenic signaling of class I PI3K isoforms. Oncogene 27, 2561–2574 (2008).
Samuels, Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004).
Fan, Q.-W. & Weiss, W. A. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr. Top. Microbiol. Immunol. 347, 279–296 (2010).
Manning, B. D. & Toker, A. AKT/PKB signaling: navigating the network. Cell 169, 381–405 (2017).
Vivanco, I. & Sawyers, C. L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 (2002).
Parikh, C. et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc. Natl Acad. Sci. USA 109, 19368–19373 (2012).
Mange, A. et al. FKBP4 connects mTORC2 and PI3K to activate the PDK1/Akt-dependent cell proliferation signaling in breast cancer. Theranostics 9, 7003–7015 (2019).
Lu, Z. et al. RICTOR/mTORC2 affects tumorigenesis and therapeutic efficacy of mTOR inhibitors in esophageal squamous cell carcinoma. Acta Pharm. Sin. B 10, 1004–1019 (2020).
Song, M. S., Salmena, L. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 (2012).
Salmena, L., Carracedo, A. & Pandolfi, P. P. Tenets of PTEN tumor suppression. Cell 133, 403–414 (2008).
Fusco, N. et al. PTEN alterations and their role in cancer management: are we making headway on precision medicine? Genes 11, 719 (2020).
Kong, Y. et al. Analysis of mTOR gene aberrations in melanoma patients and evaluation of their sensitivity to PI3K–AKT–mTOR pathway inhibitors. Clin. Cancer Res. 22, 1018–1027 (2016).
Foster, K. G. & Fingar, D. C. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J. Biol. Chem. 285, 14071–14077 (2010).
Tian, T., Li, X. & Zhang, J. mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 20, 755 (2019).
Singh, S. S. et al. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: a reality for personalized medicine? World J. Gastroenterol. 21, 12261–12273 (2015).
Schmelzle, T. & Hall, M. N. TOR, a central controller of cell growth. Cell 103, 253–262 (2000).
Garami, A. et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466 (2003).
Graff, J. R., Konicek, B. W., Carter, J. H. & Marcusson, E. G. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 68, 631–634 (2008).
Huang, J. & Manning, B. D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 37, 217–222 (2009).
Yang, W., Raufi, A. & Klempner, S. J. Targeted therapy for gastric cancer: molecular pathways and ongoing investigations. Biochim. Biophys. Acta 1846, 232–237 (2014).
Evangelisti, C., Chiarini, F., Paganelli, F., Marmiroli, S. & Martelli, A. M. Crosstalks of GSK3 signaling with the mTOR network and effects on targeted therapy of cancer. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118635 (2020).
Hermida, M. A., Dinesh Kumar, J. & Leslie, N. R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 65, 5–15 (2017).
Dal Col, J. et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood 111, 5142–5151 (2008).
Wang, X. et al. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 68, 7409–7418 (2008).
Breuleux, M. et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther. 8, 742–753 (2009).
Tapia, O. et al. The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 465, 25–33 (2014).
Velho, S. et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur. J. Cancer 41, 1649–1654 (2005).
Li, V. S. W. et al. Mutations of PIK3CAin gastric adenocarcinoma. BMC Cancer 5, 1–6 (2005).
Nosho, K. et al. Association of microRNA-31 with BRAF mutation, colorectal cancer survival and serrated pathway. Carcinogenesis 35, 776–783 (2014).
Boger, C. et al. Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann. Oncol. 28, 1005–1014 (2017).
Polom, K. et al. PIK3CA mutation in gastric cancer and the role of microsatellite instability status in mutations of exons 9 and 20 of the PIK3CA gene. Adv. Clin. Exp. Med. 27, 963–969 (2018).
Jung, E. H. et al. Efficient, stable and scalable perovskite solar cells using poly (3-hexylthiophene). Nature 567, 511–515 (2019).
Harada, Y. et al. The JRA-55 reanalysis: representation of atmospheric circulation and climate variability. J. Meteorol. Soc. Jpn Ser. II 94, 269–302 (2016).
Ito, A. & Budke, C. M. The echinococcoses in Asia: the present situation. Acta Trop. 176, 11–21 (2017).
Shi, J. et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer 12, 1–11 (2012).
Kobayashi, I., Semba, S., Matsuda, Y., Kuroda, Y. & Yokozaki, H. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology 73, 8–17 (2006).
Li, H. et al. APY0201 represses tumor growth through inhibiting autophagy in gastric cancer cells. J. Clin. Oncol. 2022, 1–16 (2022).
Altomare, D. A. & Testa, J. R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 (2005).
Manning, B. D. & Cantley, L. C. AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 (2007).
Almhanna, K., Strosberg, J. & Malafa, M. Targeting AKT protein kinase in gastric cancer. Anticancer Res. 31, 4387–4392 (2011).
Sukawa, Y. et al. Alterations in the human epidermal growth factor receptor 2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer. World J. Gastroenterol. 18, 6577–6586 (2012).
Gelaro, R. et al. The modern-era retrospective analysis for research and applications, version 2 (MERRA-2). J. Clim. 30, 5419–5454 (2017).
Brar, S. S. et al. Processes of care in the multidisciplinary treatment of gastric cancer: results of a RAND/UCLA expert panel. JAMA Surg. 149, 18–25 (2014).
Zhou, X. D. et al. Protein kinase B phosphorylation correlates with vascular endothelial growth factor A and microvessel density in gastric adenocarcinoma. J. Int. Med. Res. 40, 2124–2134 (2012).
Chen, H. et al. Lymphangiogenesis in gastric cancer regulated through Akt/mTOR-VEGF-C/VEGF-D axis. BMC Cancer 15, 1–7 (2015).
Yang, Q. & Guan, K.-L. Expanding mTOR signaling. Cell Res. 17, 666–681 (2007).
Smith, C. R. et al. Transcatheter versus surgical aortic-valve replacement in high-risk patients. N. Engl. J. Med. 364, 2187–2198 (2011).
Steelman, L. S. et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 22, 686–707 (2008).
Meric-Bernstam, F. et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin. Cancer Res. 18, 1777–1789 (2012).
Feng, W. et al. Morphoproteomic profile of mTOR, Ras/Raf kinase/ERK, and NF-kappaB pathways in human gastric adenocarcinoma. Ann. Clin. Lab Sci. 38, 195–209 (2008).
Byeon, S.-J., Han, N., Choi, J., Kim, M. A. & Kim, W. H. Prognostic implication of TSC1 and mTOR expression in gastric carcinoma. J. Surg. Oncol. 109, 812–817 (2014).
Yu, G. et al. Overexpression of phosphorylated mammalian target of rapamycin predicts lymph node metastasis and prognosis of chinese patients with gastric cancer. Clin. Cancer Res. 15, 1821–1829 (2009).
An, J. Y. et al. Prognostic role of p-mTOR expression in cancer tissues and metastatic lymph nodes in pT2b gastric cancer. Int. J. Cancer 126, 2904–2913 (2010).
Wong, H. & Yau, T. Targeted therapy in the management of advanced gastric cancer: are we making progress in the era of personalized medicine? Oncologist 17, 346–358 (2012).
Hofseth, L. J., Hussain, S. P. & Harris, C. C. p53: 25 years after its discovery. Trends Pharmacol. Sci. 25, 177–181 (2004).
Ingaramo, M. C., Sanchez, J. A. & Dekanty, A. Regulation and function of p53: a perspective from Drosophila studies. Mech. Dev. 154, 82–90 (2018).
Gupta, A., Shah, K., Oza, M. J. & Behl, T. Reactivation of p53 gene by MDM2 inhibitors: a novel therapy for cancer treatment. Biomed. Pharmacother. 109, 484–492 (2019).
Ciciarello, M. et al. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 276, 19205–19213 (2001).
Koutsodontis, G., Tentes, I., Papakosta, P., Moustakas, A. & Kardassis, D. Sp1 plays a critical role in the transcriptional activation of the human cyclin-dependent kinase inhibitor p21(WAF1/Cip1) gene by the p53 tumor suppressor protein. J. Biol. Chem. 276, 29116–29125 (2001).
Ohki, R. et al. Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J. Biol. Chem. 275, 22627–22630 (2000).
Cerda-Opazo, P. et al. Inverse expression of survivin and reprimo correlates with poor patient prognosis in gastric cancer. Oncotarget 9, 12853–12867 (2018).
Kubota, E. et al. Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 13, 2129–2137 (2014).
Zhang, X. et al. MicroRNA-181a functions as an oncomir in gastric cancer by targeting the tumour suppressor gene ATM. Pathol. Oncol. Res. 20, 381–389 (2014).
Zhang, X. et al. MicroRNA-650 targets ING4 to promote gastric cancer tumorigenicity. Biochem. Biophys. Res. Commun. 395, 275–280 (2010).
Wang, M. et al. miR-17-5p/20a are important markers for gastric cancer and murine double minute 2 participates in their functional regulation. Eur. J. Cancer 49, 2010–2021 (2013).
Bou Kheir, T. et al. miR-449 inhibits cell proliferation and is down-regulated in gastric cancer. Mol. Cancer 10, 29 (2011).
Busuttil, R. A. et al. Role of p53 in the progression of gastric cancer. Oncotarget 5, 12016–12026 (2014).
Bockerstett, K. A. et al. Single-cell transcriptional analyses identify lineage-specific epithelial responses to inflammation and metaplastic development in the gastric corpus. Gastroenterology 159, 2116–2129 (2020). e2114.
Shimizu, T. et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology 147, 407–417 (2014). e403.
Belyavskaya, V. A. et al. Genetic status of p53 in stomach cancer: somatic mutations and polymorphism of codon 72. Bull. Exp. Biol. Med. 141, 243–246 (2006).
Pizzi, M. P. et al. Identification of DNA mutations in gastric washes from gastric adenocarcinoma patients: possible implications for liquid biopsies and patient follow-up. Int. J. Cancer 145, 1090–1098 (2019).
Stachler, M. D. et al. Detection of mutations in Barrett’s esophagus before progression to high-grade dysplasia or adenocarcinoma. Gastroenterology 155, 156–167 (2018).
Fu, H. et al. Curcumin regulates proliferation, autophagy, and apoptosis in gastric cancer cells by affecting PI3K and P53 signaling. J. Cell. Physiol. 233, 4634–4642 (2018).
Cao, D.-Z. et al. Effects of folic acid on epithelial apoptosis and expression of Bcl-2 and p53 in premalignant gastric lesions. World J. Gastroenterol. 11, 1571–1576 (2005).
Wada, Y. et al. Helicobacter pylori induces somatic mutations in TP53 via overexpression of CHAC1 in infected gastric epithelial cells. FEBS Open Bio 8, 671–679 (2018).
Wei, J. et al. Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology 139, 1333–1343 (2010).
Buti, L. et al. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl Acad. Sci. USA 108, 9238–9243 (2011).
Meek, D. W. Tumour suppression by p53: a role for the DNA damage response? Nat. Rev. Cancer 9, 714–723 (2009).
Eischen, C. M. Genome stability requires p53. Cold Spring Harb. Perspect. Med. 6, a026096 (2016).
Coombs, N. et al. Helicobacter pylori affects the cellular deubiquitinase USP7 and ubiquitin-regulated components TRAF6 and the tumour suppressor p53. Int. J. Med. Microbiol. 301, 213–224 (2011).
Senchukova, M. A., Tomchuk, O. & Shurygina, E. I. Helicobacter pylori in gastric cancer: features of infection and their correlations with long-term results of treatment. World J. Gastroenterol. 27, 6290–6305 (2021).
Bussiere, F. I. et al. H. pylori-induced promoter hypermethylation downregulates USF1 and USF2 transcription factor gene expression. Cell. Microbiol. 12, 1124–1133 (2010).
Chi, T. F. et al. Loss of USF2 promotes proliferation, migration and mitophagy in a redox-dependent manner. Redox Biol. 37, 101750 (2020).
Baron, Y. et al. USF-1 is critical for maintaining genome integrity in response to UV-induced DNA photolesions. PLoS Genet. 8, e1002470 (2012).
Bouafia, A. et al. p53 requires the stress sensor USF1 to direct appropriate cell fate decision. PLoS Genet. 10, e1004309 (2014).
Costa, L. et al. USF1 defect drives p53 degradation during Helicobacter pylori infection and accelerates gastric carcinogenesis. Gut 69, 1582–1591 (2020).
Fu, J. et al. HGF/c-MET pathway in cancer: from molecular characterization to clinical evidence. Oncogene 40, 4625–4651 (2021).
Uchikawa, E., Chen, Z., Xiao, G.-Y., Zhang, X. & Bai, X.-C. Structural basis of the activation of c-MET receptor. Nat. Commun. 12, 4074 (2021).
Wang, H. et al. The function of the HGF/c-Met axis in hepatocellular carcinoma. Front. Cell. Dev. Biol. 8, 55 (2020).
Zhang, Y. et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 17, 45 (2018).
Suzuki, M. et al. Des-γ-carboxy prothrombin is a potential autologous growth factor for hepatocellular carcinoma. J. Biol. Chem. 280, 6409–6415 (2005).
Corso, S. & Giordano, S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov. 3, 978–992 (2013).
Ariyawutyakorn, W., Saichaemchan, S. & Varella-Garcia, M. Understanding and targeting MET signaling in solid tumors-are we there yet? J. Cancer 7, 633 (2016).
Faiella, A., Riccardi, F., Carteni, G., Chiurazzi, M. & Onofrio, L. The emerging role of c-Met in carcinogenesis and clinical implications as a possible therapeutic target. J. Oncol. 2022, 5179182 (2022).
Dai, L. et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. Blood 126, 2821–2831 (2015).
Toiyama, Y. et al. Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. Int. J. Cancer 130, 2912–2921 (2012).
Pereira, M. A. et al. RhoA, Claudin 18, and c-MET in gastric cancer: clinicopathological characteristics and prognostic significance in curative resected patients. Med. Sci. 10, 4 (2021).
Wang, C. et al. The prognostic value of HGF-c-MET signaling pathway in Gastric Cancer: a study based on TCGA and GEO databases. Int. J. Med. Sci. 17, 1946–1955 (2020).
Marano, L. et al. c-Met targeting in advanced gastric cancer: an open challenge. Cancer Lett. 365, 30–36 (2015).
Yu, S. et al. C-Met as a prognostic marker in gastric cancer: a systematic review and meta-analysis. PLoS ONE 8, e79137 (2013).
Ha, S. Y. et al. MET overexpression assessed by new interpretation method predicts gene amplification and poor survival in advanced gastric carcinomas. Mod. Pathol. 26, 1632–1641 (2013).
Lee, J. et al. Gastrointestinal malignancies harbor actionable MET exon 14 deletions. Oncotarget 6, 28211–28222 (2015).
Kim, H. S. et al. MET in gastric cancer with liver metastasis: the relationship between MET amplification and Met overexpression in primary stomach tumors and liver metastasis. J. Surg. Oncol. 117, 1679–1686 (2018).
Graziano, F. et al. Clinical impact of the HGF/MET pathway activation in patients with advanced gastric cancer treated with palliative chemotherapy. Pharmacogenomics J. 14, 418–423 (2014).
Ito, N., Tsujimoto, H., Ueno, H., Xie, Q. & Shinomiya, N. Helicobacter pylori-mediated immunity and signaling transduction in gastric cancer. J. Clin. Med. 9, 3699 (2020).
Che, Y. et al. Helicobacter pylori-induced exosomal MET educates tumour-associated macrophages to promote gastric cancer progression. J. Cell. Mol. Med. 22, 5708–5719 (2018).
Liu, L. et al. Helicobacter pylori infection enhances heparanase leading to cell proliferation via mitogenactivated protein kinase signalling in human gastric cancer cells. Mol. Med. Rep. 18, 5733–5741 (2018).
Hao, N.-B. et al. Hepatocyte growth factor (HGF) upregulates heparanase expression via the PI3K/Akt/NF-kappaB signaling pathway for gastric cancer metastasis. Cancer Lett. 361, 57–66 (2015).
Chen, B. et al. G6PD-NF-kappaB-HGF signal in gastric cancer-associated mesenchymal stem cells promotes the proliferation and metastasis of gastric cancer cells by upregulating the expression of HK2. Front. Oncol. 11, 648706 (2021).
Koh, S. A. & Lee, K. H. Function of hepatocyte growth factor in gastric cancer proliferation and invasion. Yeungnam Univ. J. Med. 37, 73–78 (2020).
Liu, J. et al. c-Met-dependent phosphorylation of RhoA plays a key role in gastric cancer tumorigenesis. J. Pathol. 249, 126–136 (2019).
Cheng, Y. et al. The chemokine receptor CXCR4 and c-MET cooperatively promote epithelial-mesenchymal transition in gastric cancer cells. Transl. Oncol. 11, 487–497 (2018).
Han, C. et al. MicroRNA-1 (miR-1) inhibits gastric cancer cell proliferation and migration by targeting MET. Tumour Biol. 36, 6715–6723 (2015).
Liu, J. et al. MicroRNA-144 inhibits the metastasis of gastric cancer by targeting MET expression. J. Exp. Clin. Cancer Res. 34, 35 (2015).
Wei, B., Huang, Q. Y., Huang, S. R., Mai, W. & Zhong, X. G. MicroRNA34a attenuates the proliferation, invasion and metastasis of gastric cancer cells via downregulation of MET. Mol. Med. Rep. 12, 5255–5261 (2015).
Zhang, L. et al. Activation of PAX3-MET pathways due to miR-206 loss promotes gastric cancer metastasis. Carcinogenesis 36, 390–399 (2015).
Liu, D. et al. Identification of HGF as a novel target of miR-15a/16/195 in gastric cancer. Invest. New Drugs 38, 922–933 (2020).
Chen, L. et al. IL10 secreted by cancerassociated macrophages regulates proliferation and invasion in gastric cancer cells via cMet/STAT3 signaling. Oncol. Rep. 42, 595–604 (2019).
Zhang, Z. et al. Functional genetic approach identifies MET, HER3, IGF1R, INSR pathways as determinants of lapatinib unresponsiveness in HER2-positive gastric cancer. Clin. Cancer Res. 20, 4559–4573 (2014).
Chen, C.-T. et al. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol. Cancer Ther. 11, 660–669 (2012).
Ebert, K., Mattes, J., Kunzke, T., Zwingenberger, G. & Luber, B. MET as resistance factor for afatinib therapy and motility driver in gastric cancer cells. PLoS ONE 14, e0223225 (2019).
Shen, B. et al. Crizotinib-resistant MET mutations in gastric cancer patients are sensitive to type II tyrosine kinase inhibitors. Future Oncol. 15, 2585–2593 (2019).
Huang, G. et al. CircRNA hsa_circRNA_104348 promotes hepatocellular carcinoma progression through modulating miR-187-3p/RTKN2 axis and activating Wnt/beta-catenin pathway. Cell Death Dis. 11, 1065 (2020).
Wu, C. et al. USP20 positively regulates tumorigenesis and chemoresistance through beta-catenin stabilization. Cell Death Differ. 25, 1855–1869 (2018).
Li, Y. et al. Gastrin-17 induces gastric cancer cell epithelial-mesenchymal transition via the Wnt/beta-catenin signaling pathway. J. Physiol. Biochem. 77, 93–104 (2021).
Xue, J. et al. Tumour suppressor TRIM33 targets nuclear beta-catenin degradation. Nat. Commun. 6, 6156 (2015).
Umaru, B. A. et al. Ligand bound fatty acid binding protein 7 (FABP7) drives melanoma cell proliferation via modulation of Wnt/beta-catenin signaling. Pharm. Res. 38, 479–490 (2021).
Zhang, L. et al. H19 knockdown suppresses proliferation and induces apoptosis by regulating miR-148b/WNT/beta-catenin in ox-LDL -stimulated vascular smooth muscle cells. J. Biomed. Sci. 25, 11 (2018).
Huang, L., Xiang, M., Ye, P., Zhou, W. & Chen, M. Beta-catenin promotes macrophage-mediated acute inflammatory response after myocardial infarction. Immunol. Cell Biol. 96, 100–113 (2018).
MacDonald, B. T. & He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect. Biol. 4, a007880 (2012).
Liang, T. et al. FAM46B inhibits cell proliferation and cell cycle progression in prostate cancer through ubiquitination of beta-catenin. Exp. Mol. Med. 50, 1–12 (2018).
Li, N. et al. miR-188 inhibits glioma cell proliferation and cell cycle progression through targeting beta-catenin. Oncol. Res. 26, 785–794 (2018).
Ren, Y., Guo, T., Xu, J., Liu, Y. & Huang, J. The novel target of esophageal squamous cell carcinoma: lncRNA GASL1 regulates cell migration, invasion and cell cycle stagnation by inactivating the Wnt3a/beta-catenin signaling. Pathol. Res. Pract. 217, 153289 (2021).
Hua, Y. et al. Oligomerization of Frizzled and LRP5/6 protein initiates intracellular signaling for the canonical WNT/beta-catenin pathway. J. Biol. Chem. 293, 19710–19724 (2018).
Yu, Z. et al. A novel UBE2T inhibitor suppresses Wnt/beta-catenin signaling hyperactivation and gastric cancer progression by blocking RACK1 ubiquitination. Oncogene 40, 1027–1042 (2021).
Yang, X.-Z. et al. LINC01133 as ceRNA inhibits gastric cancer progression by sponging miR-106a-3p to regulate APC expression and the Wnt/beta-catenin pathway. Mol. Cancer 17, 126 (2018).
Seidlitz, T. et al. Mouse models of human gastric cancer subtypes with stomach-specific CreERT2-mediated pathway alterations. Gastroenterology 157, 1599–1614 (2019). e1592.
Wu, Y., Hu, G., Wu, R. & Gong, N. High expression of miR-135b predicts malignant transformation and poor prognosis of gastric cancer. Life Sci. 257, 118133 (2020).
Mao, J. et al. Roles of Wnt/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 5, e1039 (2014).
Cheng, X.-X. et al. Correlation of Wnt-2 expression and beta-catenin intracellular accumulation in Chinese gastric cancers: relevance with tumour dissemination. Cancer Lett. 223, 339–347 (2005).
Kurayoshi, M. et al. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 66, 10439–10448 (2006).
Zhong, M. et al. Ubiquitin-specific protease 15 contributes to gastric cancer progression by regulating the Wnt/beta-catenin signaling pathway. World J. Gastroenterol. 27, 4221–4235 (2021).
Lian, G. et al. The screening and analysis of protein signatures and signaling associated with chemoresistance based on Protein Pathway Array technology in gastric cancer. Oncol. Rep. 39, 307–315 (2018).
Wang, X. et al. Caveolin-1 promotes chemoresistance of gastric cancer cells to cisplatin by activating WNT/beta-catenin pathway. Front. Oncol. 10, 46 (2020).
Wang, Y. et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. (2022).
Luo, Y. et al. Long noncoding RNA LINC01606 protects colon cancer cells from ferroptotic cell death and promotes stemness by SCD1-Wnt/beta-catenin-TFE3 feedback loop signalling. Clin. Transl. Med. 12, e752 (2022).
Yao, W. et al. Circular RNA circPVT1 inhibits 5-fluorouracil chemosensitivity by regulating ferroptosis through mir-30a-5p/FZD3 axis in esophageal cancer cells. Front. Oncol. 11, 780938 (2021).
Han, P. et al. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR-181a-5p-mediated regulation of Wnt/beta-catenin signaling. Mol. Cancer 16, 9 (2017).
Shang, W. et al. SETDB1 promotes gastric carcinogenesis and metastasis via upregulation of CCND1 and MMP9 expression. J. Pathol. 253, 148–159 (2021).
Takeuchi, Y. et al. Highly immunogenic cancer cells require activation of the WNT pathway for immunological escape. Sci. Immunol. 6, eabc6424 (2021).
Dholakia, J. et al. Sequential modulation of the Wnt/beta-catenin signaling pathway enhances tumor-intrinsic MHC I expression and tumor clearance. Gynecol. Oncol. 164, 170–180 (2022).
Li, X. et al. WNT/beta-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front. Immunol. 10, 2293 (2019).
Yang, D., Zhao, D. & Chen, X. MiR-133b inhibits proliferation and invasion of gastric cancer cells by up-regulating FBN1 expression. Cancer Biomark. 19, 425–436 (2017).
Li, J. et al. Disruption of Wnt/beta-catenin pathway elevates the sensitivity of gastric cancer cells to PD-1 antibody. Curr. Mol. Pharmacol. 15, 557–569 (2022).
Zhang, H. et al. Blocking Wnt/beta-catenin signal amplifies anti-PD-1 therapeutic efficacy by inhibiting tumor growth, migration, and promoting immune infiltration in glioblastomas. Mol. Cancer Ther. 20, 1305–1315 (2021).
Ji, L. et al. Blockade of beta-catenin-induced CCL28 suppresses gastric cancer progression via inhibition of Treg cell infiltration. Cancer Res. 80, 2004–2016 (2020).
Qu, Y. et al. The effects of TNF-alpha/TNFR2 in regulatory T cells on the microenvironment and progression of gastric cancer. Int. J. Cancer 150, 1373–1391 (2022).
Chen, J., Xie, Z.-R. & Wu, Y. Computational modeling of the interplay between cadherin-mediated cell adhesion and Wnt signaling pathway. PLoS ONE 9, e100702 (2014).
Czyzewska, J., Guzinska-Ustymowicz, K., Ustymowicz, M., Pryczynicz, A. & Kemona, A. The expression of E-cadherin-catenin complex in patients with advanced gastric cancer: role in formation of metastasis. Folia Histochem. Cytobiol. 48, 37–45 (2010).
Howard, S., Deroo, T., Fujita, Y. & Itasaki, N. A positive role of cadherin in Wnt/beta-catenin signalling during epithelial-mesenchymal transition. PLoS ONE 6, e23899 (2011).
Tian, S. et al. SERPINH1 regulates EMT and gastric cancer metastasis via the Wnt/beta-catenin signaling pathway. Aging 12, 3574–3593 (2020).
Zali, M. R. et al. Clinicopathological significance of E-cadherin, beta-catenin and p53 expression in gastric adenocarinoma. J. Res. Med. Sci. 14, 239–247 (2009).
Zhao, L. et al. JMJD2B promotes epithelial-mesenchymal transition by cooperating with beta-catenin and enhances gastric cancer metastasis. Clin. Cancer Res. 19, 6419–6429 (2013).
Neumann, M. & Naumann, M. Beyond IκBs: alternative regulation of NF‐KB activity. FASEB J. 21, 2642–2654 (2007).
Gilmore, T. D. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25, 6680–6684 (2006).
Rothwarf, D. M., Zandi, E., Natoli, G. & Karin, M. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 395, 297–300 (1998).
Brown, K., Gerstberger, S., Carlson, L., Franzoso, G. & Siebenlist, U. Control of IκB-α proteolysis by site-specific, signal-induced phosphorylation. Science 267, 1485–1488 (1995).
Brockman, J. A. et al. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol. Cell. Biol. 15, 2809–2818 (1995).
Kwon, H.-C. et al. Clinicopathologic significance of expression of nuclear factor-kappaB RelA and its target gene products in gastric cancer patients. World J. Gastroenterol. 18, 4744–4750 (2012).
Lee, B. L. et al. Nuclear factor-kappaB activation correlates with better prognosis and Akt activation in human gastric cancer. Clin. Cancer Res. 11, 2518–2525 (2005).
Yamashita, M. & Passegue, E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell 25, 357–372 (2019). e357.
Borghi, A., Verstrepen, L. & Beyaert, R. TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem. Pharmacol. 116, 1–10 (2016).
Luqman, S. & Pezzuto, J. M. NFkappaB: a promising target for natural products in cancer chemoprevention. Phytother. Res. 24, 949–963 (2010).
Soukhtanloo, M. et al. Natural products as promising targets in glioblastoma multiforme: a focus on NF-kappaB signaling pathway. Pharmacol. Rep. 72, 285–295 (2020).
Zhu, B. et al. Stem cell-derived exosomes prevent aging-induced cardiac dysfunction through a novel exosome/lncRNA MALAT1/NF-kappaB/TNF-alpha signaling pathway. Oxid. Med. Cell. Longev. 2019, 9739258 (2019).
Chao, X., Zao, J., Xiao-Yi, G., Li-Jun, M. & Tao, S. Blocking of PI3K/AKT induces apoptosis by its effect on NF-kappaB activity in gastric carcinoma cell line SGC7901. Biomed. Pharmacother. 64, 600–604 (2010).
Li, O. et al. UBAP2L promotes gastric cancer metastasis by activating NF-kappaB through PI3K/AKT pathway. Cell Death Discov. 8, 123 (2022).
Sokolova, O. & Naumann, M. NF-kappaB signaling in gastric cancer. Toxins 9, 119 (2017).
Zhang, J.-X. et al. LINC01410-miR-532-NCF2-NF-kB feedback loop promotes gastric cancer angiogenesis and metastasis. Oncogene 37, 2660–2675 (2018).
Stamp, D. H. Bile acids aided by acid suppression therapy may be associated with the development of esophageal cancers in westernized societies. Med. Hypotheses 66, 154–157 (2006).
Chang, M. S. et al. Cell-cycle regulators, bcl-2 and NF-kappaB in Epstein-Barr virus-positive gastric carcinomas. Int. J. Oncol. 27, 1265–1272 (2005).
Yang, Q., Tian, S., Liu, Z. & Dong, W. Knockdown of RIPK2 inhibits proliferation and migration, and induces apoptosis via the NF-kappaB signaling pathway in gastric cancer. Front. Genet. 12, 627464 (2021).
Fan, H., Zhang, S., Zhang, Y., Liang, W. & Cao, B. FERMT1 promotes gastric cancer progression by activating the NF-kappaB pathway and predicts poor prognosis. Cancer Biol. Ther. 21, 815–825 (2020).
Nam, S. Y. et al. A hypoxia-dependent upregulation of hypoxia-inducible factor-1 by nuclear factor-kappaB promotes gastric tumour growth and angiogenesis. Br. J. Cancer 104, 166–174 (2011).
Zhang, C., Tian, W., Meng, L., Qu, L. & Shou, C. PRL-3 promotes gastric cancer migration and invasion through a NF-kappaB-HIF-1alpha-miR-210 axis. J. Mol. Med. 94, 401–415 (2016).
Liu, L.-Y., Han, Y.-C., Wu, S.-H. & Lv, Z.-H. Expression of connective tissue growth factor in tumor tissues is an independent predictor of poor prognosis in patients with gastric cancer. World J. Gastroenterol. 14, 2110–2114 (2008).
Mao, Z. et al. Connective tissue growth factor enhances the migration of gastric cancer through downregulation of E-cadherin via the NF-kappaB pathway. Cancer Sci. 102, 104–110 (2011).
Song, Z.-B. et al. Connective tissue growth factor as an unfavorable prognostic marker promotes the proliferation, migration, and invasion of gliomas. Chin. Med. J. 133, 670–678 (2020).
Hochrainer, K. et al. The ubiquitin ligase HERC3 attenuates NF-kappaB-dependent transcription independently of its enzymatic activity by delivering the RelA subunit for degradation. Nucleic Acids Res. 43, 9889–9904 (2015).
Andela, V. B., Schwarz, E. M., Puzas, J. E., O’Keefe, R. J. & Rosier, R. N. Tumor metastasis and the reciprocal regulation of prometastatic and antimetastatic factors by nuclear factor kappaB. Cancer Res. 60, 6557–6562 (2000).
Basak, C. et al. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J. Biol. Chem. 280, 4279–4288 (2005).
Xu, X. et al. Upregulation of miRNA301a3p promotes tumor progression in gastric cancer by suppressing NKRF and activating NFkappaB signaling. Int. J. Oncol. 57, 522–532 (2020).
Park, B., Lim, J. W. & Kim, H. Lycopene treatment inhibits activation of Jak1/Stat3 and Wnt/beta-catenin signaling and attenuates hyperproliferation in gastric epithelial cells. Nutr. Res. 70, 70–81 (2019).
Song, X. et al. Mechanism underlying Polygonum capitatum effect on Helicobacter pylori-associated gastritis based on network pharmacology. Bioorg. Chem. 114, 105044 (2021).
Jang, J. et al. Sorbaria kirilowii ethanol extract exerts anti-inflammatory effects in vitro and in vivo by targeting Src/Nuclear Factor (NF)-kappaB. Biomolecules 10, 741 (2020).
Zhu, Y. et al. Research on the efficacy of Celastrus Orbiculatus in suppressing TGF-beta1-induced epithelial-mesenchymal transition by inhibiting HSP27 and TNF-alpha-induced NF-kappa B/Snail signaling pathway in human gastric adenocarcinoma. BMC Complement. Alter. Med. 14, 433 (2014).
Ji, A.-J., Liu, S.-L., Ju, W.-Z. & Huang, X.-E. Anti-proliferation effects and molecular mechanisms of action of tetramethypyrazine on human SGC-7901 gastric carcinoma cells. Asian Pac. J. Cancer Prev. 15, 3581–3586 (2014).
Morikawa, M., Derynck, R. & Miyazono, K. TGF-beta and the TGF-beta family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 8, a021873 (2016).
Hata, A. & Chen, Y.-G. TGF-beta signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061 (2016).
Bernard, K. et al. Glutaminolysis is required for transforming growth factor-beta1-induced myofibroblast differentiation and activation. J. Biol. Chem. 293, 1218–1228 (2018).
Goto, D. et al. Interaction between Smad anchor for receptor activation and Smad3 is not essential for TGF-beta/Smad3-mediated signaling. Biochem. Biophys. Res. Commun. 281, 1100–1105 (2001).
Klass, B. R., Grobbelaar, A. O. & Rolfe, K. J. Transforming growth factor beta1 signalling, wound healing and repair: a multifunctional cytokine with clinical implications for wound repair, a delicate balance. Postgrad. Med. J. 85, 9–14 (2009).
Glinka, Y. & Prud’homme, G. J. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 84, 302–310 (2008).
Yoo, J. et al. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 278, 43001–43007 (2003).
Yeh, Y.-Y. et al. TGF-beta1 increases motility and alphavbeta3 integrin up-regulation via PI3K, Akt and NF-kappaB-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 75, 1292–1301 (2008).
Javle, M. et al. Biomarkers of TGF-beta signaling pathway and prognosis of pancreatic cancer. PLoS ONE 9, e85942 (2014).
Ma, C. et al. Circular RNA hsa_circ_0004872 inhibits gastric cancer progression via the miR-224/Smad4/ADAR1 successive regulatory circuit. Mol. Cancer 19, 157 (2020).
Li, T. et al. TGF-beta1-SOX9 axis-inducible COL10A1 promotes invasion and metastasis in gastric cancer via epithelial-to-mesenchymal transition. Cell Death Dis. 9, 849 (2018).
Ye, J. et al. miR-4666-3p and miR-329 synergistically suppress the stemness of colorectal cancer cells via targeting TGF-beta/Smad pathway. Front. Oncol. 9, 1251 (2019).
Liu, J. et al. Epstein-Barr virus-encoded latent membrane protein 2A downregulates GCNT3 via the TGF-beta1/Smad-mTORC1 signaling axis. J. Virol. 95, e02481-20 (2021).
Saito, H. et al. Importance of human peritoneal mesothelial cells in the progression, fibrosis, and control of gastric cancer: inhibition of growth and fibrosis by tranilast. Gastric Cancer 21, 55–67 (2018).
Song, S. et al. Loss of TGF-beta adaptor beta2SP activates notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res. 73, 2159–2169 (2013).
Huang, D. et al. Long noncoding RNA SGO1-AS1 inactivates TGFbeta signaling by facilitating TGFB1/2 mRNA decay and inhibits gastric carcinoma metastasis. J. Exp. Clin. Cancer Res. 40, 342 (2021).
Willard, K. et al. Altered expression of proteoglycan, collagen and growth factor genes in a TGF-beta1 stimulated genetic risk model for musculoskeletal soft tissue injuries. J. Sci. Med. Sport 23, 695–700 (2020).
Yeh, Y.-C. et al. Transforming growth factor-β1 induces Smad3-dependent β1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am. J. Pathol. 177, 1743–1754 (2010).
Xie, X., Shirasu, T., Guo, L.-W. & Kent, K. C. Smad2 inhibition of MET transcription potentiates human vascular smooth muscle cell apoptosis. Atheroscler. Plus 44, 31–42 (2021).
Gu, H. et al. Effects and mechanisms of blocking the hedgehog signaling pathway in human gastric cancer cells. Oncol. Lett. 9, 1997–2002 (2015).
Ishimoto, T. et al. Activation of Transforming Growth Factor beta 1 signaling in gastric cancer-associated fibroblasts increases their motility, via expression of Rhomboid 5 Homolog 2, and ability to induce invasiveness of gastric cancer cells. Gastroenterology 153, 191–204 (2017). e116.
Xiao, Z. et al. TGFbeta2 is a prognostic-related biomarker and correlated with immune infiltrates in gastric cancer. J. Cell. Mol. Med. 24, 7151–7162 (2020).
Wang, G. et al. The stabilization of yes-associated protein by TGFbeta-activated kinase 1 regulates the self-renewal and oncogenesis of gastric cancer stem cells. J. Cell. Mol. Med. 25, 6584–6601 (2021).
Yang, Y. et al. Expression and function of transforming growth factor beta activated protein kinase 1 in gastric cancer. Mol. Med. Rep. 16, 3103–3110 (2017).
Morris, S. M. et al. Transforming growth factor-beta signaling promotes hepatocarcinogenesis induced by p53 loss. Hepatology 55, 121–131 (2012).
Celikel, C., Eren, F., Gulluoglu, B., Bekiroglu, N. & Turhal, S. Relation of neuroendocrine cells to transforming growth factor-alpha and epidermal growth factor receptor expression in gastric adenocarcinomas: prognostic implications. Pathol. Oncol. Res. 13, 215–226 (2007).
Guo, H. et al. TGF-beta1-induced EMT activation via both Smad-dependent and MAPK signaling pathways in Cu-induced pulmonary fibrosis. Toxicol. Appl. Pharmacol. 418, 115500 (2021).
Jin, G. et al. Arctigenin alleviates TGF-beta1-induced epithelial-mesenchymal transition and PAI-1 expression via AMPK/NF-kappaB pathway in peritoneal mesothelial cells. Biochem. Biophys. Res. Commun. 520, 413–419 (2019).
Wang, X., Pan, X. & Song, J. AMP-activated protein kinase is required for induction of apoptosis and epithelial-to-mesenchymal transition. Cell. Signal. 22, 1790–1797 (2010).
Alemohammad, H. et al. The importance of immune checkpoints in immune monitoring: A future paradigm shift in the treatment of cancer. Biomed. Pharmacother. 146, 112516 (2022).
Takaya, S., Saito, H. & Ikeguchi, M. Upregulation of immune checkpoint molecules, PD-1 and LAG-3, on CD4+ and CD8+ T cells after gastric cancer surgery. Yonago Acta Med. 58, 39–44 (2015).
Brunet, J.-F. et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature 328, 267–270 (1987).
Krummel, M. F. & Allison, J. P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 (1995).
Mimura, K. et al. Combined inhibition of PD-1/PD-L1, Lag-3, and Tim-3 axes augments antitumor immunity in gastric cancer-T cell coculture models. Gastric Cancer 24, 611–623 (2021).
Chocarro, L. et al. Understanding LAG-3 signaling. Int. J. Mol. Sci. 22, 5282 (2021).
Wang, Y., Zhao, E., Zhang, Z., Zhao, G. & Cao, H. Association between Tim3 and Gal9 expression and gastric cancer prognosis. Oncol. Rep. 40, 2115–2126 (2018).
Yu, J., Zhang, H., Sun, S., Sun, S. & Li, L. The effects of Tim-3 activation on T-cells in gastric cancer progression. Oncol. Lett. 17, 1461–1466 (2019).
He, Y. et al. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 11, 7005–7009 (2018).
Han, Y., Liu, D. & Li, L. PD-1/PD-L1 pathway: current researches in cancer. Am. J. Cancer Res. 10, 727–742 (2020).
Liu, D., Gao, S., Zhai, Y., Yang, X. & Zhai, G. Research progress of tumor targeted drug delivery based on PD-1/PD-L1. Int. J. Pharm. 616, 121527 (2022).
Vogel, I. et al. CD28/CTLA-4/B7 costimulatory pathway blockade affects regulatory T-cell function in autoimmunity. Eur. J. Immunol. 45, 1832–1841 (2015).
Fontenot, A. P. & Simonian, P. L. Murray and Nadel’s Textbook of Respiratory Medicine (eds V Courtney Broaddus et al.) 206-224.e203 (W.B. Saunders, 2016).
Wolchok, J. D. & Saenger, Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist 13, 2–9 (2008).
Carreno, B. M., Carter, L. L. & Collins, M. Therapeutic opportunities in the B7/CD28 family of ligands and receptors. Curr. Opin. Pharmacol. 5, 424–430 (2005).
Zang, K. et al. TIM-3 as a prognostic marker and a potential immunotherapy target in human malignant tumors: a meta-analysis and bioinformatics validation. Front Oncol. 11, 579351 (2021).
Li, J. et al. Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: evidence for intracellular cross talk. Oncoimmunology 5, e1200778 (2016).
Harjunpaa, H. & Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 200, 108–119 (2020).
He, W. et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res. 77, 6375–6388 (2017).
Liu, H. et al. Peritumoral TIGIT(+)CD20(+) B cell infiltration indicates poor prognosis but favorable adjuvant chemotherapeutic response in gastric cancer. Int. Immunopharmacol. 108, 108735 (2022).
Lozano, E., Dominguez-Villar, M., Kuchroo, V. & Hafler, D. A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 188, 3869–3875 (2012).
Wang, D. et al. Role of CD155/TIGIT in digestive cancers: promising cancer target for immunotherapy. Front. Oncol. 12, 844260 (2022).
Gullo, I. et al. The transcriptomic landscape of gastric cancer: insights into Epstein-Barr virus infected and microsatellite unstable tumors. Int. J. Mol. Sci. 19, 2079 (2018).
Kim, S. Y. et al. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology 148, 137–147 (2015). e139.
Derks, S. et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget 7, 32925–32932 (2016).
Hause, R. J., Pritchard, C. C., Shendure, J. & Salipante, S. J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 22, 1342–1350 (2016).
Llosa, N. J. et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpointsimmune checkpoints in human colorectal cancer. Cancer Discov. 5, 43–51 (2015).
Schwitalle, Y. et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 134, 988–997 (2008).
Wu, X. et al. Application of PD-1 blockade in cancer immunotherapy. Comput. Struct. Biotechnol. J. 17, 661–674 (2019).
Yun, T. et al. Significance of detection of the HER2 gene and PD-1/PD-L1 in gastric cancer. J. Oncol. 2020, 8678945 (2020).
Liu, X. et al. High PD-L1 expression in gastric cancer (GC) patients and correlation with molecular features. Pathol. Res Pract. 216, 152881 (2020).
Wu, Y.-Y. et al. Increased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin. Exp. Immunol. 161, 551–559 (2010).
Kang, B. W. & Chau, I. Current status and future potential of predictive biomarkers for immune checkpoint inhibitors in gastric cancer. ESMO Open 5, e000791 (2020).
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 144, 1941–1953 (2019).
Chen, L. et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 5, 5241 (2014).
Lina, T. T. et al. Helicobacter pylori cag pathogenicity island’s role in B7-H1 induction and immune evasion. PLoS ONE 10, e0121841 (2015).
Holokai, L. et al. Increased programmed death-ligand 1 is an early epithelial cell response to Helicobacter pylori infection. PLoS Pathog. 15, e1007468 (2019).
Xie, G. et al. Helicobacter Pylori promote B7-H1 expression by suppressing miR-152 and miR-200b in gastric cancer cells. PLoS ONE 12, e0168822 (2017).
Kang, S. Y. et al. Helicobacter pylori infection as an independent prognostic factor for locally advanced gastric cancer patients treated with adjuvant chemotherapy after curative resection. Int. J. Cancer 130, 948–958 (2012).
Lee, W. J. et al. Comparison between resectable gastric adenocarcinomas seropositive and seronegative for Helicobacter pylori. Br. J. Surg. 82, 802–805 (1995).
Meimarakis, G. et al. Helicobacter pylori as a prognostic indicator after curative resection of gastric carcinoma: a prospective study. Lancet Oncol. 7, 211–222 (2006).
Nishizuka, S. S. et al. Helicobacter pylori infection is associated with favorable outcome in advanced gastric cancer patients treated with S‐1 adjuvant chemotherapy. J. Surg. Oncol. 117, 947–956 (2018).
Postlewait, L. M. et al. Preoperative Helicobacter pylori infection is associated with increased survival after resection of gastric adenocarcinoma. Ann. Surg. Oncol. 23, 1225–1233 (2016).
Koizumi, Y. et al. Helicobacter pylori modulated host immunity in gastric cancer patients with S-1 adjuvant chemotherapy. J. Natl Cancer Inst. 114, 1149–1158 (2022).
Gu, L. et al. PD-L1 and gastric cancer prognosis: a systematic review and meta-analysis. PLoS ONE 12, e0182692 (2017).
Pietrantonio, F. et al. Predictive role of microsatellite instability for PD-1 blockade in patients with advanced gastric cancer: a meta-analysis of randomized clinical trials. ESMO Open 6, 100036 (2021).
Sasaki, S. et al. EBV-associated gastric cancer evades T-cell immunity by PD-1/PD-L1 interactions. Gastric Cancer 22, 486–496 (2019).
Katsurahara, K. et al. ANO9 regulates PD-L2 expression and binding ability to PD-1 in gastric cancer. Cancer Sci. 112, 1026–1037 (2021).
Wang, X. et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 38, 140 (2019).
Miliotis, C. & Slack, F. J. miR-105-5p regulates PD-L1 expression and tumor immunogenicity in gastric cancer. Cancer Lett. 518, 115–126 (2021).
Kim, W. et al. PD-1 signaling promotes tumor-infiltrating myeloid-derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology 160, 781–796 (2021).
Anjos, S. & Polychronakos, C. Mechanisms of genetic susceptibility to type I diabetes: beyond HLA. Mol. Genet. Metab. 81, 187–195 (2004).
Liu, J. et al. Lack of association between CTLA-4 genetic polymorphisms and noncardiac gastric cancer in a Chinese population. DNA Cell Biol. 38, 443–448 (2019).
Varnier, R. et al. Hyperprogressive disease after combined anti-PD-L1 and anti-CTLA-4 immunotherapy for MSI-H/dMMR gastric cancer: a case report. Front. Oncol. 11, 756365 (2021).
Shen, P. et al. Preferential Tim-3 expression on Treg and CD8(+) T cells, supported by tumor-associated macrophages, is associated with worse prognosis in gastric cancer. Am. J. Transl. Res. 8, 3419–3428 (2016).
Wang, Z. et al. Upregulation of T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) in monocytes/macrophages associates with gastric cancer progression. Immunol. Investig. 46, 134–148 (2017).
Long, B. et al. Clinical characteristics and prognostic significance of galectins for patients with gastric cancer: a meta-analysis. Int. J. Surg. 56, 242–249 (2018).
Jiang, J. et al. Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PLoS ONE 8, e81799 (2013).
Liu, X. et al. Tumor-infiltrating podoplanin(+) cells in gastric cancer: clinical outcomes and association with immune contexture. Oncoimmunology 9, 1845038 (2020).
Lu, X. et al. Tumor antigen-specific CD8(+) T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol. 313, 43–51 (2017).
Petersen, S. H., Kua, L. F., Nakajima, S., Yong, W. P. & Kono, K. Chemoradiation induces upregulation of immunogenic cell death-related molecules together with increased expression of PD-L1 and galectin-9 in gastric cancer. Sci. Rep. 11, 12264 (2021).
Ajani, J. A. et al. Galectin-3 expression is prognostic in diffuse type gastric adenocarcinoma, confers aggressive phenotype, and can be targeted by YAP1/BET inhibitors. Br. J. Cancer 118, 52–61 (2018).
Lecocq, Q., Keyaerts, M., Devoogdt, N. & Breckpot, K. The next-generation immune checkpoint LAG-3 and its therapeutic potential in oncology: third time’s a charm. Int. J. Mol. Sci. 22, 75 (2020).
Xu, D. et al. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology 225, 151915 (2020).
Matsumoto, K. et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer 106, 727–732 (2012).
Ma, J. Bioinformatics-guided analysis uncovers TIGIT as an epigenetically regulated immunomodulator affecting immunotherapeutic sensitivity of gastric cancer. Cancer Biomark. 33, 349–358 (2022).
Helsten, T., Schwaederle, M. & Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: biologic and clinical implications. Cancer Metastasis Rev. 34, 479–496 (2015).
Lengyel, C. G. et al. FGFR pathway inhibition in gastric cancer: the golden era of an old target? Life 12, 81 (2022).
Das, K. et al. Mutually exclusive FGFR2, HER2, and KRAS gene amplifications in gastric cancer revealed by multicolour FISH. Cancer Lett. 353, 167–175 (2014).
Sun, Y. et al. A comprehensive pan-cancer study of fibroblast growth factor receptor aberrations in Chinese cancer patients. Ann. Transl. Med. 8, 1290 (2020).
Zhang, J. et al. FGF18-FGFR2 signaling triggers the activation of c-Jun-YAP1 axis to promote carcinogenesis in a subgroup of gastric cancer patients and indicates translational potential. Oncogene 39, 6647–6663 (2020).
Zhang, J. et al. FGF18, a prominent player in FGF signaling, promotes gastric tumorigenesis through autocrine manner and is negatively regulated by miR-590-5p. Oncogene 38, 33–46 (2019).
Huang, T. et al. FGF7/FGFR2 signal promotes invasion and migration in human gastric cancer through upregulation of thrombospondin-1. Int. J. Oncol. 50, 1501–1512 (2017).
Siveen, K. S. et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim. Biophys. Acta 1845, 136–154 (2014).
Ashrafizadeh, M. et al. STAT3 Pathway in gastric cancer: signaling, therapeutic targeting and future prospects. Biology 9, 126 (2020).
Wang, J. et al. Feedback activation of STAT3 limits the response to PI3K/AKT/mTOR inhibitors in PTEN-deficient cancer cells. Oncogenesis 10, 8 (2021).
Pan, Y.-M. et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol. Cancer 15, 79 (2016).
Pandey, A. et al. Differentially localized survivin and STAT3 as markers of gastric cancer progression: Association with Helicobacter pylori. Cancer Rep. 1, e1004 (2018).
Wu, X. et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 8, 20741–20750 (2017).
Yang, H. & Xu, W. STAT3 promotes peritoneal metastasis of gastric cancer by enhancing mesothelial-mesenchymal transition. Biol. Chem. 402, 739–748 (2021).
Deng, P. et al. Circular RNA circVAPA promotes chemotherapy drug resistance in gastric cancer progression by regulating miR-125b-5p/STAT3 axis. World J. Gastroenterol. 27, 487–500 (2021).
Yuanyu, W. et al. MicroRNA-143 suppresses the proliferation and metastasis of human gastric cancer cells via modulation of STAT3 expression. Am. J. Transl. Res. 12, 867–874 (2020).
Yan, X.-L. et al. MicroRNA-375 reverses the expression of PD-L1 by inactivating the JAK2/STAT3 signaling pathways in gastric cancer. Clin. Res. Hepatol. Gastroenterol. 45, 101574 (2021).
Sun, B., Han, Y., Cai, H., Huang, H. & Xuan, Y. Long non-coding RNA SNHG3, induced by IL-6/STAT3 transactivation, promotes stem cell-like properties of gastric cancer cells by regulating the miR-3619-5p/ARL2 axis. Cell Oncol. 44, 179–192 (2021).
Wang, Y. et al. Circular RNAs in human cancer. Mol. Cancer 16, 1–8 (2017).
Huang, X. et al. Circular RNA AKT3 upregulates PIK3R1 to enhance cisplatin resistance in gastric cancer via miR-198 suppression. Mol. Cancer 18, 71 (2019).
Sun, G. et al. Circular RNA MCTP2 inhibits cisplatin resistance in gastric cancer by miR-99a-5p-mediated induction of MTMR3 expression. J. Exp. Clin. Cancer Res. 39, 246 (2020).
Akanji, M. A., Rotimi, D. & Adeyemi, O. S. Hypoxia-inducible factors as an alternative source of treatment strategy for cancer. Oxid. Med. Cell. Longev. 2019, 8547846 (2019).
Ke, Q. & Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 70, 1469–1480 (2006).
Li, H., Jia, Y. & Wang, Y. Targeting HIF-1alpha signaling pathway for gastric cancer treatment. Pharmazie 74, 3–7 (2019).
Chen, L. et al. HIF-1 alpha overexpression correlates with poor overall survival and disease-free survival in gastric cancer patients post-gastrectomy. PLoS ONE9, e90678 (2014).
Zhu, C.-l, Huang, Q., Liu, C.-h, Lin, X.-s & Xie, F. Prognostic value of HIF-1alpha expression in patients with gastric cancer. Mol. Biol. Rep. 40, 6055–6062 (2013).
Lin, Z. et al. Hypoxia-induced HIF-1alpha/lncRNA-PMAN inhibits ferroptosis by promoting the cytoplasmic translocation of ELAVL1 in peritoneal dissemination from gastric cancer. Redox Biol. 52, 102312 (2022).
Zhao, J. et al. The HIF-1A/miR-17-5p/PDCD4 axis contributes to the tumor growth and metastasis of gastric cancer. Signal Transduct. Target. Ther. 5, 46 (2020).
Zhao, Q. et al. HIF-1alpha induces multidrug resistance in gastric cancer cells by inducing miR-27a. PLoS ONE 10, e0132746 (2015).
Ge, X. et al. MicroRNA-421 regulated by HIF-1alpha promotes metastasis, inhibits apoptosis, and induces cisplatin resistance by targeting E-cadherin and caspase-3 in gastric cancer. Oncotarget 7, 24466–24482 (2016).
Zhang, X.-w, Bu, P., Liu, L., Zhang, X.-z & Li, J. Overexpression of long non-coding RNA PVT1 in gastric cancer cells promotes the development of multidrug resistance. Biochem. Biophys. Res. Commun. 462, 227–232 (2015).
Wang, R.-X., Ou, X.-W., Kang, M.-F. & Zhou, Z.-P. Association of HIF-1alpha and NDRG2 Expression with EMT in Gastric Cancer Tissues. Open Life Sci. 14, 217–223 (2019).
Ding, X. et al. CTHRC1 promotes gastric cancer metastasis via HIF-1alpha/CXCR4 signaling pathway. Biomed. Pharmacother. 123, 109742 (2020).
Guo, R. & Yang, B. Hypoxia-induced LXRalpha contributes to the migration and invasion of gastric cancer cells. Folia Biol. 67, 91–101 (2021).
Zhou, J. et al. Transcriptional up-regulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem. Biophys. Res. Commun. 415, 348–354 (2011).
Mu, G. et al. Calmodulin 2 facilitates angiogenesis and metastasis of gastric cancer via STAT3/HIF-1A/VEGF-A mediated macrophage polarization. Front. Oncol. 11, 727306 (2021).
Tang, E., Wang, Y., Liu, T. & Yan, B. Gastrin promotes angiogenesis by activating HIF-1alpha/beta-catenin/VEGF signaling in gastric cancer. Gene 704, 42–48 (2019).
Berman, D. M. et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425, 846–851 (2003).
Xu, Y., Song, S., Wang, Z. & Ajani, J. A. The role of hedgehog signaling in gastric cancer: molecular mechanisms, clinical potential, and perspective. Cell Commun. Signal. 17, 157 (2019).
Merchant, J. L. & Ding, L. Hedgehog signaling links chronic inflammation to gastric cancer precursor lesions. Cell. Mol. Gastroenterol. Hepatol. 3, 201–210 (2017).
Koh, V. et al. Hedgehog transcriptional effector GLI mediates mTOR-Induced PD-L1 expression in gastric cancer organoids. Cancer Lett. 518, 59–71 (2021).
Abdel-Rahman, O. Hedgehog pathway aberrations and gastric cancer; evaluation of prognostic impact and exploration of therapeutic potentials. Tumour Biol. 36, 1367–1374 (2015).
Doheny, D., Manore, S. G., Wong, G. L. & Lo, H.-W. Hedgehog signaling and truncated GLI1 in cancer. Cells 9, 2114 (2020).
Lu, Y. et al. MiR-144-3p inhibits gastric cancer progression and stemness via directly targeting GLI2 involved in hedgehog pathway. J. Transl. Med. 19, 432 (2021).
Saze, Z. et al. Activation of the sonic hedgehog pathway and its prognostic impact in patients with gastric cancer. Dig. Surg. 29, 115–123 (2012).
Kim, J.-Y. et al. Prognostic value of sonic hedgehog protein expression in gastric cancer. Jpn J. Clin. Oncol. 42, 1054–1059 (2012).
Samadani, A. A. & Akhavan-Niaki, H. Interaction of sonic hedgehog (SHH) pathway with cancer stem cell genes in gastric cancer. Med. Oncol. 32, 48 (2015).
Hibdon, E. S. et al. Notch and mTOR signaling pathways promote human gastric cancer cell proliferation. Neoplasia 21, 702–712 (2019).
Cui, Y. et al. NOTCH3 is a prognostic factor and is correlated with immune tolerance in gastric cancer. Front. Oncol. 10, 574937 (2020).
Ma, J. et al. LncRNA FENDRR inhibits gastric cancer cell proliferation and invasion via the miR-421/SIRT3/Notch-1 axis. Cancer Manag. Res. 13, 9175–9187 (2021).
Xue, D., Li, D., Dou, C. & Li, J. A comprehensive bioinformatic analysis of NOTCH pathway involvement in stomach adenocarcinoma. Dis. Markers 2021, 4739868 (2021).
Sun, Y. et al. Differential Notch1 and Notch2 expression and frequent activation of Notch signaling in gastric cancers. Arch. Pathol. Lab Med. 135, 451–458 (2011).
Guo, L.-Y. et al. Notch2 regulates matrix metallopeptidase 9 via PI3K/AKT signaling in human gastric carcinoma cell MKN-45. World J. Gastroenterol. 18, 7262–7270 (2012).
Hu, J. et al. Notch1/2/3/4 are prognostic biomarker and correlated with immune infiltrates in gastric cancer. Aging 12, 2595–2609 (2020).
Yao, F. et al. Recurrent fusion genes in gastric cancer: CLDN18-ARHGAP26 induces loss of epithelial integrity. Cell Rep. 12, 272–285 (2015).
Gunzel, D. & Yu, A. S. L. Claudins and the modulation of tight junction permeability. Physiol. Rev. 93, 525–569 (2013).
Cao, W. et al. Claudin18. 2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark. Res. 10, 1–21 (2022).
Ungureanu, B. S. et al. Clinicopathologic relevance of Claudin 18.2 expression in gastric cancer: a meta-analysis. Front. Oncol. 11, 643872 (2021).
Moon, S. Y. & Zheng, Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 13, 13–22 (2003).
Ushiku, T. et al. RHOA mutation in diffuse-type gastric cancer: a comparative clinicopathology analysis of 87 cases. Gastric Cancer 19, 403–411 (2016).
Zhang, H. et al. Gain-of-function RHOA mutations promote focal adhesion kinase activation and dependency in diffuse gastric cancer. Cancer Discov. 10, 288–305 (2020).
Lenárt, S. et al. Trop2: Jack of all trades, master of none. Cancers 12, 3328 (2020).
Lipinski, M., Parks, D. R., Rouse, R. V. & Herzenberg, L. A. Human trophoblast cell-surface antigens defined by monoclonal antibodies. Proc. Natl Acad. Sci. USA 78, 5147–5150 (1981).
Shaffer, C. Trop2 deal heats up antibody-drug conjugate space in cancer. Nat. Biotechnol. 39, 128–130 (2021).
Cubas, R., Zhang, S., Li, M., Chen, C. & Yao, Q. Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol. Cancer 9, 253 (2010).
Zhao, W. et al. Trop2 is overexpressed in gastric cancer and predicts poor prognosis. Oncotarget 7, 6136–6145 (2016).
Kushiyama, S. et al. Clinicopathologic significance of TROP2 and phospho-TROP2 in gastric cancer. Mol. Clin. Oncol. 14, 105 (2021).
Zhao, W. et al. The role and molecular mechanism of Trop2 induced epithelial-mesenchymal transition through mediated beta-catenin in gastric cancer. Cancer Med. 8, 1135–1147 (2019).
García-Cano, J. et al. p38MAPK and chemotherapy: we always need to hear both sides of the story. Front. Cell Dev. Biol. 4, 69 (2016).
Zou, X. & Blank, M. Targeting p38 MAP kinase signaling in cancer through post-translational modifications. Cancer Lett. 384, 19–26 (2017).
Gurtner, A. et al. Mutant p53-induced up-regulation of mitogen-activated protein kinase kinase 3 contributes to gain of function. J. Biol. Chem. 285, 14160–14169 (2010).
Baldari, S., Ubertini, V., Garufi, A., D’orazi, G. & Bossi, G. Targeting MKK3 as a novel anticancer strategy: molecular mechanisms and therapeutical implications. Cell Death Dis. 6, e1621–e1621 (2015).
Wang, J. et al. Distinct genomic landscapes of gastroesophageal adenocarcinoma depending on PD-L1 expression identify mutations in RAS–MAPK pathway and TP53 as potential predictors of immunotherapy efficacy. Ann. Oncol. 32, 906–916 (2021).
Seto, M. et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol. Carcinog. 48, 703–712 (2009).
Pietrobono, S., Gagliardi, S. & Stecca, B. Non-canonical hedgehog signaling pathway in cancer: activation of GLI transcription factors beyond smoothened. Front. Genet. 10, 556 (2019).
Chakrabarti, J. et al. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 9, 37439–37457 (2018).
Zhao, R. et al. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 52, e12571 (2019).
Wu, L. et al. PD-1/PD-L1 enhanced cisplatin resistance in gastric cancer through PI3K/AKT mediated P-gp expression. Int. Immunopharmacol. 94, 107443 (2021).
Siegel, P. M., Shu, W., Cardiff, R. D., Muller, W. J. & Massagué, J. Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl Acad. Sci. USA 100, 8430–8435 (2003).
Seton-Rogers, S. E. et al. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl Acad. Sci. USA 101, 1257–1262 (2004).
Lehmann, K. et al. Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev. 14, 2610–2622 (2000).
Yue, J. & Mulder, K. M. Requirement of Ras/MAPK pathway activation by transforming growth factor β for transforming growth factor β1production in a Smad-dependent pathway. J. Biol. Chem. 275, 30765–30773 (2000).
Lei, S., Dubeykovskiy, A., Chakladar, A., Wojtukiewicz, L. & Wang, T. C. The murine gastrin promoter is synergistically activated by transforming growth factor-β/Smad and Wnt signaling pathways. J. Biol. Chem. 279, 42492–42502 (2004).
Yanai, K. et al. Crosstalk of hedgehog and Wnt pathways in gastric cancer. Cancer Lett. 263, 145–156 (2008).
Ding, X. et al. HGF-mediated crosstalk between cancer-associated fibroblasts and MET-unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 9, 867 (2018).
Gao, S. et al. Crosstalk of mTOR/PKM2 and STAT3/c‐Myc signaling pathways regulate the energy metabolism and acidic microenvironment of gastric cancer. J. Cell. Biochem. 120, 1193–1202 (2019).
Dan, L., Jian, D., Na, L. & Xiaozhong, W. Crosstalk between EGFR and integrin affects invasion and proliferation of gastric cancer cell line, SGC7901. Onco Targets Ther. 5, 271 (2012).
Kazmi, H. R., Kumari, S., Tiwari, S., Khanna, A. & Narayan, G. Epigenetic mechanisms and events in gastric cancer-emerging novel biomarkers. Pathol. Oncol. Res. 24, 757–770 (2018).
Luo, C., Hajkova, P. & Ecker, J. R. Dynamic DNA methylation: in the right place at the right time. Science 361, 1336–1340 (2018).
Michigami, Y. et al. Long-term effects of H. pylori eradication on epigenetic alterations related to gastric carcinogenesis. Sci. Rep. 8, 14369 (2018).
Canale, M. et al. Epigenetic mechanisms in gastric cancer: potential new therapeutic opportunities. Int. J. Mol. Sci. 21, 5500 (2020).
Ebrahimi, V. et al. Epigenetic modifications in gastric cancer: focus on DNA methylation. Gene 742, 144577 (2020).
Oliveira, C. et al. Quantification of epigenetic and genetic 2nd hits in CDH1 during hereditary diffuse gastric cancer syndrome progression. Gastroenterology 136, 2137–2148 (2009).
Yu, J. et al. Promoter methylation of the Wnt/β‐catenin signaling antagonist Dkk‐3 is associated with poor survival in gastric cancer. Cancer 115, 49–60 (2009).
Du, W. et al. ADAMTS9 is a functional tumor suppressor through inhibiting AKT/mTOR pathway and associated with poor survival in gastric cancer. Oncogene 32, 3319–3328 (2013).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Sudo, T. et al. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 26, 777–782 (2011).
Xia, G. et al. Helicobacter pylori regulates p21WAF1 by histone H4 acetylation. Biochem. Biophys. Res. Commun. 369, 526–531 (2008).
Park, Y. S. et al. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann. Surg. Oncol. 15, 1968–1976 (2008).
Song, Y. et al. The Wnt/beta-catenin and PI3K/Akt signaling pathways promote EMT in gastric cancer by epigenetic regulation via H3 lysine 27 acetylation. Tumour Biol. 39, 1010428317712617 (2017).
Liu, X. et al. AURKA induces EMT by regulating histone modification through Wnt/beta-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget 7, 33152–33164 (2016).
Bilgic, F. et al. Potential role of chromatin remodeling factor genes in atrophic gastritis/gastric cancer risk. Turk. J. Gastroenterol. 29, 427–435 (2018).
Wiegand, K. C. et al. Loss of BAF250a (ARID1A) is frequent in high‐grade endometrial carcinomas. J. Pathol. 224, 328–333 (2011).
Zang, Z. J. et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 44, 570–574 (2012).
Zhang, Q. et al. Chromatin remodeling gene AT-rich interactive domain-containing protein 1A suppresses gastric cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget 7, 46127–46141 (2016).
Cheng, J. et al. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin. Chim. Acta 412, 1621–1625 (2011).
Cheng, J. et al. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 315, 12–17 (2012).
Ameli Mojarad, M., Ameli Mojarad, M., Shojaee, B. & Nazemalhosseini-Mojarad, E. piRNA: A promising biomarker in early detection of gastrointestinal cancer. Pathol. Res. Pract. 230, 153757 (2022).
Raei, N. et al. Crosstalk between lncRNAs and miRNAs in gastrointestinal cancer drug resistance. Life Sci. 284, 119933 (2021).
Bure, I. V. & Nemtsova, M. V. Methylation and noncoding RNAs in gastric cancer: everything is connected. Int. J. Mol. Sci. 22, 5683 (2021).
Davis, J. L. & Ripley, R. T. Postgastrectomy syndromes and nutritional considerations following gastric surgery. Surg. Clin. North Am. 97, 277–293 (2017).
Hiki, N., Nunobe, S., Kubota, T. & Jiang, X. Function-preserving gastrectomy for early gastric cancer. Ann. Surg. Oncol. 20, 2683–2692 (2013).
Cunningham, D. et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N. Engl. J. Med. 355, 11–20 (2006).
Sasako, M. et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J. Clin. Oncol. 29, 4387–4393 (2011).
Park, S. H. et al. A randomized phase III trial comparing adjuvant single-agent S1, S-1 with oxaliplatin, and postoperative chemoradiation with S-1 and oxaliplatin in patients with node-positive gastric cancer after D2 resection: the ARTIST 2 trial☆. Ann. Oncol. 32, 368–374 (2021).
Kang, Y.-K. et al. PRODIGY: a phase III study of neoadjuvant docetaxel, oxaliplatin, and S-1 plus surgery and adjuvant S-1 versus surgery and adjuvant S-1 for resectable advanced gastric cancer. J. Clin. Oncol. 39, 2903–2913 (2021).
Yoshida, K. et al. Addition of docetaxel to oral fluoropyrimidine improves efficacy in patients with stage III gastric cancer: interim analysis of JACCRO GC-07, a randomized controlled trial. J. Clin. Oncol. 37, 1296–1304 (2019).
Soetikno, R., Kaltenbach, T., Yeh, R. & Gotoda, T. Endoscopic mucosal resection for early cancers of the upper gastrointestinal tract. J. Clin. Oncol. 23, 4490–4498 (2005).
Hasuike, N. et al. A non-randomized confirmatory trial of an expanded indication for endoscopic submucosal dissection for intestinal-type gastric cancer (cT1a): the Japan Clinical Oncology Group study (JCOG0607). Gastric Cancer 21, 114–123 (2018).
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. T. & Lordick, F. Gastric cancer. Lancet 396, 635–648 (2020).
Japanese, G. C. A. Japanese gastric cancer treatment guidelines 2018 (5th edition). Gastric Cancer 24, 1–21 (2021).
Wu, C.-W. et al. Nodal dissection for patients with gastric cancer: a randomised controlled trial. Lancet Oncol. 7, 309–315 (2006).
Biondi, A. et al. Does a minimum number of 16 retrieved nodes affect survival in curatively resected gastric cancer? Eur. J. Surg. Oncol. 41, 779–786 (2015).
Wang, F.-H. et al. The Chinese Society of Clinical Oncology (CSCO): clinical guidelines for the diagnosis and treatment of gastric cancer, 2021. Cancer Commun. 41, 747–795 (2021).
Katai, H. et al. Survival outcomes after laparoscopy-assisted distal gastrectomy versus open distal gastrectomy with nodal dissection for clinical stage IA or IB gastric cancer (JCOG0912): a multicentre, non-inferiority, phase 3 randomised controlled trial. Lancet Gastroenterol. Hepatol. 5, 142–151 (2020).
Kim, H.-H. et al. Effect of laparoscopic distal gastrectomy vs open distal gastrectomy on long-term survival among patients with stage I gastric cancer: the KLASS-01 randomized clinical trial. JAMA Oncol. 5, 506–513 (2019).
Cats, A. et al. Chemotherapy versus chemoradiotherapy after surgery and preoperative chemotherapy for resectable gastric cancer (CRITICS): an international, open-label, randomised phase 3 trial. Lancet Oncol. 19, 616–628 (2018).
Yu, J. et al. Effect of laparoscopic vs open distal gastrectomy on 3-year disease-free survival in patients with locally advanced gastric cancer: the CLASS-01 randomized clinical trial. JAMA 321, 1983–1992 (2019).
Jin Hyung, W. et al. Long-term outcomes of laparoscopic distal gastrectomy for locally advanced gastric cancer: the KLASS-02-RCT randomized clinical trial. J. Clin. Oncol. 38, 3304–3313 (2020).
Wang, X. et al. The protocol of a prospective, multicenter, randomized, controlled phase III study evaluating different cycles of oxaliplatin combined with S-1 (SOX) as neoadjuvant chemotherapy for patients with locally advanced gastric cancer: RESONANCE-II trial. BMC Cancer 21, 20 (2021).
Sumpter, K. et al. Report of two protocol planned interim analyses in a randomised multicentre phase III study comparing capecitabine with fluorouracil and oxaliplatin with cisplatin in patients with advanced oesophagogastric cancer receiving ECF. Br. J. Cancer 92, 1976–1983 (2005).
Li, Z.-Y. et al. Neoadjuvant chemotherapy with FOLFOX: improved outcomes in Chinese patients with locally advanced gastric cancer. J. Surg. Oncol. 105, 793–799 (2012).
Al-Batran, S.-E. et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet 393, 1948–1957 (2019).
van Hagen, P. et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N. Engl. J. Med. 366, 2074–2084 (2012).
Song, Z., Wu, Y., Yang, J., Yang, D. & Fang, X. Progress in the treatment of advanced gastric cancer. Tumour Biol. 39, 1010428317714626 (2017).
Li, R. et al. Chemoradiation improves survival compared with chemotherapy alone in unresected nonmetastatic gastric cancer. J. Natl Compr. Canc. Netw. 16, 950–958 (2018).
Tepper, J. et al. Phase III trial of trimodality therapy with cisplatin, fluorouracil, radiotherapy, and surgery compared with surgery alone for esophageal cancer: CALGB 9781. J. Clin. Oncol. 26, 1086–1092 (2008).
Ajani, J. A. et al. Paclitaxel-based chemoradiotherapy in localized gastric carcinoma: degree of pathologic response and not clinical parameters dictated patient outcome. J. Clin. Oncol. 23, 1237–1244 (2005).
Liu, Y. et al. Multicenter phase 2 study of peri-irradiation chemotherapy plus intensity modulated radiation therapy with concurrent weekly docetaxel for inoperable or medically unresectable nonmetastatic gastric cancer. Int. J. Radiat. Oncol. Biol. Phys. 98, 1096–1105 (2017).
Kim, M. M. et al. Clinical benefit of palliative radiation therapy in advanced gastric cancer. Acta Oncol. 47, 421–427 (2008).
Wagner, A. D. et al. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J. Clin. Oncol. 24, 2903–2909 (2006).
Al-Batran, S.-E. et al. Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: a study of the Arbeitsgemeinschaft Internistische Onkologie. J. Clin. Oncol. 26, 1435–1442 (2008).
Glimelius, B. et al. Randomized comparison between chemotherapy plus best supportive care with best supportive care in advanced gastric cancer. Ann. Oncol. 8, 163–168 (1997).
Fuchs, C. S. et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 383, 31–39 (2014).
Wilke, H. et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 15, 1224–1235 (2014).
Kang, Y.-K. et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 2461–2471 (2017).
Lu, Z. et al. Early interdisciplinary supportive care in patients with previously untreated metastatic esophagogastric cancer: a phase III randomized controlled trial. J. Clin. Oncol. 39, 748–756 (2021).
Romond, E. H. et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N. Engl. J. Med. 353, 1673–1684 (2005).
Shitara, K. et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 382, 2419–2430 (2020).
Shi, F. et al. Disitamab vedotin: a novel antibody-drug conjugates for cancer therapy. Drug Deliv. 29, 1335–1344 (2022).
Peng, Z. et al. Efficacy and safety of a novel anti-HER2 therapeutic antibody RC48 in patients with HER2-overexpressing, locally advanced or metastatic gastric or gastroesophageal junction cancer: a single-arm phase II study. Cancer Commun. 41, 1173–1182 (2021).
Golan, T. et al. Overall Survival From The Phase 3 Polo Trial: Maintenance Olaparib For Germline Brca-mutated Metastatic Pancreatic Cancer. Report No. 0732-183X, (American Society of Clinical Oncology, 2021).
Medina, P. J. & Goodin, S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin. Ther. 30, 1426–1447 (2008).
Hecht, J. R. et al. Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: TRIO-013/LOGiC—a randomized phase III trial. J. Clin. Oncol. 34, 443–451 (2016).
Smyth, E. C. et al. Safety and efficacy of the addition of lapatinib to perioperative chemotherapy for resectable HER2-positive gastroesophageal adenocarcinoma: a randomized phase 2 clinical trial. JAMA Oncol. 5, 1181–1187 (2019).
Lordick, F. et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol. 14, 490–499 (2013).
Waddell, T. et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol. 14, 481–489 (2013).
Maron, S. B. et al. Targeted therapies for targeted populations: anti-EGFR treatment for EGFR-amplified gastroesophageal adenocarcinoma. Cancer Discov. 8, 696–713 (2018).
Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 (2011).
Ohtsu, A. et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J. Clin. Oncol. 29, 3968–3976 (2011).
Scott, L. J. Apatinib: a review in advanced gastric cancer and other advanced cancers. Drugs 78, 747–758 (2018).
Li, J. et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J. Clin. Oncol. 34, 1448–1454 (2016).
Fukuoka, S. et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open-label, dose-escalation, and dose-expansion phase Ib trial (REGONIVO, EPOC1603). J. Clin. Oncol. 38, 2053–2061 (2020).
Kawazoe, A. et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): an open-label, single-arm, phase 2 trial. Lancet Oncol. 21, 1057–1065 (2020).
Iveson, T. et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 15, 1007–1018 (2014).
Catenacci, D. V. T. et al. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 1467–1482 (2017).
Markham, A. Savolitinib: first approval. Drugs 81, 1665–1670 (2021).
Lee, J. et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discov. 9, 1388–1405 (2019).
Van Cutsem, E. et al. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann. Oncol. 28, 1316–1324 (2017).
Wainberg, Z. A. et al. Randomized double-blind placebo-controlled phase 2 study of bemarituzumab combined with modified FOLFOX6 (mFOLFOX6) in first-line (1L) treatment of advanced gastric/gastroesophageal junction adenocarcinoma (FIGHT). J. Clin. Oncol. 39, 160–160 (2021).
Sahin, U. et al. FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann. Oncol. 32, 609–619 (2021).
Qi, C. et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med. 28, 1189–1198 (2022).
Bravaccini, S. & Maltoni, R. Trop-2 therapy in metastatic triple-negative breast cancer in italy: clinical opportunity and regulatory pitfalls. J. Pers. Med. 11, 1211 (2021).
Bardia, A. et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann. Oncol. 32, 746–756 (2021).
Nakayama, A. et al. Viral loads correlate with upregulation of PD-L1 and worse patient prognosis in Epstein-Barr Virus-associated gastric carcinoma. PLoS ONE 14, e0211358 (2019).
Chen, L.-T. et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (ATTRACTION-2): 2-year update data. Gastric Cancer 23, 510–519 (2020).
Shitara, K. et al. Efficacy and safety of pembrolizumab or pembrolizumab plus chemotherapy vs chemotherapy alone for patients with first-line, advanced gastric cancer: the KEYNOTE-062 phase 3 randomized clinical trial. JAMA Oncol. 6, 1571–1580 (2020).
Janjigian, Y. Y. et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 398, 27–40 (2021).
Janjigian, Y. Y. et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 600, 727–730 (2021).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Bang, Y.-J. et al. Efficacy of sequential ipilimumab monotherapy versus best supportive care for unresectable locally advanced/metastatic gastric or gastroesophageal junction cancer. Clin. Cancer Res. 23, 5671–5678 (2017).
Akeso. NMPA (China) approves cadonilimab injection for the treatment of relapsed or metastatic cervical cancer. <https://www.medthority.com/news/2022/7/nmpa-china-approves-cadonilimab-injection-for-the-treatment-of-relapsed-or-metastatic-cervical-cancer.-akeso-inc/> (2022).
Ji, J. et al. A phase Ib/II, multicenter, open-label study of AK104, a PD-1/CTLA-4 bispecific antibody, combined with chemotherapy (chemo) as first-line therapy for advanced gastric (G) or gastroesophageal junction (GEJ) cancer. J. Clin. Oncol. 40, 308–308 (2022).
Tawbi, H. A. et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 386, 24–34 (2022).
Zhang, Q. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 19, 723–732 (2018).
Cho, B. C. et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 23, 781–792 (2022).
Sterner, R. C. & Sterner, R. M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11, 69 (2021).
Dawson, J. C., Serrels, A., Stupack, D. G., Schlaepfer, D. D. & Frame, M. C. Targeting FAK in anticancer combination therapies. Nat. Rev. Cancer 21, 313–324 (2021).
Wu, L. et al. A phase Ib study of IN10018 in combination with pegylated liposomal doxorubicin (PLD) in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 40, 5567–5567 (2022).
Cocco, E., Scaltriti, M. & Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 15, 731–747 (2018).
Marcus, L. et al. FDA approval summary: entrectinib for the treatment of NTRK gene fusion solid tumors. Clin. Cancer Res. 27, 928–932 (2021).
Shinozaki-Ushiku, A. et al. The first case of gastric carcinoma with NTRK rearrangement: identification of a novel ATP1B-NTRK1 fusion. Gastric Cancer 23, 944–947 (2020).
Zhu, G. et al. Expression and role of Dickkopf-1 (Dkk1) in tumors: from the cells to the patients. Cancer Manag. Res. 13, 659–675 (2021).
Leap therapeutics announces FDA fast track designation granted to DKN-01 for the treatment of gastric and gastroesophageal junction cancer. <https://investors.leaptx.com/news-releases/news-release-details/leap-therapeutics-announces-fda-fast-track-designation-granted> (2020).
Chao, J. et al. Trial in progress: a phase I study of AMG 199, a half-life extended bispecific T-cell engager (HLE BiTE) immune therapy, targeting MUC17 in patients with gastric and gastroesophageal junction (G/GEJ) cancer. J. Clin. Oncol. 38, TPS4649–TPS4649 (2020).
Schneider, B. J. et al. Phase I study of epigenetic priming with azacitidine prior to standard neoadjuvant chemotherapy for patients with resectable gastric and esophageal adenocarcinoma: evidence of tumor hypomethylation as an indicator of major histopathologic response. Clin. Cancer Res. 23, 2673–2680 (2017).
Yoo, C. et al. Vorinostat in combination with capecitabine plus cisplatin as a first-line chemotherapy for patients with metastatic or unresectable gastric cancer: phase II study and biomarker analysis. Br. J. Cancer 114, 1185–1190 (2016).
Sanoff, H. K. Improving treatment approaches for rectal cancer. N. Engl. J. Med. 386, 2425–2426 (2022).
Park, J. J. H., Hsu, G., Siden, E. G., Thorlund, K. & Mills, E. J. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J. Clin. 70, 125–137 (2020).
Vlachogiannis, G. et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018).
Ganesh, K. et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nat. Med. 25, 1607–1614 (2019).
Ooft, S. N. et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 11, eaay2574 (2019).
Xiao, X. et al. The anti-tumor effect of Nab-paclitaxel proven by patient-derived organoids. Onco Targets Ther. 13, 6017–6025 (2020).
Zhang, S.-W. et al. An efficient and user-friendly method for cytohistological analysis of organoids. J. Tissue Eng. Regen. Med. 15, 1012–1022 (2021).
Liu, G. et al. Organoids from mucinous appendiceal adenocarcinomas as high-fidelity models for individual therapy. Front. Med. 9, 829033 (2022).
Daryabari, S. S. et al. Overexpression of CFL1 in gastric cancer and the effects of its silencing by siRNA with a nanoparticle delivery system in the gastric cancer cell line. J. Cell Physiol. 235, 6660–6672 (2020).
Liu, J. et al. Long noncoding RNA PANDAR blocks CDKN1A gene transcription by competitive interaction with p53 protein in gastric cancer. Cell Death Dis. 9, 168 (2018).
Xiao, Y. et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 13, 758 (2022).
Huang, R. J. et al. An approach to the primary and secondary prevention of gastric cancer in the United States. Clin. Gastroenterol. Hepatol. 20, 2218–2228.e2 (2022).
Syahniar, R. & Kharisma, D. S. Vaccine Development (ed Desheva Y.) (IntechOpen, 2021).
Acknowledgements
This work is funded by the National Natural Science Foundation of China #U20A20379, the National Key Research and Development Program of China #2018YFA0902801, the 100 Top Talents Program of Sun Yat-Sen University (ZSQYBRJH0001), Guangdong Basic and Applied Basic Research Foundation #2021A1515010117, and Guangdong Provincial Key Laboratory of Digestive Cancer Research #2021B1212040006. Z.N.L., Q.X.T. and Y.X. appreciate the scholarship from the Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John’s University.
Author information
Authors and Affiliations
Contributions
Z.S.C., Y.P. and Y.H. designed and wrote the manuscript. Z.N.L., W.C., Q.X.T., Q.T., Y.X. and L.Z. did literature search and wrote the manuscript. Z.N.L., W.C., Q.X.T. and Q.T. prepared the table and figures. Z.S.C., Y.P., K.W. and Q.Z. reviewed and revised the manuscript. All authors listed have made a substantial contribution to the work. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lei, ZN., Teng, QX., Tian, Q. et al. Signaling pathways and therapeutic interventions in gastric cancer. Sig Transduct Target Ther 7, 358 (2022). https://doi.org/10.1038/s41392-022-01190-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01190-w
This article is cited by
-
Exosome circATP8A1 induces macrophage M2 polarization by regulating the miR-1-3p/STAT6 axis to promote gastric cancer progression
Molecular Cancer (2024)
-
Metformin suppresses proliferation and glycolysis of gastric cancer by modulating ADAMTS12
Genes and Environment (2024)
-
Circular RNA CircSLC22A23 Promotes Gastric Cancer Progression by Activating HNRNPU Expression
Digestive Diseases and Sciences (2024)
-
Deciphering cellular and molecular mechanism of MUC13 mucin involved in cancer cell plasticity and drug resistance
Cancer and Metastasis Reviews (2024)
-
Integrated Analysis Construct a Tumor-Associated Macrophage Novel Signature with Promising Implications in Predicting the Prognosis and Immunotherapeutic Response of Gastric Cancer Patients
Digestive Diseases and Sciences (2024)
