
Abstract
The term “circadian rhythms” describes endogenous oscillations with ca. 24-h period associated with the earth’s daily rotation and light/dark cycle. Such rhythms reflect the existence of an intrinsic circadian clock that temporally orchestrates physiological processes to adapt the internal environment with the external cues. At the molecular level, the circadian clock consists of multiple sets of transcription factors resulting in autoregulatory transcription-translation feedback loops. Notably, in addition to their primary role as generator of circadian rhythm, the biological clock plays a key role in controlling physiological functions of almost all tissues and organs. It regulates several intracellular signaling pathways, ranging from cell proliferation, DNA damage repair and response, angiogenesis, metabolic and redox homeostasis, to inflammatory and immune response. In this review, we summarize findings showing the crosstalk between the circadian molecular clock and some key intracellular pathways, describing a scenario wherein their reciprocal regulation impinges upon several aspects of mammalian physiology. Moreover, based on evidence indicating that circadian rhythms can be challenged by environmental factors, social behaviors, as well as pre-existing pathological conditions, we discuss implications of circadian misalignment in human pathologies, such as cancer and inflammatory diseases. Accordingly, disruption of circadian rhythm has been reported to affect several physiological processes that are relevant to human diseases. Expanding our understanding of this field represents an intriguing and transversal medicine challenge in order to establish a circadian precision medicine.
Similar content being viewed by others

The rhythm around the molecular clock
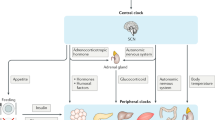
The term circadian rhythm was originally coined by Halberg to indicate the near-24-hour (h) endogenous oscillations of biological processes in organisms associated with the earth’s daily rotation cycle1. Such endogenous rhythms, observed in organisms ranging from photosynthetic prokaryotes to higher eukaryotes, reflect the existence of an intrinsic circadian clock that temporally orchestrates physiological and behavioral processes2 with the specific function of coordinating and adapting the internal environment with the external cues3. The entrainment of circadian rhythms, which consists of the alignment of the endogenous circadian oscillator to external stimuli, relies on external cues, such as the light pattern and food intake. In particular, the daily light–dark cycle represents the primary external synchronizer of circadian rhythms. In mammals, light is processed through the eye and transmitted through the retinohypothalamic tract to hypothalamic suprachiasmatic nucleus (SCN), the master internal pacemaker. In the retina, the intrinsically photoreceptive retinal ganglion cell (ipRGC) expressing the photopigment melanopsin, which renders them photosensitive to short-wavelength irradiation, together with the retinal rod and cone photoreceptors, conveys photic information to entrain SCN clocks4. Notably, SCN is composed of bilateral nuclei containing approximately 10,000 neurons, each of them displaying a cell-autonomous circadian oscillator. A number of papers have demonstrated that the SCN is both necessary and sufficient for the generation of circadian rhythms in rodents5. SNC neurons have distinct and topographically organized coupling mechanisms that allow them to remain synchronized to one another. They generate a pronounced circadian rhythm of neuronal firing frequency, which allows them to synchronize other cells throughout the body.
Notably, in the brain, beside the master clock in SCN operating as self-sustaining clocks, other functional nuclei have been found to act as semiautonomous clocks (i.e. olfactory bulb, dorsomedial hypothalamus, arcuate nucleus, habenula) or as slave oscillators (i.e. bed nucleus of the stria terminalis, amygdala, preoptic area, paraventricular nucleus, nucleus accumbens), both coordinated by the SCN.
The central pacemaker clock synchronizes multiple peripheral clocks expressed in nearly every mammalian organ (i.e. such as lungs, liver, heart, and skeletal muscle). Noteworthy, while in the SCN circadian rhythms are similar between diurnal and nocturnal species, a phase shift, matched to their active period, in the rhythms has been observed in their peripheral tissues6. The synchronization of peripheral clocks is not only hierarchically and vertically controlled by the hypothalamic master clock through the peripheral nervous system, but it also horizontally achieved through both humoral and non-humoral pathways. In particular, SCN controls peripheral oscillators through the autonomic innervation of peripheral tissues, endocrine signaling (glucocorticoids), body temperature, and feeding-related cues. The neural control of peripheral oscillators requires both sympathetic and parasympathetic pathways7. SCN projections, through the paraventricular nucleus–superior cervical ganglia (PVN-SCG) pathway, deliver the entraining signal for the submandibular salivary glands7,8. Autonomic pathways derived from the SCN provide photic information to oscillators in the adrenal gland and liver9. Moreover, sympathetic pathways modulate the sensitivity of the adrenal to adrenocorticotropic hormone (ACTH) and the release of glucocorticoids9,10. In the adrenal cortex and medulla, cellular oscillators respond to neural inputs deriving from the SCN9,11. Furthermore, glucocorticoids are humoral entraining signal for peripheral clocks entraining signals for peripheral oscillators. In particular, since glucocorticoids-response elements (GREs) are present in promoter regions of the core clock components glucocorticoids regulate the transcriptional activation of clock genes and clock-related genes12,13,14.
The circadian autoregulatory feedback loop
At the molecular level, the circadian clock consists of multiple sets of transcription factors resulting in autoregulatory transcription-translation feedback loops (TTFLs) that represent the core mechanism of the circadian clock in mammals. A central role in the regulation of this loop is played by the heterodimeric partnership between two transcription factors, i.e. the brain and muscle Arnt-like protein 1 (ARNTL, also known as BMAL1) and the circadian locomotor output cycles kaput (CLOCK). In particular, transcription of BMAL1 and CLOCK genes, or its related gene NPAS2 (neuronal PAS domain containing protein-2), which is mainly expressed in the forebrain, leads to the heterodimerization in the cytoplasm of the BMAL1:CLOCK complex, which translocates into the nucleus where it binds to canonical Enhancer Box (E-Box)-sequences containing the consensus sequence CACGTG or noncanonical E-Boxes of clock-regulated genes. Among the different target genes, BMAL1 and CLOCK also promote the expression of the components of the negative arm of the molecular clock, such as period (PER1, PER2, PER3) and cryptochrome (CRY1, CRY2). PER and CRY form a complex in the cytoplasm that translocates into the nucleus. Following their translation and nuclear accumulation, PER and CRY inhibit the transcriptional activity of BMAL1:CLOCK complex. At post-transcriptional level, the stability of PER and CRY proteins is regulated by Skp1-Cullin-F-box protein (SCF) E3 ubiquitin ligase complexes, involving β-TrCP (β-Transducin Repeat Containing E3 Ubiquitin Protein Ligase) and F-Box and Leucine Rich Repeat Protein 3 (FBXL3), respectively. The casein kinase 1ε/δ (CK1ε/δ) and adenosine 3’,5’-monophosphate (AMP) kinase (AMPK) phosphorylate PER and CRY proteins, respectively, thus promoting polyubiquitination by their respective E3 ubiquitin ligase complexes, which tag PER and CRY proteins for degradation by the 26 S proteasome complex. Decrease in PER and CRY protein levels relieves the suppression of BMAL1:CLOCK activity, thereby permitting to establish a new oscillatory cycle. In addition to such core loop, further key regulators of the circadian clock, such as the nuclear receptors REV-ERBα (also known as nuclear receptor subfamily 1, group D, member 1, NR1D1) and REV-ERBβ (also known as nuclear receptor subfamily 1, group D, member 2, NR1D2) (REV-ERBs), as well as the retinoic acid orphan receptor (ROR) (RORα, RORβ, and RORγ) establish another feedback loop. In particular, while REV-ERBs act as transcriptional repressors of BMAL1 expression, RORs positively regulate the expression of BMAL1 by binding to sites Retinoic acid receptor-related Orphan Receptor Element (RORE) elements in the BMAL1 gene promoter.
On the other hand, the biological clock is not only based on transcriptional mechanisms, but also, membrane depolarization, intracellular calcium flux, and activation of cyclic AMP (cAMP) signaling appear to be important regulators of the mammalian transcriptional clock. Accordingly, intracellular calcium is fundamental for neuronal firing rhythms in SCN slices15 and sufficient membrane depolarization, periodic calcium influx, and daily activation of cAMP signaling are required for the rhythmic expression of the core clock components in SCN neurons16,17,18. Notably, such effects are mediated by the phosphorylation-dependent activation of the calcium/cAMP response element binding protein (CREB) that binds to calcium/cAMP regulatory elements (CREs) on DNA. In particular, CRE sequences have been found in the promoters of several clock genes, such as PER1 and PER218,19. Since membrane potential20, calcium flux15, and activation of cAMP signaling16,18 have been reported to be also rhythmic themselves in SCN, they represent both outputs of and inputs to the transcriptional clock, by possibly establishing positive feedback loops participating in rhythm generation.
However, the mechanisms underlying the molecular clock cannot be simplified in such a reductionist fashion. Indeed, clock genes integrate a plethora of different signals to produce an integrated output over the 24-h cycle, in preparation for the diverse tasks related to periods of light or darkness, wherein gene expression changes in a non-linear manner during the transition hours between activity and rest periods with a gating system enhancing or softening signal transduction to avoid interferences of misaligned cycles21.
Notably, endogenous cellular clocks drive the rhythmic expression of genes. In particular, approximately up to 20% of the genome is under circadian regulation22,23,24. Accordingly, by assessing the transcriptome atlas of a primate across the major tissues and brain regions, Mure et al. demonstrated that, in baboons, daily expression rhythms occurs in >80% of protein-coding genes, responsible for different biochemical and cellular functions, and that such rhythmic activity represents the largest regulatory mechanism capable to integrate diverse biochemical functions within and across cell types. Moreover, 82.2% of genes coding for proteins that are identified as druggable targets by the US Food and Drug Administration display cyclic oscillations in transcription6.
Circadian regulation of physiological processes: reciprocal signaling between the clock and other regulatory networks
In addition to their primary role as generator of circadian rhythm, the biological clock serves a key role in controlling physiological functions of almost all tissues and organs in two major ways. First, central outputs from the SCN and/or local outputs from cell-autonomous peripheral oscillators drive circadian rhythms in several physiological pathways in a rhythmic fashion. Second, the core clock components have been observed to act as key molecular players in many intracellular pathways, serving additional physiological roles. Examples of circadian-regulated physiological pathways includes cell growth, DNA repair and damage, angiogenesis, apoptosis, metabolism, redox state, as well as immune and inflammatory processes25,26,27,28. Thus, in the following sections, we will critically discuss the mechanistic connection between the circadian molecular clock, which introduces a temporal variable into cellular functions, and other cellular networks and biological pathways.
Circadian clock and cell cycle machinery
Among the diverse biological processes controlled by the circadian clock, the regulation of the cell cycle machinery is the most elusive. However, substantial evidence highlights that the molecular architecture of the circadian oscillator displays several analogies with the complex intracellular machinery regulating cell division and that several regulators of cell cycle progression are expressed in a circadian manner, as elegantly reviewed by Hunt and Sassone-Corsi, 2007. Accordingly, both the circadian clock and the cell cycle are intracellular clocks consisting of sequential phases of transcription-translation and based on the conceptual framework of interlocked autoregulatory loops29. Noteworthy, accumulating evidence suggests that the circadian clock affect the timing of cell divisions in vivo25. Moreover, most of cell-cycle genes involved in G2-M or G1-S transitions contains E-box regulatory elements in their promoters, transcriptionally activated by CLOCK:BMAL1 heterodimer29. Accordingly, Matsuo et al. demonstrated that the cell-cycle-related gene Wee-1, which contains three B-boxes in its promoter and encodes a protein kinase that inactivates the CDC2/Cyclin B1 complex, thereby delaying or preventing entry into mitosis, is under the direct transcriptional control of CLOCK:BMAL125. (Fig. 1) The authors found that the diurnal fluctuations in Wee1 mRNA expression are reflected by its kinase activity, target phosphorylation, and CDC2/Cyclin B1 kinase activity, thus suggesting that the circadian clock–Wee1 pathway may act as a cell cycle regulator in vivo25. Then, another molecular link between clock and cell cycle genes comes from Gréchez-Cassiau et al. work showing that clock genes control G1/S progression by transcriptionally regulating Cdkn1a—a gene encoding for the cyclin/Cdk inhibitor p12WAF1/CIP1—and that alteration of circadian clock led to aberrant p21 expression and altered cellular proliferation30. Furthermore, CLOCK:BMAL1 complex has been shown to transcriptionally control the genes encoding c-Myc, which regulates cell cycle entry by responding to mitogenic stimuli, and Cyclin D1, thereby modulating cell cycle31 (Fig. 1). In this regard, Repouskou et al. demonstrated the in vivo binding of BMAL1 on the human c-Myc promoter and that c-Myc overexpression hindered PER1 transactivation by BMAL1/CLOCK by targeting E-box sequences31. The ensemble of this study depicts a scenario wherein the biological clock induces oscillation in Myc expression and, on the other hand, Myc hinders E-box-driven expression of PER1, when its protein abundance allows interaction with BMAL1.

Molecular interaction between the circadian core clock and cell cycle components. The CLOCK:BMAL1 complex transcriptionally activates genes containing E-box regulatory elements in their regulatory regions, such as clock genes and cell-cycle genes. A CLOCK:BMAL1 complex directly controls the transcription of the cell-cycle-related gene Wee-1 contains three B-boxes in its promoter and encodes a protein kinase that inactivates the CDC2/Cyclin B1 complex, thus regulating G2-M transition and cell-cycle progression. B Transcriptional activation of the genes encoding Cyclin D1 and C-MYC by CLOCK:BMAL1 affects cell proliferation and differentiation. C PER1 can complex with the ATM kinase and the checkpoint kinase Chk2, thus impinging upon DNA repair, cell cycle arrest and/or apoptosis. D Both physiological and stress-induced p53 binds to p53 response element in PER2 promoter, which overlaps with the BMAL1/CLOCK-binding site, thereby inhibiting CLOCK:BMAL1-mediated transcription of PER2. (BioRender.com has been used to create the figure)
Within this context, the circadian clock has been implicated in DNA damage response (DDR), including cell cycle checkpoints and DNA repair. DNA damage-induced cell cycle checkpoints are signal transduction pathways driven by DNA damage, acting as a genome surveillance mechanism to ensure genomic integrity32. Notably, the CLOCK:BMAL1 complex has been found to control DDR by modulating the transcription of genes in genotoxic responses26. Moreover, besides a transcriptional regulation, the core clock factors have been also reported to directly interact with key cell cycle checkpoint pathways and, in particular, with DDR factors, thereby maintaining genome integrity. Consistently, PER1 has been reported to complex with the ataxia-telangiectasia-mutated (ATM), a kinase involved in the cellular response to ionizing radiations and DNA double-strand-break-inducing events, as well as with the checkpoint kinases CHK233 (Fig. 1). Moreover, it has been demonstrated that, upon UV damage, CRY1 modulated the ATR (ATM- and Rad3-Related)-mediated DNA damage checkpoint response by interacting with TIMELESS (TIM) in a time-of-day-dependent manner, thereby generating circadian oscillation of ATR activity34. Interestingly, Shafi et al. recently demonstrated that androgen receptor-induced CRY1 was stabilized by genotoxic insult and regulated DDR by directly binding to promoters of homologous recombination factors, thereby modulating genome integrity and promoting castration-resistant prostate cancer growth27. As such, CRY1 emerges as a pro-tumorigenic factor that rhythmically controls DNA repair mechanisms and cell survival through temporal transcriptional regulation27. Therefore, it is tempting to speculate that failure in such intimate crosstalk between clock and DDR factors may cause genomic instability and tumorigenesis.
Another link between DNA damage response and circadian clock relies on the regulatory feedback loop involving the tumor suppressor and checkpoint component p53 and PER2. In particular, wild-type p53 has been shown to negatively regulate PER2 expression. Mechanistically, Miki et al. demonstrated that wild-type p53 controls the circadian behavior of mice by competing with BMAL1:CLOCK for binding to the PER2 promoter, thereby repressing the expression of this latter35. In unstressed cells, temporal oscillations of p53 levels have been reported to be inversely correlated with PER2 levels, whereas, under stress conditions, accumulation of p53 levels to block PER2 transcription. Moreover, PER2 has been reported to form a trimeric complex with p53 and the oncogenic mouse double minute-2 homolog protein (MDM2), the p53’s negative regulator, thereby increasing p53 stability by blocking Mdm2-dependent ubiquitination and transcription of p53 target genes36. Based on the relevance of p53 in check-point signaling, such findings indicate that PER2 association with p53 might be a regulatory module that influences p53’s downstream response to genotoxic stress. Moreover, also the core clock component BMAL1 has been reported to bind to p53 promoter, thereby transcriptionally triggering the downstream tumor suppressor pathway37.
Therefore, such evidence reporting the molecular interactions between the core clock proteins and key cell-cycle regulators supports the notion of a link between circadian clock and cell cycle and that a circadian oscillator may establish temporal windows, thereby enabling or suppressing cell cycle transitions. Future investigations on how the biological clock governs cell proliferation and growth and on the potential implications for the onset of human diseases are warranted. Indeed, the temporal gating of cell division by the molecular clocks may be of relevance in the pathogenesis of cancer, since dysregulation of cell cycle represents a crucial driver of enhanced cell proliferative capacity. As a proof of concept, disruption of the regulatory loop linking p53, the cell cycle, and clock genes has been correlated to cancer in mouse models38. As an example, it has been demonstrated that a mutation (S662G) in PER2, which is known to be responsible for the familial advanced sleep phase syndrome, increased resistance to apoptosis and E1A- and RAS-driven oncogenic transformation, as well as affected tumorigenesis in p53R172H/+ mice, with a cancer-sensitized genetic background38. Noteworthy, the relative phases between p21 and Cyclin D expression profiles were significantly altered in PER2 allele mutant mouse embryonic fibroblasts, thus indicating that alteration of p21 phase, due to mutation, might impact on cell phase locking, thereby triggering cell cycle progression.
Circadian clock, angiogenesis, and hypoxia
Although the role of circadian clock in controlling vascular development is largely unknown, evidence from the literature indicates that the core clock components participate in the regulation of angiogenesis, an essential process required for all tissue growth. Consistently, in developing zebrafish embryos, circadian regulatory genes, such as BMAL1 and PER2, have been reported to critically regulate vascular development, by modulating the expression of the vascular endothelial growth factor (VEGF)28. Disruption of the circadian clock by exposure to constant light and genetic manipulation of the core clock genes impaired developmental angiogenesis. Notably, the circadian regulator BMAL1 has been found to directly target and activate the promoter region of the VEGF gene via E-boxes, thereby promoting VEGF expression28. In addition, genetic deletion of BMAL1 impaired Notch-inhibition-induced vascular sprouting. Based on these data, it can be speculated that these findings unraveling mechanistic insights on the role of the circadian clock in controlling developmental angiogenesis may be also extended to other types of physiological angiogenesis (i.e. mammalian developmental angiogenesis), as well as to pathological angiogenesis in humans. In line with such hypothesis, the molecular clockwork has been found to regulate the expression of VEGF in hypoxic tumor cells39. In particular, the negative limbs of the molecular clock, i.e. PER2 and CRY1, have been reported to suppress the activity of VEGF promoter induced by hypoxia in the luciferase reporter gene analysis, thereby contributing to the circadian fluctuations of VEGF mRNA expression39.
Another master regulator of angiogenesis that has been shown to crosstalk with the molecular clock is the central regulator of oxygen homeostasis, i.e. the hypoxia-inducible factor 1α (HIF1α). Accordingly, HIF1α has been reported to bind to the E-Box and BMAL1, thereby promoting the expression of circadian genes40,41,42. Moreover, in U20S cells, E-box sequence in the promoter region of HIF1α gene as well as a direct binding of BMAL1:CLOCK complex to HIF1α have been observed41. As recently reviewed by O’Connell et al, 202043, cell line- and tissue-specific bidirectional interactions between HIF and the core clock components have been found to regulate important biological functions. In this regard, Peek et al. demonstrated that the molecular clock is involved in the regulation of anerobic glycolysis via HIF1α signaling and that circadian control of HIF1α affects glucose metabolism in a context-dependent manner44. Indeed, while BMAL1−/− liver displayed an enhanced anaerobic glycolytic gene expression45, BMAL1−/− myotubes showed a decrease in anaerobic glycolysis, mitochondrial respiration, as well as transcription of HIF1α targets, including VEGFa44, thus suggesting a tissue-specific role of clock/ HIF1α interplay. Interestingly, the authors first demonstrated a time-of-day gating of HIF1α activity, with a greater activation during the active period (i.e. daytime in humans), suggesting that circadian control of HIF1α activity allows us to respond more robustly to hypoxia when demand for strenuous activity is likely to be higher.
Interestingly, in line with data showing a circadian/hypoxia pathway crosstalk, evidence from the literature indicates the acquisition of genetic signatures involving key players both in hypoxia and circadian pathways by long-term high-altitude populations living under hypobaric hypoxia conditions46,47. As an example, by using single-nucleotide polymorphism genotype data from Ethiopian populations, a strongest signal of selection was observed in the basic helix-loop-helix family member E41 (BHLHE41) gene that serves a major role both in the regulation of hypoxia-sensing pathway and circadian clock47. Of note, mutation in this gene has been associated with short sleep phenotype48.
Furthermore, hypoxia serves a key role in several diseases, including cancer, cardiovascular, lung, and metabolic diseases49. Therefore, the molecular mechanisms underpinning HIF and clock crosstalk both in physiological oxygen fluctuations and hypoxic conditions within different cells and tissues require further investigations. Moreover, studies are needed to understand the regulatory effects of the clock on the transcriptional hypoxic stress response, which is known to promote vessel growth by upregulating multiple pro-angiogenic factors. Such information may be of key relevance for understanding angiogenesis in pathological contexts such as vascular diseases and tumorigenesis.
Circadian clock and immunity
Almost all the components of the immune system, involved in adaptive and innate immunity, have been observed to exhibit circadian variations (for a comprehensive review on the topic see50,51 For example, rhythmic oscillations occur in the trafficking of hematopoietic stem cells and leukocytes, in the expression of recognition receptors and their downstream signaling pathways, and finally in the production of cytokines and chemokines. As an example, Méndez et al demonstrated rhythmic Cxcl12 oscillations regulated by clock genes through the circadian regulation of noradrenaline secretion. In particular, the activation of β3-adrenergic receptors resulted in a reduction in Cxcl12 mRNA levels in bone marrow stromal cells, thus stimulating the mobilization of hematopoietic stem cells/progenitor52. Moreover, evidence from the literature indicates that the main components of immunity, including macrophages and natural killer (NK) cells, display a cell-autonomous circadian clock53. These endogenous clocks impose temporal gating across a range of functions, such as phagocytosis, cytokine/chemokine production, and antibacterial and antiviral activity53.
The interplay between circadian clock and inflammatory pathways
The circadian clock has been suggested to serve a key role in the rhythmic regulation of basal inflammatory responses by temporally gating them. Accordingly, the circadian oscillators have been reported to prevent the synchronous activation of the different components of the immune system, by temporally limiting various aspects of the innate immunity, as well as the expression of inflammatory genes and the trafficking of innate immune cells to sites of inflammation, to specific phases of the circadian cycle (as comprehensively reviewed by Man et al54. As far as the clock-controlled expression of inflammatory mediators, BMAL1 has been demonstrated to generate basal oscillations in the expression of chemokines, such as CCL2, via interaction with the Polycomb repressor complex 2 (PRC2) in myeloid cells55. In particular, BMAL1 has been found to recruit PRC2 complex to repress chemokine gene expression (Fig. 2). Accordingly, its deletion resulted in an enhanced expression of CCL2, CCL8, and S100a8 in monocytes and peritoneal macrophages55. Moreover, by using pharmacological and genetic approaches in human macrophages, REV-ERBα has been shown to act as a pivotal intermediary between the molecular clock and inflammatory pathways, by regulating the expression of genes involved in human innate immunity, such as IL-656, and repressing CCL2 gene expression directly through a RORE in the CCL2 promoter region57 (Fig. 2). Furthermore, recruitment of REV-ERB repressor complexes to inflammatory genes has been reported to temporally repress their expression by inhibiting enhancer-specific transcription58.

Circadian control of basal and inducible expression of inflammatory mediators in immune cells. a Circadian control of basal gene expression. Interaction between PRC2 and the CLOCK:BMAL1 complex rhythmically represses the expression of chemokine genes, such as Ccl2. b Circadian control of inducible gene expression. CLOCK acetylates the p65 subunit of NF-κB, thereby inducing the expression of TNFα. Moreover, recruitment of REV-ERB repressor complexes to inflammatory genes, such as Il6, rhythmically suppresses their expression. Finally, the cellular clock is fundamental for recruitment of GR complexes to the glucocorticoid binding site (GBS) on Cxcl5 to repress its transcription. (BioRender.com has been used to create the figure)
Besides a direct regulation of the expression of inflammatory mediators by clock genes, another mechanism by which the molecular clock establishes a temporal gating of inflammation involves the glucocorticoid receptor (GR). Endogenous glucocorticoids regulate the circadian expression of a number of inflammatory cytokines through binding to the intracellular GR, which suppresses the expression of inflammatory mediators by interacting with GREs and by directly regulating transcription factors such as NF-κB, Activator protein-1 (AP-1) as transrepression59 (Fig. 2). It has been demonstrated that the molecular clock establishes a temporal gating of inflammation via GR. In this regard, Gibbs et al. reported a regulatory mechanism coupling the local molecular clock within the pulmonary epithelial cells with the GR–driven repressive effects on the expression of inflammatory chemokines60. Specifically, they found that, in bronchiolar cells, CXCL5 expression was synergistically regulated by the local bronchiolar clock and the systemic repressive glucocorticoid signals of adrenal origin and that, in mice, genetic deletion of BMAL1 in bronchiolar cells impaired GR occupancy at the CXCL5 locus and increased CXCL5 expression despite normal corticosteroid secretion. These findings indicate that the recruitment of GR by the core clock gene BMAL1 may represent a mechanism to temporally repress the expression of inflammatory genes60, thus suggesting a key role for the adrenal axis in circadian control of inflammatory responses.
Furthermore, glucocorticoids have been shown to directly promote clock gene expression in both animal61 and human studies62. GREs have been described in the promoter regions of PER and dexamethasone has been observed to selectively induce PER1 expression at concentrations consistent with the nighttime nadir of human cortisol63. Moreover, GR has been found to directly suppress REV-ERBα expression by interacting with CLOCK:BMAL1 complex in mouse liver64. In turn, CLOCK has been shown to physically interact with GR and to suppress its binding to DNA recognition sequences, by acetylating multiple lysine residues located in its hinge region in HCT116 and HeLa cells65. Such a result suggests that CLOCK:BMAL1 may antagonize biological actions of diurnally fluctuating circulating glucocorticoids. Lamia et al. also demonstrated that CRY mediate rhythmic expression of GR66. In particular, CRY 1 and 2 associate with a GRE in the phosphoenolpyruvate carboxykinase 1 (Pck1) promoter in a hormone-dependent manner, and dexamethasone-induced transcription of the Pck1 gene has been found increased in CRY-deficient livers66.
Taken together, such evidence converges to indicate that the molecular clock directly exerts a regulatory control over inflammatory processes by finely tuning different intracellular mechanisms. However, this coupling between the circadian clock and inflammation has been suggested to be reciprocal, with also the circadian clock subjected to inflammation-driven reprogramming. In line with this hypothesis, Poolman et al. reported a significant circadian reprogramming due to chronic inflammation in patients with rheumatoid arthritis, with an increased circadian rhythmicity and expansion of the repertoire of rhythmic processes due to chronic joint inflammation67. These results reveal that, in chronic inflammatory disease, circadian mechanisms might be co-opted in order to set up a novel homeostatic oscillatory state, with an extensive reorganization of the circadian processes and de novo genesis of alternative cycling pathways. As an example, Wang et al. demonstrated that, in mice with dextran sodium sulfate (DSS)-induced colitis, colonic inflammation was associated with the dysregulation of clock genes (i.e. NR1D1/REV-ERBα, CLOCK, BMALl1, PER2, CRY1, NPAS2, NR1D2/REV-ERBβ, RORα, and DBP) expression68. However, whether this circadian reprogramming due to chronic inflammation may represent a disease-sustaining step is still unclear.
Notably, since circadian disruption has been correlated to the onset of several pathological conditions, understanding how the molecular clock intersects with inflammatory pathways, may provide novel insights into the etiopathogenesis of inflammatory conditions and reveal new therapeutic targets. Therefore, in the following sections, we will critically discuss the crosstalk between the circadian molecular clock, which introduces a temporal variable into cellular functions, and NF-κB transcription factor that have been reported to act as key molecular nodes integrating inflammation and circadian rhythmicity.
The crosstalk between NF-κB and the core clock components
Data from the literature provided evidence for an existing molecular crosstalk between the transcription factor NF-κB and the molecular components of the circadian clock. In this regard, a mechanistic framework for understanding the link between circadian rhythm, inflammatory processes, and NF-κB comes from the work by Narasimamurthy et al69. Indeed, the authors demonstrated that the absence of CRY protein(s), which act as transcriptional repressors, induce a constitutive increase in proinflammatory cytokines in a cell-autonomous manner. In particular, an increased phosphorylation of p65 at S276 residue due to increased protein kinase A (PKA) signaling activity has been found in Cry1−/− and Cry2−/− cells69. The absence of CRY has been hypothesized to release its inhibition on cAMP production, thereby increasing cAMP levels and PKA activation and, subsequently, PKA-mediated phosphorylation of the p65 subunit of NF-κB, leading to its constitutive activation. Furthermore, CRY mutation has been reported to promote the tumor necrosis factor α (TNFα)-initiated extrinsic apoptosis by interfering with NF-κB signaling in p53 mutant and oncogenically transformed cells70. Noteworthy, the core circadian clock activity has been shown to regulate the NF-κΒ pathway and apoptosis upon cytokine stimulation in p53-impaired cells, with the negative arm of the clock, i.e. CRY, inhibiting and the positive arm, i.e. BMAL1, stimulating apoptosis in this genetic background70. Accordingly, it was observed that, while stimulation with TNFα and IL-1β induced the transcription of NF-κB target genes, as well as its binding to their promoters, in p53KO cells, the cytokine-driven transcriptional activation of NF-κB was abolished in p53KO CryDKO cells. Interestingly, the effect of CRY mutation on the expression of NF-κB targets genes and NF-κB biding to their promoters was reversed by BMAL1 down-regulation.
Besides a basal modulation of inflammatory processes, a rhythmic regulation of inducible gene expression has been suggested to rely on its crosstalk with NF-κB. In this regard, Spengler et al. demonstrated a direct molecular link between the transcription factors CLOCK and NF-κB. In particular, they found that, in the absence of BMAL1, NF-κB-driven transcription was upregulated by the core circadian protein CLOCK, whereas BMAL1 hindered the CLOCK-induced increase in the activation of NF-κB-regulated genes71. Furthermore, CLOCK was found in protein complexes with the p65 subunit of NF-κB, and CLOCK overexpression was associated with enhanced phosphorylated and acetylated transcriptionally active forms of p6571. In addition, NF-κB response was significantly reduced in CLOCK-deficient cells and in the tissues of CLOCK-knockout mice71, thus supporting the notion that CLOCK might act as a positive regulator of NF-κB transcriptional activity.
Another clock gene acting as a gatekeeper of inflammation is the nuclear hormone receptor REV-ERBα, a known repressor of NF-κB activity57. In murine macrophages, REV-ERBα has been shown to repress IL-6 expression indirectly through an NF-κB binding motif72. Consistently, it has been demonstrated to exert an anti-inflammatory activity in a murine model of colitis, by specifically repressing NF-κB and Nlrp3 expression, thereby downregulating Nlrp3 inflammasome activity68.
Furthermore, besides such evidence demonstrating a regulatory role of the molecular clock on NF-κB signaling pathway, it has been also observed that, vice versa, the activation of NF-κB directly impacts on the transcriptional activity of the core clock components. Accordingly, Maury et al. recently demonstrated that overactivation of NF-κB prevents BMAL1 binding to the promoter region of PER2 and, consequently, its transcription in human omental fat73. In particular, BMAL1 has been found to bind in close proximity to NF-κB consensus motifs in human omental adipocyte precursors, revealing that IKKβ/NF-κB pathway may play a role in this repositioning. These findings indicate that inflammatory conditions associated with the overactivation of NF-κB may misalign the molecular clock, thereby altering the transcription of several target genes and contributing to metabolic inflammation through direct transcriptional reprogramming. As a proof of concept, obese mice with adipocyte-specific deletion of IKKβ displayed an improvement of adipose clock properties together with an improvement of metabolic inflammation73. In line with these findings, a previous study by Hong et al. reported that activation of NF-κB upon inflammatory stimuli markedly inhibited the clock repressors (i.e. PER, CRY, and REV-ERB) and relocated the clock components CLOCK/BMAL1 genome-wide to sites convergent with those bound by NF-κB74. These data indicate that NF-κB-driven transcriptional repression of the clock feedback limb might represent the triggering cause of circadian misalignment in response to inflammation and highlight a molecular mechanism through which immune activation may affect circadian rhythmicity. Moreover, conditional inactivation of IKKβ in mouse CNS and peripheral tissues impaired rhythmic activity behavior, thus revealing that IKKβ/NF-κB pathway controls circadian behavioral and molecular rhythms in vivo to preserve circadian homeostasis74. Therefore, it is tempting to speculate that basal level of NF-κB activity is fundamental for the maintenance of circadian homeostasis also in its latent state of immune signaling74.
Circadian clock, metabolism, and redox homeostasis
In recent years, the existence of an extensive network of interactions between the circadian clock and the cellular redox state has emerged. As an example, reduced forms of the cofactors NAD(H) and NADP(H) have been shown to promote DNA-binding activity of BMAL1 and CLOCK, whereas their oxidized forms to hinder it, with minute changes in redox state strongly affecting the binding activity of circadian transcriptional75. Furthermore, another molecular redox-clockwork interaction involves Heme, a protein cofactor acting as a sensor of cellular redox state that has been reported to affect circadian control by binding to clock proteins. Consistently, it has been found to bind to REV-ERBs in a redox-state dependent manner76 to NPAS2, a CLOCK paralog, thereby modulating its DNA binding activity, as well as to inhibit binding of CLOCK to its E-box DNA target77. However, the mechanisms of regulation are largely unknown. Furthermore, the expression of nicotinamide phosphoribosyltransferase (NAMPT), a rate-limiting enzyme in the NAD+ salvage pathway, has been reported to be regulated by the BMAL1:CLOCK complex via the E-boxes present in its promoter in a time-dependent manner, thereby driving a rhythmic production of NAD+ 78,79. This latter activates NAD+-dependent histone deacetylases, such as sirtuin 1 (SIRT1), which has been shown to be fundamental for circadian transcription of several clock genes (e.g. BMAL1, PER2, and CRY1), as well as to bind BMAL1:CLOCK complex, and to promote the deacetylation and degradation of PER280. Thus, since SIRT1 deacetylase activity relies on NAMPT-mediated NAD+ biosynthesis, SIRT1 has been hypothesized to link cellular metabolism to the circadian clockwork. Hence, NAD+ rhythmic production has been proposed to finely tune the daily cycles of energy storage and utilization, as well as to align such processes with the rest-activity cycle. In addition, a further crucial cellular energy sensor activated by a high AMP/ATP ratio, the AMP-activated protein kinase (AMPK), has been reported to mediate CRY and Casein kinases I phosphorylation, thereby modulating the negative molecular arm of the circadian clock by proteolytic degradation81. Then, the core clock gene BMAL1 has been shown to act as a negative regulator of the target of rapamycin complex 1 (mTORC1), which acts as a master switch between cell anabolic and catabolic programs, thereby suppressing anabolism82. Taken together, the above-mentioned examples demonstrate that redox state and metabolic pathways work in concert with molecular circadian timekeeping by exhibiting both input and output roles.
Besides these molecular interactions, circadian oscillation of redox state has been also linked to SCN neuronal excitability. In this regard, Wang et al. first demonstrated that redox state displays a daily rhythm in the brain83. By performing ratiometric redox fluorometry by two-photon microscopy of SCN organotypic slices, they observed a near-24-hour oscillation of redox state in SCN rat and mouse tissues83. Notably, circadian oscillation of global redox state in rat SCN was detected, with a marked oxidized state in the early night, when neuronal activity is at its lowest, compared to a reduced state during the daytime. The circadian redox rhythm was not observed in arrhythmic BMAL1−/− mice, indicating that redox oscillations rely on a functional circadian clock83. Thus, such results suggest a potential mechanism by which the molecular clock influences SCN electrical activity. Moreover, since redox state has been described as a contributor to tissue-specific rhythmicity, it is tempting to speculate that circadian redox oscillations might extend beyond the SCN to other brain regions, as well as to all the excitable cells in the body, thereby allowing a nuanced fluctuation of cell excitability.
The role of Nrf2 as a central player integrating redox, immune and circadian signals
The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a transcription factor regulating the expression of about 250 genes encoding a network of cooperating enzymes involved in endobiotic and xenobiotic biotransformation reactions, antioxidant metabolism, protein degradation, and regulation of inflammation84. By governing such complex transcriptional networks, Nrf2 coordinates a multifaceted response to various forms of stress, maintaining a homeostatic intracellular environment. Under unstressed conditions, Nrf2 is retained in the cytoplasm by its negative repressor Keap1 and rapidly subjected to ubiquitination and proteasomal degradation, mediated by the binding of Keap1 to the Cul3/Rbx1 E1 ubiquitin ligase complex85. After exposure to oxidative and/or electrophilic stimuli, Nrf2 is released from structurally modified Keap1 and translocates into the nucleus, forms a heterodimer with one of the small musculoaponeurotic fibrosarcoma (Maf) proteins, and activates the ARE-mediated expression of cytoprotective genes. Nrf2 is known to exert a pivotal role within the innate immune system, by specifically limiting inflammatory response via reactive oxygen species (ROS) suppression and preventing proinflammatory cytokine (i.e. interleukin-1β (IL-1β) and interleukin-6 (IL-6)) production through direct binding of Nrf2 to promoter regulatory regions86,87. Notably, a number of pharmacological and genetic studies demonstrates the existence of a functional crosstalk between NF-κB and Nrf2, with a range of cell-type- and tissue-dependent molecular interactions, fundamental for well-coordinated responses in inflammatory processes88.
Interestingly, several lines of evidence couple Nrf2 with the main core clock components. Rey et al. demonstrated that genetic or pharmacological inhibition of the pentose phosphate pathway (PPP) influenced circadian period length in a Nrf2-dependent manner89. In particular, they found that perturbation of PPP significantly increased Nrf2 DNA binding to NR1D1 in U2OS cells, thus indicating that the Nrf2 activation may relay redox signals to circadian clock through NR1D1. Such results corroborate the hypothesis that Nrf2 represents a key nodal player between redox and circadian oscillations and provide a molecular mechanism explaining how redox imbalance, which is a key feature in many pathology (e.g. cancer, neurodegenerative and cardiovascular diseases), may disrupt circadian rhythmicity. A further study by Wible et al. substantiated these findings by showing that electrophilic or oxidative activation of Nrf2 signaling pathway influenced clock gene expression and circadian rhythmicity90. In particular, Nrf2 has been observed to repress its own transcription through the regulation of CRY2 and subsequent repression of CLOCK/BMAL1, thus forming an interlocking loop that integrates cellular redox signals with the core molecular circadian mechanism90.
Moreover, Nrf2 expression and activity have been reported to be under the circadian control in lung tissue. Accordingly, the circadian transcriptions factors CLOCK and BMAL1 exert a transcriptional control of Nrf2 expression via E-box elements in the Nrf2 gene promoter, thereby regulating Nrf2 protein accumulation in circadian manner and triggering a rhythmic expression of its antioxidant target genes91. Interestingly, the molecular clock has been revealed to regulate Nrf2 levels and activity also in innate immune cells. Indeed, Early et al. demonstrated that, in macrophages, BMAL1 modulated the mRNA expression of Nrf2 via direct E-box binding to its promoter and that its deletion disrupted Nrf2 activity and, consequently, stimulated the production of ROS and proinflammatory cytokines, such as IL-1β. Such evidence provides a mechanism by which the molecular clock controls aspects of innate immunity and highlight the crucial role of the core clock component BMAL1 in regulating the Nrf2-mediated antioxidant response in myeloid cells92.
Taken together, these results demonstrate that the Nrf2 gene is directly regulated by the core clock via conserved E-box element in its promoter and also that Nrf2 transcriptionally regulates BMAL1 and CRY2 expression, thereby establishing a feedback loop and providing a possible molecular mechanism by which oxidative signals might input into the circadian clock.
Circadian disruption: implication for human health and diseases
Circadian rhythms, when misaligned, become pivotal biological imperatives that might be challenged by environmental factors, social behaviors (e.g. exposure to artificial light, shift work, jet travel, irregular sleep, and eating schedules), as well as pre-existing pathological conditions. The subsequent circadian misalignment has been related to several pathologies, such as circadian rhythm sleep-wake disorders (e.g. insomnia, jet lag), cancer, obesity, arthritis, atherosclerosis, and mood disorders93,94,95,96. Moreover, although it will be not discussed herein, it cannot be discounted that also neuropsychiatric conditions, including, but not limited, depression, sundown syndrome in demented patients, and some very peculiar conditions (e.g. fatal familial insomnia) have been reported to be sensitive to external synchronization and correlated with alteration of circadian rhythm. In the following sections, we will specifically discuss circadian disruption in cancer and inflammatory diseases.
Cancer
As discussed above, the molecular clockwork tightly regulates crucial cancer-related pathways, ranging from cell cycle progression to p53-mediated apoptosis. However, clock dysfunction has been either associated with pro- or anti-tumorigenic effects depending on model in a context-dependent manner, as recently reviewed by Stephenson et al, 202197. Although the underpinning molecular mechanisms are not fully elucidated, epidemiological and clinical data provide evidence of a link between circadian misalignment/disruption and increased incidence of specific cancers. Notably, an increasing number of studies of carcinogenic risk factors suggested that disruptions in circadian rhythms play a more central role in tumor progression. Recent studies have revealed alteration in the status of post-translational modifications, such as phosphorylation and methylation, in the promoters of the core clock genes in cancerous tissues, leading to the deregulation of clock gene expression98,99. Based on such evidence, in 2007, shift work leading to a disruption in circadian rhythm has been listed by the International Agency for Research on Cancer (IARC) as a probable human carcinogen (Group 2A7).
Accordingly, preclinical data correlate the environmental disruption of the biological clock, due to shift work and light exposure at night, with hormone-dependent cancers, such as breast and prostate cancer100,101,102. As an example, the Nurses’ Health Study and case-controlled studies revealed that Norwegian nurses working night shifts for less than 30 years displayed a moderate increased risk for breast cancer, which was further increased upon working 30 or more years of rotating shift work100. Furthermore, evidence from the literature suggests an increased risk of prostate cancer in night-shift workers, further enhanced with a longer duration of shift work101,103.
Circadian rhythm alteration is also known to have a significant impact on the development of endocrine tumors104,105. Of note, compared to normal tissues, the expression of clock genes is altered in many solid human tumors and it has been identified an altered pattern in cyclic genes in human breast cancer cell line compared to normal breast epithelial cell line106,107. These observations point to a global reprogramming of circadian gene expression, rather than a disruption that may confer a physiological advantage to tumor cells and prompt to suggest that clock dysfunction could be considered a pro-tumoral event, thus indicating clock dysfunction as a potential hallmark of cancer. Indeed, in recent years, the hallmarks of cancer have been well established such as proliferative signaling, resisting cell death, evading growth suppressors, angiogenesis, activating invasion and metastasis, reprogramming of energy metabolism and evading immune destruction. Therefore, given the importance of clock-regulation, as described in the previous paragraphs in terms of cellular proliferation, in the regulation of the immune system and metabolic reprogramming, an impaired molecular clockwork may represent an emerging hallmark of cancer. Moreover, not only the circadian deregulation could be considered a hallmark of cancer, but also the circadian machinery has the capability itself to control and alter other cancer hallmarks. For example, as previously described, evidence from different eukaryotic model systems reveals a tight connection between circadian clock and cell cycle progression, DDR and DNA repair. Many genes crucial for cell cycle and division are under the control of clock genes, aberrantly expressed in several tumor tissues. Moreover, in order to maintain genome stability, the circadian clock system influences G1 to S phase transition (e.g. by regulating p21), preserves DDR, NER, and PARP1 activities, all of them dysregulated in cancer30. Indeed, the deregulation in any of these pathways has been found to trigger replication stress which, in turn, alters the DNA repair capacity, leading to genome instability and, finally, to cancer development. However, the signaling pathways and molecules inter-linking DDR, DNA repair and circadian clock genes are unclear.
Altogether, one of the main challenges in cancer field consists in translating the preclinical findings to clinics, therefore trying to unravel the relationship between clock genes expression, DNA repair capacity and treatment outcome. The comprehension of the expression levels of clock-controlled genes, proliferation markers, DNA repair and DDR genes derived from different cancer tissues represents an essential issue, since the common chemotherapy used to treat several cancers is mainly based on drugs that target cancer cell proliferation by inducing DNA break.
The circadian clock machinery regulates tumor immune microenvironment
It is now well accepted that the tumor microenvironment and, in particular, immune environment plays a crucial role in controlling tumor progression and, to date, several therapeutics (e.g. pembrolizumab; ipilimumab) are known to act on it. In this context, circadian machinery alteration not only affects tumoral cells but also the interaction between cancer cells and with other stromal components, thus affecting tumor development and metastatic colonization108. In support to this, it is now well accepted that most immune cell types, such as monocytes/macrophages55,109, dendritic cells110, neutrophils/MDSCs111, NK112, T and B cells110,113, have intrinsic circadian clock, and several of their characteristics/functions (e.g. plasticity, migratory capacity) are under circadian regulation.
The circadian aspect of many immune features is well documented in the contexts of infection and inflammation. However, the effect of circadian rhythms on tumor immune microenvironment is still nascent. Notably, tumor associated macrophages (TAMs)—the most abundant immune cells in solid cancers regulating immunosuppression, tumor growth, and metastasis formation—have a robust circadian rhythm and at least 10% of their transcriptional activity has been reported to be under circadian regulation114. Therefore, it is tempting to speculate that dysregulation of the circadian clocks in TAMs may have consequences on cancer progression and metastatization. For example, the deletion of BMAL1 in myeloid cells induced myeloid infiltration in the tumor in parallel with an increased inflammatory response (e.g. increase in CCL2–8, IL-1β, IL-6 levels)92,115. In addition, the silence of BMAL1 (e.g. BMAL1−/− macrophages) favors the production of matrix metallopeptidase 9 (MMP-9)116 and the recruitment of CXCR2+ myeloid cells to the lungs60, events that are associated with the development of a pre-metastatic niche and metastatic dissemination/colonization. Moreover, long exposure to chronic jetlag of mice induced the development of spontaneous mammary tumors by an enrichment of TAMs and a reduction of anti-tumor cytotoxic CD8+ T cells in the tumor, therefore contributing to the development of immune-suppressive microenvironment a mechanism based on BMAL1-driven metabolic changes117. Thus, these studies suggest that the intimate interconnection between intrinsic circadian clock and cellular metabolism in the macrophages may play a critical role in shaping the tumor immune microenvironment.
Other key players of tumor immune environment are tumor associated neutrophils (TENs) that affect tumor progression by promoting angiogenesis, metastasis and by attenuating anti-tumor immunity118. The specific deletion of BMAL1 in TENs has been described to result in an impairment of homeostatic neutrophil clearance and enhanced migration to inflamed tissues, therefore suggesting a critical role of BMAL1/circadian machinery for the control of neutrophil immune responses119,120. In the context of tumor immune microenvironment, these findings suggest that circadian disruption potentially increases NETs formation, which has been shown to interfere with anti-tumor T and NK cell cytotoxicity121.
Among the other myeloid lineage, also dendritic cells (DCs), which are heterogenous antigen-presenting cells with a critical role in the activation of NL and T cell cytotoxicity, are affected by the clock machinery, even if the real mechanism at the basis is less understood. For example, BMAL1−/− DCs had impaired capacity to induce Th1 immune responses due to the downregulation of several cytokines (e.g. IL-12, IL-23, or IL-27)122. Moreover, BMAL1−/− DCs display also a reduced migration into the spleen, resulting in significantly reduced CD8 T cell expansion, a mechanism that is essential for the antitumor immunity response against cancer123. These data suggest that circadian disruption may alter DC-induced T cell responses either through the alteration of their cytokine production or through their migratory capacities.
Finally, also T and B lymphocyte seem to follow the circadian rhythm. For example, it has been demonstrated that their migration from the lymph nodes is regulated by the expression of CCR7 and S1PR1, a mechanism that is fine dependent on BMAL1. Furthermore, the secretion of cytokines (e.g. IL-2, IFNγ) from CD4+ T cells follows diurnal rhythms113. However, Treg cells do not seem to have a functional circadian machinery124. The general idea is that the circadian machinery may significantly impair the adaptive anti-tumor immune responses through the attenuation of T cell homing to the lymph nodes and the reduction of APC-T cell interaction with consequent reduction in T cell activation, an event essential for killing cancer cells.
Noteworthy, mutations in several components of the molecular clock have been linked to chemoresistance125. In this context, immunotherapy (e.g. anti-PD-L1, anti-CTLA4) has been used in recent years to overcome therapy resistance. However, many endocrine cancers, in which the clock genes are deregulated, are immunologically ‘cold’. Of note, data on the role of circadian machinery in anti-tumor immunity suggest a possible role of circadian machinery in the development of cold tumors through the accumulation of immune-suppressive cells in the microenvironment and through the regulation of the expression of PDL1 and CTLA4, two major targets of the current immunotherapy126. All these data suggest that circadian clock components also affect tumor escape mechanisms and, therefore, could be responsible for the induction of cold tumors and the failure for current immunotherapies.
Altogether the existing data suggest that clock genes are essential regulator of the anti-tumor immunity and the alteration of their expression leads to an immunosuppressive environment, leading to cancer progression, metastatization and the development of immune checkpoint escape mechanisms.
Inflammatory diseases
Strong diurnal variations in the symptomatic expression and the severity of several chronic inflammatory diseases have been observed127. Given the role of the molecular clock in regulating immunity and inflammatory pathways under both homeostatic conditions and upon inflammatory challenge, deregulation of such rhythmic control has been suggested to promote an altered immune and inflammatory response. As an example, in mouse models of inflammatory arthritis, disruption of the molecular clock both in vivo, by constant light and genetic approaches, and in vitro, by pharmacological manipulation, altered clock gene expression in the joints, exacerbated joint inflammation, and enhanced arthritis score, thus indicating a direct role of the clock in disease progression127. However, while a robust link between circadian disruption and a number of inflammatory diseases is well documented, understanding of the underpinning molecular mechanisms is nascent. Indeed, although evidence from the literature discussed above reveals connections among the molecular clock, the immune system, and inflammation, as yet, little is known concerning the influence of the circadian clock on immune mechanisms underlying inflammatory disorders and several questions remain unanswered. First, circadian disruption and inflammation have been linked to these diseases independently; how an improper inflammatory response contributes to the onset of pathologies by dysregulating circadian rhythmicity and, vice versa, how circadian disruption triggers pathological inflammatory conditions remains largely unclear. Moreover, it is not known how a pathological condition in a given tissue may affect systemic circadian homeostasis in different tissues. Interestingly, Masri et al. demonstrated that lung adenocarcinoma contributed to the distal remodeling of circadian gene expression and metabolism in the liver via pro-inflammatory mediators, without affecting the hepatic core clock components128. Since the clock components of the liver were not altered, the authors speculated that the tumor may not function as a classical zeitgeber, but rather that it acts on the liver by rewiring circadian metabolic control, thereby dictating the pathophysiological dimension of a distal tissue128.
Therefore, further investigations are needed to decode the molecular mechanisms by which the circadian clock intersects with inflammatory signaling pathways in order to open new avenue for the treatment of inflammatory disorders.
Time as a crucial dimension to medicine
Mounting evidence from the literature indicates that time represents a crucial dimension to medicine for establishing effective treatments in several pathological contexts. In particular, it is widely accepted that a breakthrough in the field of chronotherapy relies on establishing a circadian precision medicine based on treatments delivered in harmony with target physiology. Consistently, rhythmic oscillations ranging from drug absorption, distribution, metabolism, and excretion to the expression of drug targets have been shown to strongly impact on drug pharmacokinetics and pharmacodynamics. A proof of concept comes from clinical trials on patients affected by advanced stage ovarian cancer where the combination therapy, in which doxorubicin was administered at 6 a.m. and cisplatin given 12 h later, led to a fourfold increase in survival of patients, in comparison with patients treated with the same dose of both drugs at the same time (6 p.m)129,130. Moreover, the incidence of some pathological events has been reported to be time of the day dependent. Among the best-known examples, the incidence of myocardial infarction and stroke related to a rapid rise of blood pressure has been observed in the early morning131,132. Moreover, while rheumatoid arthritis patients experience joint stiffness and pain in the early morning, osteoarthritic patients experience pain towards the afternoon. In addition, asthmatic subjects are more prone to nocturnal worsening mainly due to the overnight recruitment of inflammatory cells into the airways133. In this regard, dosing of prednisone in the afternoon (3 p.m.) showed an increased efficacy in counteracting the inflammatory milieu and spirometric decline associated with nocturnal worsening of asthma, compared to its administration in the morning or later in the evening133.
Furthermore, Zhang et al. found that 56 of the top 100 best-selling drugs in the United States target products of a circadian gene24. Moreover, a meta-analysis of clinical trials comparing at least two different times of drug administration schedules revealed that, in the 75%, drug efficacy and/or toxicity relied on the time of the administration, across a number of conditions, such as hypertension, cancer, asthma, and arthritis134.
Targeting circadian rhythm in pathology: pharmacological modulation of circadian machinery
Various therapeutic strategies targeting circadian rhythm are currently under investigation. In particular, besides ongoing clinical trials, not reviewed herein, testing the effects of behavioral and environmental modifications (e.g. light therapy, sleep interventions, and time-restricted feeding) on human health (Table 1), several attempts have been directed toward developing candidate drug molecules that specifically target the core clock components (e.g. REV-ERBs, RORs, PERs, CRYs), or other key regulators of the molecular oscillator (e.g. CK1), as detailed in Table 2.
Small molecules directly targeting the clock proteins offer the potential to directly manipulate endogenous circadian rhythm to improve clock-regulated output processes, as well as to treat diseases associated with clock misalignment (for a comprehensive review on the topic see135. As an example, in in vivo studies, synthetic REV-ERBα/β agonists, such as SR9009 and SR9011, showed remarkable effects in metabolic diseases, cancer, as well as mood disorders136,137,138. In this regard, Solt et al. demonstrated that, in diet-induced obese mice, the administration of a REV-ERBα/β agonists reduced obesity by decreasing fat mass and significantly ameliorating dyslipidaemia and hyperglycemia, thus indicating a potential applicability for the treatment of metabolic diseases136. In addition, Sulli et al. observed that the pharmacological activation of REV-ERBs by SR9009 and SR9011 selectively induced cytotoxicity in leukemia and several solid tumors with different tumorigenesis drivers137. In particular, SR9009 and SR9011 induced apoptosis in cancer cells and oncogene-induced senescent cells, including melanocytic naevi, without affecting the viability of normal cells or tissues, by inactivating two key cancer hallmarks, such as de novo lipogenesis and autophagy137.
Furthermore, several RORs synthetic ligands have been optimized and tested in in vivo studies in different pathological contexts, including autoimmune disorders139,140. Consistently, RORα/RORγ ligands (i.e. SR1001) have been reported to suppress the differentiation of T-helper cells that produce interleukin-17 (Th17 cells), crucial effector cells implicated in the pathology of numerous autoimmune diseases, and to reduce cytokines expression, thereby alleviating autoimmune disease symptoms in animal models of multiple sclerosis139. In line with such results, also non-toxic digoxin-like synthetic derivatives (20,22-dihydrodigoxin-21,23-diol and digoxin-21-salicylidene) have been shown to specifically block IL-17 expression in human CD4+ T cells by inhibiting RORγt activity141.
Another potential therapeutic route is based on the use of CRY-stabilizing compounds142,143. In this regard, the CRY1- and CRY2-stabilizing carbazole small molecule KL001 was first identified in a cell-based screen to lengthen period and reduce amplitude144. Structural studies further demonstrated that KL001 binds to the FAD-binding pocket of CRY and interferes with its recognition by FBXL3, thus hindering CRY ubiquitination and the consequent proteasomal degradation144. Interestingly, analysis of The Cancer Genome Atlas (TCGA) revealed that higher levels of mRNAs encoding the circadian clock repressor CRY2 were associated with improved survival in patients with glioma, thus supporting the hypothesis that the use of CRY-stabilizing compounds might be a promising strategy for glioblastoma therapy in humans145. Within this context, among the developed CRY stabilizers, the most promising compound SHP656, derived from KL001, has been found to specifically inhibit the growth of patient-derived glioblastoma stem cells in vitro, without impacting on differentiated glioblastoma cells or non-malignant epilepsy-derived neural cells145. Moreover, in the mouse model of glioblastoma, treatment with SHP656 reduced tumor growth and prolonged mouse survival145. Recently, isoform-selective compounds (i.e. KL101 and TH301, selectively stabilizing CRY1 and CRY2, respectively) have been developed, thereby offering valuable tools to test in order to assess the potential similarities and differences of CRY1 and CRY2 selective stabilization146.
Based on the growing number of clock modulators with promising pharmacokinetic properties and efficacies in different mouse models of diseases, extending their preclinical efficacy to human trials represent a major future challenge to assess the therapeutic potential of circadian manipulation. In particular, given the role of the core components in controlling key aspects of immune response (e.g. immune cell development, function, and trafficking), the pharmacological modulators of circadian proteins may represent a novel approach to reprogram the tumor microenvironment by promoting anti-tumor immunity and, consequently, impinging upon cancer immune surveillance.
Conclusions
The core clock components represent key molecular players temporally gating many intracellular signaling pathways, ranging from cell growth, DNA repair and DDR, angiogenesis, apoptosis, metabolism, redox state, to immune and inflammatory processes. In this regard, based on the complex scenario depicted by evidence from the literature discussed above, we applied a model-based network approach for the Homo Sapiens proteome recapitulating the mechanistic connection between the molecular clock and the biological pathways herein investigated (Fig. 3). The protein-protein interaction (PPI) network was built starting from a small group of proteins selected from the literature showing a significative evidence to be functionally correlated with the circadian molecular clock. Subsequently, the output generated by STRING (Ver 11.5)147 was manually filtered in order to clearly summarize the interconnection between the core clock components and different cellular pathways.

Protein–protein interaction (PPI) network between the core clock components and clock-related proteins. STRING Protein–Protein Interaction database (Ver 11.5)147 has been used to build the PPI network. The network contains 48 nodes and the edges represent the crosstalk between the core clock components and proteins belonging to other key intracellular pathways. Line thickness reflects the strength of data support for protein-protein interaction that derives from databases, experiments, or based on computational predictions. The network is clustered based on a specified “MCL inflation parameter” and on a customized clustering coefficient of 0.300. The different biological processes investigated are shown in the network according to the color legend
A detailed analysis of the network reported in Fig. 3 shows an intricate and highly interconnected PPI network between the core clock components and several proteins belonging to other key intracellular pathways. In particular, there is peculiar distribution of the proteins into the network in which the clock genes are uniformly grouped forming a dense cluster. The clock-related proteins belonging to different pathways strongly interact each other and concomitantly with all the core clock components, increasing significantly the complexity of the PPI network. Taking into account the complexity of these multiple intra- and interconnections, the circadian clock emerges as a crucial regulator of key physiological processes and its misalignment as potential triggering factor and/or pathological outcome in several human diseases, such as cancer and inflammatory diseases. Although a detailed description of PPI is beyond the aim of our review, such network partially recapitulates the peculiar crosstalk and close association between the intrinsic biological clock and the intracellular signaling pathways described above, with relevant implications in the therapeutic management of diseases. Furthermore, the PPI network shown in Fig. 3 is a clear example of how different intracellular pathways, which appear to be clearly grouped and separated from each other, are functionally interconnected both between the different groups and also between the different members of each group.
Overall, to deep inside the molecular and mechanistic knowledge of circadian pathways, their relationship in pathophysiological processes, as well as the development of novel chronotherapeutics, represent an intriguing and transversal medicine challenge.
References
Halberg, F. Physiologic 24-hour periodicity; general and procedural considerations with reference to the adrenal cycle. Int. Z. Vitaminforsch. Beih. 10, 225–296 (1959).
Harmer, S. L., Panda, S. & Kay, S. A. Molecular bases of circadian rhythms. Annu. Rev. Cell Dev. Biol. 17, 215–253 (2001).
Jagannath, A., Taylor, L., Wakaf, Z., Vasudevan, S. R. & Foster, R. G. The genetics of circadian rhythms, sleep and health. Hum. Mol. Genet. 26, R128–R138 (2017).
Chen, S.-K., Badea, T. C. & Hattar, S. Photoentrainment and pupillary light reflex are mediated by distinct populations of ipRGCs. Nature 476, 92–95 (2011).
Webb, A. B., Angelo, N., Huettner, J. E. & Herzog, E. D. Intrinsic, nondeterministic circadian rhythm generation in identified mammalian neurons. Proc. Natl Acad. Sci. USA 106, 16493–16498 (2009).
Mure, L. S. et al. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359, eaao0318 (2018).
Ueyama, T. et al. Suprachiasmatic nucleus: a central autonomic clock. Nat. Neurosci. 2, 1051–1053 (1999).
Vujovic, N., Davidson, A. J. & Menaker, M. Sympathetic input modulates, but does not determine, phase of peripheral circadian oscillators. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R355–360 (2008).
Buijs, R. M. et al. Anatomical and functional demonstration of a multisynaptic suprachiasmatic nucleus adrenal (cortex) pathway. Eur. J. Neurosci. 11, 1535–1544 (1999).
Kaneko, M., Kaneko, K., Shinsako, J. & Dallman, M. F. Adrenal sensitivity to adrenocorticotropin varies diurnally. Endocrinology 109, 70–75 (1981).
Mahoney, C. E., Brewer, D., Costello, M. K., Brewer, J. M. & Bittman, E. L. Lateralization of the central circadian pacemaker output: a test of neural control of peripheral oscillator phase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R751–761 (2010).
Reddy, A. B. et al. Glucocorticoid signaling synchronizes the liver circadian transcriptome. Hepatology 45, 1478–1488 (2007).
Yamamoto, T. et al. Acute physical stress elevates mouse period1 mRNA expression in mouse peripheral tissues via a glucocorticoid-responsive element. J. Biol. Chem. 280, 42036–42043 (2005).
So, A. Y.-L., Bernal, T. U., Pillsbury, M. L., Yamamoto, K. R. & Feldman, B. J. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc. Natl Acad. Sci. USA 106, 17582–17587 (2009).
Ikeda, M. et al. Circadian dynamics of cytosolic and nuclear Ca2+ in single suprachiasmatic nucleus neurons. Neuron 38, 253–263 (2003).
Lundkvist, G. B., Kwak, Y., Davis, E. K., Tei, H. & Block, G. D. A calcium flux is required for circadian rhythm generation in mammalian pacemaker neurons. J. Neurosci. 25, 7682–7686 (2005).
Nahm, S.-S., Farnell, Y. Z., Griffith, W. & Earnest, D. J. Circadian regulation and function of voltage-dependent calcium channels in the suprachiasmatic nucleus. J. Neurosci. 25, 9304–9308 (2005).
O’Neill, J. S., Maywood, E. S., Chesham, J. E., Takahashi, J. S. & Hastings, M. H. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science 320, 949–953 (2008).
Travnickova-Bendova, Z., Cermakian, N., Reppert, S. M. & Sassone-Corsi, P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc. Natl Acad. Sci. USA 99, 7728–7733 (2002).
de Jeu, M., Hermes, M. & Pennartz, C. Circadian modulation of membrane properties in slices of rat suprachiasmatic nucleus. Neuroreport 9, 3725–3729 (1998).
Minnetti, M. et al. Fixing the broken clock in adrenal disorders: focus on glucocorticoids and chronotherapy. J. Endocrinol. 246, R13–R31 (2020).
Panda, S. et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109, 307–320 (2002).
Storch, K.-F. et al. Extensive and divergent circadian gene expression in liver and heart. Nature 417, 78–83 (2002).
Zhang, R., Lahens, N. F., Ballance, H. I., Hughes, M. E. & Hogenesch, J. B. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl Acad. Sci. USA 111, 16219–16224 (2014).
Matsuo, T. et al. Control mechanism of the circadian clock for timing of cell division in vivo. Science 302, 255–259 (2003).
Kondratov, R. V. & Antoch, M. P. Circadian proteins in the regulation of cell cycle and genotoxic stress responses. Trends Cell Biol. 17, 311–317 (2007).
Shafi, A. A. et al. The circadian cryptochrome, CRY1, is a pro-tumorigenic factor that rhythmically modulates DNA repair. Nat. Commun. 12, 401 (2021).
Jensen, L. D. et al. Opposing effects of circadian clock genes bmal1 and period2 in regulation of VEGF-dependent angiogenesis in developing zebrafish. Cell Rep. 2, 231–241 (2012).
Hunt, T. & Sassone-Corsi, P. Riding tandem: circadian clocks and the cell cycle. Cell 129, 461–464 (2007).
Gréchez-Cassiau, A., Rayet, B., Guillaumond, F., Teboul, M. & Delaunay, F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J. Biol. Chem. 283, 4535–4542 (2008).
Repouskou, A. & Prombona, A. c-MYC targets the central oscillator gene Per1 and is regulated by the circadian clock at the post-transcriptional level. Biochim. Biophys. Acta 1859, 541–552 (2016).
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kaçmaz, K. & Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 (2004).
Gery, S. et al. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 22, 375–382 (2006).
Kang, T.-H. & Leem, S.-H. Modulation of ATR-mediated DNA damage checkpoint response by cryptochrome 1. Nucleic Acids Res. 42, 4427–4434 (2014).
Miki, T., Matsumoto, T., Zhao, Z. & Lee, C. C. p53 regulates Period2 expression and the circadian clock. Nat. Commun. 4, 2444 (2013).
Gotoh, T., Vila-Caballer, M., Liu, J., Schiffhauer, S. & Finkielstein, C. V. Association of the circadian factor Period 2 to p53 influences p53’s function in DNA-damage signaling. Mol. Biol. Cell 26, 359–372 (2015).
Jiang, W. et al. The circadian clock gene Bmal1 acts as a potential anti-oncogene in pancreatic cancer by activating the p53 tumor suppressor pathway. Cancer Lett. 371, 314–325 (2016).
Gu, X. et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ. 19, 397–405 (2012).
Koyanagi, S. et al. A molecular mechanism regulating circadian expression of vascular endothelial growth factor in tumor cells. Cancer Res. 63, 7277–7283 (2003).
Takahata, S. et al. Transcriptionally active heterodimer formation of an Arnt-like PAS protein, Arnt3, with HIF-1a, HLF, and clock. Biochem. Biophys. Res. Commun. 248, 789–794 (1998).
Wu, Y. et al. Reciprocal regulation between the circadian clock and hypoxia signaling at the genome level in mammals. Cell Metab. 25, 73–85 (2017).
Hogenesch, J. B., Gu, Y. Z., Jain, S. & Bradfield, C. A. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc. Natl Acad. Sci. USA 95, 5474–5479 (1998).
O’Connell, E. J., Martinez, C.-A., Liang, Y. G., Cistulli, P. A. & Cook, K. M. Out of breath, out of time: interactions between HIF and circadian rhythms. Am. J. Physiol. Cell Physiol. 319, C533–C540 (2020).
Peek, C. B. et al. Circadian clock interaction with HIF1α mediates oxygenic metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab. 25, 86–92 (2017).
Peek, C. B. et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417 (2013).
Ilardo, M. & Nielsen, R. Human adaptation to extreme environmental conditions. Curr. Opin. Genet. Dev. 53, 77–82 (2018).
Huerta-Sánchez, E. et al. Genetic signatures reveal high-altitude adaptation in a set of Ethiopian populations. Mol. Biol. Evol. 30, 1877–1888 (2013).
He, Y. et al. The transcriptional repressor DEC2 regulates sleep length in mammals. Science 325, 866–870 (2009).
Semenza, G. L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365, 537–547 (2011).
Scheiermann, C., Kunisaki, Y. & Frenette, P. S. Circadian control of the immune system. Nat. Rev. Immunol. 13, 190–198 (2013).
Carroll, R. G., Timmons, G. A., Cervantes-Silva, M. P., Kennedy, O. D. & Curtis, A. M. Immunometabolism around the Clock. Trends Mol. Med. 25, 612–625 (2019).
Méndez-Ferrer, S., Lucas, D., Battista, M. & Frenette, P. S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 (2008).
Hayashi, M., Shimba, S. & Tezuka, M. Characterization of the molecular clock in mouse peritoneal macrophages. Biol. Pharm. Bull. 30, 621–626 (2007).
Man, K., Loudon, A. & Chawla, A. Immunity around the clock. Science 354, 999–1003 (2016).
Nguyen, K. D. et al. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science 341, 1483–1488 (2013).
Gibbs, J. E. et al. The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl Acad. Sci. USA 109, 582–587 (2012).
Sato, S. et al. A circadian clock gene, Rev-erbα, modulates the inflammatory function of macrophages through the negative regulation of Ccl2 expression. J. Immunol. 192, 407–417 (2014).
Eichenfield, D. Z. et al. Tissue damage drives co-localization of NF-κB, Smad3, and Nrf2 to direct Rev-erb sensitive wound repair in mouse macrophages. Elife 5, e13024 (2016).
Ratman, D. et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol. Cell. Endocrinol. 380, 41–54 (2013).
Gibbs, J. et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat. Med. 20, 919–926 (2014).
Balsalobre, A. et al. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289, 2344–2347 (2000).
Yurtsever, T. et al. The acute and temporary modulation of PERIOD genes by hydrocortisone in healthy subjects. Chronobiol. Int. 33, 1222–1234 (2016).
Reddy, T. E. et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19, 2163–2171 (2009).
Murayama, Y. et al. Glucocorticoid receptor suppresses gene expression of Rev-erbα (Nr1d1) through interaction with the CLOCK complex. FEBS Lett. 593, 423–432 (2019).
Nader, N., Chrousos, G. P. & Kino, T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB. J. Publ. Fed. Am. Soc. Exp. Biol. 23, 1572–1583 (2009).
Lamia, K. A. et al. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature 480, 552–556 (2011).
Poolman, T. M. et al. Rheumatoid arthritis reprograms circadian output pathways. Arthritis Res. Ther. 21, 47 (2019).
Wang, S. et al. REV-ERBα integrates colon clock with experimental colitis through regulation of NF-κB/NLRP3 axis. Nat. Commun. 9, 4246 (2018).
Narasimamurthy, R. et al. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc. Natl Acad. Sci. USA 109, 12662–12667 (2012).
Lee, J. H. & Sancar, A. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc. Natl Acad. Sci. USA 108, 12036–12041 (2011).
Spengler, M. L. et al. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc. Natl Acad. Sci. USA 109, E2457–E2465 (2012).
Sato, S. et al. Direct and indirect suppression of interleukin-6 gene expression in murine macrophages by nuclear orphan receptor REV-ERBα. ScientificWorldJournal 2014, 685854 (2014).
Maury, E., Navez, B. & Brichard, S. M. Circadian clock dysfunction in human omental fat links obesity to metabolic inflammation. Nat. Commun. 12, 2388 (2021).
Hong, H.-K. et al. Requirement for NF-κB in maintenance of molecular and behavioral circadian rhythms in mice. Genes Dev. 32, 1367–1379 (2018).
Rutter, J., Reick, M., Wu, L. C. & McKnight, S. L. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293, 510–514 (2001).
Carter, E. L., Gupta, N. & Ragsdale, S. W. High affinity heme binding to a heme regulatory motif on the nuclear receptor Rev-erbβ leads to its degradation and indirectly regulates its interaction with nuclear receptor corepressor. J. Biol. Chem. 291, 2196–2222 (2016).
Freeman, S. L. et al. Heme binding to human CLOCK affects interactions with the E-box. Proc. Natl Acad. Sci. USA 116, 19911–19916 (2019).
Ramsey, K. M. et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654 (2009).
Nakahata, Y., Sahar, S., Astarita, G., Kaluzova, M. & Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657 (2009).
Asher, G. et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328 (2008).
Lee, Y. & Kim, E.-K. AMP-activated protein kinase as a key molecular link between metabolism and clockwork. Exp. Mol. Med. 45, e33 (2013).
Khapre, R. V. et al. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 6, 48–57 (2014).
Wang, T. A. et al. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science 337, 839–842 (2012).
Hayes, J. D. & Dinkova-Kostova, A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218 (2014).
Niture, S. K., Khatri, R. & Jaiswal, A. K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 66, 36–44 (2014).
Kobayashi, E. H. et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 7, 11624 (2016).
Mills, E. L. et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018).
Wardyn, J. D., Ponsford, A. H. & Sanderson, C. M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 43, 621–626 (2015).
Rey, G. et al. The pentose phosphate pathway regulates the circadian clock. Cell Metab. 24, 462–473 (2016).
Wible, R. S. et al. NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. Elife 7, e31656 (2018).
Pekovic-Vaughan, V. et al. The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 28, 548–560 (2014).
Early, J. O. et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl Acad. Sci. USA 115, E8460–E8468 (2018).
Shafi, A. A. & Knudsen, K. E. Cancer and the circadian clock. Cancer Res. 79, 3806–3814 (2019).
Kamdar, B. B., Needham, D. M. & Collop, N. A. Sleep deprivation in critical illness: its role in physical and psychological recovery. J. Intensive Care Med. 27, 97–111 (2012).
Pan, A., Schernhammer, E. S., Sun, Q. & Hu, F. B. Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women. PLoS Med. 8, e1001141 (2011).
Haupt, C. M. et al. The relation of exposure to shift work with atherosclerosis and myocardial infarction in a general population. Atherosclerosis 201, 205–211 (2008).
Stephenson, E. M., Usselmann, L. E. J., Tergaonkar, V., Virshup, D. M. & Dallmann, R. Cancer clocks in tumourigenesis: the p53 pathway and beyond. Endocr. Relat. Cancer 28, R95–R110 (2021).
Reszka, E. & Zienolddiny, S. Epigenetic basis of circadian rhythm disruption in cancer. Methods Mol. Biol. 1856, 173–201 (2018).
Masri, S., Kinouchi, K. & Sassone-Corsi, P. Circadian clocks, epigenetics, and cancer. Curr. Opin. Oncol. 27, 50–56 (2015).
Lie, J.-A. S. et al. Night work and breast cancer risk among Norwegian nurses: assessment by different exposure metrics. Am. J. Epidemiol. 173, 1272–1279 (2011).
Papantoniou, K. et al. Night shift work, chronotype and prostate cancer risk in the MCC-Spain case-control study. Int. J. Cancer 137, 1147–1157 (2015).
Schernhammer, E. S. et al. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl Cancer Inst. 93, 1563–1568 (2001).
Kakizaki, M. et al. Sleep duration and the risk of breast cancer: the Ohsaki Cohort Study. Br. J. Cancer 99, 1502–1505 (2008).
Urbanski, H. F. Role of circadian neuroendocrine rhythms in the control of behavior and physiology. Neuroendocrinology 93, 211–222 (2011).
Reinke, H. & Asher, G. Crosstalk between metabolism and circadian clocks. Nat. Rev. Mol. Cell Biol. 20, 227–241 (2019).
Gutiérrez-Monreal, M. A., Treviño, V., Moreno-Cuevas, J. E. & Scott, S.-P. Identification of circadian-related gene expression profiles in entrained breast cancer cell lines. Chronobiol. Int. 33, 392–405 (2016).
Ye, Y. et al. The genomic landscape and pharmacogenomic interactions of clock genes in cancer chronotherapy. Cell Syst. 6, 314–328.e2 (2018).
El-Kenawi, A., Hänggi, K. & Ruffell, B. The immune microenvironment and cancer metastasis. Cold Spring Harb. Perspect. Med. 10, a037424 (2020).
Keller, M. et al. A circadian clock in macrophages controls inflammatory immune responses. Proc. Natl Acad. Sci. USA 106, 21407–21412 (2009).
Silver, A. C., Arjona, A., Hughes, M. E., Nitabach, M. N. & Fikrig, E. Circadian expression of clock genes in mouse macrophages, dendritic cells, and B cells. Brain. Behav. Immun. 26, 407–413 (2012).
Ella, K., Csépányi-Kömi, R. & Káldi, K. Circadian regulation of human peripheral neutrophils. Brain. Behav. Immun. 57, 209–221 (2016).
Arjona, A. & Sarkar, D. K. Circadian oscillations of clock genes, cytolytic factors, and cytokines in rat NK cells. J. Immunol. 174, 7618–7624 (2005).
Bollinger, T. et al. Circadian clocks in mouse and human CD4+ T cells. PLoS ONE 6, e29801 (2011).
Trott, A. J. & Menet, J. S. Regulation of circadian clock transcriptional output by CLOCK:BMAL1. PLoS Genet. 14, e1007156 (2018).
Oishi, Y. et al. Bmal1 regulates inflammatory responses in macrophages by modulating enhancer RNA transcription. Sci. Rep. 7, 7086 (2017).
Owyong, M. et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci. Alliance 2, e201800226 (2019).
Hadadi, E. et al. Chronic circadian disruption modulates breast cancer stemness and immune microenvironment to drive metastasis in mice. Nat. Commun. 11, 3193 (2020).
Masucci, M. T., Minopoli, M. & Carriero, M. V. Tumor associated neutrophils. their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 9, 1146 (2019).
Adrover, J. M. et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity 50, 390–402.e10 (2019).
Adrover, J. M. et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 21, 135–144 (2020).
Teijeira, Á. et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity 52, 856–871.e8 (2020).
Hopwood, T. W. et al. The circadian regulator BMAL1 programmes responses to parasitic worm infection via a dendritic cell clock. Sci. Rep. 8, 3782 (2018).
Nobis, C. C. et al. The circadian clock of CD8 T cells modulates their early response to vaccination and the rhythmicity of related signaling pathways. Proc. Natl Acad. Sci. USA 116, 20077–20086 (2019).
Hand, L. E. et al. Regulatory T cells confer a circadian signature on inflammatory arthritis. Nat. Commun. 11, 1658 (2020).
Fang, L. et al. Circadian Clock Gene CRY2 Degradation Is Involved in Chemoresistance of Colorectal Cancer. Mol. Cancer Ther. 14, 1476–1487 (2015).
Zhou, J. et al. The aberrant expression of rhythm genes affects the genome instability and regulates the cancer immunity in pan-cancer. Cancer Med. 9, 1818–1829 (2020).
Hand, L. E. et al. The circadian clock regulates inflammatory arthritis. FASEB J. 30, 3759–3770 (2016).
Masri, S. et al. Lung adenocarcinoma distally rewires hepatic circadian homeostasis. Cell 165, 896–909 (2016).
Kobayashi, M., Wood, P. A. & Hrushesky, W. J. M. Circadian chemotherapy for gynecological and genitourinary cancers. Chronobiol. Int. 19, 237–251 (2002).
Lévi, F. et al. Chemotherapy of advanced ovarian cancer with 4’-O-tetrahydropyranyl doxorubicin and cisplatin: a randomized phase II trial with an evaluation of circadian timing and dose-intensity. J. Clin. Oncol. 8, 705–714 (1990).
Muller, J. E. et al. Circadian variation in the frequency of onset of acute myocardial infarction. N. Engl. J. Med. 313, 1315–1322 (1985).
Kanth, R., Ittaman, S. & Rezkalla, S. Circadian patterns of ST elevation myocardial infarction in the new millennium. Clin. Med. Res. 11, 66–72 (2013).
Beam, W. R., Weiner, D. E. & Martin, R. J. Timing of prednisone and alterations of airways inflammation in nocturnal asthma. Am. Rev. Respir. Dis. 146, 1524–1530 (1992).
Ruben, M. D., Smith, D. F., FitzGerald, G. A. & Hogenesch, J. B. Dosing time matters. Science 365, 547–549 (2019).
Chen, Z., Yoo, S.-H. & Takahashi, J. S. Development and therapeutic potential of small-molecule modulators of circadian systems. Annu. Rev. Pharmacol. Toxicol. 58, 231–252 (2018).
Solt, L. A. et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 485, 62–68 (2012).
Sulli, G. et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 553, 351–355 (2018).
Banerjee, S. et al. Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nat. Commun. 5, 5759 (2014).
Solt, L. A. et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472, 491–494 (2011).
Kumar, N. et al. The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Mol. Pharmacol. 77, 228–236 (2010).
Huh, J. R. et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 472, 486–490 (2011).
Humphries, P. S. et al. Carbazole-containing sulfonamides and sulfamides: discovery of cryptochrome modulators as antidiabetic agents. Bioorg. Med. Chem. Lett. 26, 757–760 (2016).
Humphries, P. S. et al. Carbazole-containing amides and ureas: discovery of cryptochrome modulators as antihyperglycemic agents. Bioorg. Med. Chem. Lett. 28, 293–297 (2018).
Hirota, T. et al. Identification of small molecule activators of cryptochrome. Science 337, 1094–1097 (2012).
Dong, Z. et al. Targeting glioblastoma stem cells through disruption of the circadian clock. Cancer Discov. 9, 1556–1573 (2019).
Miller, S. et al. Isoform-selective regulation of mammalian cryptochromes. Nat. Chem. Biol. 16, 676–685 (2020).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Chu, G., Zhou, X., Hu, Y., Shi, S. & Yang, G. Rev-erbα inhibits proliferation and promotes apoptosis of preadipocytes through the agonist GSK4112. Int. J. Mol. Sci. 20, 4524 (2019).
Shao, R. et al. REV-ERBα agonist GSK4112 attenuates Fas-induced acute hepatic damage in mice. Int. J. Med. Sci. 18, 3831–3838 (2021).
Guo, D.-K. et al. Pharmacological activation of REV-ERBα represses LPS-induced microglial activation through the NF-κB pathway. Acta Pharmacol. Sin. 40, 26–34 (2019).
Lee, J. et al. Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell 19, e13078 (2020).
Guo, L. et al. Targeted inhibition of Rev-erb-α/β limits ferroptosis to ameliorate folic acid-induced acute kidney injury. Br. J. Pharmacol. 178, 328–345 (2021).
De Mei, C. et al. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene 34, 2597–2608 (2015).
He, B. et al. The small molecule nobiletin targets the molecular oscillator to enhance circadian rhythms and protect against metabolic syndrome. Cell Metab. 23, 610–621 (2016).
Nohara, K. et al. Nobiletin fortifies mitochondrial respiration in skeletal muscle to promote healthy aging against metabolic challenge. Nat. Commun. 10, 3923 (2019).
Zapadka, T. E. et al. RORγt inhibitor-SR1001 halts retinal inflammation, capillary degeneration, and the progression of diabetic retinopathy. Int. J. Mol. Sci. 21, 3547 (2020).
Zhang, Q. et al. Targeting Th17-IL-17 pathway in prevention of micro-invasive prostate cancer in a mouse model. Prostate 77, 888–899 (2017).
Kumar, N. et al. Identification of SR3335 (ML-176): a synthetic RORα selective inverse agonist. ACS Chem. Biol. 6, 218–222 (2011).
Rajput, C. et al. RORα-dependent type 2 innate lymphoid cells are required and sufficient for mucous metaplasia in immature mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L983–L993 (2017).
Wang, Y., Billon, C., Walker, J. K. & Burris, T. P. Therapeutic effect of a synthetic RORα/γ agonist in an animal model of autism. ACS Chem. Neurosci. 7, 143–148 (2016).
Byun, J.-K. et al. Retinoic acid-related orphan receptor alpha reprograms glucose metabolism in glutamine-deficient hepatoma cells. Hepatology 61, 953–964 (2015).
Lee, J. et al. Digoxin ameliorates autoimmune arthritis via suppression of Th17 differentiation. Int. Immunopharmacol. 26, 103–111 (2015).
Saeed, H., Mateen, S., Moin, S., Khan, A. Q. & Owais, M. Cardiac glycoside digoxin ameliorates pro-inflammatory cytokines in PBMCs of rheumatoid arthritis patients in vitro. Int. Immunopharmacol. 82, 106331 (2020).
Gao, M. et al. Orphan nuclear receptor RORγ confers doxorubicin resistance in prostate cancer. Cell Biol. Int. 44, 2170–2176 (2020).
Chang, M. R., Lyda, B., Kamenecka, T. M. & Griffin, P. R. Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor γ is therapeutic in the collagen-induced arthritis experimental model. Arthritis Rheumatol. (Hoboken, N. J.) 66, 579–588 (2014).
Chun, S. K. et al. A synthetic cryptochrome inhibitor induces anti-proliferative effects and increases chemosensitivity in human breast cancer cells. Biochem. Biophys. Res. Commun. 467, 441–446 (2015).
Yuan, F. et al. IC261 suppresses progression of hepatocellular carcinoma in a casein kinase 1 δ/ε independent manner. Biochem. Biophys. Res. Commun. 523, 809–815 (2020).
Janovska, P. et al. Casein kinase 1 is a therapeutic target in chronic lymphocytic leukemia. Blood 131, 1206–1218 (2018).
Adler, P., Mayne, J., Walker, K., Ning, Z. & Figeys, D. Therapeutic targeting of casein kinase 1δ/ε in an Alzheimer’s disease mouse model. J. Proteome Res. 18, 3383–3393 (2019).
Acknowledgements
This work has been supported by the University of Pavia (grants from FR&G 2020, Fondo Ricerca & Giovani, to C.L.; PRIN 2017B9NCSX_003 to M.R.; an Educational Grant from Aboca S.p.A. to M.R. and S.G.).
Author information
Authors and Affiliations
Contributions
F.F. and C.L. conceived the idea. F.F., and C.L. wrote the manuscript. D.D.M. and A.R. performed network analysis. D.V. prepared the tables. F.F., D.D.M., C.T., D.V., S.G., M.R., and C.L. contributed to the critical discussion. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fagiani, F., Di Marino, D., Romagnoli, A. et al. Molecular regulations of circadian rhythm and implications for physiology and diseases. Sig Transduct Target Ther 7, 41 (2022). https://doi.org/10.1038/s41392-022-00899-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-00899-y
This article is cited by
-
Deficiency of circadian clock gene Bmal1 exacerbates noncanonical inflammasome-mediated pyroptosis and lethality via Rev-erbα-C/EBPβ-SAA1 axis
Experimental & Molecular Medicine (2024)
-
Investigation of the rhythmic recruitment of tear neutrophils to the ocular surface and their phenotypes
Scientific Reports (2024)
-
Circadian regulation of cancer stem cells and the tumor microenvironment during metastasis
Nature Cancer (2024)
-
Melatonin Attenuates Imiquimod-Induced Psoriasis-Like Inflammation and Restores the Th17/Treg Immune Balance
Inflammation (2024)
-
Combined use of multiparametric high-content-screening and in vitro circadian reporter assays in neurotoxicity evaluation
Archives of Toxicology (2024)
