
Abstract
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway was discovered more than a quarter-century ago. As a fulcrum of many vital cellular processes, the JAK/STAT pathway constitutes a rapid membrane-to-nucleus signaling module and induces the expression of various critical mediators of cancer and inflammation. Growing evidence suggests that dysregulation of the JAK/STAT pathway is associated with various cancers and autoimmune diseases. In this review, we discuss the current knowledge about the composition, activation, and regulation of the JAK/STAT pathway. Moreover, we highlight the role of the JAK/STAT pathway and its inhibitors in various diseases.
Similar content being viewed by others

Introduction
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is regarded as one of the central communication nodes in the cell function. More than 50 cytokines and growth factors have been identified in the JAK/STAT signaling pathway, such as hormones, interferons (IFN), interleukins (ILs), and colony-stimulating factors.1 JAK/STAT-mediated downstream events vary and include hematopoiesis, immune fitness, tissue repair, inflammation, apoptosis, and adipogenesis.2 Loss or mutation of JAK/STAT components is related to many diseases in humans. JAKs are noncovalently associated with cytokine receptors, mediate tyrosine phosphorylation of receptors, and recruit one or more STAT proteins. Tyrosine-phosphorylated STATs dimerize and are then transported into the nucleus through the nuclear membrane to regulate specific genes. Although STATs can be activated by partially overlapping cytokines, different STATs have nonredundant biological effects.3
The JAK/STAT signaling pathway has profoundly influenced recent understanding attained of human health and disease. Many papers have reported the importance of this pathway in malignancies and autoimmune diseases.4,5,6,7,8,9 Thus, inhibiting the JAK/STAT pathway is promising for treating various diseases. Currently, many JAK inhibitors have achieved efficacy in many clinical settings, and more medications are currently being studied.10 In this review, we aim to provide updated and comprehensive views of the JAK/STAT signaling pathway at the cellular, molecular, and genomic levels, and elucidate the relationship between JAK/STAT pathway components and human diseases. Finally, we focus on the current market-approved and preclinical medications designed to target this pathway.
Discovery of the JAK/STAT signaling pathway
The JAK/STAT signaling pathway was first discovered when studying how IFNs lead to the activation of a transcription factor.11 In 1990, the transcriptional activator interferon-stimulated gene factor 3 (ISGF3), a transcription factor that responds to IFN-α, was discovered to be composed of multiple interacting polypeptide chains (48, 84, 91, and 113 kDa).11 In 1992, Fu reported that 113, 91, and 84 kDa (ISGF3α) proteins of ISGF3 contain conserved SH2 and SH3 domains. Moreover, a specific IFN-α-induced cytoplasmic tyrosine kinase can phosphorylate and activate ISGF3α. Thus, Fu proposed a direct effector model for signal transduction induced by IFN-α, which revealed the signal transduction mode of the JAK/STAT signaling pathway.12,13 Later studies have identified that ISGF3 is comprised of STAT1, STAT2, and IRF9.14 Since then, STAT3, STAT4, STAT5a, STAT5b, and STAT6 were found in several laboratories during 1993–1995.15,16,17
The discovery of JAKs happened in 1989–1994. In 1989, Wilks et al. identified that a tyrosine kinase has a recognizable kinase domain and a pseudokinase domain. In 1991, they found a second tyrosine kinase with this feature. Wilks called them JAK1 and JAK2.18,19 The other two JAKs, tyrosine kinase 2 (TYK2) and JAK3, were identified in 1990 and 1994.20 The connection between JAKs and STATs began in 1992 when Velazquez et al. discovered that TYK2 is an essential protein in the IFN-α/β signaling pathway.21 Later, Müller et al. found that IFN-dependent signaling required JAKs to phosphorylate STATs.22 Thus, in the late 1980s to early 1990s, the components and outlines of the JAK/STAT signaling pathway were completed. Research on more proteins and functions of the JAK/STAT pathway has continued to the present, making the JAK/STAT landscape more abundant.
Composition of the JAK/STAT pathway
The JAK/STAT signaling pathway is evolutionarily conserved. It is composed of ligand-receptor complexes, JAKs, and STATs. There are 4 members in the JAK family: JAK1, JAK2, JAK3, and TYK2. The STAT family comprises seven members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. We introduce them by family.
The JAK family: JAK1, JAK2, TYK2, and JAK3
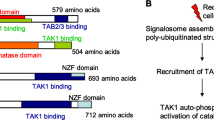
JAK family consists of non-receptor tyrosine protein kinases (Fig. 1). When cytokines bind to their receptors, JAK tyrosine kinases are activated and transmit regulatory signals. The JAK family has four main members, JAK1, JAK2, JAK3, and TYK2. JAK3 is only expressed in the bone marrow and lymphatic system, as well as endothelial cells and vascular smooth muscle cells,23,24 other members are expressed in almost all tissues.19,20,25,26,27,28,29 JAKs have seven homology domains (the JAK homology domain, JH). Starting from the carboxyl terminus, JH1 is the first JH, known as the kinase domain, and is composed of approximately 250 amino acid residues. JH1 encodes a kinase protein that constitutes the kinase structure domain that phosphorylates a substrate; JH2 is a PK domain. JH2 is structurally similar to the kinase domain but has no kinase activity. Its main function is to regulate the activity of the kinase domain. The pseudokinase domain participates in the interaction of JAK and STAT, and the PK domain can also inhibit Tyr kinase activity by binding to the kinase domain; JH3 with one-half of JH4 constitutes the Src-homology 2(SH2) domain, the combination of one-half of JH4, JH5, JH6, and JH7 constitutes the FERM domain, and the SH2 and FERM domains mainly regulate the binding of JAK and cytokine-receptor membrane-proximal box1/2 regions.19,25,30,31,32

Structure of JAKs. a Structure and conserved phosphorylation sites of the JAK family. The JAK family has four main members: JAK1, JAK2, JAK3, and TYK2. Each is composed of seven homology domains (JH), of which JH1 constitutes the kinase domain; JH2 constitutes the pseudokinase domain; a part of JH3 and JH4 together constitute the SH2 domain; and the FERM domain is composed of the JH5, JH6, and part of the JH4 domains. The conserved tyrosine phosphorylation sites in JAK1 are Y1038/Y1039; the conserved tyrosine phosphorylation sites in JAK2 are Y1007/Y1008; the conserved tyrosine phosphorylation sites in JAK3 are Y980/Y981; the conserved tyrosine phosphorylation sites in Tyk2 are Y1054/Y1055. b Structure of JAKs and targeting sites of JAK inhibitors. Created with BioRender.com
JAK1
Y1038/Y1039 in JAK1 is a conserved tyrosine that constitutes a vital part of the activation loop. The phosphorylation of a double tyrosine in the SH1 domain of each JAK results in a more favorable conformation for substrate binding.33 JAK1 is widely expressed in tissues and can phosphorylate all STATs.4 JAK1 is phosphorylated by four cytokine-receptor families: (1) Cytokine receptors with the γc receptor subunit, IL-2 receptor, IL-4 receptor, IL-7 receptor, IL-9 receptor, and IL-15 receptor; (2) class II cytokine receptors include the IFNα/β receptor, IFN-γ receptor, and IL-10 family cytokine receptors; and (3) receptors with a gp130 subunit, including the IL-6 receptor, IL-11 receptor, ciliary neurotrophic factor (CNTF) receptor, oncostatin M (OSM) receptor, leukemia inhibitory factor (LIF) receptor, and cardiotrophin-1 (CT-1) receptor.34 JAK1 can promote body haematopoietic function after being activated by IL-3, IL-5, IL-7, granulocyte–macrophage colony-stimulating factor (GM-CSF), or granulocyte colony-stimulating factor (G-CSF).35 JAK1−/− mice are perinatal dead and exhibit neurological disease and severe lymphocyte damage caused by deficient of LIF and IL-7 signal, respectively.34
JAK2
The conserved tyrosine sites in JAK2 are Y1007 and Y1008.33 Similar to JAK1, JAK2 can also be phosphorylated by members of the gp130 receptor family and class II cytokine-receptor family. It also participates in the signal transduction of the IL-3 receptor family (IL-3R, IL-5R, and GM-CSF receptor), and single-chain receptors (such as erythropoietin receptor (EPO), growth hormone (GH) receptor, prolactin receptor, and thrombopoietin (TPO) receptor).36 JAK2-knockout mice die at approximately 12 days of gestation primarily due to the impaired hematopoietic function mediated by EPO. Therefore, the embryonic lethality of JAK2-knockout mice and EPO-knockout mice is very similar.37,38 JAK2-knockout mice exhibit specific defects in IFN-γ-related biological responses, but they do not respond to IFN-α or IFN-β.
JAK3
Y980/Y98 in JAK3 are the conserved phosphorylation sites.33 JAK3 is mainly involved in the signal transduction of the IL-2 receptor, IL-4 receptor, IL-7 receptor, IL-9 receptor, IL-15 receptor, and IL-21 receptor. These receptors are γC receptors with the γ receptor chain.39 JAK3-knockout mice are defective in lymphocyte production due to the lack of γC signaling. These mice are very likely to have severe combined immunodeficiency, but JAK3-knockout mice can still survive in the absence of specific pathogens.40,41 IL-2, IL-4, and IL-7 transmit growth signals through JAK3, and autoreactive T cells in JAK3-deficient mice are permanently activated. Lack of JAK3 may lead to autosomal recessive combined immunodeficiency, indicating that JAK3 plays an important regulatory role in the negative selection of T cells and the maintenance of the normal phenotype and function of peripheral T cells.42
TYK2
Y1054/Y1055 in Tyk2 are conservative phosphorylation sites.33 Tyk2 was the first discovered member of the JAK family and was originally found to be able to transmit IFN-α/β signals.43 Later, it was discovered that Tyk2 is also involved in IL-6,44 IL-10,45 IL-12,46 IL-13,47 and IL-23 signaling.48 Interestingly, Tyk2-knockout mice do not completely lose cytokine signaling and exhibit partial defects in IFN-α, IFN-β, and IL-12 signal transduction.49 Tyk2-defective mice show an insufficient response to a small amount of IFN-α, and increasing the amount of IFN-α can restore signal transduction. Thus, Tyk2 does not seem necessary for type I interferon signal transduction.50 Moreover, Tyk2 regulates the balance of Th1 and Th2 cells in mice and regulates the allergic reaction mediated by Th2 cells.51 The symptoms of Tyk2 deficiency in human are somewhat different from those in mice. In the clinic, patients with high immunoglobulin E syndrome have defective signal transduction of IFN-α, IL-12, IL-6, and IL-10, which can be alleviated by treating with Tyk2 gene transduction therapy. In patients with Tyk2 deficiency, the phosphorylation of STAT cannot be detected even when they are treated with high concentrations of IFN-α. Tyk2-defective humans develop severe allergic phenotypes due to IFN-mediated loss of antimicrobial capacity. These studies have shown that Tyk2 plays a necessary role in human innate and acquired immunity (Table 1).50
The STAT family: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6
STAT family is composed of STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 (Fig. 2). STAT family members consist of 750–900 amino acids. From the N-terminus to the C-terminus, there are the N-terminal domain and coil, helix domain, DNA-binding domain, connection domain, SH2 domain, and transcription-activation domain. Six domains regulate different functions of STAT.12,15,17,52,53,54,55 (1) The N-terminal domain promotes the formation of STAT dimers, which enables their subsequent binding with transcription factors. Studies have also shown that the N-terminus can also promote the interaction of STAT and transcription co-activators, the PIAS family, and receptors and regulate nuclear translocation.56,57,58,59 (2) The coiled-coil domain is composed of a potentially dynamic four-helix bundle. This domain is related to regulatory proteins and participates in the control of nuclear import and export processes. It can interact with p48/IRF9, Nmi, c-Jun, StlP, etc.60,61,62,63,64,65,66 (3) The linking domain, as the name implies, structurally connects the DNA-binding domain to the SH2 domain. It is involved in the transcriptional regulation of STAT1.63,67 (4) The DNA-binding domain can recognize and bind to the DNA sequence in the regulatory region of the target gene. It also participates in the regulation of nuclear import and export. (5) The SH2 domain of STAT is very different from other SH2 domains, but this domain is very conserved in the STAT family.68 The primary function of SH2 is to recognize phosphotyrosine motifs of cytokine receptors. Moreover, the SH2 domain cooperates with activated JAK to drive the SH2 domain of STAT to interact with the tail of another STAT monomer after phosphorylation to form a homodimer or heterodimer.69,70,71,72 (6) The transcriptional activation domain is critical for DNA transcription elements and the recruitment of co-activators through a conserved serine phosphorylation site and regulating the transcription. STAT4, STAT5, and STAT6 can be used as targets for ubiquitin-dependent destruction, while STAT1, STAT2, and STAT3 are more stable, indicating that the transcriptional active region also regulates protein stability.

Structure and phosphorylation sites of the STAT protein family. The signal transduction and activator of transcription (STAT) family have six members: STAT1 (STAT1 has two splicing isoforms, STAT1α and STAT1β), STAT2, STAT3 (STAT3 has two splicing isoforms, STAT3α and STAT3β), STAT4, STAT5a, STAT5b, and STAT6. STATs are composed of 750–900 amino acids. From the N-terminus to the C-terminus, the domains are the nitrogen-terminal domain, coiled-coil domain, DNA-binding domain, junction domain, SH2 domain, and transcription-activation structure. "Y" represents a tyrosine phosphorylation site, and "S" represents a serine phosphorylation site. Created with BioRender.com
STAT1
STAT1 has two splice isomers. One is STAT1α, with a size of 91 kD. STAT1α, similar to other STATs, has a complete transcription-activation domain. The amino acid positions 701 and 727 are the two phosphorylation sites, while most of the transcriptional activation region in the protein form of STAT1β is missing. The size of STAT1β is 84 kD, and STAT1β has only one phosphorylation site, at the amino acid 701.73 Functionally, STAT1β can respond to IFN-I ligands, but its response to IFN-γ is defective and may have an antagonistic effect on STAT1α.74 STAT1 is mainly activated by IFN. In addition, other cytokines, including IL-2, IL-6, platelet-derived growth factor, epidermal growth factor (EGF), hepatocyte growth factor, tumor necrosis factor (TNF), and angiotensin II can also activate STAT1. The biological function of STAT1 includes the following aspects: (1) Inhibits cell growth. STAT1 can inhibit cell growth by regulating the expression of cell cycle-related genes, such as promoting the expression of the Cyclin-dependent kinase inhibitors P21 and P27 or inhibiting the expression of c-myc. STAT1 can also control cell growth by inhibiting the expression of cyclin.75,76 (2) Regulates cell differentiation. Phosphorylation of STAT1 can regulate the differentiation of human granulocytes and osteoblasts.77,78 (3) Promotes cell apoptosis. The expression of STAT1 can promote the expression of a series of apoptosis proteins, which is the leading way for STAT1 to promote apoptosis. For example, STAT1 induces the formation of apoptotic protein caspase1, 3, and 11 precursors, and interacts with the p53 protein.79,80,81 Moreover, STAT1 can also induce Fas, Bcl-2, and Bcl-X gene expression.82,83 (4) Inhibits tumor occurrence. In STAT1/p53-knockout mice, the spontaneous or induced tumor formation rate was higher than that in mice in which only p53 was knocked out, and the antitumor activity of IFN-α disappeared. Simultaneously, in human tumors, such as breast cancer and Wilms tumor, the expression of STAT1 is associated with a better prognosis.84 However, some studies have shown that STAT1 can also promote the occurrence of hematological tumors unrelated to IFN.85 (5) Regulates the immune system. STAT1 participates in all major histocompatibility complex (MHC)-dependent antigen presentation processes.86 In addition, STAT1 is involved in the early development of B cells.87,88 The absence of STAT1 or a mutation in the STAT1 gene increases the body’s susceptibility to parasites, bacteria, viruses, etc.89
STAT2
STAT2 has six complete domains, but STAT2 cannot form homopolymers, nor can it directly bind to DNA.90 STAT2 is different from other STATs as the only STAT that does not bind to the original gamma-activated site. STAT2 is activated by type I IFNs (including IFN-α and IFN-β). The biological functions of STAT2 are as follows. (1) Antiviral effects. Interferon stimulation genes are induced to exert the body’s antiviral effect.91 (2) Immune regulation.
In STAT2-knockout mice, the lack of type I IFNs autocrine loop and the defective response of T cells and macrophages indicate that STAT2 is essential for regulating the immune response. STAT2 may also be involved in the maintenance of memory cells upon induction by IFN-α.92 (3) Regulation of tumorigenesis. STAT2 is highly expressed in ovarian cancer. In addition, STAT2 is associated with the poor overall survival of ovarian cancer and non-small cell lung cancer.93
STAT3
Similar to STAT1, STAT3 also has two splicing isoforms (STAT3α and STAT3β) with different functions.94 STAT3α has a complete domain, while STAT3β lacks 55 amino acids at the C-terminus, replaced by seven amino acid residues.95,96 STAT3 is activated when either Y705 or S727 is phosphorylated. Nevertheless, STAT3β lacks S727 and is only activated when Y705 is phosphorylated. STAT3β has better specific DNA-binding activity than STAT3α, but in terms of transcription activity, STAT3α performs better. The transcription factor STAT3 is activated by the IL-6 family members (IL-6, IL-11, IL-31, LIF, CNTF, CT-1, OSM, etc.), IL-10 family members (IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26), and IL-21, IL-27, G-CSF, leptin, and IFN-Is.55,97,98,99 STAT3 has a more complicated role in biological functions. STAT3 is mainly involved in the negative regulation of the immune response, cell growth, differentiation and apoptosis, and tumor occurrence and metastasis. (1) Immune regulation. STAT3 can function in a series of signal transduction processes, such as membrane binding, phosphorylation, and nuclear translocation through a cycle of palmitoylation–depalmitoylation, thereby regulating the differentiation of Th17 cells.100 STAT3 also promotes the immunosuppression of tumor-associated macrophages and myeloid-derived suppressor cells.101,102 Excessive activation of STAT3 is related to immunosuppression and transformation.103,104 (2) Regulation of cell growth, differentiation, and apoptosis. Inhibition of JAK that transduces IL-6 signals to STAT3 may inhibit the expression of the apoptotic protein Bcl-xl, thereby inducing cell apoptosis. Moreover, constitutively activated STAT3 may induce the occurrence of multiple myeloma by inhibiting cell apoptosis.105 (3) Regulation of tumorigenesis. The constitutive activation of STAT3 is related to the occurrence of head and neck tumors, breast cancer, non-small-cell lung cancer, colorectal cancer, and hematological tumors. Besides, high expression of STAT3 and IL-6 are closely related to poor chemotherapy sensitivity of triple-negative or high-grade breast cancers.106 However, the two splicing isoforms of STAT3 have different functions in the regulation of tumors. STAT3α activation is believed to promote tumor occurrence, while STAT3β inhibits the occurrence of cancer and is considered an effective tumor suppressor.95,107,108,109 (4) Regulation of cancer stem cells (CSCs). The persistent activation of STAT3 maintains the stemness of breast CSCs. There are multiple pathways that promote the survival of CSCs in breast or colon cancer, which include IL-6-JAK1-STAT3, IL-6-JAK2-STAT3, hypoxia-inducible factor-1α-JAK1-STAT3, and retinol-binding protein 4-JAK2-STAT3.110
STAT4
STAT4 consists of 784 amino acids, and its protein structure is similar to that of other STATs. The cytokines that activate STAT4 mainly include type I IFN, IL-12, and IL-23.111,112 In mice and humans, STAT4 plays an essential role in the differentiation and development of Th1 cells and helper follicular T (Tfh) cells. Moreover, STAT4 promotes the germinal center response after a virus attack. Cytokines such as IL-21 produced by Tfh cells are also crucial for the maturation and development of B cells and the isotype conversion of Ig.113 Phosphorylation of STAT4 is critical for the humoral immune response.
STAT5
The transcriptional activators of STAT5 include STAT5a and STAT5b. The similarity of STAT5a and STAT5b at the amino acid level is 91%. STAT5a is composed of 794 amino acids, and the 694th amino acid is a tyrosine phosphorylation site, while STAT5b is composed of 787 amino acids.114,115 STAT5 tyrosine phosphorylation site is the 699th amino acid. Purified STAT5b has a higher DNA-binding capacity than STAT5a.116 STAT5a can form tetramers in addition to dimers, while STAT5b binds to DNA in the form of dimers.117 The cytokines that activate STAT5 mainly include IL-3, prolactin, and the IL-2 cytokine family (including IL-2, IL-4, IL-7, IL-9, and IL-15). In addition, EGF, EPO, GM-CSF, TPO, GH, and platelet-derived growth factors can also effectively activate STAT5.54,114,115,118,119 The biological functions of STAT5 include the following.120 (1) Regulation of growth and development. Since it was initially found to nullify the β-casein gene, STAT5a was originally called prolactin-induced mammary gland factor. STAT5-knockout mice present severe defects in mammary gland development and milk secretion, and STAT5b-/- mice exhibit defects in the production of GH.121 (2) Regulation of the immune system. STAT5 dimers are essential for survival, STAT5a- and STAT5b-deficient mice exhibit severe defects in lymphatic development and perinatal lethality. However, STAT5a-STAT5b tetramer-deficient mice are viable, whereas had fewer number of T cells, natural killer (NK) cells, and impaired proliferation capacity of CD8+ T cells, and impaired NK cell maturation.117,122 (3) Regulation of tumor immunity. After inoculating tumors in immunocompromised mice, the levels of STAT5a and STAT5b of T and B lymphocytes isolated from mice were significantly reduced, indicating that the levels of STAT5 are related to tumor progression.120,123 Besides, STAT5 is involved in breast tumorigenesis, and mainly participates in the early development of breast cancer.106 (4) Regulation of cell growth, differentiation, and apoptosis. Studies have found that IL-2-induced activation of STAT5 can also lead to an increase in FasL, indicating that STAT5 activation is involved in IL-2-induced activation-induced cell death.124,125
STAT6
The STAT6 gene encodes 850 amino acids, and the tyrosine phosphorylation site in the STAT6 protein at position 641 marks the activation of STAT6.126 however, studies have also pointed out that S407 may be the key phosphorylation site for virus activation of STAT6.127 In some cells and tissues, splice variants of STAT6 are evident. STAT6b has a deletion at the amino terminus, and part of the SH2 domain of STAT6c is missing.128 STAT6 is mainly involved in the transduction of IL-4 and IL-13 signals.129 IL-4 induces activation of STAT6, which is the key to Th2 cells differentiation and immunoglobulin isotypes conversion.130,131,132 Furthermore, IL-4-induced activation of STAT6 in T cells can also inhibit the expression of VAL-4, a member of the family of integrin adhesion molecules, thereby inhibiting the infiltration of CD8+ T cells into tumors.133 STAT6 can promote the proliferation and maturation of B cells, mediate the expression of MHC-II and IgE, and play an important role in mast cell activation.115 In contrast to other STATs, STAT6 can be activated by viruses without relying on JAK.127 STAT6 also induces the expression of homing-related genes in immune cells and plays an important role in innate immunity. For example, the STAT6 dimer induces the expression of CCL2 and recruits T cells, macrophages, and monocytes; CCL26 induces homing of eosinophils/basic granulocytes and NK cells; and CCR6 recruits dendritic cells, B cells, T cells, etc. (Table 2).
Activation and regulation of JAK/STAT signaling pathways
Canonical JAK/STAT signaling pathway
The classic JAK/STAT signaling is as follows (Fig. 3): the cell ligand interacts with its receptor to cause receptor dimerization. Nevertheless, gp130,134 EpoR,135,136 TNF-R1,137 IL-17R,138, IL-10R,139 and GH receptor140 etc. can pre-form inactive receptor dimers before binding to the ligands, which may facilitate rapid receptor complex assembly and signal transduction. The connection between the ligand and the receptor induces transphosphorylation of JAK. Activated JAK causes tyrosine phosphorylation of the bound receptor, forming a docking site for STATs. At this docking site, JAK phosphorylates STAT, and then STAT dissociates from the receptor and forms homodimers or heterodimers through SH2-domain–phosphotyrosine interactions. These dimers translocate to target gene promoters, regulation the transcription of the target genes.4,141 STAT usually regulates transcription through the following mechanisms: (1) STAT binds to its DNA target site to drive transcription activation. (2) STAT protein may form a transcription complex with non-STAT transcription factors to trigger the transcription mediated by STAT; (3) STAT associates with non-STAT DNA-binding elements to promote STAT-dependent transcription; (4) STAT and non-STAT transcription factors can synergistically activate transcription by binding to clusters of independent DNA-binding sites.

Activation and negative regulation of JAK/STAT signaling pathways. Black arrows indicate the activation process. Red dotted arrows indicated negative regulation. Activation of the JAK/STAT signaling pathway: (1) cytokines and growth factors bind to their corresponding receptors, leading to receptor dimerization and recruitment of related JAKs; (2) JAK activation leads to tyrosine phosphorylation of the receptors and formation of docking sites for STAT; (3) STATs are phosphorylated by tyrosine; (4) STATs dissociate from the receptor to form homodimers or heterodimers; (5) STAT dimers enter the nucleus, bind to DNA, and regulate transcription. Negative regulation of the JAK/STAT signaling pathway: There are three main types of proteins involved in the negative regulation of the JAK/STAT signaling pathway: the PIAS (protein inhibitor of activated STAT), CIS/SOCS (suppressor of cytokine signaling) family, and PTPs (protein tyrosine phosphatase). PIAS mainly interacts with STAT dimers to inhibit STAT binding to DNA, thereby blocking JAK/STAT signal transduction. The CIS/SOCS family negatively regulates the JAK/STAT pathway in three ways: (1) binding to a tyrosine kinase receptor to block the recruitment of STAT; (2) binding directly to JAK to inhibit its kinase activity; (3) forming an elongin B/C-cullin5 complex that degrades JAK or STAT bound to the SOCS protein through polyubiquitination and proteasome degradation. PTPs inhibit the JAK/STAT pathway by interacting with JAK, STAT, or receptors to (1) dephosphorylate the STAT dimer; (2) interact with the receptor to dephosphorylate the related JAK; and (3) in the case of CD45 (a transmembrane PTP) inhibits the phosphorylation of JAK. Created with BioRender.com
Noncanonical JAK/STAT signaling pathway
Studies have also shown that JAK/STAT also is involved in nonclassical signal transduction, which is more complicated. Unphosphorylated STAT3 could induce multiple STAT3 target gene expressions without S727 phosphorylation, Lys-685 acetylation and NF-κB contribute to this process. Besides, STATs can be activated not only in the cytoplasm, all STATs except STAT4 can localize to the mitochondrion and promote oxidative phosphorylation and membrane permeability, STAT3 can localize to the endoplasmic reticulum and help to resist apoptosis induced by oxidative stress.142 A portion of the unphosphorylated STAT pool is located on heterochromatin related to the allelic autosomal gene heterochromatin protein-1 (HP1) in the nucleus. JAK or other kinases induce the activation of STAT and cause HP1 to dissociate from heterochromatin, and then, phosphorylated STAT binds to cognitive sites on autosomes to regulate gene transcription. This atypical JAK/STAT signal transduction is essential for maintaining the stability of heterochromatin.143,144,145 In a study of Drosophila, it was found that the phosphorylation of STAT can cause HP1 to break away from heterochromatin, thereby destroying the stability of heterochromatin, and the instability of heterochromatin may promote the occurrence of tumors.145,146,147,148 Some researchers have discovered that atypical JAK/STAT signal transduction patterns exist in mammals. For example, MHC high-order chromosomal remodeling is caused by IFN-induced STAT1 activation;149 other studies have shown that activation of JAK3-STAT5 causes chromatin remodeling at the Ifng locus during Th1 cell differentiation.150 Interestingly, JAK protein can also be activated by tumorigenic tyrosine kinases independent of cytokine receptors.151 For example, the proto-oncogene v-Abl of Abelson murine leukemia virus constitutively activates the JAK/STAT pathway by regulating the interaction between suppressors of cytokine signaling (SOCS)-1 and JAK in the JAK/STAT pathway.152 The oncogenic fusion chimeric protein nucleophosmin-anaplastic lymphoma kinase induces the inactivation of SH2-containing protein tyrosine phosphatase-1 (SHP-1) to inhibit the degradation of JAK3 and enhance the signal transduction initiated by JAK3-STAT3, which may lead to the occurrence of anaplastic large cell lymphoma.153 The BCR-ABL fusion gene exerts anti-apoptotic effects. BCR-ABL can synergize with hematopoietic growth factors through low constitutive phosphorylation of JAK protein, thereby regulating STAT activation.154 Similarly, STAT can also be activated by other non-receptor tyrosine kinases or directly activated by receptors independent of JAK. c-Src tyrosine kinase can constitutively activate STAT3, which increases the possibility of the STAT signaling pathway regulating tumor-related gene expression.155 Epidermal growth factor receptor can directly activate STAT1, STAT3, and STAT5, furthermore, STAT5 can be directly activated by the platelet-derived growth factor receptor.99,156,157
Positive regulation of JAK/STAT signaling
In addition to the main components of the JAK/STAT signaling pathway, many related proteins play indispensable roles in STAT-dependent transcription and JAK–STAT interactions with other signaling pathways. Upon co-stimulation of glucocorticoids and prolactin, activated STAT5 and glucocorticoid receptor (GR) form a complex. GR acts as a transcriptional coactivator of STAT5 to promote STAT5-dependent transcription.158 Moreover, CBP and p300 act as auxiliary activators of STAT1α to regulate the response of JAK/STAT, but this regulation can be realized by integration of common transcripts of the JAK/STAT and other signaling pathways.159 Another cytoplasmic protein, Nmi, may promote the activation of STAT1 and STAT5 through the recruitment of STAT1 and STAT5 by CBP. In vitro GST pull-down assay results showed that STATs except STAT2 could interact with Nmi.66 Some adaptor proteins can also promote the JAK/STAT signaling pathway. The SH2 protein subfamily composed of lymphocyte adaptor protein (Lnk), SH2-B, and APS has potential adaptor functions. SH2-2B can promote the activation of JAK2 induced by GH, while APS is a negative regulator of the JAK/STAT signaling pathway.160 signal transducing adapter molecule is a transduction adapter molecule containing an SH3 domain and one ITAM domain. It can interact with JAK2 and JAK3 through its ITAM domain to enhance IL-2 and GM-CSF-mediated C-myc transcription.161
Negative regulation of JAK/STAT signaling
Many negative regulators are involved in the regulation of JAK/STAT signal transduction. They maintain the balance and steady state of the JAK/STAT pathway. There are three main types of negative regulation of the JAK/STAT signaling pathway: protein inhibitor of activated STAT (PIAS), SOCS/CIS family members, and PTPs (protein tyrosine phosphatases) (Fig. 3).
SOCS/CIS family
The SOCS protein family are intracellular proteins that include CIS, SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, and SOCS7. Activated STATs dimerize and enter the nucleus to induce SOCS expression and the SOCS proteins bind to phosphorylated JAK and its receptor to negatively regulate the JAK–STAT signaling pathway. SOCS mainly negatively regulates the JAK/STAT pathway in three ways. (1) It binds to the phosphotyrosine on the receptor to prevent STAT recruitment to the receptor; CIS binds stably to the tyrosine-phosphorylated β chain of the IL-3 receptor and the tyrosine-phosphorylated EPO receptor.162 (2) It directly and specifically binds to JAK or its receptor to inhibit the kinase activity of JAK. For example, SOCS3 binds to JAK and its receptor gp130 simultaneously. SOCS3 targets the IL-6 family cytokine-receptor complex containing the signaling receptor gp130. After the SH2 domain of SOCS3 binds to phosphorylated Tyr759 of gp130, SOCS3 Ig-like receptors (KIR) bind to gp130-related JAK in a nonphosphorylation-dependent manner. Upon SOCS3 binding, the substrate-binding groove of JAK is hidden, inhibiting the JAK/STAT signaling pathway.163 The SH2 domain of the SOCS1 protein can target the activation loop of JAKs, and SOCS1 can also directly inhibit JAK tyrosine kinase activity through KIR.164 (3) The SOCS proteins interact with the elongation protein B/C complex through its C-terminal SOCS box and simultaneously binds to the cullin5 scaffold protein to form an elongation protein-cullin-SOCS3 E3 ubiquitin-linked enzyme complex.165 This complex undergoes polyubiquitination, and the proteasome degrades signaling factors such as JAKs and STATs that bind to SOCS, thereby blocking signal transduction. Cullin5 also contains a Really Interesting New Gene (RING) domain that can bind to the protein Rbx2, which can interact with the E2 ubiquitin-conjugating enzyme. SOCS not only plays a simple negative feedback function but also plays an important role in the immune response and inflammation regulation. Experimental data in SOCS1 knockout mice showed that SOCS1 inhibits Treg cells secretion of IFN-γ by regulating STAT1, and the inhibition of IFN signaling prevented atherosclerosis.166 CIS does not interact with JAKs and therefore does not inhibit JAKs. It was initially identified as a negative feedback regulator of STAT5. Moreover, overexpression of SOCS3 in Ba/F3 cells can inhibit the activation of STAT5. SOCS3 inhibits Th1 cells and promotes Th2 production by inhibiting IL-12-mediated STAT4 activation. The loss of SOCS3 can also inhibit the production of Th1 cells and Treg cells by promoting the production of IL-10 and transforming growth factor β (TGFβ).166
PIAS
PIAS is a family of transcriptional regulators. There are four PIAS family members that are found in mammals, namely, PIAS1 (also known as PIASx, and two splice variants PIASx-α and PIASx-β), PIAS2, PIAS3 (with splice variant PIAS3b), and PIAS4 (also known as PIASy with splice variant PIASyE6). The PIAS homolog dPIAS/Zimp was identified in Drosophila,167 and PIAS-related proteins SIZ1 and SIZ2 also exist in yeast.168 PIAS was initially found to be an inhibitor of STAT, and PIAS1 and PIAS4 can interact with STAT1, PIAS3 and PIAS1 interact with STAT3 and STAT4, respectively. PIAS only interacts with STAT dimers formed after being phosphorylated by JAK, and does not interact with STAT monomers. PIAS mainly regulates transduction through the following mechanisms. (1) Blocking the DNA-binding activity of transcription factors. For example, PIAS1 and PIAS3 block JAK/STAT signal transduction by blocking STAT and DNA-binding activity.37,169 (2) Promoting transcription factor sumoylation. Research results show that PIAS1 can interact with Lys703 on STAT1.170 (3) Recruiting other co-regulatory factors, namely, PIAS1 and PIAS4, through the recruitment of the co-inhibitory molecule histone deacetylase, which prevents STAT binding to DNA and leads to transcription-activation failure.171 (4) Chelating transcription factors to form the subnuclear structures of repressor complexes to regulate transcription.172 PIAS also acts as a SUMO (small ubiquitin-related modifier) E3 ligase, which can regulate many cellular processes through protein ubiquitination; however, there is still debate on whether the SUMO E3 ligase activity of PIAS regulates STAT signaling. PIASx-α can act as an E3 ligase to modify the Lys703 SUMO of STAT1. However, interestingly, mutating Lys703 to Arg can eliminate the SUMO modification, but the activation of STAT1 and PIAS1 inhibition of STAT1 signaling is not affected.170 In contrast to these findings, Ungureanu et al. revealed that the same mutation caused an increase in IFN-γ-mediated transactivation of STAT1, leading to increased activation of STAT1.173 A large number of genetic studies have also verified the physiological role of PIAS in the gene regulation mediated by the JAK/STAT signaling pathway. JAK/STAT transduction activity is elevated when PIAS was knocked out, which leads to the formation of hematological tumors, and PIAS1 selectively regulates IFN-β and IFN-γ inducible genes by interfering with the recruitment of STAT1 to gene promotors.174 However, how the SUMO E3 ligase activity of PIAS regulates STAT activity in vivo and the physiological role of STAT-mediated gene regulation need further research and elucidation.
PTPs
The JAK/STAT signaling pathway can also be negatively regulated by PTPs. The SH domain in PTPs can bind to signaling molecules, activated receptors, and JAK to dephosphorylate a substrate. PTPs can dephosphorylate STAT and inhibit its activity, and inhibit JAK/STAT signal transduction. For example, the nuclear isoform TC45 of T-cell PTPs has been extracted from HeLa cells. Nuclear TC45 dephosphorylates and inactivates STAT dimers in the nucleus.175 SH2-containing protein tyrosine SHP-1 is also an important member of the PTP family. When it is activated by GH and transfers to the nucleus, SHP-1 can dephosphorylate STAT5b.176 PTPs not only act on activated STAT but can also dephosphorylate JAK and block the JAK/STAT signaling pathway. The transmembrane PTP CD45 can inhibit IL-3-induced JAK2 phosphorylation and negatively regulate JAK/STAT signal transduction, thereby inhibiting IL-3-mediated cell proliferation.177 PTP1B can act on specific sequences in the JAK activation loop in the cytoplasm, dephosphorylating JAK2 and TYK2, but it has also been reported that the main target of PTP1B in the suppression of JAK/STAT signaling is STAT5.178 Other PTPs can also act on ligand-receptor complexes. For example, hematopoietic protein tyrosine phosphatase SH-PTP1 can bind to pY429 in the cytoplasmic region of the EPO receptor, thereby mediating dephosphorylation and inactivation of JAK2. After adding IFN-α, SHP-1 can also reversibly bind to IFN-α receptors and selectively regulate JAK/STAT signal transduction in mice.179 SH2-containing protein tyrosine phosphatase-2 (SHP-2) can negatively regulate the cytotoxic effect of IFN on the overactivation of STAT to promote cell growth, but the specific role of SHP-2 is related to a part of the JAK/STAT signaling pathway that remains to be studied.180
Signaling cross-talk between JAK/STAT and other pathways
Cross-talk between components in the JAK/STAT pathway and those in other pathways is complex, occurs at various levels, and involves diverse molecules, such as a receptor, JAK, STAT, and gene transcription factors (Fig. 4). These cross-talk activities play vital roles in pluripotency and differentiation transcription program, immune regulation, and tumorigenesis.

Signaling cross-talk between JAK/STAT and other pathways. (1) STAT3-p300- SMAD1 forms a complex to induce astrocyte differentiation; (2) JAK1 binds to TGFβRI and activates STAT3; (3) TGF-β also activates STAT3 via SMAD-dependent manner. (4) STAT3 inhibits SMAD3–SMAD4 complex formation and suppress SMAD3-DNA binding; (5) SMAD3 inhibits STAT3 activation via recruiting PIAS3 to STAT3; (6) TGF-β blocks IL-12 mediated JAK2 and TYK2 tyrosine phosphorylation; (7) Notch signaling suppresses JAK/STAT activation by interfering with STAT translocation to the DNA domain, and signals of JAK/STAT inhibited Notch signaling conversely. (8) Hes proteins directly bind to STAT3 and induce phosphorylation by recruiting JAK2; (9) STAT5 direct interacts with PI3K; (10) STAT5 upregulates the expression of p85α (Pik3r1), p110α (Pik3ca), and AKT1; (11) STAT3 drives the hyperacetylation of RelA via interacting with p300; (12) cyclooxygenase-2, IL-17, IL-21, and IL-23 encoded by NF-κB activate STAT3; (13) NF-κB preceded ISGF3 (a complex containing STAT1, STAT2, and IRF9 subunits) at the Nos2 promoter, thus regulating nitric oxide synthase expression; (14) IRF9-STAT1-STAT2 trimeric complex induce gene expression; (15) STAT1 regulates IRF8 synthesis; (16) STAT5 suppresses IRF8 activity; (17) STAT1 and IRF1 synergistic induce IFNγ-induced gene transcription; (18) IRF8 increases IFNγ-induced gene transcription mediated by STAT1 and IRF1. Created with BioRender.com
The TGFβ signaling pathway
The TGFβ family consists of TGFβs, bone morphogenic proteins (BMPs), activins, and Nodal. The TGFβ signaling pathway regulates a wide range of biological activities in various cell types, such as embryonic development and cell homeostasis. SMAD proteins are pivotal intracellular effectors or modulators of the TGFβ family. SMADs and STATs are often combined in the same transcription complex. For example, LIF-STAT3 and BMP2-SMAD1 synergistically induce primary fetal neural progenitor cells to differentiate into astrocytes. STAT3 interacts with p300 at its amino terminus, SMAD1 interacts with p300 at its carboxyl terminus. STAT3 and SMAD1 form a complex linked by p300, which contributes to the astrocyte differentiation.181 It has been reported that the TGFβ signaling pathway regulates the JAK/STAT pathway in both a positive and negative manner, depending on the cell type and protein status.182 In pancreatic ductal carcinoma, tumor-secreted TGFβ antagonizes the upregulation of LIF induced by IL-1 by downregulating the expression of IL-1R1 and promoting the differentiation of cancer-associated fibroblasts into myofibroblasts, thereby inhibiting JAK/STAT signaling.183 In contrast, in hepatocytes, hematopoietic stem cells (HSCs), and hepatoma cells, TGFβ can potentiate IL-6 mediated STAT3 activation. In liver fibrosis, JAK1 is a constitutive TGFβRI-binding protein; thus, STAT3 is activated directly through JAK1 within minutes of TGFβ stimulation in a SMAD-independent manner. TGFβ also provokes a second phase activation of STAT3, which depends on SMADs, de novo protein synthesis, and JAK1. Activated SMAD and STAT3 bind to their respective DNA domains in the JUNB promoter to enhance the expression of TGFβ related genes.184 In addition to the cooperative function between SMAD3 and STAT3, it is reported that STAT3 can also attenuate SMAD3–SMAD4 complex formation and suppress SMAD3-DNA binding. Furthermore, SMAD3 can recruit PIAS3 to STAT3, thus inhibiting STAT3 activation.185 The phosphorylation status of SMAD3 and STAT3 determines whether the relationship between them is cooperative or antagonistic.184 In T lymphocytes, TGFβ blocked IL-12-mediated JAK2 and TYK2 tyrosine phosphorylation and STAT3 and STAT4 activation in T lymphocytes, resulting in decreased T-cell proliferation and diminished IFN-γ production.186
The MAPK signaling pathway
Mitogen-activated protein kinase (MAPK) cascades are complex signaling pathways that regulate various cellular activities, including inflammation, apoptosis, proliferation, and differentiation. There are three major subfamilies in the MAPK family: extracellular-signal-regulated kinases (ERK), c-jun N-terminal kinase or stress-activated protein kinases (JNK or SAPK), and MAPK14. As a well-known pathway participating in cellular activities, the JNK signaling pathway is involved in both apoptosis and cancer cell survival. For example, JNK mediates the compensatory cell proliferation in tumors. Nevertheless, via regulating the fos gene and pro-apoptotic hid gene, the JAK/STAT pathway and downstream effector Zfh2 promotes JNK-mediated cell survival, and inhibits JNK-mediated cell apoptosis.187
The Notch signaling pathway
The Notch signaling pathway is evolutionarily conserved and extensively controls cell processes, such as proliferation, differentiation, and cell death. The crosstalk between components in the Notch signaling pathway and those in the JAK/STAT signaling pathway is mainly studied in organ development. In Drosophila posterior foregut development, analysis of gene expression mediated by a model Notch response element revealed that the JAK/STAT signaling pathway is required for the expression of Notch-dependent genes in the foregut, which indicated the function of the JAK/STAT pathway controlling cell movement during embryonic foregut development, the molecular mechanism remains to be determined.188 In Drosophila intestinal stem cells (ISCs), researchers found that the JAK/STAT pathway regulated ISCs proliferation, and that this effect may have been inhibited by Notch signaling at the transcriptional level. This type of antagonistic relationship ensures a balance between proliferation and differentiation of ISCs.189 In the development of drosophila follicle cell patterning, Notch signaling suppresses JAK/STAT activation by interfering with STAT translocation to the DNA domain, and signals of JAK/STAT inhibited Notch signaling conversely. Thus, the continuous balance of two pathways specifies the identity of different types of follicle cells.190 In the development of the central nervous system, Hes proteins, downstream effectors of Notch, directly bind to STAT3 and induce phosphorylation by recruiting JAK2, indicating that Hes proteins may be non-receptor scaffold proteins that facilitate JAK2 phosphorylation of STAT3.191 In breast cancer, the Notch signaling pathway is often hyperactivated, noncanonical Notch signaling upregulates IL-6 expression, then activates downstream JAK/STAT, and Notch-mediated IL-6 upregulation occurs only when p53 was mutated or lost. In addition, activation of IL-6 by Notch required the IKKα/IKKβ function (inhibitor of NF-κB kinase subunit alpha and beta, respectively). IKKα and IKKβ are two proteins in the NF-κB signaling pathway.192
The PI3K/AKT/mTOR pathway
The phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway plays a vital role in most cellular processes, such as proliferation, adhesion, migration, and invasion. In human melanoma cells, PI3K negatively regulates STAT activity.193 In mammary epithelial cells, the JAK2/STAT5 pathway controls mammary epithelial cell survival and death through direct interaction with the p58α regulatory subunit of PI3K and upregulation of the expression of p85α (Pik3r1), p110α (Pik3ca), and AKT1.194 In cytokine-receptor-like factor 2-rearranged B-precursor acute lymphoblastic leukemia, increased pJAK2, pSTAT5, and pS6 levels were observed in patient samples. JAK inhibitors inhibited both the JAK/STAT and PI3K/mTOR pathways, which suggests an interconnection between them. Nevertheless, for full elucidation of the mechanism, additional work is needed.195
The NF-κB signaling pathway
The NF-κB family comprises five members: p50, p52, p65, c-RelA, and RelB. NF-κB dimers bind to DNA sites called κB sites to modulate gene expression. NF-κB regulates a large variety of cellular responses, especially throughout the immune system.196 The cross-talk between the JAK/STAT signaling pathway components and the NF-κB signaling pathway components is extensive. NF-κB can induce the expression of a variety of inflammatory mediators and is a core transcription factor in various immune responses. Therefore, it is believed that NF-κB can induce malignancy and antitumor immunity through simultaneous inflammation.197 Some factors regulated by STAT3 also play essential roles in the tumor microenvironment.105,198,199,200 When it was found that NF-κB and STAT3 in tumor cells were activated simultaneously, people connected the two.201 Among these factors, IL-6 is an important factor that links the NF-κB signaling pathway with STAT3. As we mentioned above, IL-6 and its ligand can effectively activate STAT3; and the target gene of NF-κB encodes IL-6. STAT3 plays a vital role in the activation of the NF-κB pathway. In cancer cells and tumor-related haematopoietic cells, constitutively activated STAT3 drives the hyperacetylation of RelA, mediated by interactions with p300, thereby prolonging NF-κB nuclear retention and promoting the activation of NF-κB.202 In addition, cyclooxygenase-2, IL-17, IL-21, and IL-23 encoded by NF-κB can activate STAT3 in various ways.200,203,204 IL-4-mediated STAT6 activation plays a crucial role in inflammatory gene inhibition, partly because STAT6 acts as an antagonist of NF-κB upon the binding of the E-selectin gene promoter.205 Moreover, NF-κB preceded ISGF3 (a complex containing STAT1, STAT2, and IRF9 subunits) at the Nos2 promoter, thus regulating nitric oxide synthase expression.206
The IRF family
The IRF family includes nine members: IRF1, IRF2, IRF3, IRF4/ICSAT/PIP/LSIRF, IRF5, IRF6, IRF7, IRF8/ICSBP, and IRF9/ISGFγ. These factors were initially identified as transcriptional regulators of type 1 interferon. Further studies revealed more functions of the IRF family in addition to their functions in the IFN system, such as immune cell development, innate immune responses, and tumor suppression.207 Cross-talk between IRFs and STATs includes both direct physical binding and indirect gene regulation. For example, IRF9 physically binds to STAT1-STAT2 heterodimer, and this trimeric complex binds to a composite DNA element comprising binding sites for both STAT1 and IRF9.208 STAT1 stimulates the transcription of IFNγ-inducible genes, and IFN consensus sequence binding protein (ICSBP/IRF8) is an IFNγ-inducible gene. Thus, STAT1 regulates IRF8 synthesis.209 Conversely, IRF8 increases IFNγ-induced gene transcription mediated by STAT1 and IRF1.210 IRF can be negatively regulated by STAT. For instance, STAT5 suppresses IRF8 during the plasmacytoid dendritic cell development.211
The JAK/STAT pathway in human diseases
The JAK/STAT pathway is a highly conserved pathway of signal transduction. It regulates multiple cellular mechanisms associated with varieties of diseases development. Dysregulation of the JAK/STAT pathway is associated with various diseases. For example, JAK2V617F mutation frequently occurs in myeloproliferative neoplasms (MPN). More frequently, the JAK/STAT pathway serves as a mediator of abnormally elevated cytokines to induce gene transcription. Furthermore, inhibitors of JAK/STAT have been effective in treating multiple diseases, such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), which shows that JAK/STAT is important in disease development (Table 3).212,213,214
Malignancies
Hematological malignancies
Abnormal amplification and recruitment of blood cells lead to hematologic malignancies. The normal actions of the JAK/STAT pathway rely on various components. Thus, basic molecular alterations, such as those caused by gain-of-function mutations in different components (JAK, STAT) and extensive expression (cytokine receptors, JAK), may result in aberrant activation of a signaling cascade. JAK2 acts as an essential mediator in HSCs by transmitting signals from TPO and activating downstream stem cell factors.215,216 JAK2 mediates myelopoietic formation at different stages through its interactions with various receptors (e. g. EPO, TPO, and GM-CSF).135 In addition, the combined actions of JAK1 and JAK2 are crucial for lymphopoiesis. Both JAK1 and JAK3 can bind to IL-2R, IL-4R, IL-7R, and IL-15R.34,217 Gain-of-function mutations in four Janus kinases play roles in hematologic malignancies. The majority of these alternations appear to be point mutations of varying frequency in different JAK members. JAK1 mutations are the most frequent in T-cell acute lymphoblastic leukemia (6.5–27%), followed by B-cell acute lymphoblastic leukemia (1.5%),218,219,220 indicating that JAK inhibitors are necessary to treat hematological disease.
Hodgkin lymphoma
Classical Hodgkin lymphoma (cHL), primarily derived from germinal central B cells, represents a case of successful treatment.221 Eighty percent of patients with Hodgkin lymphoma achieve complete remission by using recently combined modality therapies. Despite high cure rates in adolescents and young adults, treatment-related toxicity and long-term morbidity remain a significant challenge in the clinic.221
Previous studies revealed that cHL patients experience a recurrence in some genomic lesions, associated with persistent activation of the NF-kB and JAK–STAT signaling pathways with proinflammatory and anti-apoptotic features.222 Gain-of-function mutation of STAT6 is evident in most patients with cHL (~80%).223,224 Furthermore, when STAT6 is mutated, the mutant maintains tumor cell survival and growth in conjunction with unidentified SOCS1 variants by inducing an anti-apoptotic response.225 JAK2/STAT6 signaling is activated by lymphotoxin-a produced by cHL cell lines, inducing target gene expression to promote the immunosuppressant microenvironment and lineage ambiguity in cHL.225 cHL cells exhibit an aberrant cytokine level that is essential for the proliferation of Hodgkin and Reed/Sternberg cells and a favorable environment for tumor cells. Constitutive activation of the JAK/STAT pathway may be associated with increased cytokine and receptor expression in cHL. Moreover, the role of the JAK/STAT pathway in immune evasion by mediating PD-L1/L2 expression has been reported in Hodgkin lymphoma. Chromosome 9p24.1/PD-L1/PD-L2 mutation upregulates PD-1 ligands and PD-L1 on the membrane through JAK/STAT signaling.226,227,228
Natural killer/T-cell lymphoma
Current knowledge on natural killer/T-cell lymphoma (NKTCL) is insufficient to understand its molecular mechanisms well. Furthermore, few therapeutic approaches are available to patients with NKTCL. To date, simple dependence on multiagent chemotherapy and localized radiotherapy has shown poor benefits. With technical progress, more disease-related genes have been found in NKTCLs. The role of the JAK/STAT pathway in promoting the maturation of HSCs has been gradually acknowledged. Increasing evidence shows that a persistently active JAK/STAT pathway may be caused by mutations in JAK gene domains, and they probably lead to the pathogenesis of lymphocyte-related malignancies, including T-cell acute lymphoblastic lymphoma/leukemia, cutaneous TCL, mantle cell lymphoma, and acute megakaryoblastic leukemia.218,229,230,231,232,233,234 JAK3 mutation has been reported in many other cancers, such as breast, stomach, and lung cancer.219,235 Concordant with these results, the samples from patients with NKTCL tumor were found to express JAK3 mutations.236 In addition, Cornejo and colleagues showed that transplanting JAK3-mutant bone marrow cells into C57BL/6 mice induced continuous activation of the JAK/STAT signaling pathway, resulting in the generation of aggressive T-cell lymphoproliferative disorders. These data suggest that JAK3-activating mutations may be involved in the development of NKTCLs.237
Myeloproliferative neoplasm
Myeloproliferative neoplasm (MPN) refers to a group of disorders whose distinctive feature is an extensive expansion of one or more blood cell types, such as white blood cells, red blood cells, and platelets. Patients with MPN may experience thrombohemorrhagic complications. MPN may develop into myelofibrosis (MF) or acute myeloid leukemia (AML), resulting in severe symptoms and a reduced life span. JAK2V617F is the most frequent genetic alteration, whose expression is different in PV (>95%) and ET/PMF (50-60%).238,239,240,241 In cells carrying JAK2V617F, a high-frequency mutation, the inhibitory functions of the JH2 pseudokinase domain are disrupted, resulting in overactivation of the JAK/STAT pathway.242 JAK2V617F in megakaryocytes plays a vital role in maintaining the myeloproliferative state of both mutant and non-mutant hematopoietic cells. Excessive proliferation of cells can lead to increased erythropoiesis and fibrosis. The lack of megakaryocytes in JAK2V617F and MPLW515L BMT models leads to significantly alleviated polycythemia and leukocytosis,242 indicating that the activation of the JAK/STAT pathway in megakaryocytes is positively linked with myeloproliferation and promotes MPN progression. Aging patients may acquire more frequent mutations of JAK. It is hypothesized that increasing age can be a crucial risk factor for MPN progression.
A majority of patients with MPN present chronic inflammation with enhanced circulating proinflammatory cytokines. It is well-known that continued inflammation may contribute to the progression of MPN.239 Thus, the activity of the JAK/STAT pathway may be elevated in response to increases in the levels of proinflammatory cytokines.243 Previous studies showed that activated STAT3 proteins could promote cytokine production in a variety of cancers.244 Using a JAK2 inhibitor to treat mice with MPN resulted in reduced cytokine levels and attenuated systemic symptoms.245 In MPNs, abnormal activation in JAK/STAT signaling is commonly accompanied by mutations in tyrosine kinases. It is well-known that TPO stimulation activates JAK2-STAT3/5.246 With further investigation about MPN, the importance of the Lnk has been gradually realized in the field. Lnk as a member of adaptor protein has a negative effect on signaling pathways activated by TPO-R/MPL in either megakaryopoiesis or HSCs.247,248,249,250 The lack of Lnk leads to significant interference in the hematopoietic function of mice, including a threefold increase in white blood cells and platelets in the circulation, the accumulation of B cells with different states in the bone marrow and spleen, and the expansion of HSCs.247,248,251 Data from biochemical experiments implicate that in response to TPO stimulation, the SH2 domain of Lnk interacts with the phosphorylated tyrosine residue 813 (Y813) of JAK2, which makes JAK2 activation suppressed to constrain the quiescence and self-renewal of HSCs. In addition, the published studies reveal that the deficiency in Lnk has shown advanced JAK/STAT signaling in a cytokine-independent manner and the increased ability of oncogenic JAK2 to promote the expansion of myeloid progenitors both in vitro and in vivo.252 Moreover, JAK inhibitors inhibit Lnk-deficient cell lines, suggesting that the treatment of JAK2 inhibitors may be a novel choice for MPN patients with Lnk deficiency.
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) ranks third as the most common cause of cancer-related death worldwide.253,254 Various factors contribute to the pathogenesis of this cancer, including viral infection, particularly hepatitis B virus (HBV), continuous alcohol consumption, and aflatoxin B1 contaminated food.255,256 Despite acquiring the remarkable improvement in understanding risk factors of HCC, the current theories explaining the molecular mechanism of HCC have not been verified.257 Thus, deeper exploration needs to be conducted to find better treatments.
SOCS3 can block various cytokine signaling pathways, including the JAK/STAT, and NFκB signaling pathways,258,259 and sustain normal immune reactions. Hypermethylation of SOCS3 has been found in multiple malignant diseases, such as lung cancer, head and neck cancer, and prostate cancer.260,261,262 Niwa et al. reported that 33.3% of HCC tissues exhibited hypermethylated SOCS3.263 Furthermore, long noncoding RNA promotes HCC progression,264 most likely via activation of the JAK/STAT pathway. LINC00346, an intergenic lncRNA located on chromosome 13q34, is upregulated in HCC and promotes tumor cell growth by decreasing the cell apoptosis rate and increasing the cell proliferation rate, which depends on the level of LINC00346 and the activated JAK/STAT signaling pathway.265 The phosphorylated transcription factor STAT3 significantly contributes to cancer growth and recurrence. Sorafenib, a Food and Drug Administration (FDA)-approved first-line drug for advanced HCC treatment, induces HCC cell death. STAT prevents the anti-apoptosis effect of sorafenib by modulating Mcl-1 expression.266,267,268 Moreover, STAT3 partially contributes to the sensitivity of HCC cells to sorafenib-mediated cell death.269 In contrast, some cytokines that do not activate cytokine receptors negatively regulate HCC progression by inhibiting the JAK/STAT pathway. For example, angiopoietin-like protein (ANGPTL1) acts as a tumor suppressor, not only inhibiting STAT3/Bcl-2–driven anti-apoptotic signals to promote apoptosis but also downregulating certain transcription factors (e.g., SNAIL and SLUG) to suppress cell migration and invasion.270,271,272 Many studies have revealed that DNA hypermethylation in promotors can be associated with tumor suppressor gene dysfunction, leading to HCC development and progression.273,274 Thus, the downregulation of ANGPTL1 is insufficient to inhibit STAT3 signaling.275
Inflammatory and immune diseases
Systemic lupus erythematosus
SLE, caused by an aberrant autoimmune response,276 is a complex immune disorder that can result in inflammation of multiple tissues or organs in the body and in most cases affects the kidneys. A common characteristic of SLE is recognition of specific autoantigens and against which it produces multiple autoantibodies.277 Lupus nephritis is a poor prognostic indicator for patients with SLE.
Cytokines play a central role in the pathogenesis of SLE.278,279 A wide range of cytokines are considered immunopathological in the initiation and development of human SLE, such as IFNs, TNF, IL-6, IL-10, and IL-12.280,281 Increased serum IFN and expression of IFN-inducible genes mediated by the JAK/STAT pathway is thought to be pivotal in the molecular pathogenesis of SLE. JAK1 and TYK2 are downstream signals of IFN. Moreover, TYK2 polymorphism is closely linked with SLE.282 CXCR4, a vital chemokine receptor with multiple immune functions, is significantly upregulated in patients with SLE.283,284 CXCR4 endocytosis is mediated through IL-21 and B-cell receptor interactions, which are likely dependent on the JAK/STAT signaling pathway.285 Furthermore, CXCR4 undergoes tyrosine phosphorylation by JAK2 and JAK3.286 These data indicate that the activated JAK/STAT pathway is tightly associated with an abnormal increase in CXCR4 in patients with SLE. In addition, increased infiltration of immune cells in renal interstitium and glomeruli contributes to disease progression and development through overexpression of the JAK/STAT pathway. It is reported that JAK inhibitors significantly suppress the infiltration and cytokine production of T cells.287,288
Atopic dermatitis
Atopic dermatitis is a chronic inflammatory skin disease caused by aberrant autoimmune responses. The prevalence ranges from 5 to 20%.289,290 The incidence tends to be higher in children than adults. Th2 differentiation, important for the initiation and development of atopic dermatitis, may be regulated by activating the JAK/STAT pathway. Thus, the JAK/STAT pathway is linked to inflammation and pruritis in atopic dermatitis. Many therapies have been applied to improve patients’ quality of life, including phototherapy, systemic corticosteroids, systemic immunosuppressants, and monoclonal antibody dupilumab.291 Their insufficient effect and potential risks still need to be addressed.291 An increase in cytokines, such as Th2, Th22, Th1, and Th17 secreted cytokines, has been identified in atopic dermatitis skin lesions.292,293,294 The JAK/STAT pathway, as a cytokine-mediated signal transduction pathway, can exacerbate disease development.294 For instance, IL-4 has a critical role in the pathogeny of atopic dermatitis. Moreover, JAK1 and JAK3 are related to Th1 cell activation in the acute phase of atopic dermatitis.295 Multiple studies have shown that STAT6 exerts a significant effect on the immune response by regulating B-cell differentiation and contributing to IgE class switching.296 Therefore, STAT6 is a potent transductor and activator in allergic disorders. Abnormalities in Th2 immune responses are also associated with high JAK/STAT pathway activity. Moreover, they cause an increase in cytokine, chemokine, and IgE production leading to the exacerbated inflammatory reactions of atopic dermatitis.297
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a complex and chronic systemic inflammatory disease involving multiple organs and tissues that most frequently affects diarthrodial joints.298,299 Although many novel therapeutic approaches have been developed through a deeper understanding of the molecular and cellular mechanisms of rheumatoid arthritis, a series of problems remain to be resolved, including inadequate or partial responses, a lack of appropriate biomarkers, and drug-related toxicity. Cytokines have been reported to accelerate RA progression, as evidenced by a significant increase in proinflammatory cytokines, such as TNF-α, IL-1ß, IL-6, and IFN-γ. Some of these cytokines exert a profound influence on RA primarily by the JAK/STAT pathway. For example, IL-6 and IFN-γ can mediate the activation of the JAK/STAT pathway. In addition, TNF is able to activate this pathway by causing STAT3 phosphorylation.300,301 Besides, STAT4 and STAT6 polymorphisms play central roles in RA.129,302
Other diseases
Parkinson’s disease
Parkinson’s disease (PD) is an age-related disease that manifests as significantly dysfunctional dopaminergic neurons and accumulation of misfolded α-synuclein, leading to severe disease burdens for older adults. Symptoms of PD include movement disorders and poor memory. Oligomeric α-SYN produced by neurons can activate microglia and macrophages and generate proinflammatory meditators.303,304,305 Various data obtained from PD patients and animal models indicate that the toxic effect of α-SYN in neurons is potentially exacerbated via neuroinflammatory processes. Thus, inflammation may contribute to the pathogenesis and development of PD.306 Recent studies showed that aberrant expression of α-SYN was associated with the activation of the JAK/STAT pathway, resulting in dysfunction of innate and adaptive immune responses and ultimately inducing neurodegeneration. The increase in the number of microglia and macrophages plays an important role in the progression of PD. MHC Class 2 is considered a biomarker that can be used to assess the activation of microglia and macrophages and the expression of a gene induced by STAT activation. α-SYN is involved in the regulation of proinflammatory markers, including MHC Class 2, TNF, inducible nitric oxide synthase (iNOS), IL-6, and CCL2 in myeloid tissue from the midbrain.307,308 The JAK/STAT pathway is required for the transmission of signals induced by a large number of cytokines/chemokines. T cells and myeloid cells may be activated and polarized into pathogenic phenotypes when the JAK/STAT pathway is aberrantly activated.8,309 Importantly, preclinical data obtained from PD rat models confirmed that JAK inhibitors could significantly repress inflammatory cytokine production. In general, suppression of the activated JAK/STAT pathway may be a promising therapeutic target for treating patients with PD. Myeloid cells are reported as a significant risk factor related to the pathogenesis of PD.308,310 Suppressing the JAK/STAT pathway blocks myeloid cell transformation into a proinflammatory phenotype, which indirectly dampens innate immune reactions in the midbrain. IFN-γ and IL-6 are known as the most potent activators, and enhanced levels of both IFN-gamma and IL-6 have been found in patients with PD.311,312,313 JAK inhibitors can reduce IL-6 or IFN-γ serum levels in patients. These results indicate that patients can acquire clinical benefits when activation of the JAK/STAT pathway is inhibited.
Hair loss
A typical growth cycle of hair consists of three phases: (1) the growth phase (anagen), (2) the regression phase (catagen), and (3) the rest phase (telogen).314 Hair loss upon entry into the growth phase leads to complications. Several factors potentially contribute to disease pathogenesis, including hair follicle miniaturization and immune dysregulation. Recent evidence has shown that small-molecule inhibitors, such as JAK inhibitors, are effective for treating hair growth disorders. Activation of the JAK/STAT pathway can drive hair follicles into a quiescent state,315 leading to decreased hair growth capacity. Suppression of the JAK/STAT signaling pathway induces hair follicle exit from a resting state and enhances the ability of hair follicles to enter the hair cycle. Hyperactive JAK/STAT signaling in aged mice is closely associated with the suppression of hair follicle stem cell function in vitro.316 Furthermore, phosphorylated STAT5 plays a central role in regulating HF stem cells during pregnancy.317 Due to the importance of the dermal papilla (DP) in continual hair follicle cycling,318 various signaling pathways in the DP have been uncovered. STAT5a and STAT5b are significantly upregulated in the DP,319 while knockout of STAT5b in mice results in an apparently delayed entry to anagen in the early time of postnatal hair follicle growth.320 Moreover, previous studies have identified activated STAT5 as a vital switch to drive natural growth when post-developmental hair follicle cycle begins in mesenchymal cells.321 However, many functional experiments have also demonstrated that the JAK/STAT pathway may have an opposite role in the hair cycle compared with protective effects. Inhibitors blocking JAK/STAT signaling are shown to be effective for hair growth by inducing growth in the resting hair follicle.322 Various molecular mechanisms of the JAK/STAT signaling pathway have been discovered in different stages of hair growth. OSM is a negative regulator involved in hair growth as it can activate the JAK-STAT5 pathway to maintain the hair follicles in a static state.323 Data from these studies reveal the action of the JAK/STAT pathway in different conditions is not always toward a beneficial or harmful direction, thus more additive investigations need to be performed on these mechanisms. Moreover, IL-6 has a higher expression in keratinocytes, which links with the suppression of hair growth.324
Age-related diseases
Chronic inflammation is a typical symptom in aging and age-related diseases.325 The increased production of proinflammatory cytokines and chemokines is a major risk factor for many age-related diseases and cellular senescence.326,327 The cooperative effect of senescent cells and inflammation significantly contributes to age-related pathology. The JAK/STAT pathway is a common cytokine-medicated cascade and important for cytokine production.201,328 Senescent cells accumulating in adipose tissue appear to be the development of a senescence-associated secretory phenotype (SASP), which is closely associated with higher activation of the JAK/STAT pathway and inflammation. Preclinical studies have demonstrated that JAK1 and JAK2 activation in the adipose tissue of old rats was increased. STAT3 plays a central role in inducing and maintaining an inflammatory microenvironment by mediating a wide range of SASP components, including IL-6, IL-8, plasminogen activator inhibitor 1, monocyte chemoattractant protein-1 (MCP-1), and GM-CSF.201,329 These results indicate that JAK1 and JAK2 regulate the effects of the SASP. A significant reduction in activated STAT3 upon treatment with a JAK inhibitor was observed, suggesting a specific interaction between STAT3 and age-related adipose tissue inflammation. Increased IL-6 in serum is linked with low physical activity and frailty in older adults. A number of methods have been used to delay aging, such as GH receptor knockout (GHRKO) and surgical removal of visceral fat.330,331 The gene disruption of GH receptor in mice results in a longer life span and less or delayed occurrence of age-related or malignant diseases potentially via inducing metabolic changes and increasing insulin sensitivity, indicating that insulin signaling exerts an important impact on aging.330 It is reported that JAK2 activity is controlled by the interaction between insulin signaling and angiotensin II (AII) systems.332 In response to insulin-induced stimulation, JAK2 is phosphorylated and forms a complex with STAT1, named JAK2-STAT1 complex, to transmit directly insulin signal towards the nucleus.333 As GHRKO mice do not respond to GH, this is accompanied by severely reduced plasma levels of insulin-like growth factor-1 (IGF-1), suggesting IGF-1 may be involved in aging. IGF-1 can act as a growth factor through JAK/STAT pathway in many tissues, for example, IGF-1 selectively activates JAK1 but not JAK2 or TYK2. However, few studies have been performed to identify the molecular mechanism JAK/STAT pathway regulating age-related diseases. This potentially is an important insight to extend life span.
Inhibitors and clinical applications
Medications that target the JAK/STAT pathway can be classified into three types: (1) cytokine or receptor antibodies, (2) JAK inhibitors, (3) STAT inhibitors (Fig. 5). They have been applied to various cancers and autoimmune diseases. Several of these drugs have been approved for the market, such as the JAK inhibitors: tofacitinib and ruxolitinib. Most of these drugs are in preclinical and clinical trials. We will introduce them in this section.

Therapeutic targets of the JAK/STAT signaling pathway. (1) Recombinant cytokines, (2) cytokine antibodies, and (3) receptor antibodies are designed to target cytokines or receptors; (4) JAK inhibitors are designed to target JAKs; and (5) peptide inhibitors, (6) small-molecule inhibitors, (7) decoy oligonucleotides (ODNs), (8) antisense oligonucleotides (ASOs), and (9) siRNAs target STATs. Created with BioRender.com
Manipulation of activators
Upstream cytokines and receptors are crucial for determining the functions of the JAK/STAT signaling pathway. Thus, manipulating upstream activators has long been used in clinical settings, and such manipulation is performed with recombinant cytokines, cytokine–antibody fusions, and cytokine/receptor-blocking antibodies.
Engineered cytokines
As we previously discussed, a large majority of cytokines are involved in the JAK/STAT signaling pathway. Their enhancement or blockade has been applied to treat multiple diseases. For example, recombinant IL-2 is approved to treat metastatic melanoma and renal cell carcinoma, type 1 interferons are used for the treatment of Hepatitis C virus, IFNα is used for treating the viral infections, and TNF is a treatment of cancer. Research on other interleukins is ongoing. Nevertheless, adverse events and low selectivity have restrained their clinical use. Engineered cytokines such as ALKS 4320 (recombinant IL-2) and RMP16 (recombinant TNF) have been tested in preclinical studies and demonstrated relatively better efficacy and less toxicity.334,335
Cytokine–antibody fusions are novel biopharmaceuticals that increase the therapeutic index of cytokine “payloads”. They are generated by the fusion of two compounds. One of the compounds is an antibody specific to accessible markers, which can be overexpressed in certain diseases. Another is therapeutic payloads, including cytokines, these fusion proteins can dramatically increase the therapeutic index of the cytokine payload. These include ch14.18-IL-2, Hu14.18-IL-2, NHS-IL2LT, DI-Leu16-IL-2, BC1-IL-12, L19-TNF.336
Antibody-based blockade
Antibody-based blockade of cytokines and their cognate receptors has been extensively studied. For example, blockade of IL-2, IL-12, IL-17, and TNF has been successfully used to treat chronic inflammatory diseases such as RA, IBD, and psoriasis. Some of these blockades are market-approved, such as anti-IL-2Rα, anti-IL-5, anti-IL-6, anti-IL-6R, anti-IL-12, and anti-IL-23. The anti- IL-2Rα antibody, also known as daclizumab, markedly inhibited the phosphorylation of JAK1, JAK3, and STAT5a/b, thus significantly decreasing transplant rejection.337 Siltuximab is an IL-6 antagonist and has been approved for the treatment of idiopathic multicentric Castleman’s disease (iMCD). Tocilizumab, an anti-IL-6R humanized antibody, has been approved for the treatment of RA, cytokine release syndrome (CRS), and iMCD. New-generation anti-IL-6 and anti-IL-6R monoclonal antibodies improved binding affinity and specificity and reduced toxicity. They are in clinical trials focused on various diseases. For example, sarilumab, sirukumab, clazakizumab, and olokizumab target IL-6, vobarilizumab, olamkicept, satralizumab, and NI-1202 target IL-6R.338,339
IL-5 is important for the priming and survival of mature eosinophils, and it is crucial for the proliferation and maturation of eosinophil progenitors. Anti-IL-5 antibodies are used in diseases such as eosinophilic asthma, eosinophilic oesophagitis, hypereosinophilic syndrome, and eosinophilic granulomatosis with polyangiitis (EGPA).340 Mepolizumab and reslizumab act against IL-5, and benralizumab targets IL-5R. In EGPA patients, mepolizumab combined with standard treatment led to prolonged remission and less steroid use.341 IL-12 and IL-23 share the signature p40 subunit, and an anti-IL-12/23 p40 antibody (p40 mAb) interferes with Tfh cell differentiation and inhibits proinflammatory cytokine secretion. Thus, this antibody attenuates chronic graft-versus-host disease in murine models of lupus nephritis.342 P40 mAb has been tested in many diseases in preclinical studies, such as psoriasis and Crohn’s disease.343,344 The results of phase 3 clinical trial including 312 adults with active psoriatic arthritis (PsA) show that ustekinumab (45/90 mg q12 weeks) led to significant and long-term improvement of symptoms/signs, including in patients who had previously received anti-TNF treatment.345 Pegvisomant, a competitive GH receptor antagonist, is used to treat acromegaly as monotherapy or combinational therapy with somatostatin analogs. Pegvisomant is generally used as a second-line therapy and has a high curative effect but the high cost in the treatment of acromegaly.346
More cytokine/receptor antibodies are being studied, such as anti-IL-4, anti-IL-4R, anti-IL-5R, anti-IL-6R, anti-IL-9, and anti-IL-13.6
JAK inhibitors
JAK inhibitors are a group of small-molecule inhibitors with different chemical structures (Fig. 6). The therapeutic effects of JAK inhibitors are based on two factors. First, JAKs mediate various cellular activities. Inhibition of JAK function can cause immunosuppression and decrease the abnormally elevated serum proinflammatory cytokines mediated by the JAK/STAT signaling pathway. Second, in some diseases, such as in myeloproliferative diseases and cancers, identifying gain-of-function JAK mutants enables treatment through their inhibition.3 Various JAK inhibitors are being studied in preclinical and clinical studies. Tofacitinib and baricitinib are the first orally available JAK inhibitors to be approved in treating RA and other autoimmune diseases.347

Chemical structures of JAK inhibitors.
Serious adverse events are always a concern when JAK inhibitors are considered for treatment. All JAK inhibitors have similar adverse effects, including infection, hyperlipidemia, and cytopenia. The first two JAK inhibitors approved for RA treatment, tofacitinib and baricitinib, have black box warnings of severe infections and malignancies. Some preclinical studies indicated that a reduction in lymphocytes, NK cells, and neutrophils might be associated with biological differences in different subtypes of JAK inhibitors.348 In addition to clinical applications, JAK inhibitors can be powerful tools for scientific research. For example, events downstream of certain ligands have been investigated and mechanisms of immune checkpoint blockade drug resistance have been delineated.
The first-generation JAK inhibitors (tofacitinib, oclacitinib, baricitinib, and ruxolitinib) are adenosine triphosphate (ATP)-competitive compounds. They target the JAK homology 1 tyrosine kinase domain in its active conformation. The ATP-binding pocket structure is highly conserved. Thus, first-generation JAK inhibitors target more than one JAK member.30 Most next-generation JAK inhibitors are also ATP-competitive. Nevertheless, there are also some JAK inhibitors (such as Deucravacitinib) that target the JH2 domain of JAK (Table 4).349
First-generation JAK inhibitors
Tofacitinib
Tofacitinib, also named Xeljanz or CP690, 550, was the first JAK inhibitor studied in humans. Tofacitinib preferentially inhibits JAK1 and JAK3 and, to a lesser extent, JAK2 and TYK2. It is the first JAK inhibitor approved mainly to treat RA and other autoimmune diseases. Tofacitinib blocks the γc cytokine-receptor signaling pathway through JAK1 and JAK3 in T cells. Thus, it interferes with Th1 and Th2 differentiation and impairs the production of inflammatory Th17 cells. Tofacitinib also suppresses cytokine production through both innate and adaptive processes, including common γ chain cytokines IFN-γ, TNF, IL-6, IL-12, IL-17, and IL-23. Nevertheless, tofacitinib increased serum levels of IL-35 and IL-35 might be an indicator of the disease activity attenuated by tofacitinib efficacy.350,351
Tofacitinib is effective in preclinical studies and has been applied in various phase 2 and phase 3 clinical trials. Most often, it is applied to patients whose previous therapies failed. Tofacitinib is under investigation for use in various diseases, including RA, ulcerative colitis, Crohn’s disease, relapsing polychondritis, atopic dermatitis, alopecia areata, cutaneous dermatomyositis, psoriasis, psoriatic arthritis, and ankylosing spondylitis.352,353,354,355,356,357,358,359,360 In total, 5 or 10 mg of tofacitinib twice a day is the most commonly used dosage.352 Recently, tofacitinib was regarded as a candidate in treating coronavirus disease 2019 (COVID-19), although no published study showed the benefits, several clinical trials are ongoing, clinical trial identifiers, including NCT04415151, NCT04469114, NCT04390061, and NCT04332042.361
Adverse events of tofacitinib are mostly tolerable, including opportunistic infections (OIs), gastrointestinal perforation, thromboembolism, and herpes zoster.362,363 Tuberculosis (TB) was the most common OI reported thus far.364 Incidence rates of thromboembolic events were elevated in patients with baseline cardiovascular or venous thromboembolism (VTE) risk factors versus without these risk factors, indicating that VTE is an important risk factor for thromboembolism during tofacitinib treatment.365 For patients with chronic viral infections such as HBV infection, tofacitinib therapy appears safe, but physicians should be aware of the risk of HBV reactivation.366 For pregnant patients, unintentional exposure to tofacitinib does not appear to be associated with an increased risk to the fetus.367 Twenty-four weeks of tofacitinib treatment appeared to be associated with a lower risk of future major adverse cardiovascular events (MACE) risk, including myocardial infarction, stroke, cardiovascular death.368 Based on the limited data, treatment of RA with tofacitinib does not increase the incidence of malignant tumors.369 Nevertheless, more safety data are needed as tofacitinib inhibited several key cytokines and immune cells.
Baricitinib
Baricitinib is a selective oral JAK1 and JAK2 inhibitor with moderate activity against TYK2 and significantly less activity against JAK3. It is generated by modifying the structure of tofacitinib.347 Baricitinib achieved efficacy in multiple autoimmune diseases, including RA, lupus erythematosus, juvenile dermatomyositis, atopic dermatitis, and IFN-mediated autoinflammatory disease, it inhibits the worsening of symptoms and reduces inflammation. Similar to tofacitinib, baricitinib is mostly used in patients whose previous therapies failed, 2 or 4 mg once daily is recommended in most randomized controlled trials (RCTs).370,371,372,373,374,375,376,377
Moreover, baricitinib decreased albuminuria over 24 weeks of treatment in patients with diabetes and diabetic kidney disease (DKD). A possible explanation is that baricitinib treatment decreased inflammatory biomarkers associated with DKD pathophysiology, including CCL2 (MCP-1), CXCL10 (IP-10), SSA, tumor necrosis factor receptor (STNFR)1 and STNFR2, VCAM1, and intercellular adhesion molecule-1 (ICAM-1).378 Baricitinib has also been studied in many animal models to investigate its potential for broader clinical application, such as in human immunodeficiency virus (HIV)-associated neurocognitive disorders and osteoporosis.379,380
More importantly, baricitinib is predicted to be effective in the treatment of COVID-19. AP2-associated protein kinase 1 (AAK1) is one of the known regulators of endocytosis. Disruption of AAK1 might interrupt the virus from getting into lung AT2 alveolar epithelial cells via ACE2 receptor. Baricitinib is one of the six high-affinity AAK1-binding drugs, which also binds cyclin G-associated kinase, another regulator of endocytosis. Moreover, baricitinib inhibits inflammation via JAK1/2-mediated cytokine signaling.381,382 One of the most important effects was the rapid and remarkable inhibition of macrophage production of the cytokines and chemokines critical for inflammation and neutrophil recruitment.383 A double-blind, randomized, placebo-controlled trial including 1033 hospitalized adults revealed that baricitinib plus remdesivir was superior to remdesivir alone in reducing recovery time and accelerating improvement in clinical status.384 Patients with severe COVID-19 had a significant reduction in serum IL-6, IL-1β, TNF, increased antibodies against the SARS-CoV-2 spike protein, and rapid recovery of B-cell and T-cell frequencies.385
With exposure as many as 5.5 years, baricitinib has an acceptable safety profile. There is no difference in serious adverse effects such as death, adverse events leading to drug discontinuation, MACE, and malignancies.386 The most frequently reported AE was dose-dependent increased low-density lipoprotein (42.1%), followed by an increased risk of infections, including herpes zoster and TB. Baricitinib should be used with caution in patients with VTE risk factors.387
Oclacitinib
Oclacitinib is a cyclohexylamino pyrrolo [2,3-d] pyrimidine derivative that targets the JAK family. It is the most potent in inhibiting JAK1. Oclacitinib is currently used mostly to treat canine and cat pruritus and allergic skin diseases. There is no report of this drug being used to treat humans.388,389
Ruxolitinib
Ruxolitinib, also named INCB018424 or INC424, was found to inhibit JAK1 and JAK2, which is often dysregulated in myelopathies. Ruxolitinib is oral or topical administered. Clinical studies of ruxolitinib for the treatment of malignant tumors, acute graft-versus-host disease (aGVHD), MF, polycythaemia vera, alopecia areata, vitiligo, essential thrombocythemia, and COVID-19 are conducted worldwide.390,391,392,393,394,395,396,397 Ruxolitinib was first approved for the treatment of MF by the US FDA in 2011 and approved by the European Medicines Agency in 2012, followed by approval for the treatment of polycythaemia vera in 2014.398 Although ruxolitinib achieved clinical benefits in many patients with autoimmune diseases, it failed to significantly improve overall survival in patients with malignant tumors, including pancreatic cancer and colorectal cancer.390,399,400 Ruxolitinib has received much attention in the past year for its efficacy in treating COVID-19.396 Although there was no significant difference between ruxolitinib plus standard-of-care treatment and placebo, ruxolitinib improved the clinical symptoms and chest computed tomography images in COVID-19 patients.396
In 2011, ruxolitinib was the first drug approved by the US FDA to treat patients with intermediate or high-risk MF. According to previous clinical trials, the starting dose of ruxolitinib is 20 mg taken orally twice daily for patients with platelet counts higher than 200 × 109/L, and 15 mg twice daily for those with a platelet count between 100 × 109/L and 200 × 109/L. The dose was increased based on the response and a maximum of 25 mg was recommended twice daily. Ruxolitinib is not specific for the JAK2V617F mutation, and its efficacy in MF is mainly due to the attenuation of the constitutive activation of the JAK/STAT pathway and myelosuppression.398 For ruxolitinib-resistant or ruxolitinib-intolerant MF patients, another JAK2-selective inhibitor fedratinib might lead to clinical benefits and alleviate adverse events.401 However, another JAK1 and JAK2 inhibitor, momelotinib, failed to provide better clinical benefits for patients previously treated with ruxolitinib.402
The most common toxicity induced by ruxolitinib is myelosuppression, which results in anemia (64.8%), thrombocytopenia (62.0%), and neutropenia (47.9%). Other common adverse events include hypokalaemia (49.3%), peripheral edema (45.1%), and a high treatment discontinuation rate.391 The high treatment discontinuation rate is primarily caused by clinical benefit loss and drug toxicity. It has also been reported that severe withdrawal symptoms occur during MF treatment called “ruxolitinib discontinuation syndrome”. It is characterized by the acute relapse of disease symptoms and worsening splenomegaly.403 These events are related to the acute rebound of cytokine storms. Careful tapering is regarded as a preventive strategy.404 When ruxolitinib was used to treat skin depigmentation of vitiligo, adverse events also included application site pruritus and acne.405 Importantly, because JAK inhibitors interfere with many physical and pathological processes, ruxolitinib may be associated with immunosuppressive diseases such as severe and fatal infections, viral reactivation, and even Kaposi sarcoma (KS). KS was reported in an essential thrombocythemia patient. KS regressed 10 months after ruxolitinib discontinuation.406,407 When ruxolitinib was used to treat COVID-19, there were reports of severe purpuric lesions on the skin of dorsal and upper limbs, with a concomitant decrease in platelet counts. One patient displayed diffuse erythematous.408
Next-generation JAK inhibitors
JAK1 and JAK2 are critical for signal transduction by numerous cytokines, while JAK3 and TYK2 are activated by relatively few cytokines. Next-generation JAK inhibitors with higher specificity may reduce adverse events.
JAK1 inhibitors
Filgotinib: Filgotinib is a JAK1 inhibitor. Filgotinib inhibited Th1, Th2, and Th17 differentiation, and JAK1-dependent cytokines in a dose-dependent manner, including IL-2, IL-4, and IL-6, which plays vital pathological roles in chronic inflammation and autoimmune disorders.409 Filgotinib is mostly used for inflammatory and autoimmune diseases, including inflammatory bowel diseases, rheumatoid arthritis, PsA, and ankylosing spondylitis.410,411,412 In September 2020, the EU approved filgotinib for the treatment of moderate-to-severe RA patients who inadequately respond to one or more DMARD. The recommended dose for adults is 200 mg taken once daily. The same dose has been applied in a phase 2 clinical trial of moderate-to-severe Crohn’s disease, in which it led to significant clinical remission.413,414 More importantly, by combining high-throughput drug screening and the transcriptome analysis (differential analysis, gene set enrichment, and exon-intron landscape analysis), researchers found that filgotinib is not merely a JAK inhibitor, it can act as a splicing inhibitor and modulate HIV splicing, as well as inhibit T-cell activation, thus suppressing HIV-1 transcription and reducing the proliferation of HIV-infected cells. Therefore, filgotinib may be a candidate drug for use in the therapy of acquired immunodeficiency syndrome patients.415
The most common adverse events are nasopharyngitis, headache, and upper respiratory infections. There were no reports of opportunistic infections, malignancy, gastrointestinal perforation, or death. More long-term safety data are needed for this new drug.
Upadacitinib: Upadacitinib, also named ABT-494, is an orally administered JAK1 inhibitor. It potently inhibits JAK1-dependent cytokines, including IL-6, OSM, IL-2, and IFNγ. On 16 August 2019, upadacitinib was approved to treat moderate to severely active RA in patients with inadequate response or intolerance to methotrexate. The recommended dosage is 15 mg taken once daily. 30 mg taken once daily provided only a small incremental benefit. Upadacitinib can be administered alone or in combination with methotrexate or DMARDs.416 Analysis of RA patient plasma proteins suggested that treatment with upadacitinib normalizes key pathways associated with RA pathobiology, including IL-1, IL-6, IFNγ, and TNF. Upadacitinib is also associated with leukocyte activity, including cell migration and inflammatory responses.417 Compared to the first approved JAK inhibitor, tofacitinib combined with methotrexate, upadacitinib displays better outcomes as both a monotherapy and a combination therapy at 3 and 6 months.418 In addition to its use as an RA treatment, researchers are exploring other indications of upadacitinib, such as Crohn’s disease, ulcerative colitis, atopic dermatitis, psoriatic arthritis, and ankylosing spondylitis.419,420,421,422,423
The most common adverse events are infections and increases in lipid parameters, creatine phosphokinase, and hepatic aminotransferase, followed by a reduction in neutrophil and lymphocyte counts. Serious adverse events, including death, stroke, and venous thromboembolic, were rare but reported in a phase 3 clinical trial with RA patients. More extensive and longer clinical trials are required to verify the safety of upadacitinib.424
Abrocitinib: Abrocitinib, also named PF-04965842, is an oral JAK1 inhibitor. Abrocitinib is mainly used to treat atopic dermatitis. Phase1, 2, 3 clinical trials reported the clinical efficacy and acceptable tolerability, but no obvious improvements were observed between abrocitinib and dupilumab, a monoclonal antibody targeting IL-4Rα.425,426
There were no deaths or serious adverse events reported. Headache, diarrhea, nausea, upper respiratory tract infection, hematologic abnormalities, and nasopharyngitis are the most common adverse events.425
Itacitinib: Itacitinib (INCB039110) is a selective JAK1 inhibitor that has exhibited efficacy in preclinical studies of arthritis, IBD, and aGVHD.427 Moreover, itacitinib dose-dependently reduced the levels of multiple cytokines common to CRS during CAR-T therapy. Thus, itacitinib can be a prophylactic agent for CAR-T therapy-induced CRS, and a relative phase 2 clinical trial (NCT04071366) is ongoing.428 The commonly used dosage in clinical trials is 200 mg or 300 mg taken once daily, and phase 1 clinical trials preliminarily demonstrated the safety and efficacy of itacitinib. Larger-scale clinical trials are required in the future.429
JAK2 inhibitors
Fedratinib: Fedratinib is an orally administered kinase inhibitor that selectively targets both wild-type and mutated JAK2 and FMS-like tyrosine kinase 3 (FLT3), and inhibits the phosphorylation of STAT3 and STAT5. Fedratinib received approval on 16 August 2019 in the USA for the treatment of patients with intermediate- or high-risk primary or secondary MF. The recommended dosage is 400 mg taken once daily in patients with platelet counts of more than 50 × 109/L. The dosage should be one-half the recommended dose in patients with severe renal impairment or patients concomitantly receiving potent CYP3A4 inhibitors. Fedratinib prolonged survival in many murine tumor models, including prostate cancer. However, the development of fedratinib for use in treating malignant tumors has been discontinued.430
Adverse events warnings include severe to fatal encephalopathies, such as Wernicke’s encephalopathy. A putative mechanism for this adverse effect is related to the individual human thiamine transporter, which is inhibited by fedratinib. Fedratinib mediates the thiamine uptake in Caco-2 cells, and Wernicke’s encephalopathy is mediated by thiamine deficiency. Inhibition of thiamine uptake seems to be unique to fedratinib and is not a feature of other marketed JAK inhibitors.431
Pacritinib: Pacritinib inhibits both JAK2 and FLT3. Moreover, it has high selectivity against the JAK2V617F and FLT3 D835Y mutants, which are frequently found in MPN and AML. Pacritinib inhibits IRAK1 (an IL-1 receptor kinase), and IRAK1 is commonly mutated in two dysregulated hematopoiesis diseases (myelodysplastic syndromes and Fanconi anemia).432 Pacritinib is currently mainly used in MF and AML patients with a dosage of 200 mg twice a day or 400 mg once a day.433 In patients with MF and thrombocytopenia, 200 mg of pacritinib twice daily is better than 400 mg of pacritinib once daily in terms of hemoglobin and reduction in transfusion burden. Moreover, pacritinib is superior to the best available therapy (BAT), including ruxolitinib, in reducing spleen volume and symptoms.434 More importantly, pacritinib is effective at all JAK2V617F allele burden quartiles and in JAK2V617F-negative MF patients, suggesting that pacritinib may be uniquely suited for treating myelodepletive MF patients.435 Pacritinib has also been researched for treating several other types of cancers, including colon, rectal, and non-small cell lung cancer, but no objective response was observed in colorectal cancer.436,437
The most common grade 3/4 adverse events were anemia, thrombocytopenia, and diarrhea, and the most severe adverse events were anemia (5%), cardiac failure (2%), pyrexia (2%), and pneumonia (2%). Twenty-seven (12%) patients died due to serious adverse events in the pacritinib group compared with 14 (13%) in the BAT group.433
Lestaurtinib: Lestaurtinib is a multi-kinase inhibitor that targets a broad array of kinases, including JAK2, FLT3, RET, and TRK. However, lestaurtinib more effectively inhibits FLT3 than other kinases.438
Gandotinib: Gandotinib, also named LY2784544, is another JAK2 inhibitor. It inhibits JAK2V617F mutant in a dose-dependent manner and may inhibit additional JAK2 mutants in preclinical studies. Gandotinib is used to treat MPN, polycythemia vera, and essential thrombocythemia in phase 1/2 clinical trials. It demonstrated acceptable toxicity and a maximum tolerated dose of 120 mg taken daily.439,440
JAK3 inhibitors
Decernotinib: Decernotinib is a newly developed JAK inhibitor that potently inhibits JAK3 with limited or no measurable potency against the other three JAKs or non-JAK kinases. Decernotinib is effective in animal models in reducing ankle swelling T-cell-mediated inflammatory response in the skin.441 Three phase 2 clinical trials demonstrated that decernotinib can reduce the signs and symptoms of RA patients when it was administered monotherapy or in combination with DMARD or methotrexate.442,443,444 Adverse events include neutropenia, lymphopenia, hyperlipidaemia, and elevated hepatic transaminases. Lymphopenia may be associated with JAK3-associated cytokines, including IL-7 and IL-15.445 More clinical data are needed to verify the efficacy and safety of decernotinib treatment of more diseases.
Peficitinib: Peficitinib, also named Smyraf, ASP015K, and JNJ-54781532, is an orally administered JAK3-selective inhibitor. It inhibits IL-2-induced T-cell proliferation and STAT5 phosphorylation. Peficitinib was developed in Japan for the treatment of RA and received approval in Japan and Korea to treat RA patients inadequately responding to conventional therapies.446 The peficitinib pharmacokinetic profile is altered in subjects with moderate-to-severe hepatic impairment. Peficitinib exposure and adverse effects are similar to or without renal impairment.447,448 The recommended dosage is 150 or 100 mg once daily and 50 mg once daily for patients with moderate liver dysfunction. It is contraindicated in patients with severe liver dysfunction. Peficitinib is mainly investigated for treating RA. In addition to RA, peficitinib has been investigated for its efficacy in treating other autoimmune diseases, including psoriasis and ulcerative colitis.
The most frequent adverse events are nasopharyngitis, herpes zoster infection, a plasma creatine kinase increase, and lymphopenia, followed by pneumonia, pharyngitis, epipharyngitis, upper respiratory tract infection, bronchitis, influenza, and cystitis. The rare severe adverse events are gastrointestinal perforation and sepsis.446 Peficitinib does not have a significant effect on the pharmacokinetics of rosuvastatin, a statin.449
Pan-JAK inhibitors
Momelotinib: Momelotinib, formerly named CYT387, is an oral selective ATP-competitive inhibitor of JAK1, wild-type and mutated JAK2, and activin A receptor type 1.450 Momelotinib induced growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines when added between 0.5 and 1.5 μM, without affecting nonhematopoietic cells. In murine models, momelotinib is unable to completely eliminate JAK2-dependent cells, and MPN often reappears, suggesting that it is not curative and is better used in combinational therapy.451 In clinical studies, Momelotinib is effective in treating MF patients at a dosage of 200 mg twice a day or 300 mg once daily. In the patients with the JAK2V617F mutation, momelotinib significantly reduced the allele burden (21.1%).452 In a 7-year follow-up of 100 MF patients, momelotinib had been discontinued in 91% of patients after a median treatment of 1.4 years, suggesting that momelotinib is well-tolerated and induces long-term benefits. More importantly, in contrast to most other JAK2 inhibitors, momelotinib improved anemia in a substantial fraction of patients, which may be attributed to the inhibitory effects of momelotinib against ALK2-mediated hepcidin expression.453 In patients with previous ruxolitinib failure, momelotinib was not superior to the BAT in reducing spleen volume, which was reduced by 35% compared with the baseline volume. There is no evidence that JAK2 inhibitors are effective in reversing MF or inducing cytogenetic or molecular remission, and the efficacy of momelotinib contributes to the nonspecific inhibition of inflammatory cytokines.402 Momelotinib combined with trametinib does not perform better than single-agent trametinib in KRAS-mutated non-small cell lung cancer.454
The most frequent adverse events of momelotinib are diarrhea, cough, and nausea in patients with MF.455 Grade 3/4 adverse events include anemia, neutropenia, thrombocytopenia, and liver/pancreatic test abnormalities.453,455 A significant adverse event of momelotinib is treatment-emergent peripheral neuropathy (TE-PN), which has been documented with a 44% (44/100) incidence rate, and TE-PN is significantly associated with prolonged survival due to treatment response.456
Gusacitinib: Gusacitinib, also named ASN002, is a multi-target JAK inhibitor that targets JAK2, JAK3, TYK2, with a lesser extent inhibit JAK1. Gusacitinib also inhibits spleen tyrosine kinase (SYK). Both JAK and SYK are involved in the pathogenesis of atopic dermatitis. In two phase 1b clinical trials on atopic dermatitis, gusacitinib (40 mg or 80 mg daily) achieved efficacy rapidly and downregulated several biomarkers involved in systematic inflammation, such as E selectin.457 There were no serious adverse events that happened, and changes in serum cholesterol and blood pressure were observed.458
Delgocitinib: Delgocitinib, also named JTE-052, inhibits all for members of the JAK family. Delgocinib is developed in Japan for the treatment of autoimmune disorders and hypersensitivity. On 23 January 2020, topical delgocinib 0.5% ointment received its first approval for the treatment of atopic dermatitis in Japan. Delgocitinib achieved efficacy in atopic dermatitis, alopecia areata, and chronic hand eczema. Clinical trials on inverse psoriasis and discoid lupus erythematosus are ongoing.459 Common adverse events include mild-to-moderate nasopharyngitis (25.9%), contact dermatitis (4.5%), and acne (4.3%). Seven serious adverse events were reported, one being Kaposi’s varicelliform eruption.460
Cerdulatinib: Cerdulatinib, also known as PRT062070, inhibits JAK1, JAK2, TYK2, and SYK. Preclinical studies revealed cerdulatinib potently inhibited the proliferation of B-cell lymphoma cell lines.461 A phase 1 study revealed that cerdulatinib was well-tolerated and demonstrated promising antitumor effects in B-cell or T-cell non-Hodgkin lymphoma.462 More clinical data on cerdulatinib are needed.
Comparisons between JAK inhibitors
As we discussed before, dozens of JAK inhibitors are used in various diseases. Thus, comparisons between JAK inhibitors are clinically meaningful.
In RA, there are six JAK inhibitors that have received market approval or are undergoing clinical trials. They are tofacitinib, baricitinib, filgotinib, upadacitinib, decernotinib, and peficitinib. For patients who are refractory to conventional RA treatment, All JAK inhibitors achieved efficacy in ACR20 (American College of Rheumatology 20% response) and DAS28 (Disease Activity Score in 28 joints). Increasing the dose of baricitinib (4 mg versus 2 mg), tofacitinib (10 mg versus 5 mg), upadacitinib (30 mg versus 15 mg) does not provide significant additional benefits.463 Moreover, compared to biological DMARDS, JAK inhibitors have a much shorter half-life, indicating that they are suitable for RA patients with comorbidities, such as heart diseases. For a certain index, in CRP-DAS28 (C-reactive protein) for LDA (low disease activity) and remission, upadacitinib is superior to other JAK inhibitors. In ESR-DAS28 (Erythrocyte sedimentation) for remission, tofacitinib achieved the best efficacy. For safety data, there were 11 deaths reported in tofacitinib and more serious infections in upacitinib.464
In IBD, all four JAKs are involved in the signal transduction of proinflammatory cytokine, and four JAK transcripts are significantly upregulated in the intestinal mucosa of patients with active ulcerative colitis.465 Thus, pan-JAK inhibitors may be particularly suitable for treating IBD. Various JAK inhibitors are undergoing clinical trials, including tofacitinib, filgotinib, upadacitinib, peficitinib, itacitinib, TD-1473. A systematic review compared tofacitinib, filgotinib, peficitinib, and TD-1473. Treatment with four JAK inhibitors can increase the clinical remission rate of Crohn’s disease by 38% and the clinical remission rate of ulcerative colitis by more than threefold. Similar therapeutic effects were observed in patients naive to TNF antagonists compared to patients with previous exposure, and tofacitinib ranked the highest remission in patients with previous exposure to TNF antagonists.466,467 For adverse events, mortality was not increased in JAK inhibitor treatment compared to placebo. Nevertheless, JAK inhibitors increase infection risk, especially herpes infection, which could be mitigated by the injection of a vaccine.468 There are several clinical trials completed in the past 2 years, an updated meta-analysis could be meaningful.
In alopecia areata, tofacitinib, ruxolitinib, and baricitinib are used in clinical trials. Oral JAK inhibitors were associated with four times higher odds of achieving response compared with topical JAK inhibitors, with no difference between tofacitinib, ruxolitinib, and baricitinib.469 More studies are needed to identify the role of JAK inhibitors in the therapy of other types of hair loss, such as Androgenetic alopecia and cicatricial alopecia.
In COVID-19, there are three JAK inhibitors undergoing phase 2/3 clinical trials, and they are tofacitinib, baricitinib, and ruxolitinib. Baricitinib and ruxolitinib were associated with a reduced risk of mortality.470 They reduced the use of invasive mechanical ventilation and had a borderline impact on the admission rate of the intensive care unit (ICU) and the incidence of acute respiratory distress syndrome (ARDS). Nonetheless, none of them decreased the length of hospitalization. Besides, the high cost and adverse events may limit the application of JAK inhibitors in COVID-19.382 More data are needed to illustrate the timing of JAK inhibitors treatment during the course of COVID-19 may affect the outcome.471
In atopic dermatitis, seven JAK inhibitors are undergoing clinical studies. Four (baricitinib, upadacitinib, abrocitinib, gusacitinib) were orally administered, the remaining three (tofacitinib, ruxolitinib, delgocitinib) were topically administered. A meta-analysis of 15 RCTs showed that JAK inhibitors were more effective in achieving eczema area and severity index-75 (EASI-75), Investigator’s Global Assessment (IGA), and itching-NRS responses than placebo. For the subgroup analysis, gusacitinib seems unlikely to achieve EASI-75, IGA responses, and topical delgocitinib had higher rates of achieving EASI- 75, while topical tofacitinib and ruxolitinib had higher rates of achieving IGA and pruritus-NRS. Ruxolitinib and delgocitinib have fewer TEAEs. A head-to-head meta-analysis may be essential for more data about the comparisons between JAK inhibitors in atopic dermatitis.472,473
STAT inhibitors
JAK inhibitors can prevent phosphorylation and activation of STATs. However, other signaling pathways can also be inhibited. More adverse events may ensue from the inhibition of upstream tyrosine kinases. Thus, STAT inhibitors seem to be more specific with fewer adverse effects. Among all seven STATs, inhibitors targeting STAT3 and STAT5 have been the most widely studied.474 However, STATs do not have intrinsic catalytic activity, thus, drug research for STATs is challenging. Most studies are based on preclinical research, and few drugs are in clinical trials or market-approved because high concentrations are required for them to be effective. Most STAT inhibitors focus on restricting STAT phosphorylation and/or dimerization by peptidomimetic approaches, virtual or library screening. STAT activity can be inhibited by drugs that are not pathway-specific, such as resveratrol and curcumin. Other approaches include non-peptide small molecules and oligonucleotide-based STAT inhibitors specific to the STAT–DNA-binding domain. Antisense oligonucleotides (ASOs) interfere with STAT mRNA.
Peptides and peptidomimetics
The STAT3 SH2 domain is required for STAT dimerization. Thus, inhibitors targeting pTyr-SH2 interactions have been developed. The first SH2-binding peptide and peptidomimetic, named PY*LKTK (Y = phosphotyrosine), was developed in 2001. It is a phosphotyrosyl protein that binds to the native C-terminal STAT3 SH2 domain, inhibits STAT1 to a lesser extent and has no effect on STAT5. PY*LKTK and its tripeptides PY*L and AY*L disrupt STAT3:STAT3 dimerization, and blocks STAT3-mediated DNA-binding activity and gene regulation.474,475 In addition, no further studies of PY*LKTK were found. Peptidomimetics have better pharmacokinetic properties than peptides. With XpTL as the basic structural scaffold for developing peptidomimetic compounds, ISS 610 and S31-M2001 showed superior pharmacokinetic profiles.476 Through amide coupling to the Leu residue’s free acid, newly modified ISS 610 was found to have reduced selectivity against STAT3 and greater selectivity for STAT1. This new peptidomimetic is called ISS 840. ISS 840 inhibited STAT1 or STAT3 homodimerization, with 20-fold higher inhibition of STAT1 dimerization compared to STAT3 dimerization.477 PM-73G is a cell-permeable, phosphatase-stable phosphopeptide mimic. It targets the STAT3 SH2 domain and inhibits the phosphorylation of STAT3 at Tyr705. PM-73G exhibited antitumor efficacy in a breast cancer murine model, inhibiting VEGF production and reducing vessel density. These findings indicate the role of PM-73G as a novel anti-angiogenesis drug.476 Several other peptidomimetic molecules were developed from the basic scaffold of Ac-PYLPQTV-NH2, such a CJ-1383.478 All the aforementioned peptidomimetics need to be further studied in vivo to improve their metabolic susceptibility and cellular permeability prior to clinical applications.479
PIAS3 protein serves as a negative regulator of STAT3. It has been reported that ~89% of human glioblastoma samples demonstrate low expression of PIAS3 and elevated STAT3 expression, and the ectopic addition of PIAS3 to glioblastoma cells results in inhibition of pSTAT3 activity.480 rPP-C8, a derivate of PIAS3, is a polypeptide derived from the C-terminal acidic region of PIAS3. It is derived from the interacting domains of STAT3 and PIAS3. A protein transduction domain consisting of 9 arginine residues was added to the primary sequence of rPP-C8 to improve its cellular permeability. This modified rPP-C8 slowed cell growth and inhibited the migration of breast and brain cancer cell lines.481
Small-molecule inhibitors
STAT3 inhibitors
Small-molecule inhibitors constitute the largest portion of STAT inhibitors. Most of them have been identified through computational modeling, docking studies, and virtual screening of chemical libraries. Stattic (STAT Three Inhibitory Compound) was discovered by high-throughput screening of chemical libraries consisting of 17298 substances. It is the first non-peptide small-molecule inhibitor that targets STAT3. Stattic selectively inhibits the activation, dimerization, and nuclear translocation of STAT3.482
STA-21 (also named NSC628869) and LLL-3 are structural analogs developed by the same group. They were identified by screening the National Cancer Institute (NCI) chemical library. Both bind to STAT3 SH2 domain and inhibit STAT3 dimerization. Notably, LLL-3 has better cellular permeability than STA-21.483 In a glioblastoma murine model, LLL-3 inhibited STAT3 activation and tumor progression.484 LLL12 is generated by replacing the acetyl group of LLL-3 with sulfonamide. It specifically inhibits STAT3 phosphorylation. LLL12 combined with chemotherapy better inhibited the cell viability, cell migration, and cell growth of several ovarian cancer cell lines, such as A2780, SKOV3, CAOV-3, and OVCAR5 cells.485 LLL12 has also been studied in several other diseases, including non-small cell lung cancer, osteosarcoma, multiple myeloma, acute lung injury, pancreatic cancer, rhabdomyosarcoma, hepatocellular cancer, medulloblastoma, and glioblastoma. LLL12 has shown promise for use in clinical settings.486,487,488,489,490,491 Recently, a study showed that XZH-5 blocks STAT3 phosphorylation at Try705, and inhibition of STAT3 signaling by XZH-5 could induce the apoptosis of human breast and pancreatic cancer cells.492 OPB-31121 inhibited both constitutively activated and IL-6-induced STAT3 activation in gastric cancer models. Moreover, OPB-31121 showed synergistic activity with 5-FU and cisplatin.493 A related phase 1 clinical trial with 25 patients demonstrated the feasibility of using OPB-31121 to treat advanced solid tumors, with two patients exhibiting tumor shrinkage. Common adverse events were gastrointestinal events.494,495 In 2011, Turkson and his colleagues found that compounds 10, 11, and 12 inhibit STAT3-DNA-binding activity via structural-based high-throughput screening of NCI chemical libraries. S3I-201 (compound 10) is the strongest inhibitor of STAT3-DNA binding. It is effective in treating several diseases in murine models, such as autism, dry eye, liver fibrosis, obstructive nephropathy, and tumors.496,497,498,499,500,501
Other small-molecule inhibitors, including S3I-201.1066, S3I-1757, STX-0119, BP-1-102, OPB-11077, Napabucasin (BBI608), Pyrimethamine (GLG-801), TTI-101 (C188-9), Nitazoxanide, WP1066, and BP-5-087 have also been investigated to target STAT3. In general, the applications of STAT3 inhibitors are extensive, and more clinical data are needed to verify their safety and efficacy.
Other STAT inhibitors (STAT1, STAT2, STAT4, STAT5, and STAT6): Compared to STAT3 inhibitors, inhibitors of STAT1, STAT2, STAT4, STAT5, and STAT6 are reported much less frequently, and most of these compounds are natural products. Few small-molecule inhibitors have been reported. However, pravastatin repressed IFNγ-mediated STAT1 activation to prevent aortic atherosclerosis. Pravastatin was developed to decrease plasma cholesterol by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase.502 Recently, pravastatin was found to repress the IL-6/STAT3 signaling pathway in rats with preeclampsia, alleviate oxidative stress and decrease the apoptosis of placental trophoblastic cells.503 Pimozide belongs to the diphenylbutylpiperidine class of drugs. When pimozide was used to treat schizophrenia, researchers found that the treated patients had a lower incidence of some types of cancers, including respiratory tract, prostate, and bladder cancer. Further studies have revealed that pimozide induces apoptosis and suppressed metastasis in vivo and ex vivo.504 Pimozide treatment reduced prolactinoma growth and increased apoptosis and cell cycle arrest in bromocriptine-resistant prolactinoma via the inhibition of the STAT5/Bcl-xL and STAT5/cyclin D1 pathway.505 There are several other molecules that inhibit STAT5 or STAT6, such as chromone-derived nicotinylhydrazone, BP-1108, BP-1075, leflunomide, and niflumic acid.
Natural products and derivatives
Natural products achieved therapeutic effects by influencing multiple biological processes, some of which inhibit the JAK/STAT pathway, and most of them target more than one component of the JAK/STAT pathway, including JAK, STAT, and inhibitory proteins. It should be noted that their effects might be indirect. Dozens of natural products are comprehensively reviewed in the previous articles, such as emodin, aloin, capsaicin, avicin D, celastrol, withaferin A, thymoquinone, caffeic acid, vinorelbine, paclitaxel, evodiamine, cryptotanshinone, honokiol, berbamine, cinnamon bark, and indirubin.506,507,508,509 Most of them are in the preclinical research stage, few are widely studied in clinic, we will introduce the most widely clinic studied natural products in the following.
Curcumin
Curcumin is a naturally occurring nutraceutical compound extensively found in the rhizome plant Curcuma longa. Curcumin inhibits STAT3 and induces the apoptosis of human glioblastoma and squamous carcinoma cells.510,511 In addition to direct inhibition of STAT3, curcumin diminishes the expression of STAT3 and STAT6 by upregulating SOCS1, SOCS3, and PIAS3.512,513 FLLL11, FLLL12, FLLL32, and FLLL62 are generated from curcumin and possess better biochemical properties than curcumin. These derivatives inhibit STAT3 phosphorylation at Tyr705 and induce caspase-dependent apoptosis of melanoma cells without abrogating IFNγ-induced STAT1 phosphorylation or gene expression.514 HCT-15 cells were co-cultured with lymphocytes from 20 stage III colon cancer patients/healthy donors. FLLL32 inhibited PD-L1 expression, decreased the number of Treg cells, and promoted Th1-protective immune responses.515 Besides, FLLL32 inhibited STAT3 phosphorylation induced by IFNα and IL-6 in breast cancer cells and retarded tumor growth in chicken embryo and mouse models.516
Resveratrol
Resveratrol, a natural polyphenolic stilbenoid, is found in grapes, mulberries, peanuts, rhubarb, etc. At the molecular level, resveratrol targets inflammatory cytokines, nuclear factor-κB, sirtuin, adenosine monophosphate kinase, and antioxidant enzymes.517 Resveratrol regulates immune responses by suppressing the phosphorylation of STAT1, STAT3, and NF-κB signaling pathways.518 In leukemia (Jurkat, SUP-B15, and Kasumi-1 cell lines), resveratrol inhibited IL-6-mediated STAT3 activation, induced apoptosis, and cell cycle arrest. Resveratrol also prolonged the survival period of tumor-bearing mice.519 However, resveratrol has poor bioavailability. LTR71 (6-methyl-2-propylimino-6, 7-dihydro-5H-benzo[1,3]-oxathiol-4-one), a derivative of resveratrol, suppressed RANTS induced STAT3 activation in breast cancer cells. Moreover, LTR71 inhibited the expression and activity of MMP-9 and prolonged the survival of murine models.520 Furthermore, the substitution of hydroxyl groups with methoxy groups improved the therapeutic versatility of resveratrol, and relative derivatives include pterostilbene, trimethoxystilbene, tetramethoxystilbene, and pentamethoxystilbene. In another method, 4-hydroxy group in the trans conformation is added to the 4- and 4’-positions in the stilbene structure. These derivatives include dihydroxystilbene, tetrahydroxystilbene, and hexahydroxystilbene. Piceatannol is a hydroxylated derivative that shows similar functions to resveratrol. It is regarded as a promising drug in treating aging-related diseases and various cancers.521 The third group of resveratrol derivatives is generated from the addition of halogen groups to the stilbene structure, such as 2-bromo-resveratrol and 2-chloro-resveratrol.522
Oleanolic acid
Oleanolic acid, a pentacyclic triterpenoid compound, exists in over 2000 plant species, especially the Oleaceae family. It is reported that oleanolic acid interfered with TYK2-STAT1/3 signaling, significantly decreased STAT3 dimers formation, and promoted the expression of SOCS3.523,524 To promote the clinical application, oleanolic acid can be structurally modified on the A ring, c-28, and C ring. Its derivatives include novel 3,5-disubstituted isoxazoles derivatives, acyloxyimino derivatives, 3-acetylated derivatives, acetate, ester derivatives, and oximes derivatives. Bardoxolone is one of the most widely studied derivatives. Moreover, nanometer preparation could improve the biochemical properties of oleanolic acid.525
Catechins
Catechins, also named flavanes, belong to the class of flavonoids. Catechins are the major constituent of green tea. Catechins are well-known for their antioxidant, anti-inflammation, and anticancer effects. Catechins affect multiple signal pathways, such as JAK/STAT, MAPK, PI3K/AKT, Wnt, and Notch.526 Catechins exhibited the chemoprotective effect during cancer therapy, partly due to inhibiting STAT1 and STAT3, then decreasing the expression of iNOS and ICAM-1.527
Artemisinins
Artemisinins are extracted from the sweet worm-wood (Artemisia annua). Artemisinin is used to treat fever in Chinese history, and it is famous for treating malaria. Besides, artemisinins also have anticancer, anti-virus, anti-schistosomes, anti-inflammatory properties, and artemisinins are effective in ocular diseases.528,529,530,531 Derivatives of artemisinin are collectively termed artemisinins.532 In preadipocytes and cancer cells, artemisinins interfered with STAT3 dimerization by binding to the SH2 domain, then inhibited STAT3 target gene expression.533,534
Clinical application and limitations of natural products
There are many clinical trials that have been completed or are in progress. For example, clinical studies on Curcumin are more than 250 on www.clinicaltrials.gov. Besides, their application covers a wide range of diseases. We will briefly introduce the clinical applications of curcumin, resveratrol, oleanolic acid, catechins, and artemisinins in the following.
Clinical trials of curcumin cover cancers, oral diseases, autoimmune diseases, metabolic diseases, cardiovascular diseases, neurological diseases, such as oral submucous fibrosis, periodontitis, osteoarthritis, nonalcoholic fatty liver disease, diabetes, IBD, polycystic ovary syndrome, radiation dermatitis, migraine, psychiatric disorders, and COVID-19. Most of them reported positive results, indicating that curcumin was well-tolerated and biologically active. Adjuvant therapy of curcumin may reduce fatigue in cancer patients.535 Curcumin and its derivatives can produce various dermatological effects, such as antioxidant, anti-inflammatory, reducing axillary hair growth, and increasing skin moisture. Further studies could explore the application of curcumin in dermatological diseases.536
Clinical trials regarding resveratrol achieved benefits in many human diseases, such as diabetes, obesity, cardiovascular diseases, neurodegeneration, muscular dystrophies, and cancers. Notably, resveratrol contributes to improve physical functions. For example, resveratrol and curcumin promote the recovery of bone and muscle mass in chronic kidney disease patients.537 Resveratrol combined with exercise in the elderly could improve physical functions and mitochondrial function.538
In clinical trials, oleanolic acid is studied in metabolic diseases including diabetes, obesity, and hyperlipidemia. Based on the published data, Oleanolic acid has anti-inflammatory, anticancer, antiosteoporotic, antioxidant, antiaging, neuroprotective, and hepatoprotective effects.539 Based on the preclinical data, future clinical studies should focus more on cancer, neurological diseases, cardiovascular diseases, oral diseases, and hepatitis.
Clinical trials of catechins revealed their therapeutic efficacy in diabetes, hyperlipidemia, hypertension, and obesity. Several clinical studies focus on multiple sclerosis, hepatitis, acute radiation-induced esophagitis. Notably, catechin may have regional analgesic efficacy for pain relief after surgery.540 Besides, Catechin combined with xanthan gum protect against upper respiratory infections.541
The clinical application of artemisinins is mainly for malaria. In the past 10 years, there have been few reports of clinical trials of artemisinin in the treatment of schistosomiasis. A few clinical trials have revealed that artemisinins are well-tolerated in cancer patients. A larger scale of clinical trials in cancer or metabolic diseases is needed to identify the optimal dose and efficacy of artemisinins.
Nevertheless, there are multiple limitations of natural products. First, many of them simultaneously destroy abnormally proliferating cancer cells and normal cells, partly due to the broad-spectrum inhibition of biological processes. Second, many natural products are underutilized owing to intrinsic pharmacokinetics, including short half-life, low bioavailability, inadequate biological stability, and poor aqueous solubility. Third, some natural products have hepatotoxicity, kidney toxicity, and reproductive toxicity. New developments aim to overcome these obstacles, such as synthesizing derivatives of natural products and utilizing nanoparticles, exosomes, liposomes, and phospholipids.
Nucleotide-based agents
Decoy oligonucleotides
Oligodeoxynucleotide (ODN) decoys are specific DNA-binding domain inhibitors. They compete with endogenous promotor sequences for binding to active STAT-STAT dimers, thus suppressing gene expression.506 For example, the STAT1 decoy ODN effectively inhibited murine antigen-induced arthritis and acute rejection of mouse/rat heart allografts, and as verified by in vitro study, nuclear extracts from synoviocytes were inhibited by STAT1 decoy ODN, as determined through electrophoretic mobility shift analysis.542,543,544 Decoy ODN targets both STAT1 and STAT3 to decrease allergic inflammation in rat asthma.545 In summary, most decoy ODNs targeting STAT are in the preclinical investigation stage of development. More studies are needed to further promote their application.
Antisense oligonucleotide
ASO targets a complementary coding sequence of mRNA and downregulates STAT expression at the transcriptional level. ASO is used in the inhibition of STAT and blocks cells from malignant transformation. Limitations of ASO include elevated liver enzymes, splenomegaly, and thrombocytopenia.546,547 AZD9150, a STAT3 ASO, was evaluated in diffuse large B-cell lymphoma patients and was well-tolerated and effective.548
STAT-targeting small interfering RNAs (siRNAs)
siRNAs are RNA duplexes of 19–22 bp. They incorporate their guide strands into a structure named the RNA-induced silencing complex. This process naturally occurs and is termed RNA interference.549 For example, STAT5B expression that was blocked by siRNA enhanced the chemosensitivity of gastric cancer cells, contributing to increased mitochondrial pathway-mediated apoptosis.550
Peripheral neuropathy and increased mortality are toxicities of concern in siRNA therapy. New modifications enable lower dosing and exposure levels while maintaining efficacy and will hopefully show fewer adverse effects.549
New developments and challenges of JAK/STAT inhibition
Some JAK inhibitors are newly developed and/or rarely reported, including pan-JAK inhibitor: TD-1473; JAK1 inhibitor: AZD4205, INCB-047986, INCB039110, INCB-052793; JAK2 inhibitor: AZD1480, AC-410; JAK3 inhibitor: Ritlecitinib (PF-06651600); TYK2 inhibitor: Deucravacitinib (BMS-986165), brepocitinib (PF-06700841), PF-06826647.93,551,552,553 These newly developed JAK inhibitors are expected to have increased selectivity and reduced adverse events. For example, TD-1473 is a gut-selective pan-JAK inhibitor used in ulcerative colitis. The novelty of TD-1473 is that it can locally inhibit proinflammatory cytokines in the gastrointestinal tract while minimizing systematic exposure.553 Moreover, multi-kinase inhibitors may achieve unexpected effects. Gusacitinib and cerdulatinib inhibit JAK and SYK, both JAK and SYK are involved in the pathogenesis of atopic dermatitis. Newly developed ritlecitinib inhibits JAK3 and the tyrosine kinase expressed in hepatocellular carcinoma kinase family, and ritlecitinib has achieved efficacy in RA patients.554
There are still many unanswered questions and challenges in the clinical application of JAK inhibition. First, optimizing dosing and formulation is important. For short half-lives JAK/STAT inhibitors, pulsatile high doses may result in more effective targeting, fewer adverse effects, and less resistance.555 Besides, similar to steroids, patients could be given a bolus dose of JAK/STAT inhibitor to induce remission and gradually reduce the dose for maintenance. Second, in clinical trials, JAK/STAT inhibitors elevated physical signs and symptoms in many autoimmune diseases. Nevertheless, rare significant anticancer effects are observed except for hematological tumors. This is partly due to the complex immune microenvironment in cancer patients. JAK/STAT pathway can both promote tumor progression and mediate anticancer effects. Moreover, JAK/STAT inhibitors may inhibit the function of cytotoxic T cells and NK cells.556 Cross-talk between JAK/STAT pathway and other pathways, post-translational control, epigenetic modification, and noncanonical signal transduction make the mechanisms more complicated. Third, for patients with liver impairment, renal impairment, risk of thromboembolism, and chronic virus infection, such as HBV, the dose should be reduced according to the condition.365,366,447,448 Forth, although most adverse events are well-tolerated, there is a possibility of life-threatening adverse events, such as severe infections and malignancies, ruxolitinib discontinuation syndrome, and Wernicke’s encephalopathy.403,406,407,431 Fifth, combinational therapy may provide additional benefits. For example, hsp90 inhibitors or histone deacetylase inhibitors promote JAK2 degradation.557 In addition, it is reported that many JAK inhibitors, such as baricitinib and fedratinib, downregulated the PD-L1 expression. Thus, combinational therapy of JAK inhibitor and immune checkpoint blockers may augment therapeutic efficacy.556
The most important limitation of STAT inhibitors is their toxicity, which comes from several aspects. First, there is considerable homology between different STATs, thus it is difficult to design highly specific STAT inhibitors, which can lead to off-target toxicity. Second, STATs participate in biological processes in the mitochondrion and endoplasmic reticulum. Therefore, STAT inhibitors may interfere with these processes.142 Third, the function of a certain STAT could be compensated by other STATs. For example, STAT3-deficient cells still respond to IL-6 stimulation through STAT1 transduction.5
Conclusion and future directions
The JAK/STAT pathway is central to extracellular cytokine-activated receptor-mediated signal transduction, which is involved in cellular proliferation and differentiation, organ development, and immune homeostasis. In this review, we discussed the composition and function of the JAK/STAT pathway and discussed the role of JAK/STAT in various diseases. Dysregulation of the JAK/STAT signaling pathway is recognized as a major contributor to various diseases, especially malignant tumors, and autoimmune diseases.
Diseases characterized by hyperactivated JAK/STAT pathway, elevated serum JAK-dependent cytokines, and mutated JAK/STAT are thought to respond well to JAK/STAT inhibitors. JAK/STAT inhibitors are currently applied in autoimmune diseases, malignant tumors, GVHD, and infectious diseases. Research into more clinical indications is ongoing, including IL-6 driven diseases: large-vessel vasculitis, type I IFN-related diseases: monogenic interferonopathies, scleroderma, myositis, and primary Sjogren’s syndrome.
There are still some unanswered questions about signal transduction. First, although JAK/STAT signal transduction is usually presented in a simple way, decades of research have shown that it is full of complexity. For example, the functions of STATs in organelles need further to be studied. In addition, JAKs and STATs are regulators of the epigenetic landscape, conversely, they are regulated by the epigenetic landscape, via promoting permissive marks and limiting repressive marks. For instance, JAK2 phosphorylates histone H3 at tyrosine 41, thus decrease the affinity of H3 to HP1α, promote tumorigenesis.557 Second, STAT can directly bind to DNA, but where and how they bind is an ongoing issue of debate, deep sequencing and chromatin immunoprecipitation (ChIP-seq) may help construct a comprehensive and unbiased STAT–DNA-binding map. Third, more research is needed to explain how STAT-mediated transcriptional inhibition works, either through direct binding of genomic locales or through the induction of secondary agents, such as inhibitory transcription factors and miRNA. Fourth, STATs can be activated by different cytokines, conversely, a cytokine can activate multiple STATs. Different cytokines are seen as different signals, a putative explanation is that different cytokines activate different phosphorylation levels of various STAT and other signal modules. More studies are needed to support the hypothesis. Fifth, how JAK/STAT pathway participates in the pathogenesis of diseases is not fully elucidated. For example, in the case of JAK2V617Fmutation of MPN, how does the JAK/STAT pathway go wrong?558 Sixth, most diseases result from multiple genetic abnormities, the cross-talk between JAK/STAT pathway components and other pathway components has not been fully elucidated.
Future studies should offer transformative insights into the underlying mechanisms of the JAK/STAT pathway effects and disease development. Moreover, we should aim to maximize efficacy and minimize adverse effects in patients in different stages of certain diseases and to explore biomarkers that predict efficacy and offer prognoses.
References
Darnell, J. E. Jr STATs and gene regulation. Science 277, 1630–1635 (1997).
Owen, K. L., Brockwell, N. K. & Parker, B. S. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers 11, 2002 (2019).
Aittomäki, S. & Pesu, M. Therapeutic targeting of the Jak/STAT pathway. Basic Clin. Pharm. Toxicol. 114, 18–23 (2014).
O’Shea, J. J. et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med. 66, 311–328 (2015).
Banerjee, S., Biehl, A., Gadina, M., Hasni, S. & Schwartz, D. M. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77, 521–546 (2017).
Villarino, A. V., Kanno, Y. & O’Shea, J. J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18, 374–384 (2017).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
O’Shea, J. J. & Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36, 542–550 (2012).
Aaronson, D. S. & Horvath, C. M. A road map for those who don’t know JAK-STAT. Science 296, 1653–1655 (2002).
Xin, P. et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 80, 106210 (2020).
Fu, X. Y., Kessler, D. S., Veals, S. A., Levy, D. E. & Darnell, J. E. Jr ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl Acad. Sci. USA 87, 8555–8559 (1990).
Fu, X. Y. A transcription factor with SH2 and SH3 domains is directly activated by an interferon alpha-induced cytoplasmic protein tyrosine kinase(s). Cell 70, 323–335 (1992).
Fu, X. Y. A direct signaling pathway through tyrosine kinase activation of SH2 domain-containing transcription factors. J. Leukoc. Biol. 57, 529–535 (1995).
Shuai, K., Stark, G. R., Kerr, I. M. & Darnell, J. E. Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 261, 1744–1746 (1993).
Zhong, Z., Wen, Z. & Darnell, J. E. Jr Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc. Natl Acad. Sci. USA 91, 4806–4810 (1994).
Liu, X., Robinson, G. W., Gouilleux, F., Groner, B. & Hennighausen, L. Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc. Natl Acad. Sci. USA 92, 8831–8835 (1995).
Hou, J. et al. An interleukin-4-induced transcription factor: IL-4 Stat. Science 265, 1701–1706 (1994).
Wilks, A. F. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc. Natl Acad. Sci. USA 86, 1603–1607 (1989).
Wilks, A. F. et al. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol. Cell. Biol. 11, 2057–2065 (1991).
Krolewski, J. J., Lee, R., Eddy, R., Shows, T. B. & Dalla-Favera, R. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene 5, 277–282 (1990).
Velazquez, L., Fellous, M., Stark, G. R. & Pellegrini, S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 70, 313–322 (1992).
Müller, M. et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature 366, 129–135 (1993).
Lai, K. S. et al. A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells. J. Biol. Chem. 270, 25028–25036 (1995).
Verbsky, J. W. et al. Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells. J. Biol. Chem. 271, 13976–13980 (1996).
Gurniak, C. B. & Berg, L. J. Murine JAK3 is preferentially expressed in hematopoietic tissues and lymphocyte precursor cells. Blood 87, 3151–3160 (1996).
Harpur, A. G., Andres, A. C., Ziemiecki, A., Aston, R. R. & Wilks, A. F. JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 7, 1347–1353 (1992).
Kawamura, M. et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc. Natl Acad. Sci. USA 91, 6374–6378 (1994).
Rane, S. G. & Reddy, E. P. JAK3: a novel JAK kinase associated with terminal differentiation of hematopoietic cells. Oncogene 9, 2415–2423 (1994).
Takahashi, T. & Shirasawa, T. Molecular cloning of rat JAK3, a novel member of the JAK family of protein tyrosine kinases. FEBS Lett. 342, 124–128 (1994).
Ferrao, R. & Lupardus, P. J. The janus kinase (JAK) FERM and SH2 domains: bringing specificity to JAK-receptor interactions. Front. Endocrinol. 8, 71 (2017).
Frank, S. J. et al. Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor. J. Biol. Chem. 270, 14776–14785 (1995).
Velazquez, L. et al. Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-alpha/beta and for signal transduction. J. Biol. Chem. 270, 3327–3334 (1995).
Leonard, W. J. & O’Shea, J. J. Jaks and STATs: biological implications. Annu Rev. Immunol. 16, 293–322 (1998).
Rodig, S. J. et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 93, 373–383 (1998).
Ihle, J. N., Witthuhn, B. A., Quelle, F. W., Yamamoto, K. & Silvennoinen, O. Signaling through the hematopoietic cytokine receptors. Annu Rev. Immunol. 13, 369–398 (1995).
Schindler, C. & Strehlow, I. Cytokines and STAT signaling. Adv. Pharm. 47, 113–174 (2000).
Wu, H., Liu, X., Jaenisch, R. & Lodish, H. F. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 83, 59–67 (1995).
Neubauer, H. et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93, 397–409 (1998).
Russell, S. M. et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science 266, 1042–1045 (1994).
Cao, X. et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity 2, 223–238 (1995).
Park, S. Y. et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity 3, 771–782 (1995).
Saijo, K., Park, S. Y., Ishida, Y., Arase, H. & Saito, T. Crucial role of Jak3 in negative selection of self-reactive T cells. J. Exp. Med 185, 351–356 (1997).
Mogensen, K. E. et al. Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-α/β and for signal transduction. J. Biol. Chem. 270, 3327–3334 (1995).
Stahl, N. et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 263, 92–95 (1994).
Finbloom, D. S. & Winestock, K. D. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J. Immunol. 155, 1079–1090 (1995).
Bacon, C. M. et al. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J. Exp. Med. 181, 399–404 (1995).
Welham, M. J., Learmonth, L., Bone, H. & Schrader, J. W. Interleukin-13 signal transduction in lymphohemopoietic cells: similarities and differences in signal transduction with interleukin-4 and insulin. J. Biol. Chem. 270, 12286–12296 (1995).
Watford, W. T. & O’Shea, J. J. Human tyk2 kinase deficiency: another primary immunodeficiency syndrome. Immunity 25, 695–697 (2006).
Karaghiosoff, M. et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity 13, 549–560 (2000).
Minegishi, Y. et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 25, 745–755 (2006).
Seto, Y. et al. Enhanced Th2 cell-mediated allergic inflammation in Tyk2-deficient mice. J. Immunol. 170, 1077–1083 (2003).
Fu, X. Y., Schindler, C., Improta, T., Aebersold, R. & Darnell, J. E. Jr The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. Proc. Natl Acad. Sci. USA 89, 7840–7843 (1992).
Hou, J., Schindler, U., Henzel, W. J., Wong, S. C. & McKnight, S. L. Identification and purification of human Stat proteins activated in response to interleukin-2. Immunity 2, 321–329 (1995).
Mui, A. L., Wakao, H., O’Farrell, A. M., Harada, N. & Miyajima, A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 14, 1166–1175 (1995).
Zhong, Z., Wen, Z. & Darnell, J. E. Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98 (1994).
Horvath, C. M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 25, 496–502 (2000).
Murphy, T. L., Geissal, E. D., Farrar, J. D. & Murphy, K. M. Role of the Stat4 N domain in receptor proximal tyrosine phosphorylation. Mol. Cell Biol. 20, 7121–7131 (2000).
Shuai, K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene 19, 2638–2644 (2000).
Vinkemeier, U., Moarefi, I., Darnell, J. E. Jr & Kuriyan, J. Structure of the amino-terminal protein interaction domain of STAT-4. Science 279, 1048–1052 (1998).
Begitt, A., Meyer, T., van Rossum, M. & Vinkemeier, U. Nucleocytoplasmic translocation of Stat1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc. Natl Acad. Sci. USA 97, 10418–10423 (2000).
Collum, R. G., Brutsaert, S., Lee, G. & Schindler, C. A Stat3-interacting protein (StIP1) regulates cytokine signal transduction. Proc. Natl Acad. Sci. USA 97, 10120–10125 (2000).
Horvath, C. M., Stark, G. R., Kerr, I. M. & Darnell, J. E. Jr Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol. Cell Biol. 16, 6957–6964 (1996).
Kisseleva, T., Bhattacharya, S., Braunstein, J. & Schindler, C. W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285, 1–24 (2002).
Zhang, J. G. et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl Acad. Sci. USA 96, 2071–2076 (1999).
Zhang, T., Kee, W. H., Seow, K. T., Fung, W. & Cao, X. The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell Biol. 20, 7132–7139 (2000).
Zhu, M., John, S., Berg, M. & Leonard, W. J. Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNgamma-mediated signaling. Cell 96, 121–130 (1999).
Yang, E., Wen, Z., Haspel, R. L., Zhang, J. J. & Darnell, J. E. Jr The linker domain of Stat1 is required for gamma interferon-driven transcription. Mol. Cell Biol. 19, 5106–5112 (1999).
Kawata, T. et al. SH2 signaling in a lower eukaryote: a STAT protein that regulates stalk cell differentiation in Dictyostelium. Cell 89, 909–916 (1997).
Barahmand-Pour, F., Meinke, A., Groner, B. & Decker, T. Jak2-Stat5 interactions analyzed in yeast. J. Biol. Chem. 273, 12567–12575 (1998).
Chen, X. et al. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 93, 827–839 (1998).
Gupta, S. et al. The SH2 domains of Stat1 and Stat2 mediate multiple interactions in the transduction of IFN-alpha signals. EMBO J. 15, 1075–1084 (1996).
Shuai, K. et al. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell 76, 821–828 (1994).
Schindler, C., Fu, X. Y., Improta, T., Aebersold, R. & Darnell, J. E. Jr Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc. Natl Acad. Sci. USA 89, 7836–7839 (1992).
Müller, M. et al. Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. EMBO J. 12, 4221–4228 (1993).
Dimberg, A., Karlberg, I., Nilsson, K. & Öberg, F. Ser727/Tyr701-phosphorylated Stat1 is required for the regulation of c-Myc, cyclins, and p27Kip1 associated with ATRA-induced G0/G1 arrest of U-937 cells. Blood 102, 254–261 (2003).
Schlee, M. et al. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt’s lymphoma. Int. J. Cancer 120, 1387–1395 (2007).
Dimberg, A., Nilsson, K. & Öberg, F. Phosphorylation-deficient Stat1 inhibits retinoic acid-induced differentiation and cell cycle arrest in U-937 monoblasts. Blood 96, 2870–2878 (2000).
Takayanagi, H., Kim, S., Koga, T. & Taniguchi, T. Stat1-mediated cytoplasmic attenuation in osteoimmunology. J. Cell. Biochem. 94, 232–240 (2005).
Lee, C.-K., Smith, E., Gimeno, R., Gertner, R. & Levy, D. E. STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-γ. J. Immunol. 164, 1286–1292 (2000).
Wold, W. S. & Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 13, 421–433 (2013).
Gu, W. & Roeder, R. G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 (1997).
Xu, X., Fu, X. Y., Plate, J. & Chong, A. S. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 58, 2832–2837 (1998).
Stephanou, A., Brar, B. K., Knight, R. A. & Latchman, D. S. Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-x promoters. Cell Death Differ. 7, 329–330 (2000).
Dovhey, S. E., Ghosh, N. S. & Wright, K. L. Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res. 60, 5789–5796 (2000).
Kovacic, B. et al. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 10, 77–87 (2006).
Chatterjee-Kishore, M., Kishore, R., Hicklin, D. J., Marincola, F. M. & Ferrone, S. Different requirements for signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 in the regulation of low molecular mass polypeptide 2 and transporter associated with antigen processing 1 gene expression. J. Biol. Chem. 273, 16177–16183 (1998).
Najjar, I. et al. STAT1-dependent IgG cell-surface expression in a human B cell line derived from a STAT1-deficient patient. J. Leukoc. Biol. 87, 1145–1152 (2010).
Yoshimoto, T. et al. Induction of IgG2a class switching in B cells by IL-27. J. Immunol. 173, 2479–2485 (2004).
Dupuis, S. et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33, 388–391 (2003).
Schindler, C. & Plumlee, C. Inteferons pen the JAK–STAT pathway. Semin. Cell Dev. Biol. 19, 311–318 (2008).
Wang, Y. et al. A virus-induced conformational switch of STAT1-STAT2 dimers boosts antiviral defenses. Cell Res. 31, 206–218 (2021).
Park, C., Li, S., Cha, E. & Schindler, C. Immune response in Stat2 knockout mice. Immunity 13, 795–804 (2000).
Qureshy, Z., Johnson, D. E. & Grandis, J. R. Targeting the JAK/STAT pathway in solid tumors. J. Cancer Metastasis Treat 6 (2020).
Maritano, D. et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 5, 401–409 (2004).
Caldenhoven, E. et al. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 271, 13221–13227 (1996).
Schaefer, T. S., Sanders, L. K. & Nathans, D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc. Natl Acad. Sci. USA 92, 9097–9101 (1995).
Darnell, J. E. Jr, Kerr, I. M. & Stark, G. R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421 (1994).
Heinrich, P. C., Behrmann, I., Müller-Newen, G., Schaper, F. & Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochemical J. 334(Pt 2), 297–314 (1998).
Ruff-Jamison, S. et al. Epidermal growth factor and lipopolysaccharide activate Stat3 transcription factor in mouse liver. J. Biol. Chem. 269, 21933–21935 (1994).
Zhang, M. et al. A STAT3 palmitoylation cycle promotes TH17 differentiation and colitis. Nature 586, 434–439 (2020).
Kortylewski, M. et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321 (2005).
Ostrand-Rosenberg, S. & Sinha, P. Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182, 4499–4506 (2009).
Bromberg, J. F. et al. Stat3 as an oncogene. Cell 98, 295–303 (1999).
Yu, H., Kortylewski, M. & Pardoll, D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51 (2007).
Catlett-Falcone, R. et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10, 105–115 (1999).
Shao, F., Pang, X. & Baeg, G. H. Targeting the JAK/STAT signaling pathway for breast cancer. Curr. Med. Chem. https://doi.org/10.2174/0929867328666201207202012 (2020).
Avalle, L., Camporeale, A., Camperi, A. & Poli, V. STAT3 in cancer: a double edged sword. Cytokine 98, 42–50 (2017).
Niu, G. et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 59, 5059–5063 (1999).
Xu, G., Zhang, C. & Zhang, J. Dominant negative STAT3 suppresses the growth and invasion capability of human lung cancer cells. Mol. Med Rep. 2, 819–824 (2009).
Yang, L. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8 (2020).
Miyagi, T. et al. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204, 2383–2396 (2007).
Thieu, V. T. et al. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29, 679–690 (2008).
Weinstein, J. S. et al. STAT4 and T-bet control follicular helper T cell development in viral infections. J. Exp. Med. 215, 337–355 (2018).
Murray, P. J. The JAK-STAT signaling pathway: input and output integration. J. Immunol. 178, 2623–2629 (2007).
Schindler, C., Levy, D. E. & Decker, T. JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063 (2007).
Soldaini, E. et al. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol. Cell Biol. 20, 389–401 (2000).
Lin, J. X. et al. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat. Commun. 8, 017–01477 (2017).
Azam, M. et al. Interleukin-3 signals through multiple isoforms of Stat5. EMBO J. 14, 1402–1411 (1995).
Wakao, H., Gouilleux, F. & Groner, B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 14, 854–855 (1995).
Lin, J. X. & Leonard, W. J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 19, 2566–2576 (2000).
Liu, X. et al. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 11, 179–186 (1997).
Lin, J. X. et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 36, 586–599 (2012).
Pericle, F. et al. Immunocompromised tumor-bearing mice show a selective loss of STAT5a/b expression in T and B lymphocytes. J. Immunol. 159, 2580–2585 (1997).
Refaeli, Y., Van Parijs, L., London, C. A., Tschopp, J. & Abbas, A. K. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8, 615–623 (1998).
Van Parijs, L. et al. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity 11, 281–288 (1999).
Mikita, T., Daniel, C., Wu, P. & Schindler, U. Mutational analysis of the STAT6 SH2 domain. J. Biol. Chem. 273, 17634–17642 (1998).
Chen, H. et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147, 436–446 (2011).
Patel, B. K., Pierce, J. H. & LaRochelle, W. J. Regulation of interleukin 4-mediated signaling by naturally occurring dominant negative and attenuated forms of human Stat6. Proc. Natl Acad. Sci. USA 95, 172–177 (1998).
Duetsch, G. STAT6 as an asthma candidate gene: polymorphism-screening, association and haplotype analysis in a Caucasian sib-pair study. Hum. Mol. Genet. 11, 613–621 (2002).
Zhu, J. & Paul, W. E. CD4 T cells: fates, functions, and faults. Blood 112, 1557–1569 (2008).
Shimoda, K. et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 380, 630–633 (1996).
Kaplan, M. H., Schindler, U., Smiley, S. T. & Grusby, M. J. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 4, 313–319 (1996).
Sasaki, K. et al. Stat6 signaling suppresses VLA-4 expression by CD8+ T cells and limits their ability to infiltrate tumor lesions in vivo. J. Immunol. 181, 104–108 (2008).
Tenhumberg, S. et al. gp130 dimerization in the absence of ligand: preformed cytokine receptor complexes. Biochem. Biophys. Res. Commun. 346, 649–657 (2006).
Livnah, O. et al. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283, 987–990 (1999).
Remy, I., Wilson, I. A. & Michnick, S. W. Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993 (1999).
Naismith, J. H., Devine, T. Q., Brandhuber, B. J. & Sprang, S. R. Crystallographic evidence for dimerization of unliganded tumor necrosis factor receptor. J. Biol. Chem. 270, 13303–13307 (1995).
Kramer, J. M. et al. Evidence for ligand-independent multimerization of the IL-17 receptor. J. Immunol. 176, 711–715 (2006).
Krause, C. D. et al. Interactions among the components of the interleukin-10 receptor complex. Biochem. Biophys. Res. Commun. 340, 377–385 (2006).
Brooks, A. J. et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344, 1249783 (2014).
Durham, G. A., Williams, J. J. L., Nasim, M. T. & Palmer, T. M. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol. Sci. 40, 298–308 (2019).
Bharadwaj, U., Kasembeli, M. M., Robinson, P. & Tweardy, D. J. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol. Rev. 72, 486–526 (2020).
Li, W. X. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 18, 545–551 (2008).
Shi, S. et al. JAK signaling globally counteracts heterochromatic gene silencing. Nat. Genet. 38, 1071–1076 (2006).
Shi, S. et al. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat. Cell Biol. 10, 489–496 (2008).
Ogawa, H., Ishiguro, K., Gaubatz, S., Livingston, D. M. & Nakatani, Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296, 1132–1136 (2002).
Braig, M. et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660–665 (2005).
Narita, M. et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716 (2003).
Christova, R. et al. P-STAT1 mediates higher-order chromatin remodelling of the human MHC in response to IFN. J. Cell Sci. 120, 3262–3270 (2007).
Shi, M., Lin, T. H., Appell, K. C. & Berg, L. J. Janus-kinase-3-dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity 28, 763–773 (2008).
Lai, S. Y. & Johnson, F. M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist. Updat. 13, 67–78 (2010).
Limnander, A. & Rothman, P. B. Abl oncogene bypasses normal regulation of Jak/STAT activation. Cell Cycle 3, 1486–1488 (2004).
Han, Y. et al. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 108, 2796–2803 (2006).
Coppo, P. et al. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 134, 171–179 (2006).
Yu, C. L. et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269, 81–83 (1995).
Fu, X. Y. & Zhang, J. J. Transcription factor p91 interacts with the epidermal growth factor receptor and mediates activation of the c-fos gene promoter. Cell 74, 1135–1145 (1993).
Ruff-Jamison, S., Chen, K. & Cohen, S. Epidermal growth factor induces the tyrosine phosphorylation and nuclear translocation of Stat 5 in mouse liver. Proc. Natl Acad. Sci. USA 92, 4215–4218 (1995).
Stöcklin, E., Wissler, M., Gouilleux, F. & Groner, B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature 383, 726–728 (1996).
Horvai, A. E. et al. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl Acad. Sci. USA 94, 1074–1079 (1997).
Wakioka, T. et al. APS, an adaptor protein containing Pleckstrin homology (PH) and Src homology-2 (SH2) domains inhibits the JAK-STAT pathway in collaboration with c-Cbl. Leukemia 13, 760–767 (1999).
Tanaka, N. et al. Possible involvement of a novel STAM-associated molecule "AMSH" in intracellular signal transduction mediated by cytokines. J. Biol. Chem. 274, 19129–19135 (1999).
Yoshimura, A. et al. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 14, 2816–2826 (1995).
Kershaw, N. J. et al. SOCS3 binds specific receptor–JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 20, 469–476 (2013).
Yasukawa, H. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 18, 1309–1320 (1999).
Okumura, F., Joo-Okumura, A., Nakatsukasa, K. & Kamura, T. The role of cullin 5-containing ubiquitin ligases. Cell Div. 11, 1 (2016).
Tamiya, T., Kashiwagi, I., Takahashi, R., Yasukawa, H. & Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb. Vasc. Biol. 31, 980–985 (2011).
Mohr, S. E. & Boswell, R. E. Zimp encodes a homologue of mouse Miz1 and PIAS3 and is an essential gene in Drosophila melanogaster. Gene 229, 109–116 (1999).
Takahashi, Y., Kahyo, T., Toh-e, A., Yasuda, H. & Kikuchi, Y. Yeast Ull1/Siz1 is a novel SUMO1/Smt3 ligase for septin components and functions as an adaptor between conjugating enzyme and substrates. J. Biol. Chem. 276, 48973–48977 (2001).
Sonnenblick, A., Levy, C. & Razin, E. Interplay between MITF, PIAS3, and STAT3 in mast cells and melanocytes. Mol. Cell. Biol. 24, 10584–10592 (2004).
Rogers, R. S., Horvath, C. M. & Matunis, M. J. SUMO modification of STAT1 and its role in PIAS-mediated inhibition of gene activation. J. Biol. Chem. 278, 30091–30097 (2003).
Tussié-Luna, M. I., Bayarsaihan, D., Seto, E., Ruddle, F. H. & Roy, A. L. Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxbeta. Proc. Natl Acad. Sci. USA 99, 12807–12812 (2002).
Sachdev, S. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15, 3088–3103 (2001).
Ungureanu, D. et al. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood 102, 3311–3313 (2003).
Liu, B. et al. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 5, 891–898 (2004).
ten Hoeve, J. et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell Biol. 22, 5662–5668 (2002).
Ram, P. A. & Waxman, D. J. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J. Biol. Chem. 272, 17694–17702 (1997).
Irie-Sasaki, J. et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409, 349–354 (2001).
Myers, M. P. et al. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 276, 47771–47774 (2001).
Klingmuller, U., Lorenz, U., Cantley, L. C., Neel, B. G. & Lodish, H. F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 80, 729–738 (1995).
You, M., Yu, D. H. & Feng, G. S. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell Biol. 19, 2416–2424 (1999).
Nakashima, K. et al. Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 284, 479–482 (1999).
Luo, K. Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harb. Perspect. Biol. 9, a022137 (2017).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Tang, L. Y. et al. Transforming growth factor-β (TGF-β) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 292, 4302–4312 (2017).
Wang, G. et al. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene 35, 4388–4398 (2016).
Bright, J. J. & Sriram, S. TGF-beta inhibits IL-12-induced activation of Jak-STAT pathway in T lymphocytes. J. Immunol. 161, 1772–1777 (1998).
Wu, Q. et al. JNK signaling in cancer cell survival. Medicinal Res. Rev. 39, 2082–2104 (2019).
Josten, F., Fuss, B., Feix, M., Meissner, T. & Hoch, M. Cooperation of JAK/STAT and Notch signaling in the Drosophila foregut. Dev. Biol. 267, 181–189 (2004).
Liu, W., Singh, S. R. & Hou, S. X. JAK-STAT is restrained by Notch to control cell proliferation of the Drosophila intestinal stem cells. J. Cell Biochem. 109, 992–999 (2010).
Assa-Kunik, E., Torres, I. L., Schejter, E. D., Johnston, D. S. & Shilo, B. Z. Drosophila follicle cells are patterned by multiple levels of Notch signaling and antagonism between the Notch and JAK/STAT pathways. Development 134, 1161–1169 (2007).
Kamakura, S. et al. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 6, 547–554 (2004).
Jin, S. et al. Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKα/IKKβ. Oncogene 32, 4892–4902 (2013).
Krasilnikov, M., Ivanov, V. N., Dong, J. & Ronai, Z. ERK and PI3K negatively regulate STAT-transcriptional activities in human melanoma cells: implications towards sensitization to apoptosis. Oncogene 22, 4092–4101 (2003).
Rädler, P. D., Wehde, B. L. & Wagner, K. U. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol. Cell Endocrinol. 451, 31–39 (2017).
Tasian, S. K. et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 120, 833–842 (2012).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Baud, V. & Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40 (2009).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Kortylewski, M. et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15, 114–123 (2009).
Wang, L. et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 206, 1457–1464 (2009).
Yu, H., Pardoll, D. & Jove, R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 (2009).
Lee, H. et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 15, 283–293 (2009).
Chen, Z. & O’Shea, J. J. Th17 cells: a new fate for differentiating helper T cells. Immunol. Res. 41, 87–102 (2008).
Dalwadi, H. et al. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin. Cancer Res. 11, 7674–7682 (2005).
Bennett, B. L., Cruz, R., Lacson, R. G. & Manning, A. M. Interleukin-4 suppression of tumor necrosis factor alpha-stimulated E-selectin gene transcription is mediated by STAT6 antagonism of NF-kappaB. J. Biol. Chem. 272, 10212–10219 (1997).
Farlik, M. et al. Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity 33, 25–34 (2010).
Savitsky, D., Tamura, T., Yanai, H. & Taniguchi, T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 59, 489–510 (2010).
Levy, D. E. & Darnell, J. E. Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 (2002).
Contursi, C. et al. IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macrophages. Proc. Natl Acad. Sci. USA 97, 91–96 (2000).
Platanitis, E. & Decker, T. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front. Immunol. 9, 2542 (2018).
Esashi, E. et al. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 28, 509–520 (2008).
O'Shea, J. J. et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annual review of medicine 66, 311–328 (2015).
Clark, J. D., Flanagan, M. E. & Telliez, J. B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 57, 5023–5038 (2014).
Thomas, S. et al. Effect of methotrexate on JAK/STAT pathway activation in myeloproliferative neoplasms. Lancet 385(Suppl 1), S98 (2015).
Akada, H. et al. Critical role of Jak2 in the maintenance and function of adult hematopoietic. Stem Cells Stem Cells 32, 1878–1889 (2014).
Radosevic, N. et al. JAK2 contributes to the intrinsic capacity of primary hematopoietic cells to respond to stem cell factor. Exp. Hematol. 32, 149–156 (2004).
Thomis, D. C., Gurniak, C. B., Tivol, E., Sharpe, A. H. & Berg, L. J. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science 270, 794–797 (1995).
Flex, E. et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 205, 751–758 (2008).
Jeong, E. G. et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin. Cancer Res. 14, 3716–3721 (2008).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
van Leeuwen, F. E. & Ng, A. K. Long-term risk of second malignancy and cardiovascular disease after Hodgkin lymphoma treatment. Hematol. Am. Soc. Hematol. Educ. Program 2016, 323–330 (2016).
Küppers, R. New insights in the biology of Hodgkin lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2012, 328–334 (2012).
Skinnider, B. F. et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 99, 618–626 (2002).
Guiter, C. et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood 104, 543–549 (2004).
Tiacci, E. et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 131, 2454–2465 (2018).
Curiel, T. J. et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 9, 562–567 (2003).
Chikamatsu, K. et al. Immunosuppressive activity of CD14+ HLA-DR- cells in squamous cell carcinoma of the head and neck. Cancer Sci. 103, 976–983 (2012).
Han, S. J. et al. Gamma interferon-mediated superinduction of B7-H1 in PTEN-deficient glioblastoma: a paradoxical mechanism of immune evasion. Neuroreport 20, 1597–1602 (2009).
Walters, D. K. et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell 10, 65–75 (2006).
Bercovich, D. et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet 372, 1484–1492 (2008).
Elliott, N. E. et al. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood 118, 3911–3921 (2011).
Krejsgaard, T. et al. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia 20, 1759–1766 (2006).
Yared, M. A., Khoury, J. D., Medeiros, L. J., Rassidakis, G. Z. & Lai, R. Activation status of the JAK/STAT3 pathway in mantle cell lymphoma. Arch. Pathol. Lab. Med. 129, 990–996 (2005).
Kiyoi, H., Yamaji, S., Kojima, S. & Naoe, T. JAK3 mutations occur in acute megakaryoblastic leukemia both in Down syndrome children and non-Down syndrome adults. Leukemia 21, 574–576 (2007).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008).
Koo, G. C. et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov. 2, 591–597 (2012).
Cornejo, M. G. et al. Constitutive JAK3 activation induces lymphoproliferative syndromes in murine bone marrow transplantation models. Blood 113, 2746–2754 (2009).
Baxter, E. J. et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054–1061 (2005).
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005).
Levine, R. L. et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397 (2005).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
Lacout, C. et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 108, 1652–1660 (2006).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Zhang, Q. et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015).
Stivala, S. et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Investig. 129, 1596–1611 (2019).
Oh, S. T. et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 116, 988–992 (2010).
Bersenev, A., Wu, C., Balcerek, J. & Tong, W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Investig. 118, 2832–2844 (2008).
Buza-Vidas, N. et al. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev. 20, 2018–2023 (2006).
Tong, W. & Lodish, H. F. Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J. Exp. Med. 200, 569–580 (2004).
Seita, J. et al. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc. Natl Acad. Sci. USA 104, 2349–2354 (2007).
Ema, H. et al. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev. Cell 8, 907–914 (2005).
Bersenev, A. et al. Lnk constrains myeloproliferative diseases in mice. J. Clin. Investig. 120, 2058–2069 (2010).
Hoshida, Y. Risk of recurrence in hepatitis B-related hepatocellular carcinoma: impact of viral load in late recurrence. J. Hepatol. 51, 842–844 (2009).
Brown, Z. J., Heinrich, B. & Greten, T. F. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 15, 536–554 (2018).
Costentin, C. E., Bababekov, Y. J., Zhu, A. X. & Yeh, H. Is it time to reconsider the Milan criteria for selecting patients with hepatocellular carcinoma for deceased-donor liver transplantation? Hepatology 69, 1324–1336 (2019).
Farazi, P. A. & DePinho, R. A. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev. Cancer 6, 674–687 (2006).
Sangro, B. et al. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 72, 320–341 (2020).
Dhar, K., Rakesh, K., Pankajakshan, D. & Agrawal, D. K. SOCS3 promotor hypermethylation and STAT3-NF-κB interaction downregulate SOCS3 expression in human coronary artery smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 304, H776–H785 (2013).
Iwahori, K. et al. Overexpression of SOCS3 exhibits preclinical antitumor activity against malignant pleural mesothelioma. Int. J. Cancer 129, 1005–1017 (2011).
He, B. et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl Acad. Sci. USA 100, 14133–14138 (2003).
Li, Y. et al. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 59, 227–235 (2010).
Pierconti, F. et al. Epigenetic silencing of SOCS3 identifies a subset of prostate cancer with an aggressive behavior. Prostate 71, 318–325 (2011).
Zhang, X. et al. An integrated analysis of SOCS1 down-regulation in HBV infection-related hepatocellular carcinoma. J. Viral Hepat. 21, 264–271 (2014).
Qiu, L., Tang, Q., Li, G. & Chen, K. Long non-coding RNAs as biomarkers and therapeutic targets: recent insights into hepatocellular carcinoma. Life Sci. 191, 273–282 (2017).
Yin, Y. Z., Zheng, W. H., Zhang, X., Chen, Y. H. & Tuo, Y. H. LINC00346 promotes hepatocellular carcinoma progression via activating the JAK-STAT3 signaling pathway. J. Cell Biochem. 121, 735–742 (2020).
Blechacz, B. R. et al. Sorafenib inhibits signal transducer and activator of transcription-3 signaling in cholangiocarcinoma cells by activating the phosphatase shatterproof 2. Hepatology 50, 1861–1870 (2009).
Isomoto, H. et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 42, 1329–1338 (2005).
Inuzuka, H. et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109 (2011).
Xie, L. et al. Chemical and genetic inhibition of STAT3 sensitizes hepatocellular carcinoma cells to sorafenib induced cell death. Int. J. Biol. Sci. 14, 577–585 (2018).
Xu, Y., Liu, Y. J. & Yu, Q. Angiopoietin-3 inhibits pulmonary metastasis by inhibiting tumor angiogenesis. Cancer Res. 64, 6119–6126 (2004).
Galaup, A. et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc. Natl Acad. Sci. USA 103, 18721–18726 (2006).
Sasaki, H. et al. Angioarrestin mRNA expression in early-stage lung cancers. Eur. J. Surg. Oncol. 29, 649–653 (2003).
Matsuda, Y., Ichida, T., Matsuzawa, J., Sugimura, K. & Asakura, H. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology 116, 394–400 (1999).
Lim, S. O. et al. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology 135, 2128–2140 (2008). 2140.e2121-2128.
Wong, R. S. Apoptosis in cancer: from pathogenesis to treatment. J. Exp. Clin. Cancer Res. 30, 87 (2011).
Tsokos, G. C. Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 (2011).
Bagavant, H. & Fu, S. M. Pathogenesis of kidney disease in systemic lupus erythematosus. Curr. Opin. Rheumatol. 21, 489–494 (2009).
Pathak, S. & Mohan, C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res. Ther. 13, 241 (2011).
Uhm, W. S. et al. Cytokine balance in kidney tissue from lupus nephritis patients. Rheumatology 42, 935–938 (2003).
Lettre, G. & Rioux, J. D. Autoimmune diseases: insights from genome-wide association studies. Hum. Mol. Genet. 17, R116–R121 (2008).
Kuhn, A., Wenzel, J. & Weyd, H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin. Rev. Allergy Immunol. 47, 148–162 (2014).
Sigurdsson, S. et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet. 76, 528–537 (2005).
Wang, A. et al. CXCR4/CXCL12 hyperexpression plays a pivotal role in the pathogenesis of lupus. J. Immunol. 182, 4448–4458 (2009).
Chong, B. F. & Mohan, C. Targeting the CXCR4/CXCL12 axis in systemic lupus erythematosus. Expert Opin. Ther. Targets 13, 1147–1153 (2009).
Deng, X. M., Yan, S. X. & Wei, W. IL-21 acts as a promising therapeutic target in systemic lupus erythematosus by regulating plasma cell differentiation. Cell Mol. Immunol. 12, 31–39 (2015).
Vila-Coro, A. J. et al. The chemokine SDF-1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 13, 1699–1710 (1999).
Peled, A. et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood 95, 3289–3296 (2000).
Usui, F. et al. Interleukin-17 deficiency reduced vascular inflammation and development of atherosclerosis in Western diet-induced apoE-deficient mice. Biochem. Biophys. Res. Commun. 420, 72–77 (2012).
Deckers, I. A. et al. Investigating international time trends in the incidence and prevalence of atopic eczema 1990-2010: a systematic review of epidemiological studies. PLoS ONE 7, 11 (2012).
Hay, R. J. et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J. Investig. Dermatol. 134, 1527–1534 (2014).
Kim, B. S. et al. Basophils promote innate lymphoid cell responses in inflamed skin. J. Immunol. 193, 3717–3725 (2014).
Seltmann, J., Roesner, L. M., von Hesler, F. W., Wittmann, M. & Werfel, T. IL-33 impacts on the skin barrier by downregulating the expression of filaggrin. J. Allergy Clin. Immunol. 135, 1659–1661.e1654 (2015).
Cole, C. et al. Filaggrin-stratified transcriptomic analysis of pediatric skin identifies mechanistic pathways in patients with atopic dermatitis. J. Allergy Clin. Immunol. 134, 82–91 (2014).
Palmer, C. N. et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 38, 441–446 (2006).
Ferreira, S., Guttman-Yassky, E. & Torres, T. Selective JAK1 inhibitors for the treatment of atopic dermatitis: focus on upadacitinib and abrocitinib. Am. J. Clin. Dermatol. 21, 783–798 (2020).
Noda, S. et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J. Allergy Clin. Immunol. 136, 1254–1264 (2015).
Boniface, K. et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 174, 3695–3702 (2005).
Firestein, G. S. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J. Clin. Rheumatol.: Practical Rep. Rheum. Musculoskelet. Dis. 11, S39–S44 (2005).
Firestein, G. S. Evolving concepts of rheumatoid arthritis. Nature 423, 356–361 (2003).
Kaul, A. et al. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2, 16039 (2016).
O’Shea, J. J., Holland, S. M. & Staudt, L. M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 368, 161–170 (2013).
Remmers, E. F. et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 357, 977–986 (2007).
Zhang, W. et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542 (2005).
Béraud, D. & Maguire-Zeiss, K. A. Misfolded α-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat. Disord. 18(Suppl 1), S17–S20 (2012).
Fellner, L. et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360 (2013).
McGeer, P. L., Itagaki, S., Boyes, B. E. & McGeer, E. G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291 (1988).
Gray, G. K., McFarland, B. C., Nozell, S. E. & Benveniste, E. N. NF-κB and STAT3 in glioblastoma: therapeutic targets coming of age. Expert Rev. Neurotherapeutics 14, 1293–1306 (2014).
Moehle, M. S. & West, A. B. M1 and M2 immune activation in Parkinson’s Disease: Foe and ally? Neuroscience 302, 59–73 (2015).
Liu, Y. et al. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J. Immunol. 192, 59–72 (2014).
Allen Reish, H. E. & Standaert, D. G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinson’s Dis. 5, 1–19 (2015).
Mount, M. P. et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337 (2007).
Sherer, T. B. Biomarkers for Parkinson’s disease. Sci. Transl. Med. 3, 79ps14 (2011).
Chen, H., O’Reilly, E. J., Schwarzschild, M. A. & Ascherio, A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am. J. Epidemiol. 167, 90–95 (2008).
Fuchs, E. Scratching the surface of skin development. Nature 445, 834–842 (2007).
Lin, K. K., Chudova, D., Hatfield, G. W., Smyth, P. & Andersen, B. Identification of hair cycle-associated genes from time-course gene expression profile data by using replicate variance. Proc. Natl Acad. Sci. USA 101, 15955–15960 (2004).
Doles, J., Storer, M., Cozzuto, L., Roma, G. & Keyes, W. M. Age-associated inflammation inhibits epidermal stem cell function. Genes Dev. 26, 2144–2153 (2012).
Goldstein, J. et al. Calcineurin/Nfatc1 signaling links skin stem cell quiescence to hormonal signaling during pregnancy and lactation. Genes Dev. 28, 983–994 (2014).
Fuchs, E., Merrill, B. J., Jamora, C. & DasGupta, R. At the roots of a never-ending cycle. Dev. Cell 1, 13–25 (2001).
Biernaskie, J. et al. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell 5, 610–623 (2009).
Udy, G. B. et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl Acad. Sci. USA 94, 7239–7244 (1997).
Legrand, J. M. D. et al. STAT5 activation in the dermal papilla is important for hair follicle growth phase induction. J. Investig. Dermatol. 136, 1781–1791 (2016).
Harel, S. et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci. Adv. 1, e1500973 (2015).
Wang, E. C. E., Dai, Z., Ferrante, A. W., Drake, C. G. & Christiano, A. M. A subset of TREM2(+) dermal macrophages secretes oncostatin M to maintain hair follicle stem cell quiescence and inhibit hair growth. Cell Stem Cell 24, 654–669.e656 (2019).
Turksen, K., Kupper, T., Degenstein, L., Williams, I. & Fuchs, E. Interleukin 6: insights to its function in skin by overexpression in transgenic mice. Proc. Natl Acad. Sci. USA 89, 5068–5072 (1992).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995).
Harris, T. B. et al. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 106, 506–512 (1999).
Bruunsgaard, H., Andersen-Ranberg, K., Hjelmborg, J., Pedersen, B. K. & Jeune, B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am. J. Med. 115, 278–283 (2003).
Meyer, S. C. & Levine, R. L. Molecular pathways: molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 20, 2051–2059 (2014).
Richard, A. J. & Stephens, J. M. Emerging roles of JAK-STAT signaling pathways in adipocytes. Trends Endocrinol. Metab.: TEM 22, 325–332 (2011).
Zhou, Y. et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl Acad. Sci. USA 94, 13215–13220 (1997).
Barzilai, N. et al. Surgical removal of visceral fat reverses hepatic insulin resistance. Diabetes 48, 94–98 (1999).
Velloso, L. A. et al. Cross-talk between the insulin and angiotensin signaling systems. Proc. Natl Acad. Sci. USA 93, 12490–12495 (1996).
Velloso, L. A., Carvalho, C. R., Rojas, F. A., Folli, F. & Saad, M. J. Insulin signalling in heart involves insulin receptor substrates-1 and -2, activation of phosphatidylinositol 3-kinase and the JAK 2-growth related pathway. Cardiovasc Res. 40, 96–102 (1998).
Lopes, J. E. et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J. Immunother. Cancer 8, 2020–000673 (2020).
Ma, Y. et al. A novel recombinant slow-release TNF α-derived peptide effectively inhibits tumor growth and angiogensis. Sci. Rep. 5, 1–17 (2015).
Murer, P. & Neri, D. Antibody-cytokine fusion proteins: a novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N. Biotechnol. 52, 42–53 (2019).
Tkaczuk, J. et al. Effect of anti-IL-2Ralpha antibody on IL-2-induced Jak/STAT signaling. Am. J. Transpl. 2, 31–40 (2002).
Uciechowski, P. & Dempke, W. C. M. Interleukin-6: a masterplayer in the cytokine network. Oncology 98, 131–137 (2020).
Kampan, N. C. et al. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: challenges and opportunities. Curr. Med Chem. 25, 4785–4806 (2018).
Hassani, M. & Koenderman, L. Immunological and hematological effects of IL-5(Rα)-targeted therapy: an overview. Allergy 73, 1979–1988 (2018).
Isozaki, T., Homma, T., Sagara, H. & Kasama, T. Role of cytokines in EGPA and the possibility of treatment with an anti-IL-5 antibody. J. Clin. Med. 9, 3890 (2020).
Gao, Y. et al. Anti-IL-12/23 p40 antibody attenuates chronic graft-versus-host disease with lupus nephritis via inhibiting Tfh cell in mice. Biomed. Pharmacother. 129, 21 (2020).
Takahashi, T., Koga, Y. & Kainoh, M. Anti-IL-12/IL-23p40 antibody ameliorates dermatitis and skin barrier dysfunction in mice with imiquimod-induced psoriasis-like dermatitis. Eur. J. Pharm. 828, 26–30 (2018).
Davies, S. C. et al. Anti-IL-12/23p40 antibodies for maintenance of remission in Crohn’s disease. Cochrane Database Syst. Rev. 12 (2019).
Ritchlin, C. et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann. Rheum. Dis. 73, 990–999 (2014).
Neggers, S. J., Muhammad, A. & van der Lely, A. J. Pegvisomant treatment in acromegaly. Neuroendocrinology 103, 59–65 (2016).
Taylor, P. C. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology 58, i17–i26 (2019).
Singh, J. A. Filgotinib, a JAK1 inhibitor, for treatment-resistant rheumatoid arthritis. J. Am. Med. Assoc. 322, 309–311 (2019).
Burke, J. R. et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 11, eaaw1736 (2019).
Ghoreschi, K. et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 186, 4234–4243 (2011).
Li, Y. et al. Changes in serum cytokines may predict therapeutic efficacy of tofacitinib in rheumatoid arthritis. Mediators Inflamm. 24 (2019).
Burmester, G. R. et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 381, 451–460 (2013).
Panés, J. et al. Tofacitinib in patients with ulcerative colitis: health-related quality of life in phase 3 randomised controlled induction and maintenance studies. J. Crohns Colitis 12, 145–156 (2018).
Panés, J. et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: results of two phase IIb randomised placebo-controlled trials. Gut 66, 1049–1059 (2017).
Meshkov, A. D., Novikov, P. I., Zhilyaev, E. V., Ilevsky, I. D. J. & Moiseev, S. V. Tofacitinib in steroid-dependent relapsing polychondritis. Ann. Rheum Dis. 78, e72 (2019).
Bissonnette, R. et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br. J. Dermatol. 175, 902–911 (2016).
Liu, L. Y., Craiglow, B. G., Dai, F. & King, B. A. Tofacitinib for the treatment of severe alopecia areata and variants: A study of 90 patients. J. Am. Acad. Dermatol. 76, 22–28 (2017).
Kurtzman, D. J. et al. Tofacitinib citrate for refractory cutaneous dermatomyositis:an alternative treatment. JAMA Dermatol. 152, 944–945 (2016).
Mease, P. et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N. Engl. J. Med. 377, 1537–1550 (2017).
van der Heijde, D. et al. Tofacitinib in patients with ankylosing spondylitis: a phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann. Rheum. Dis. 76, 1340–1347 (2017).
Luo, W. et al. Targeting JAK-STAT signaling to control cytokine release syndrome in COVID-19. Trends Pharmacol. Sci. 41, 531–543 (2020).
Curtis, J. R., Xie, F., Yun, H., Bernatsky, S. & Winthrop, K. L. Real-world comparative risks of herpes virus infections in tofacitinib and biologic-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 75, 1843–1847 (2016).
Xie, F., Yun, H., Bernatsky, S. & Curtis, J. R. Brief report: risk of gastrointestinal perforation among rheumatoid arthritis patients receiving tofacitinib, tocilizumab, or other biologic treatments. Arthritis Rheumatol. 68, 2612–2617 (2016).
Winthrop, K. L. et al. Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 75, 1133–1138 (2016).
Mease, P. et al. Incidence of venous and arterial thromboembolic events reported in the tofacitinib rheumatoid arthritis, psoriasis and psoriatic arthritis development programmes and from real-world data. Ann. Rheum. Dis. 79, 1400–1413 (2020).
Chen, Y. M. et al. Reactivation of hepatitis B virus infection in patients with rheumatoid arthritis receiving tofacitinib: a real-world study. Ann. Rheum. Dis. 77, 780–782 (2018).
Clowse, M. E. et al. Pregnancy outcomes in the tofacitinib safety databases for rheumatoid arthritis and psoriasis. Drug Saf. 39, 755–762 (2016).
Charles-Schoeman, C. et al. Risk factors for major adverse cardiovascular events in phase III and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 71, 1450–1459 (2019).
Maneiro, J. R., Souto, A. & Gomez-Reino, J. J. Risks of malignancies related to tofacitinib and biological drugs in rheumatoid arthritis: Systematic review, meta-analysis, and network meta-analysis. Semin Arthritis Rheum. 47, 149–156 (2017).
Fautrel, B. et al. Effect of baricitinib and adalimumab in reducing pain and improving function in patients with rheumatoid arthritis in low disease activity: exploratory analyses from RA-BEAM. J. Clin. Med. 8, 1394 (2019).
Smolen, J. S. et al. Patient-reported outcomes from a randomised phase III study of baricitinib in patients with rheumatoid arthritis and an inadequate response to biological agents (RA-BEACON). Ann. Rheum. Dis. 76, 694–700 (2017).
Taylor, P. C. et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N. Engl. J. Med. 376, 652–662 (2017).
Tanaka, Y. et al. Clinical outcomes in patients switched from adalimumab to baricitinib due to non-response and/or study design: phase III data in patients with rheumatoid arthritis. Ann. Rheum. Dis. 78, 890–898 (2019).
Wallace, D. J. et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 392, 222–231 (2018).
Papadopoulou, C., Hong, Y., Omoyinmi, E., Brogan, P. A. & Eleftheriou, D. Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain 142, e8 (2019).
Guttman-Yassky, E. et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J. Am. Acad. Dermatol. 80, 913–921 (2019).
Sanchez, G. A. M. et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Investig. 128, 3041–3052 (2018).
Tuttle, K. R. et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a phase 2 randomized controlled clinical trial. Nephrol. Dial. Transpl. 33, 1950–1959 (2018).
Gavegnano, C. et al. Baricitinib reverses HIV-associated neurocognitive disorders in a SCID mouse model and reservoir seeding in vitro. J. Neuroinflammation 16, 019–1565 (2019).
Murakami, K. et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS ONE 12, e0181126 (2017).
Richardson, P. et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395, e30–e31 (2020).
Seif, F. et al. JAK inhibition as a new treatment strategy for patients with COVID-19. Int. Arch. Allergy Immunol. 181, 467–475 (2020).
Hoang, T. N. et al. Baricitinib treatment resolves lower-airway macrophage inflammation and neutrophil recruitment in SARS-CoV-2-infected rhesus macaques. Cell 10, 31466–31465 (2020).
Kalil, A. C. et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N. Engl. J. Med. 11, 795–807 (2020).
Bronte, V. et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J. Clin. Investig. 130, 6409–6416 (2020).
Smolen, J. S. et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J. Rheumatol. 46, 7–18 (2019).
Honda, S. & Harigai, M. The safety of baricitinib in patients with rheumatoid arthritis. Expert Opin. Drug Saf. 19, 545–551 (2020).
Gonzales, A. J. et al. Oclacitinib (APOQUEL(®)) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J. Vet. Pharm. Ther. 37, 317–324 (2014).
Lopes, N. L. et al. A blinded, randomized, placebo-controlled trial of the safety of oclacitinib in cats. BMC Vet. Res. 15, 019–1893 (2019).
Fogelman, D. et al. Randomized, double-blind, phase two study of ruxolitinib plus regorafenib in patients with relapsed/refractory metastatic colorectal cancer. Cancer Med. 7, 5382–5393 (2018).
Jagasia, M. et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood 135, 1739–1749 (2020).
Cervantes, F. & Pereira, A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 129, 832–837 (2017).
McKeage, K. Ruxolitinib: a review in polycythaemia vera. Drugs 75, 1773–1781 (2015).
Olsen, E. A., Kornacki, D., Sun, K. & Hordinsky, M. K. Ruxolitinib cream for the treatment of patients with alopecia areata: A 2-part, double-blind, randomized, vehicle-controlled phase 2 study. J. Am. Acad. Dermatol. 82, 412–419 (2020).
Ezzedine, K., Eleftheriadou, V., Whitton, M. & van Geel, N. Vitiligo. Lancet 386, 74–84 (2015).
Cao, Y. et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): a multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 146, 137–146 (2020).
Harrison, C. N. et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamide. Blood 130, 1889–1897 (2017).
Ajayi, S. et al. Ruxolitinib. Recent Results Cancer Res. 212, 119–132 (2018).
Hurwitz, H. I. et al. Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J. Clin. Oncol. 33, 4039–4047 (2015).
Hurwitz, H. et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. N. Drugs 36, 683–695 (2018).
Harrison, C. N. et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 4, e317–e324 (2017).
Harrison, C. N. et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 5, e73–e81 (2018).
Tefferi, A. & Pardanani, A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin. Proc. 86, 1188–1191 (2011).
Palandri, F. et al. Ruxolitinib discontinuation syndrome: incidence, risk factors, and management in 251 patients with myelofibrosis. Blood Cancer J. 11, 4 (2021).
Rosmarin, D. et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet 396, 110–120 (2020).
Sant’Antonio, E., Bonifacio, M., Breccia, M. & Rumi, E. A journey through infectious risk associated with ruxolitinib. Br. J. Haematol. 187, 286–295 (2019).
Loscocco, G. G. et al. Kaposi sarcoma in a patient treated with ruxolitinib. Ann. Oncol. 28, 1670–1671 (2017).
Gaspari, V., Zengarini, C., Greco, S., Vangeli, V. & Mastroianni, A. Side effects of ruxolitinib in patients with SARS-CoV-2 infection: two case reports. Int. J. Antimicrob Agents. 56, 106023 (2020).
Van Rompaey, L. et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J. Immunol. 191, 3568–3577 (2013).
White, J. R. et al. Review article: novel oral-targeted therapies in inflammatory bowel disease. Aliment Pharm. Ther. 47, 1610–1622 (2018).
Orbai, A. M. et al. Effect of filgotinib on health-related quality of life in active psoriatic arthritis: a randomized phase 2 trial (EQUATOR). Rheumatology 59, 1495–1504 (2020).
van der Heijde, D. et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): results from a randomised, placebo-controlled, phase 2 trial. Lancet 392, 2378–2387 (2018).
Dhillon, S. & Keam, S. J. Filgotinib: first approval. Drugs 80, 1987–1997 (2020).
Vermeire, S. et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet 389, 266–275 (2017).
Yeh, Y. J. et al. Filgotinib suppresses HIV-1-driven gene transcription by inhibiting HIV-1 splicing and T cell activation. J. Clin. Investig. 130, 4969–4984 (2020).
Nader, A. et al. Exposure-response analyses of upadacitinib efficacy and safety in phase II and III studies to support benefit-risk assessment in rheumatoid arthritis. Clin. Pharm. Ther. 107, 994–1003 (2020).
Duggan, S. & Keam, S. J. Upadacitinib: first approval. Drugs 79, 1819–1828 (2019).
Edwards, C. J. et al. A matching-adjusted indirect comparison of upadacitinib versus tofacitinib in adults with moderate-to-severe rheumatoid arthritis. Rheumatol. Ther. 26, 020–00257 (2020).
Sandborn, W. J. et al. Efficacy and safety of upadacitinib in a randomized trial of patients with Crohn’s disease. Gastroenterology 158, 2123–2138 (2020).
Sandborn, W. J. et al. Efficacy of upadacitinib in a randomized trial of patients with active ulcerative colitis. Gastroenterology 158, 2139–2149 (2020).
Guttman-Yassky, E. et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 145, 877–884 (2020).
Mease, P. J. et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann. Rheum. Dis. 3, 2020–218870 (2020).
van der Heijde, D. et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet 394, 2108–2117 (2019).
Rubbert-Roth, A. et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N. Engl. J. Med. 383, 1511–1521 (2020).
Crowley, E. L., Nezamololama, N., Papp, K. & Gooderham, M. J. Abrocitinib for the treatment of atopic dermatitis. Expert Rev. Clin. Immunol. 16, 955–962 (2020).
Bieber, T. et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N. Engl. J. Med. 384, 1101–1112 (2021).
Covington, M. et al. Preclinical characterization of itacitinib (INCB039110), a novel selective inhibitor of JAK1, for the treatment of inflammatory diseases. Eur. J. Pharm. 885, 28 (2020).
Huarte, E. et al. Itacitinib (INCB039110), a JAK1 inhibitor, reduces cytokines associated with cytokine release syndrome induced by CAR T-cell therapy. Clin. Cancer Res. 26, 6299–6309 (2020).
Schroeder, M. A. et al. A phase 1 trial of itacitinib, a selective JAK1 inhibitor, in patients with acute graft-versus-host disease. Blood Adv. 4, 1656–1669 (2020).
Liu, L. W., Hsieh, Y. Y. & Yang, P. M. Bioinformatics data mining repurposes the JAK2 (Janus kinase 2) inhibitor fedratinib for treating pancreatic ductal adenocarcinoma by reversing the KRAS (Kirsten Rat Sarcoma 2 Viral Oncogene Homolog)-driven gene signature. J. Pers. Med. 10, 130 (2020).
Zhang, Q. et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab. Disposition: Biol. Fate Chem. 42, 1656–1662 (2014).
Beauverd, Y., McLornan, D. P. & Harrison, C. N. Pacritinib: a new agent for the management of myelofibrosis? Expert Opin. Pharmacother. 16, 2381–2390 (2015).
Mesa, R. A. et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. 4, e225–e236 (2017).
Mascarenhas, J. et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 4, 652–659 (2018).
Tremblay, D. et al. Pacritinib demonstrates spleen volume reduction in patients with myelofibrosis independent of JAK2V617F allele burden. Blood Adv. 4, 5929–5935 (2020).
Regenbogen, T. et al. Pacritinib to inhibit JAK/STAT signaling in refractory metastatic colon and rectal cancer. J. Gastrointest. Oncol. 8, 985–989 (2017).
Ochi, N. et al. Synergistic effect of pacritinib with erlotinib on JAK2-mediated resistance in epidermal gowth factor receptor mutation-positive non-small cell lung Cancer. Exp. Cell Res. 344, 194–200 (2016).
Wu, M., Li, C. & Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 11, 018–0675 (2018).
Berdeja, J. et al. Phase 2 study of gandotinib (LY2784544) in patients with myeloproliferative neoplasms. Leuk. Res. 71, 82–88 (2018).
Verstovsek, S. et al. A phase 1 study of the Janus kinase 2 (JAK2)(V617F) inhibitor, gandotinib (LY2784544), in patients with primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Leuk. Res. 61, 89–95 (2017).
Mahajan, S. et al. VX-509 (decernotinib) is a potent and selective janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune disease. J. Pharm. Exp. Ther. 353, 405–414 (2015).
Genovese, M. C., Yang, F., Østergaard, M. & Kinnman, N. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patients with rheumatoid arthritis: clinical and MRI findings. Ann. Rheum. Dis. 75, 1979–1983 (2016).
Genovese, M. C., van Vollenhoven, R. F., Pacheco-Tena, C., Zhang, Y. & Kinnman, N. VX-509 (decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol. 68, 46–55 (2016).
Fleischmann, R. M. et al. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. 67, 334–343 (2015).
Gadina, M., Schwartz, D. M. & O'Shea, J. J. Decernotinib: a next-generation jakinib. Arthritis Rheumatol. 68, 31–34 (2016).
Markham, A. & Keam, S. J. Peficitinib: first global approval. Drugs 79, 887–891 (2019).
Miyatake, D. et al. Pharmacokinetics and safety of a single oral dose of peficitinib (ASP015K) in Japanese subjects with normal and impaired hepatic function. Clin. Pharmacol. Drug Dev. 9, 699–708 (2020).
Miyatake, D. et al. Pharmacokinetics and safety of a single oral dose of peficitinib (ASP015K) in Japanese subjects with normal and impaired renal function. Clin. Drug Investig. 40, 149–159 (2020).
Zhu, T. et al. Drug interactions between peficitinib, an orally administered, once-daily janus kinase inhibitor, and rosuvastatin in healthy subjects. Clin. Pharmacokinet. 56, 747–757 (2017).
Monaghan, K. A., Khong, T., Burns, C. J. & Spencer, A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia 25, 1891–1899 (2011).
Tyner, J. W. et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 115, 5232–5240 (2010).
Gupta, V. et al. A phase 1/2, open-label study evaluating twice-daily administration of momelotinib in myelofibrosis. Haematologica 102, 94–102 (2017).
Tefferi, A. et al. Momelotinib therapy for myelofibrosis: a 7-year follow-up. Blood Cancer J. 8, 018–0067 (2018).
Barbie, D. A. et al. Phase 1B study of momelotinib combined with trametinib in metastatic, kirsten rat sarcoma viral oncogene homolog-mutated non-small-cell lung cancer after platinum-based chemotherapy treatment failure. Clin. Lung Cancer 19, e853–e859 (2018).
Oh, S. T. et al. ACVR1/JAK1/JAK2 inhibitor momelotinib reverses transfusion dependency and suppresses hepcidin in myelofibrosis phase 2 trial. Blood Adv. 4, 4282–4291 (2020).
Abdelrahman, R. A. et al. Momelotinib treatment-emergent neuropathy: prevalence, risk factors and outcome in 100 patients with myelofibrosis. Br. J. Haematol. 169, 77–80 (2015).
Pavel, A. B. et al. Oral Janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J. Allergy Clin. Immunol. 144, 1011–1024 (2019).
Bissonnette, R. et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: results from a randomized double-blind placebo-controlled study. Br. J. Dermatol. 181, 733–742 (2019).
Dubin, C., Del Duca, E. & Guttman-Yassky, E. Drugs for the treatment of chronic hand eczema: successes and key challenges. Therapeutics Clin. Risk Manag. 16, 1319–1332 (2020).
Dhillon, S. Delgocitinib: first approval. Drugs 80, 609–615 (2020).
Coffey, G. et al. The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer. J. Pharmacol. Exp. Therapeut. 351, 538–548 (2014).
Hamlin, P. A. et al. Efficacy and safety of the dual SYK/JAK inhibitor cerdulatinib in patients with relapsed or refractory B-cell malignancies: Results of a phase I study. Am. J. Hematol. 94, E90–e93 (2019).
Weng, C. et al. Comparative efficacy and safety of Janus kinase inhibitors and biological disease-modifying antirheumatic drugs in rheumatoid arthritis: a systematic review and network meta-analysis. Therapeutic Adv. Musculoskelet. Dis. 13, 1759720x21999564 (2021).
Rocha, C. M. et al. Current jakinibs for the treatment of rheumatoid arthritis: a systematic review. Inflammopharmacology https://doi.org/10.1007/s10787-021-00822-x (2021).
Salas, A. et al. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 17, 323–337 (2020).
Ma, C. et al. Systematic review with meta-analysis: efficacy and safety of oral Janus kinase inhibitors for inflammatory bowel disease. Alimentary Pharmacol. Therapeut. 50, 5–23 (2019).
Singh, S., Fumery, M., Sandborn, W. J. & Murad, M. H. Systematic review with network meta-analysis: first- and second-line pharmacotherapy for moderate-severe ulcerative colitis. Alimentary Pharmacol. Therapeut. 47, 162–175 (2018).
Olivera, P. A., Lasa, J. S., Bonovas, S., Danese, S. & Peyrin-Biroulet, L. Safety of Janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology 158, 1554–1573.e1512 (2020).
Phan, K. & Sebaratnam, D. F. JAK inhibitors for alopecia areata: a systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. : JEADV 33, 850–856 (2019).
Wijaya, I. et al. The use of Janus Kinase inhibitors in hospitalized patients with COVID-19: systematic review and meta-analysis. Clin. Epidemiol. Glob. health 11, 100755 (2021).
Chen, C. X. et al. JAK-inhibitors for coronavirus disease-2019 (COVID-19): a meta-analysis. Leukemia 1–5, https://doi.org/10.1038/s41375-021-01266-6 (2021).
Tsai, H. R., Lu, J. W., Chen, L. Y. & Chen, T. L. Application of Janus Kinase Inhibitors in Atopic Dermatitis: An Updated Systematic Review and Meta-Analysis of Clinical Trials. J. Personal. Med. 11, https://doi.org/10.3390/jpm11040279 (2021).
Zhou, S., Qi, F., Gong, Y., Zhang, J. & Zhu, B. Biological therapies for atopic dermatitis: a systematic review. Dermatology 1–11, https://doi.org/10.3390/jpm11040279 (2021).
Turkson, J. et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 3, 261–269 (2004).
Turkson, J. et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 276, 45443–45455 (2001).
Auzenne, E. J. et al. A phosphopeptide mimetic prodrug targeting the SH2 domain of Stat3 inhibits tumor growth and angiogenesis. J. Exp. Ther. Oncol. 10, 155–162 (2012).
Gunning, P. T. et al. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg. Med Chem. Lett. 17, 1875–1878 (2007).
Chen, J. et al. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem. Lett. 1, 85–89 (2010).
Furqan, M. et al. STAT inhibitors for cancer therapy. J. Hematol. Oncol. 6, 1756–8722 (2013).
Brantley, E. C. et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin. Cancer Res. 14, 4694–4704 (2008).
Borghouts, C. et al. The intracellular delivery of a recombinant peptide derived from the acidic domain of PIAS3 inhibits STAT3 transactivation and induces tumor cell death. Mol. Cancer Res. 8, 539–553 (2010).
Schust, J., Sperl, B., Hollis, A., Mayer, T. U. & Berg, T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13, 1235–1242 (2006).
Song, H., Wang, R., Wang, S. & Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl Acad. Sci. USA 102, 4700–4705 (2005).
Fuh, B. et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 100, 106–112 (2009).
Zhang, R., Chen, X., Fu, S., Xu, L. & Lin, J. A small molecule STAT3 inhibitor, LLL12, enhances cisplatin‑ and paclitaxel‑mediated inhibition of cell growth and migration in human ovarian cancer cells. Oncol. Rep. 44, 1224–1232 (2020).
Nie, Y., Li, Y. & Hu, S. A novel small inhibitor, LLL12, targets STAT3 in non-small cell lung cancer in vitro and in vivo. Oncol. Lett. 16, 5349–5354 (2018).
Onimoe, G. I. et al. Small molecules, LLL12 and FLLL32, inhibit STAT3 and exhibit potent growth suppressive activity in osteosarcoma cells and tumor growth in mice. Investig. N. Drugs 30, 916–926 (2012).
Lin, L. et al. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int. J. Cancer 130, 1459–1469 (2012).
Wei, C. C. et al. Two small molecule compounds, LLL12 and FLLL32, exhibit potent inhibitory activity on STAT3 in human rhabdomyosarcoma cells. Int. J. Oncol. 38, 279–285 (2011).
Zuo, M., Li, C., Lin, J. & Javle, M. LLL12, a novel small inhibitor targeting STAT3 for hepatocellular carcinoma therapy. Oncotarget 6, 10940–10949 (2015).
Ball, S., Li, C., Li, P. K. & Lin, J. The small molecule, LLL12, inhibits STAT3 phosphorylation and induces apoptosis in medulloblastoma and glioblastoma cells. PLoS ONE 6, 0018820 (2011).
Liu, A. et al. XZH-5 inhibits STAT3 phosphorylation and enhances the cytotoxicity of chemotherapeutic drugs in human breast and pancreatic cancer cells. PLoS ONE 7, 3 (2012).
Kim, M. J. et al. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 335, 145–152 (2013).
Oh, D. Y. et al. Phase I study of OPB-31121, an oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res. Treat. 47, 607–615 (2015).
Okusaka, T. et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol. Res. 45, 1283–1291 (2015).
Ahmad, S. F. et al. The Stat3 inhibitor, S3I-201, downregulates lymphocyte activation markers, chemokine receptors, and inflammatory cytokines in the BTBR T(+) Itpr3(tf)/J mouse model of autism. Brain Res. Bull. 152, 27–34 (2019).
Qu, M. et al. Therapeutic effects of STAT3 inhibition on experimental murine dry eye. Investig. Ophthalmol. Vis. Sci. 60, 3776–3785 (2019).
Wang, Z., Li, J., Xiao, W., Long, J. & Zhang, H. The STAT3 inhibitor S3I-201 suppresses fibrogenesis and angiogenesis in liver fibrosis. Lab Investig. 98, 1600–1613 (2018).
Pang, M. et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 78, 257–268 (2010).
Zhang, X. et al. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharm. 79, 1398–1409 (2010).
Ashizawa, T. et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of a temozolomide-resistant glioblastoma cell line. Int. J. Oncol. 45, 411–418 (2014).
Zhou, X. X., Gao, P. J. & Sun, B. G. Pravastatin attenuates interferon-gamma action via modulation of STAT1 to prevent aortic atherosclerosis in apolipoprotein E-knockout mice. Clin. Exp. Pharm. Physiol. 36, 373–379 (2009).
Wang, G. J., Yang, Z., Huai, J. & Xiang, Q. Q. Pravastatin alleviates oxidative stress and decreases placental trophoblastic cell apoptosis through IL-6/STAT3 signaling pathway in preeclampsia rats. Eur. Rev. Med. Pharm. Sci. 24, 12955–12962 (2020).
Shaw, V., Srivastava, S. & Srivastava, S. K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 68, 75–83 (2021).
Xiao, Z. et al. Pimozide augments bromocriptine lethality in prolactinoma cells and in a xenograft model via the STAT5/cyclin D1 and STAT5/Bcl‑xL signaling pathways. Int. J. Mol. Med. 47, 113–124 (2021).
Miklossy, G., Hilliard, T. S. & Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 12, 611–629 (2013).
Garg, M. et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Medicinal Res. Rev. https://doi.org/10.1002/med.21761 (2020).
Bose, S. et al. Targeting the JAK/STAT signaling pathway using phytocompounds for cancer prevention and therapy. Cells 9, https://doi.org/10.3390/cells9061451 (2020).
Mohan, C. D. et al. Targeting STAT3 signaling pathway in cancer by agents derived from mother nature. Semin. Cancer Biol. https://doi.org/10.1016/j.semcancer.2020.03.016 (2020).
Wu, J., Lu, W. Y. & Cui, L. L. Inhibitory effect of curcumin on invasion of skin squamous cell carcinoma A431 cells. Asian Pac. J. Cancer Prev. 16, 2813–2818 (2015).
Wu, B., Yao, X., Nie, X. & Xu, R. Epigenetic reactivation of RANK in glioblastoma cells by curcumin: involvement of STAT3 inhibition. DNA Cell Biol. 32, 292–297 (2013).
Porro, C., Cianciulli, A., Trotta, T., Lofrumento, D. D. & Panaro, M. A. Curcumin regulates anti-inflammatory responses by JAK/STAT/SOCS signaling pathway in BV-2 microglial cells. Biology 8, 51 (2019).
Zhao, H. M. et al. Curcumin suppressed activation of dendritic cells via JAK/STAT/SOCS signal in mice with experimental colitis. Front. Pharmacol. 7, 455 (2016).
Bill, M. A. et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol. Cancer 9, 1476–4598 (2010).
Jahangiri, A., Dadmanesh, M. & Ghorban, K. STAT3 inhibition reduced PD-L1 expression and enhanced antitumor immune responses. J. Cell Physiol. 235, 9457–9463 (2020).
Lin, L. et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. 70, 2445–2454 (2010).
Malaguarnera, L. Influence of resveratrol on the immune response. Nutrients 11, 946 (2019).
Ma, C., Wang, Y., Dong, L., Li, M. & Cai, W. Anti-inflammatory effect of resveratrol through the suppression of NF-κB and JAK/STAT signaling pathways. Acta Biochim. Biophys. Sin. 47, 207–213 (2015).
Li, T., Wang, W., Chen, H. & Ye, L. Evaluation of anti-leukemia effect of resveratrol by modulating STAT3 signaling. Int. Immunopharmacol. 10, 18–25 (2010).
Kim, J. E. et al. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp. Mol. Med. 40, 514–522 (2008).
Zhang, Y., Zhang, L. H., Chen, X., Zhang, N. & Li, G. Piceatannol attenuates behavioral disorder and neurological deficits in aging mice via activating the Nrf2 pathway. Food Funct. 9, 371–378 (2018).
Nawaz, W. et al. Therapeutic versatility of resveratrol derivatives. Nutrients 9, 1188 (2017).
Zhang, Y. L. et al. Oleanolic acid inhibiting the differentiation of neural stem cells into astrocyte by down-regulating JAK/STAT signaling pathway. Am. J. Chin. Med. 44, 103–117 (2016).
Kim, H. S. et al. Oleanolic acid suppresses resistin induction in adipocytes by modulating Tyk-STAT signaling. Nutr. Res. 33, 144–153 (2013).
Feng, A. et al. Development and evaluation of oleanolic acid dosage forms and its derivatives. BioMed. Res. Int. 2020, 1308749 (2020).
Singh, B. N., Shankar, S. & Srivastava, R. K. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 82, 1807–1821 (2011).
Senggunprai, L., Kukongviriyapan, V., Prawan, A. & Kukongviriyapan, U. Quercetin and EGCG exhibit chemopreventive effects in cholangiocarcinoma cells via suppression of JAK/STAT signaling pathway. Phytother. Res. 28, 841–848 (2014).
Wong, Y. K. et al. Artemisinin as an anticancer drug: recent advances in target profiling and mechanisms of action. Medicinal Res. Rev. 37, 1492–1517 (2017).
Shi, C., Li, H., Yang, Y. & Hou, L. Anti-inflammatory and immunoregulatory functions of artemisinin and its derivatives. Mediators Inflamm. 2015, 435713 (2015).
Ho, W. E., Peh, H. Y., Chan, T. K. & Wong, W. S. Artemisinins: pharmacological actions beyond anti-malarial. Pharmacol. Therapeutics 142, 126–139 (2014).
Lu, B. W. & Xie, L. K. Potential applications of artemisinins in ocular diseases. Int. J. Ophthalmol. 12, 1793–1800 (2019).
Khanal, P. Antimalarial and anticancer properties of artesunate and other artemisinins: current development. Monatshefte fur chemie. 1–14, https://doi.org/10.1007/s00706-021-02759-x (2021).
Jang, B. C. Artesunate inhibits adipogeneis in 3T3-L1 preadipocytes by reducing the expression and/or phosphorylation levels of C/EBP-α, PPAR-γ, FAS, perilipin A, and STAT-3. Biochem. Biophys. Res. Commun. 474, 220–225 (2016).
Ilamathi, M., Santhosh, S. & Sivaramakrishnan, V. Artesunate as an anti-cancer agent targets Stat-3 and favorably suppresses hepatocellular carcinoma. Curr. Top. Medicinal Chem. 16, 2453–2463 (2016).
Saghatelyan, T. et al. Efficacy and safety of curcumin in combination with paclitaxel in patients with advanced, metastatic breast cancer: A comparative, randomized, double-blind, placebo-controlled clinical trial. Phytomedicine: Int. J. Phytother. Phytopharmacology 70, 153218 (2020).
Barbalho, S. M. et al. Dermatological effects of Curcuma species: a systematic review. Clin. Exp. Dermatol. 46, 825–833 (2021).
Murillo Ortiz, B. O. et al. Recovery of bone and muscle mass in patients with chronic kidney disease and iron overload on hemodialysis and taking combined supplementation with curcumin and resveratrol. Clin. Interventions Aging 14, 2055–2062 (2019).
Harper, S. A. et al. Resveratrol and exercise combined to treat functional limitations in late life: a pilot randomized controlled trial. Exp. Gerontol. 143, 111111 (2021).
Sen, A. Prophylactic and therapeutic roles of oleanolic acid and its derivatives in several diseases. World J. Clin. Cases 8, 1767–1792 (2020).
Zhang, L. et al. Assessing the analgesic efficacy of oral epigallocatechin-3-gallate on epidural catheter analgesia in patients after surgical stabilisation of multiple rib fractures: a prospective double-blind, placebo-controlled clinical trial. Pharm. Biol. 58, 741–744 (2020).
Furushima, D. et al. Prevention of acute upper respiratory infections by consumption of catechins in healthcare workers: a randomized, placebo-controlled trial. Nutrients 12, https://doi.org/10.3390/nu12010004 (2019).
Stojanovic, T. et al. STAT-1 decoy oligodeoxynucleotide inhibition of acute rejection in mouse heart transplants. Basic Res. Cardiol. 104, 719–729 (2009).
Hückel, M. et al. Attenuation of murine antigen-induced arthritis by treatment with a decoy oligodeoxynucleotide inhibiting signal transducer and activator of transcription-1 (STAT-1). Arthritis Res. Ther. 8, 1–13 (2006).
Stadlbauer, T. H. et al. AP-1 and STAT-1 decoy oligodeoxynucleotides attenuate transplant vasculopathy in rat cardiac allografts. Cardiovasc Res. 79, 698–705 (2008).
Lührmann, A. et al. Decoy oligodeoxynucleotide against STAT transcription factors decreases allergic inflammation in a rat asthma model. Exp. Lung Res. 36, 85–93 (2010).
Sen, M. & Grandis, J. R. Nucleic acid-based approaches to STAT inhibition. Jakstat 1, 285–291 (2012).
Engelhard, H. H. Antisense oligodeoxynucleotide technology: potential use for the treatment of malignant brain tumors. Cancer Control 5, 163–170 (1998).
Reilley, M. J. et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J. Immunother. Cancer 6, 018–0436 (2018).
Chi, X., Gatti, P. & Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 22, 823–833 (2017).
Sun, T., Jia, Y. & Xiao, D. Interference of STAT 5b expression enhances the chemo-sensitivity of gastric cancer cells to gefitinib by promoting mitochondrial pathway-mediated cell apoptosis. Oncol. Rep. 34, 227–234 (2015).
Norman, P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin. Investig. Drugs 23, 1067–1077 (2014).
Jo, C. E., Gooderham, M. & Beecker, J. TYK 2 inhibitors for the treatment of dermatologic conditions: the evolution of JAK inhibitors. Int. J. Dermatol. https://doi.org/10.1111/ijd.15605 (2021).
Sandborn, W. J. et al. Development of gut-selective Pan-Janus kinase inhibitor TD-1473 for ulcerative colitis: a translational medicine programme. J. Crohn’s Colitis 14, 1202–1213 (2020).
Robinson, M. F. et al. Efficacy and safety of PF-06651600 (ritlecitinib), a novel JAK3/TEC inhibitor, in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 72, 1621–1631 (2020).
Sansone, P. & Bromberg, J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 30, 1005–1014 (2012).
Hosseini, A. et al. Janus kinase inhibitors: a therapeutic strategy for cancer and autoimmune diseases. J. Cell. Physiol. 235, 5903–5924 (2020).
Quintás-Cardama, A. & Verstovsek, S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin. Cancer Res. 19, 1933–1940 (2013).
Morris, R., Kershaw, N. J. & Babon, J. J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 27, 1984–2009 (2018).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2020YFC0860200), and Key Research and Development Program of Sichuan Province (2020YFS0008), and the National Natural Science Foundation of China (No. 81902662). Images were created with Biorender.com.
Author information
Authors and Affiliations
Contributions
X.Y.H., J.L., and M.R.F. collected references and wrote the manuscript and tables. X.Y.H. drew the figures. X.Z. and W.W. provided valuable guidance and revised the paper. All authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hu, X., li, J., Fu, M. et al. The JAK/STAT signaling pathway: from bench to clinic. Sig Transduct Target Ther 6, 402 (2021). https://doi.org/10.1038/s41392-021-00791-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00791-1
This article is cited by
-
Functional CRISPR screens in T cells reveal new opportunities for cancer immunotherapies
Molecular Cancer (2024)
-
Paclitaxel combined with Compound K inducing pyroptosis of non-small cell lung cancer cells by regulating Treg/Th17 balance
Chinese Medicine (2024)
-
Causal relationship between multiple sclerosis and cortical structure: a Mendelian randomization study
Journal of Translational Medicine (2024)
-
Clinicopathological and molecular genetic alterations in monomorphic–epitheliotropic intestinal T-cell lymphoma of the small intestine
European Journal of Medical Research (2024)
-
The oncogenic mechanisms of the Janus kinase-signal transducer and activator of transcription pathway in digestive tract tumors
Cell Communication and Signaling (2024)
