
Abstract
Background
Diagnosis of rare diseases possesses a great challenge in pediatric hepatology because expert knowledge in the field is extremely insufficient. The study aims to explore new findings and collect diagnostic experience from pediatric rare liver diseases.
Methods
The large-sample case analysis study included pediatric patients who had liver-involved rare diseases. All cases underwent liver biopsy and/or gene sequencing.
Results
A total of 1158 pediatric patients were identified. Liver-based genetic diseases were most frequent (737 cases), followed by liver damages involved in extrahepatic or systemic disorders (151 cases) and cryptogenic hepatobilliary abnormalities (123 cases). Of note, diagnoses of 16 patients were re-evaluated according to genetic results combined with clinical pointers. In addition, 101 patients who underwent gene sequencing remained undiagnosed. Of them, 55 had negative genetic findings, 30 harbored mutations that failed to meet their typically pathogenic condition, and 16 had detected variants that were inconsistent with clinical pointers.
Conclusions
As a study involving known largest number of children with rare hepatobiliary disorders, it allows us to accumulate information (especially new findings) on the etiology and diagnosis of these disorders. The results can help to improve the diagnostic quality in the population.
Impact
-
Liver-based genetic diseases were most frequent in clinical profiles of pediatric rare liver diseases.
-
Some novel variants in cases with genetic diseases (for example, two variants of c.3638G>T and c.1435G>C in a patient with progressive familial intrahepatic cholestasis type 2) were identified.
-
As a study involving known largest number of pediatric cases with rare hepatobiliary disorders, it allows us to accumulate information on the etiology and diagnosis of these disorders.
-
The study can help to optimize the diagnostic process and significantly improve the diagnostic quality in the field of pediatric hepatology. Given that clinical variability often exists within rare genetic disease entities and not all rare disorders are genetic, clinicians should not over-depend on the genetic results in the diagnosis.
Similar content being viewed by others

Introduction
Pediatric liver diseases remain a great challenge worldwide.1 Much of pediatric hepatology practice involves considering rare diseases in the differential diagnosis of children presenting with both common and uncommon clinical symptoms.2 For example, as a common condition of jaundice in an infant, there are >100 diagnostic considerations, the majority of which are rare liver disorders. Therefore, accurate diagnosis is the most critical course for the management of pediatric liver diseases, especially rare diseases. Without an accurate diagnosis, clinicians can neither identify the cause nor design an effective treatment strategy to suppress or ameliorate the condition.3 However, recognition of rare diseases is often insufficient in pediatric clinical practice, which can put children in a potentially life-threatening situation. Lack of data and experience in pediatric rare liver diseases has led to limited progress achieved in their etiologies, diagnoses, and treatments.4,5,6
The majority of rare disorders in pediatric hepatology practice may have genetic causes and are intractable to clinical diagnosis by classical approaches.7 During recent years, the use of next-generation sequencing (NGS) has largely facilitated a better understanding of diseases caused by genetic defects. The introduction of NGS has accomplished the simultaneous analysis of a large number of genes, up to whole-exome sequencing (WES) or even whole-genome sequencing.8 However, sequencing still uncovers many rare variants for which the functional impact is not known, and interpretation can be difficult as healthy individuals often carry alleles that would be deleterious or lethal when homozygous.3 Moreover, not all rare disorders are genetic. Therefore, inconsistencies between clinical manifestations and genetic findings are often encountered by clinicians. Comprehensive evaluations become crucial in real-world medical practice. Herein, we performed the present large-sample case analysis study to investigate a clinical spectrum of pediatric rare liver diseases and collect novel findings and relevant experience in the pathogenesis and diagnosis of these disorders.
Methods
Study population
Pediatric patients (<=16 years) with hepatobiliary abnormalities associated with rare diseases (a prevalence of <50 per 100,000 of the population9) from May 1999 to May 2019 were retrospectively identified and included in the large-sample case analysis (Fig. 1). All cases underwent liver biopsy and/or gene sequencing. Diagnostic evidence in the majority of included cases was rechecked according to recent perspectives or guidelines.10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28 Cases with diagnosis or suspected diagnosis of genetic disorders were pathologically or genetically re-evaluated. Informed consent was obtained at admission from parents or guardians of all included patients and assent from older children as appropriate. The study was approved by the ethics committees of the Fifth Medical Center (formerly Beijing 302 Hospital) of Chinese PLA General Hospital.

Diagnostic flowchart of the study.
Laboratory tests
Laboratory parameters, including biochemical and hematological indicators, viral profiles (including hepatitis viruses A, B, C, D, E, Epstein–Barr virus, cytomegalovirus (CMV), and human immunodeficiency virus), autoantibody spectrum, immunoglobulins, C-reactive protein, ceruloplasmin, copper concentration, ferritin, iron concentration, free thyroxine, free triiodothyronine, thyrotropin, blood ammonia, alpha-fetoprotein, alpha-1 antitrypsin, etc., were measured.
Imaging examinations
Imaging examinations, including ultrasonography, X-ray, computed tomography, magnetic resonance imaging, and magnetic resonance cholangiopancreatography, were carried out wherever necessary.
Screening for metabolic disorders
For suspected cases with hereditary metabolic disorders, a dried blood spot liquid chromatography–tandem mass spectrometry assay and a urinary gas chromatography–mass spectrometry assay were performed according to reported methods.29,30
Liver biopsy
Liver biopsy was performed in cases having normal prothrombin time and normal or mildly reducing platelet count and undertaken with all aseptic precautions. Biopsy specimens were examined and evaluated by experienced pathologists.
Whole-exome sequencing
Genomic DNA was isolated from peripheral blood of the patients. Qualified DNA samples were fragmented into 200–300 base pairs (bp) segments to construct a library. Subsequently, WES was performed using the Agilent SureSelect™ Human All Exon V5 Kit for exome enrichment and the Illumina HiSeq2500 platform with a paired-end reads of 100 bp protocol for sequencing. Allele frequencies were determined using the Single Nucleotide Polymorphism Database and the Genome Aggregation Database (the 1000 Genomes Project and Exome Aggregation Consortium). Variants with minor allele frequency of >1% in these databases were excluded. In silico analysis of the variants was conducted using MutationTaster (www.mutationtaster.org) to predict their functions. The identified causal variants were further confirmed by means of Sanger sequencing. Co-segregation analysis was carried out to verify the variants using samples from other family members. Variant pathogenicity was evaluated according to the American College of Medical Genetics and Genomics standards and guidelines.31
Results
Clinical spectrum
A total of 1158 pediatric patients, including 763 boys and 395 girls, were eventually identified (median age, 6 years (range, 1 month–16 years)). Among these patients, 923 underwent liver biopsies and 409 underwent genetic tests.
Liver-based genetic diseases were most frequent in the population (737, 63.64%), followed by liver damages involved in extrahepatic or systemic disorders (151, 13.04%) and cryptogenic hepatobilliary abnormalities (123, 10.62%). Autoimmune, miscellaneous, vascular, infectious, and neoplastic etiologies were, respectively, seen in 71 (6.13%), 34 (2.94%), 25 (2.16%), 9 (0.78%), and 8 (0.69%) cases. Notably, Wilson disease predominated in the clinical spectrum amounting to 338 cases (29.19%). Table 1 shows the details.
Undiagnosed cases
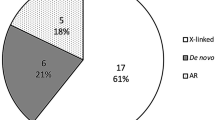
Among the cases without definitive diagnosis, 22 patients did not undergo genetic tests but underwent one or more histological examinations; 19 patients did not undergo histological examinations but underwent genetic tests; 82 patients underwent both histological examinations and genetic tests. Totally, 101 cases undergoing genetic tests remained undiagnosed. Among these cases, 55 had negative genetic findings, 30 harbored mutations that failed to meet their typically pathogenic condition (for example, patients with a monoallelic mutation unfitting a recessive model), and 16 had detected variants that were inconsistent with clinical pointers (Fig. 2). Cases with negative genetic findings predominated, accounting for 54.46%.

Reasons for undiagnosis of patients undergoing genetic test.
Diagnostic experience
Sixteen cases with histories of misdiagnosis and close follow-up are listed in Table 2, which summarizes their demographic features, prior diagnosis, genetic findings, current diagnosis, and outcomes at follow-up. Of note, two cases deserve special attention.
Case 5 was a 6-year-old boy with persistent elevation of aminotransferases for 18 months. At referral to our hospital, he was still undiagnosed. His family history was non-contributory and his two elder brothers were both healthy. His viral profiles and autoantibody spectrum were negative. His serum copper and ceruloplasmin concentrations were within normal limits and 24-h urinary copper excretion showed minimal increase. Screening for metabolic diseases did not display clinically relevant alterations. Imaging examinations of the liver and brain did not reveal any positive findings. Liver biopsy showed mild portal inflammation and severe hepatic steatosis (Fig. 3a). The copper staining indicated no positive results (Fig. 3b). According to these results, Wilson disease was not considered at initial diagnosis. After common diseases were excluded one by one, the WES was performed to investigate whether rare genetic disorders existed. Unexpectedly, two heterozygous variants (chr13:52520505, c.2975C>T, p.Pro992Leu; chr13:52524434, c.2549C>T, p.Thr850Ile) in ATP7B gene were observed, both of which were reported pathogenic. Sanger sequencing confirmed the variants in the proband and further pedigree analysis showed that his mother harbored the variant of c.2549C>T and his father harbored the other. The patient was eventually diagnosed as Wilson disease. As of now, his condition has been gradually improved by decoppering therapy.

a Photomicrograph of the liver biopsy shows mild portal inflammation and severe hepatic steatosis (hematoxylin & eosin staining, ×200). b The copper staining shows negativity (×400).
Case 12 was a 7-month-old girl with increased aminotransferases (aspartate transaminase and alanine transaminase >10 times upper limit of normal), progressive conjugated hyperbilirubinemia, cholestasis and hepatosplenomegaly. Her gamma-glutamyltransferase (GGT) level was normal. Of note, the patient had multiple skin scratches because of pruritus. Her autoantibodies and viral profiles, except CMV and immunoglobulin M, were negative. The screening test results for inherited metabolic diseases were normal. Liver biopsy showed giant cell transformation of hepatocyte, canalicular cholestasis (Fig. 4a), and fibrosis (Fig. 4b). After a tentative treatment with ganciclovir, her condition was not significantly improved, and therefore a genetic condition was suspected. Genetic test revealed two heterozygous variants (chr2:169781294, c.3638G>T, p.Gly1213Val and chr2:169828560, c.1435G>C, p.Val479Leu) in ABCB11 gene, both of which were predicted to be damaging by MutationTaster. Sanger sequencing confirmed the variants in the patient’s genomic DNA. Pedigree analysis revealed that her mother was a heterozygous carrier of p.Gly1213Val and her father was a heterozygous carrier of p.Val479Leu (Fig. 4c). The two variants had never been reported in Human Gene Mutation Database, Leiden Open Variation Database, and published works. The diagnosis of progressive familial intrahepatic cholestasis type 2 (PFIC-2) in this patient became a challenge until her younger sister was born and found to have similar clinical symptoms (at 2 months of age) and identical variants in ABCB11 gene (Fig. 4c, d). Finally, the patient and her sister were both diagnosed as PFIC-2. The patient is now alive, with persistent cholestasis and no transplantation, but her sister died of posttransplantation complications at 23 months of age.

a, b Liver biopsy shows giant cell transformation of hepatocyte, canalicular cholestasis (hematoxylin & eosin staining, ×400) and fibrosis (fiber staining, ×100). c Sanger sequencing chromatograms of the patient, her sister and parents (ABCB11 gene). d Pedigree charts in the family (black solid circle representing female affected subject, circle with black half-solid representing female carrier and square with black half-solid representing male carrier).
Discussion
Liver diseases in children represent a rising problem with significant effects on public health.32 Because of enhanced awareness with technical advances, real increase in their prevalence and decrease in incidence of viral hepatitis, pediatric rare liver disorders are diagnosed more frequently than in the past.33,34 Pediatric hepatology appears to be a very specific field of pediatrics that deals mainly with rare diseases. Within our study, up to 56 rare disorders or syndromes were diagnosed, which related to a variety of etiologies. Liver-based hereditary metabolic disorders were most frequent amounting to 629 cases (54.32%) and possessed complicated disease entities that involved deficiencies or impairments of various enzymes associated with the production, breakdown, or transport of protein, carbohydrate, fatty acids, and so on. This result seems not unexpected, because the liver is the main metabolizing organ in human. Changes in gene expression can alter the liver’s physiological and biochemical functions; subsequently, pathologic conditions emerge. In these cases, feeding history is crucial for the diagnosis, which requires more attention. For example, children with citrin deficiency that is caused by a mutation in the SLC25A13 gene encoding citrin have dietary predilections for protein- and fat-rich food.
In addition to these genetic diseases that solely or mainly affect the liver, hepatobiliary abnormalities involved in extrahepatic or systemic disorders affected many cases in our study. For this group of patients, pseudohypertrophy muscular dystrophy was evidently predominant (94/151). A serum elevation of creatine phosphokinase accompanying increased aminotransferases was helpful in orienting diagnosis of the disorder. Particularly, the less common diseases, like Shwachman–Diamond syndrome, immune dysregulation, polyendocrinopathy, enteropathy, and X-linked syndrome, were diagnosed with great difficulties and required specific concerns.
Wilson disease, a disorder caused by a defect in the biliary excretion of copper by the transporter ATP7B, predominated in the study. As a “common” rare disorder, Wilson disease is proteiform, which brings great difficulties in clinical diagnosis. Even in histological examination, histochemistry results may be negative because of uneven accumulation of copper in the liver.35 However, early diagnosis of Wilson disease is crucial because early diagnosis and initiation of anti-copper therapy can prevent the development of symptoms and reduce the risk of disease progression.36 Low ceruloplasmin is generally prominent in Wilson disease and indicative of the diagnosis. For cases with normal ceruloplasmin, a delayed or mistaken diagnosis often occurs, especially combined with negative clinical signs (Kayser–Fleischer rings, etc) and histological copper staining. In the study, Case 5 with these features had an 18-month undiagnosed history before acquiring definite diagnosis in our hospital through genetic sequencing. This case demonstrates an important contribution of genetic sequencing to confirmation of diagnostically difficult Wilson disease.
Diagnosis between affected siblings with hereditary disorders sometimes can be mutually authenticated. This is an important cue in diagnostically difficult pediatric cases, especially in patients harboring variants of uncertain significance. Case 12 was an infant harboring novel variants in ABCB11 gene. Mutations in ABCB11, the gene encoding the bile salt export pump, induce rare autosomal-recessive hereditary disorders: PFIC-2 or benign recurrent intrahepatic cholestasis type 2 (BRIC-2), but the phenotypes of PFIC-2 and BRIC-2 differ. PFIC-2 is featured with progressive liver damage, is more severe than BRIC-2, and usually requires liver transplantation.37 In Case 12, the patient displayed the characteristics of progressive aggravation of jaundice and cholestasis and was accordingly diagnosed as PFIC-2. Coincidentally, her younger sister had similar symptoms after birth, including neonatal cholestasis, jaundice, and abnormal liver functions with a normal GGT level, and genetic test revealed the same mutations as the patient. Therefore, PFIC-2 in the siblings was finally diagnosed. Alike with the diagnostic experience in the two sisters, Case 7 and his younger brother shared the same novel mutations in ABCB4 gene and similar clinical symptoms. They were both diagnosed as PFIC-3.
Though the technique of NGS, such as WES, brings revolutionary changes in the diagnosis of pediatric rare diseases, it has some pitfalls and limitations. For example, exome sequencing has generally been underpowered to identify deleterious alleles.38,39 Beside these technical issues that are increasingly overcome, the interpretative challenges become more relevant, especially with the increase of the number of genes explored.8 Within our study, a number of patients who had undergone WES and pedigree analysis remained undiagnosed because of the complexity of genetic background and limitations of current methodologies. Among these cases, the majority had negative genetic findings, which might be attributed to methodological shortcomings or etiological diversities. After all, not all rare pediatric disorders are genetic in origin.7 In this sense, clinicians should not over-depend on NGS, which will never replace biochemical and pathological tests in the management of rare diseases.
Emphatically, autoimmune liver disease in the list should not be neglected. It remains difficult to diagnose till date. In children, it can present with wide variation, including autoimmune hepatitis (AIH), primary biliary cirrhosis, primary sclerosing cholangitis (PSC), and the “overlap syndrome” of AIH-PSC, also known as autoimmune sclerosing cholangitis. These liver disorders are thought to be immune mediated, but their etiology remains unclear. They are not secondary to inherited or acquired diseases and they are not associated with any drugs, so they can only be diagnosed if these other diseases or conditions are excluded. Because there is considerable commonality in the clinical presentation of these diseases but differences in their management, appropriate treatment may be delayed, increasing the risk for liver transplantation.17 Consequently, studies on early and accurate diagnosis of autoimmune liver diseases in children needs to be encouraged.
Case analysis studies represent one of the most practical methods to study rare diseases in real-world settings, which play a critical role in the accumulation of clinical knowledge and experience. However, because of the retrospective design of the study, the incidence of individual rare liver diseases cannot be acquired.
Conclusion
Collectively, our large-sample case study on pediatric rare liver diseases has allowed us to accumulate important information (especially novel findings) on the etiology and diagnosis of these disorders. In addition to the absolute majority of rare genetic disease entities, disorders with unusual non-inherited cause in the spectrum should be recognized in pediatric hepatology. WES is a powerful tool for the diagnosis of rare genetic diseases but many challenges remain to be faced. Given that clinical variability often exists within rare genetic disease entities and not all rare disorders are genetic, clinicians should perform detailed and comprehensive analysis in the diagnosis of pediatric rare diseases.
References
Taylor-Phillips, S. et al. Association between use of systematic reviews and national policy recommendations on screening newborn babies for rare diseases: systematic review and meta-analysis. BMJ 361, k1612 (2018).
Sokol, R. J. Reloading against rare liver diseases. J. Pediatr. Gastroenterol. Nutr. 50, 9–10 (2010).
Wangler, M. F. et al. Model organisms facilitate rare disease diagnosis and therapeutic research. Genetics 207, 9–27 (2017).
Németh, A. The coming of age of a young subspecialty: paediatric hepatology. Acta Paediatr. 106, 1742–1746 (2017).
Bourgeois, F. T. & Hwang, T. J. Improving the study of new medicines for children with rare diseases. JAMA Pediatr. 172, 7–9 (2018).
Dhawan, A., Samyn, M. & Joshi, D. Young adults with paediatric liver disease: future challenges. Arch. Dis. Child. 102, 8–9 (2017).
Wright, C. F., FitzPatrick, D. R. & Firth, H. V. Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 19, 253–268 (2018).
Nicastro, E. & D’Antiga, L. Next generation sequencing in pediatric hepatology and liver transplantation. Liver Transpl. 24, 282–293 (2018).
Jones, D. E. J., Sturm, E. & Lohse, A. W. Access to care in rare liver diseases: new challenges and new opportunities. J. Hepatol. 68, 577–585 (2018).
Hooks, K. B. et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology 68, 89–102 (2018).
Mitchell, E., Gilbert, M. & Loomes, K. M. Alagille syndrome. Clin. Liver Dis. 22, 625–641 (2018).
Wagner, K. H. et al. Diagnostic criteria and contributors to Gilbert’s syndrome. Crit. Rev. Clin. Lab. Sci. 55, 129–139 (2018).
Ebrahimi, A. & Rahim, F. Crigler-Najjar syndrome: current perspectives and the application of clinical genetics. Endocr. Metab. Immune Disord. Drug Targets 18, 201–211 (2018).
Chong, S. C. et al. Molecular and clinical characterization of citrin deficiency in a cohort of Chinese patients in Hong Kong. Mol. Genet. Metab. Rep. 17, 3–8 (2018).
Bull, L. N. & Thompson, R. J. Progressive familial intrahepatic cholestasis. Clin. Liver Dis. 22, 657–669 (2018).
Togawa, T. et al. Clinical, pathologic, and genetic features of neonatal Dubin-Johnson syndrome: a multicenter study in Japan. J. Pediatr. 196, 161.e1–167.e1 (2018).
Mataya, L., Patel, N. & Azzam, R. K. Autoimmune liver diseases in children. Pediatr. Ann. 47, e452–e457 (2018).
Hashemian, S. et al. Niemann-Pick diseases: the largest Iranian cohort with genetic analysis. Iran. J. Child Neurol. 13, 155–162 (2019).
Zimran, A. & Szer, J. Recent advances and future challenges in Gaucher disease. Blood Cells Mol. Dis. 68, 9–13 (2018).
Terziroli Beretta-Piccoli, B., Vergani, D. & Mieli-Vergani, G. Autoimmune sclerosing cholangitis: evidence and open questions. J. Autoimmun. 95, 15–25 (2018).
Gerhard, G. S., Paynton, B. V. & DiStefano, J. K. Identification of genes for hereditary hemochromatosis. Methods Mol. Biol. 1706, 353–365 (2018).
Hernández-Gea, V. et al. Idiopathic portal hypertension. Hepatology 68, 2413–2423 (2018).
Iida, T., Yamano, H. & Nakase, H. Systemic amyloidosis with gastrointestinal involvement: diagnosis from endoscopic and histological views. J. Gastroenterol. Hepatol. 33, 583–590 (2018).
Peters, C., van Trotsenburg, A. S. P. & Schoenmakers, N. Diagnosis of endocrine disease: congenital hypothyroidism: update and perspectives. Eur. J. Endocrinol. 179, R297–R317 (2018).
Ciepiela, O. Old and new insights into the diagnosis of hereditary spherocytosis. Ann. Transl. Med. 6, 339 (2018).
Jaime-Pérez, J. C. et al. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J. Blood Med. 9, 171–184 (2018).
Bezzerri, V. & Cipolli, M. Shwachman-Diamond syndrome: molecular mechanisms and current perspectives. Mol. Diagn. Ther. 23, 281–290 (2019).
Bacchetta, R., Barzaghi, F. & Roncarolo, M. G. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann. NY Acad. Sci. 1417, 5–22 (2018).
Kimura, M. et al. A sensitive and simplified method to analyze free fatty acids in children with mitochondrial beta oxidation disorders using gas chromatography/mass spectrometry and dried blood spots. Clin. Chim. Acta 316, 117–121. (2002).
Kimura, M. & Yamaguchi, S. Screening for fatty acid beta oxidation disorders. Acylglycine analysis by electron impact ionization gas chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 731, 105–110. (1999).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Della Corte, C. et al. Pediatric liver diseases: current challenges and future perspectives. Expert Rev. Gastroenterol. Hepatol. 10, 255–265 (2016).
Bernts, L. H. P. et al. Position statement on access to care in rare liver diseases: advancements of the European reference network (ERN) RARE-LIVER. Orphanet J. Rare Dis. 14, 169 (2019).
Zhu, S., Dong, Y., Wang, L., Liu, W. & Zhao, P. Early initiation of antiviral therapy contributes to a rapid and significant loss of serum HBsAg in infantile-onset hepatitis B. J. Hepatol. 71, 871–875 (2019).
Karadag, N. et al. Effect of copper staining in Wilson disease: a liver explant study. Exp. Clin. Transpl. 15, 542–546 (2017).
Mohr, I. & Weiss, K. H. Biochemical markers for the diagnosis and monitoring of Wilson disease. Clin. Biochem. Rev. 40, 59–77 (2019).
Imagawa, K. et al. Clinical phenotype and molecular analysis of a homozygous ABCB11 mutation responsible for progressive infantile cholestasis. J. Hum. Genet. 63, 569–577 (2018).
Locke, A. E. et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 572, 323–328 (2019).
Niroula, A. & Vihinen, M. Variation interpretation predictors: principles, types, performance, and choice. Hum. Mutat. 37, 579–597 (2016).
Acknowledgements
We thank all the pediatric patients in the study. They are all angels. We thank Hongfei Zhang, Min Zhang, Rongmu Luo, Zhiqiang Xu, Yanling Sun, Muruo Zhao, Gang Chen, Huijuan Liu, Yu Gan, Jindou Gong, Chao Dong, Zhenhua Cao, Dawei Chen, Limin Wang, and Fuchuan Wang. Some data in Table 1 were once presented in Chinese.
Author information
Authors and Affiliations
Contributions
P.Z. contributed to the design of the study. Y.D., P.Z., and S.Z. were responsible for data collection. Y.D., P.Z., H.Z., and C.W. analyzed the data. P.Z., J.W., and Y.D. checked and re-evaluated the gene sequencing results. P.Z. wrote the manuscript. P.Z. and C.W. revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Patient consent
Informed consent was obtained at admission from parents or guardians of all included patients.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dong, Y., Wang, J., Zhu, S. et al. Clinical profiles and diagnostic challenges in 1158 children with rare hepatobiliary disorders. Pediatr Res 89, 238–245 (2021). https://doi.org/10.1038/s41390-020-0888-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-0888-4
This article is cited by
-
Follow-Up of Newborns with Hepatitis B Antigenemia
Infectious Diseases and Therapy (2022)
