
Abstract
Synaptic plasticity configures interactions between neurons and is therefore likely to be a primary driver of behavioral learning and development. How this microscopic-macroscopic interaction occurs is poorly understood, as researchers frequently examine models within particular ranges of abstraction and scale. Computational neuroscience and machine learning models offer theoretically powerful analyses of plasticity in neural networks, but results are often siloed and only coarsely linked to biology. In this review, we examine connections between these areas, asking how network computations change as a function of diverse features of plasticity and vice versa. We review how plasticity can be controlled at synapses by calcium dynamics and neuromodulatory signals, the manifestation of these changes in networks, and their impacts in specialized circuits. We conclude that metaplasticity—defined broadly as the adaptive control of plasticity—forges connections across scales by governing what groups of synapses can and can’t learn about, when, and to what ends. The metaplasticity we discuss acts by co-opting Hebbian mechanisms, shifting network properties, and routing activity within and across brain systems. Asking how these operations can go awry should also be useful for understanding pathology, which we address in the context of autism, schizophrenia and Parkinson’s disease.
Similar content being viewed by others
Introduction
Synaptic plasticity leads a double life. A great deal of research has addressed the biological substrates of plasticity, under the working hypothesis that changes in inter-neuronal communication subtend behavioral adaptation. While this connection has often been directly demonstrated, the mechanisms linking microscopic (e.g., synaptic) to macroscopic (behavioral and network) change have generally remained obscure. How are the diverse pairwise interactions between neurons related to network function? Which changes in these interactions bear on network computation and which don’t? How are different network, cellular, and sub-cellular needs and goals balanced via adaptation?
Answering these questions is complicated by the different approaches biological and computational researchers take to investigating plasticity. Biologically, diverse neuronal changes can directly impact how strongly the activity of one cell influences another. These include both pre- and postsynaptic modification, as well as intracellular alterations that interact with extracellular signals (e.g., neuromodulation via G-protein coupled receptors). More specifically, for example, cell responsiveness can be influenced by axonal changes [1, 2], spatial and electrotonic dendritic arbor remodelling [3,4,5,6], spine modification [5, 7,8,9,10,11,12,13,14], active-zone expansion and shrinkage [15], AMPA tetramer modification [16,17,18,19,20], calcium channel modifications [21,22,23], and many more mechanisms. From a computational perspective, in contrast, synapses are often reduced to single "weights", which are idealized as edges in graphs representing pairwise influence between neurons, and plasticity is cast as change of influence [24,25,26,27,28]. The forms of plasticity used in artificial neural networks are also largely selected to optimize the performance of certain functions, such as image recognition [28, 29], memory formation [30,31,32,33,34,35], or reinforcement learning [36]. Thus, while plasticity is increasingly well understood in both biological and computational terms, these literatures are often relatively siloed.
In this review, we examine relationships between the two, applying the general principle that integrating across scales and levels of analysis can facilitate progress in neuroscience [37]. We decompose the underlying problem into two sub-problems, examining (i) how cellular models of plasticity determine network changes, and (ii) how these changes interact with the functional, computational properties of those networks. The first is probably most well understood from the perspective of unsupervised mathematical models of plasticity. The latter is more well understood in terms of mathematical gradients of error landscapes. Considering these along with the biology of plasticity suggests a significant role for metaplasticity, or "the plasticity of plasticity" in multi-scale adaptation. Specifically, the expression of, criteria for, and circumstances inducing plasticity can be adaptively controlled [38,39,40], and this flexibility theoretically allows metaplasticity to align adaptation across scales in the service of functional outcomes. This further suggests that the specific mechanisms of metaplasticity in different neural circuits and contexts will reflect network function.
Concretely, the logic of our review proceeds as follows: (1) Calcium and related signalling cascades are key regulators of Hebbian post-synaptic plasticity, and diverse processes impact both. This makes those processes, in part, plasticity controllers, and we review some of their key elements; (2) Hebbian learning algorithms have long been addressed by computational theories, which characterize their impacts on networks. Analyses of such learning rules and the neuromodulation thereof have increasingly indicated how controlling Hebbian change can produce functional network outcomes, so we review these points; Lastly, (3) different brain areas are specialized and this should be reflected in terms of the metaplasticity they express. Scale also introduces forms of metaplasticity such as activity routing, which conditions neural population activity. These observations should be integrated with the former two points, so we review some potential connections between them. We conclude by discussing applications of these ideas to pathology, specifically Parkinson’s disease, autism, and schizophrenia.
Diverse signals converge on Ca2+ as a plasticity controller
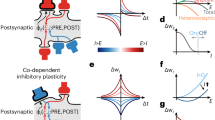
Experimentally, synaptic plasticity can be induced by many means, not all of which are naturalistic. Protocols that are plausibly similar to in-vivo conditions include burst induction in presynaptic afferents, which can mimic endogenous hippocampal activity, spike-pair protocols, which can mimic correlative spiking, and spike-burst protocols, which can mimic diffuse drive generating strong responses, for example [41,42,43,44,45,46,47,48,49,50] (Fig. 1a, b). Plasticity can also be induced and manipulated chemically, and with sub-threshold membrane currents. In-vivo and naturalistic manipulations are increasingly the norm however, and technical progress has improved control of key quantities, such as trans-membrane currents, over time (see e.g. [51]). Synapse potentiating protocols generally require considerable depolarization of post-synaptic cells, whereas depressing protocols require lesser depolarization.

a A pyramidal neuron with an afferent axon impinging on a dendrite, with two probes shown for stimulation and/or recording. b Three example stimulation protocols. Top: Burst inducing stimulation of an afferent connection (teal). Middle: STDP protocol, afferent stimulation (teal) paired with subsequent soma stimulation (purple). Bottom: A spike-burst protocol. Many other protocols exist, e.g. using different current injections, repetition timings and numbers, etc. c Phosphorylation of AMPARs changes their membrane densities and relative compositions of GluA1 to GluA2 subunits (shown in red and blue), mediating EPSC amplitudes. Similar changes in NR2A vs NR2B (as opposed to NR1) subunits of NMDARs modify their relative calcium permeability. Colors are visual guides. d Plasticity vs. Ca2+ concentration in canonical metaplasticity. A small amount of Ca2+ entering the postsynaptic cell induces LTD; larger amounts induce LTP. Changes in Ca2+ permeability change the amount of calcium delivered for a given depolarization, acting like a changeable ("floating") threshold, enforcing homeostasis, and inducing competitive learning. More realistic models are more sophisticated (see e.g., [56]) but this is a well established and reasonable first approximation.
There are many biological mechanisms by which neural activity (such as that generated by induction protocols) can potentiate or depress synapses. These can be roughly classified by their occurrence either pre- or post-synaptically, by the time-scales at which they occur, and by the signals that activate them. They include changes in vesicle count and content, spine properties, active zone surface areas, receptor densities, or dendritic function and morphology, for example [52]. Lasting, input dependent forms of plasticity generate "long term potentiation" (LTP) or depression (LTD) of synapses, which are measured as changes in post-synaptic trans-membrane current elicited by pre-synaptic activity [15]. The induction protocols discussed above generally explore these types of plasticity.
The most studied varieties of LTD and LTP occur post-synaptically in glutamatergic synapses [15]. When a presynaptic neuron fires an action potential, glutamate traverses the synapse and binds to postsynaptic AMPA and NMDA receptors, opening ion channels permeable to sodium and calcium. Whereas sodium influx primarily depolarizes the post-synaptic cell, calcium ions initiate intra-cellular processes that produce lasting changes. These include modifying NMDA and AMPA receptor densities and subunit compositions, directly impacting future glutamatergic transmission [15, 19]. Both the direction (LTP or LTD) and the amount of plasticity depend on the amount of calcium that enters the cell (Fig. 1d) [15, 53,54,55,56]. While it is increasingly recognized that the location of calcium entry can be critical for different processes, and that synapses vary in their exact properties, this is a widely accepted first approximation [56].
There are many other molecules and phenomena that interact with both post-synaptic calcium concentration and the machinery it engages, however. Related findings can be loosely organized according to whether they mainly addresses trans-membrane elements (AMPARs, NMDARs, VGCCs, GPCRs), primary signalling molecules other than glutamate (DA, ACh, NE, 5-HT, BDNF, TNF, eCBs), intracellular molecular players (Ca2+, cAMP, CaMKII, PKA, PKC, PKMζ, IP3), or modulation by other features of neurons, such dendrites (bAPs, spine clustering, endogenous spiking, electrotonic remodelling). This list is certainly not comprehensive, but it is diverse, and as we discuss below, many of the noted items modulate or are required for plasticity in particular circuits. The following several subsections discuss these roles, but also present a basic synthetic challenge to neuroscientists: What logic governs the mechanisms operating at any given synapse? Why are there so many "cooks in the kitchen" when it comes to calcium? Fortunately, the approximate common currency of post-synaptic calcium is compatible with ideas about synaptic plasticity arising from computational theories. The work we review therefore provides a cellular and molecular basis for comparison with abstract plasticity models (discussed in later sections), with the latter being theoretically linked to network functional and computational properties.
Ca2+, AMPARs, and NMDARs mediate canonical glutamatergic plasticity
As noted above, post-synaptic depolarization, the starting point for activity-dependent plasticity, is usually generated by inward Na+ and Ca2+ currents. In canonical LTP, Ca2+ currents contribute little to membrane depolarization but control plasticity, and they arise when NMDA receptors are (1) sufficiently depolarized, and (2) activated by co-agonists such as glycine (e.g., [15, 57,58,59,60]). Calcium entry leads to a number of signalling cascades, three of which we refer to here as the CaM–CaMKII pathway, AC–cAMP–PKA pathway, and the PLC–DAG–PKC pathways [15, 19, 61, 62]. Each molecule in these cascades is involved in multiple processes, but the kinases CaMKII, PKA, and PKC contribute to plasticity in significant part by phosphorylating AMPARs and initiating structural changes to preserve the consequences of AMPAR trafficking (see e.g., [15, 19, 63,64,65,66]). AMPARs are tetramers comprised of GluA1, GluA2, GluA3, or GluA4 subunits, with variable properties and molecular interactions based on composition [67]. CaMKII, PKA, and PKC differentially phosphorylate these subunits, leading to receptor exo- and endocytosis, as well broader trafficking changes, and these signalling cascades ultimately modify membrane AMPAR density and composition (Fig. 1c) [15, 19, 66, 68].
Long-timescale changes in synaptic activity can also modify the amount of glutamatergic transmission needed to initiate plasticity, thereby instantiating a type of "metaplasticity", or plasticity-of-plasticity [53,54,55,56]. Specifically, NMDARs are also tetramers, and are generally composed of two GluN1 subunits and two GluN2 family (GluN2A, GluN2B, GluN2C, or GluN2D) subunits [69]. As with AMPARs, different subunit compositions confer different properties on the resulting receptors. A key property that differs by composition is Ca2+ permeability, with the result that different post-synaptic receptor distributions will require greater or lesser activation in order to achieve a given integrated calcium flux. Importantly, NMDAR composition itself, and in particular the ratio of GluN2A to GluN2B subunits (also known as NR2A and NR2B units) can be modified by use-dependent post-synaptic signalling cascades. This determines how use impacts Ca2+ concentrations, and thereby AMPAR plasticity (Fig. 1c) [70,71,72,73,74,75,76,77,78,79]. Functionally, it produces a type of "floating threshold" for plasticity induction, which was notably predicted by computational theories of plasticity [53,54,55]. This threshold, along with the basic phenomenon of lesser and greater calcium influx producing LTD and LTP, is illustrated in Fig. 1d. One resulting function is homeostatic, in that depression becomes easier to induce in stronger synapses, whereas potentiation is favored by weaker synapses. As we discuss below, more general forms of metaplasticity have many additional computational implications.
Many processes converge on Ca2+ signalling
Beyond solely providing a floating threshold over long timescales, Ca2+, PKA, PKC, and CaMKII related pathways have been increasingly recognized as flexibly altering plasticity on the basis of non-NMDAR mediated signals, which likely serve important computational ends as well. Specifically, various other trans-membrane players directly impact calcium, perhaps most notably voltage gated calcium channels (VGCCs) and calcium-permeable AMPA receptors (CP-AMPARs). In addition, G-protein coupled receptors (GPCRs) also modulate elements of the CaM–CaMKII, AC–cAMP–PKA, and PLC–DAG–PKC pathways, and this family includes receptors that respond to all the major neuromodulators. Muscarinic acetylcholine receptors (mAChRs), α - and β-adrenergic receptors (β–ARs), dopamine receptors (DARs), and serotonin receptors (5HTRs), are all GPCRs impacting these pathways, as are metabotropic glutamate receptors (mGluRs) and metabotropic GABA receptors (GABABRs) [80, 81]. As a result, neuromodulators have diverse impacts on plasticity. An important question is therefore: Given broader theories of neuromodulators and the regulation of calcium-permeable channels, how are these impacts orchestrated in the service of computational goals? The most well understood case is probably dopaminergic modulation of plasticity for reinforcement learning, for example, but more generally, answers for particular circuits will require synthesizing functional observations across scales. To this end, we review some of the molecular aspects here.
From a computational perspective, coordinated, fine-grained spatial control of post-synaptic calcium is important because it can theoretically direct plasticity to specific stimuli or sets of post-synaptic inputs. VGCCs are strong candidates for mediating this capacity, (along with other dendritic parameters) because of their roles in dendritic processing and regulation of Ca2+. Voltage-gated calcium channels form several subfamilies based on their pore-forming proteins (α subunits) [22]. The Cav1 family (Cav1.1, Cav1.2, Cav1.3, and Cav1.4) conduct L-type calcium channels, and Cav1.2 and Cav1.3 are generally located postsynaptically in dendrites and cell bodies [82,83,84]. Cav2.1, Cav2.2, and Cav2.3 conduct P/Q-, N- and R-type currents and are primarily located presynaptically. Cav2.1 and Cav2.2 are involved in vesicle exocytosis and Cav2.1 participates in short-term synaptic facilitation and depression. Cav2.3 channels also appear to be located postsynaptically in some areas [85, 86]. The Cav3 family (Cav3.1, Cav3.2, and Cav3.3) conduct T-type calcium currents, which are involved in rhythmic and burst firing, particularly in the thalamus [22]. Of these, one therefore expects relatively direct effects of Cav1.2 and Cav1.3 channels on post-synaptic glutamatergic LTP induction, via calcium currents, and potentially less direct effects of Cav3 family channels via bursting related back-propagating action potentials (bAPs).
In line with these predictions, L-type calcium currents contribute to plasticity in a number of ways, several of which go beyond floating threshold effects. Cav1.2 contributes to LTP in (hippocampal) Schaffer collaterals, for example [87,88,89,90]. Blocking these currents in several circumstances either reduces or abolishes LTP that would have occurred otherwise. Cav1.2 channels also form highly localized signalling complexes with β2–adrenergic receptors and several members of the CaM–CaMKII and AC–cAMP-PKA cascades, such that adrenergic signalling up-regulates channel conductance and increases protein-based second messenger activity [90, 91]. This promotes LTP under joint, weak, theta-burst and adrenergic stimulation, which occur naturally in the hippocampus during exploratory behavior [92]. Moreover, NMDARs and VGCCs appear to regulate one another, with chronic increases in Cav1.2 L-currents lowering NMDAR Ca2+ flux for example [83, 86, 93]. Finally, L-type calcium currents also appear to contribute to (slow) spike after-hyperpolarization and to frequency-based adaptation, processes that regulate cell excitability and thereby plasticity [94,95,96,97]. Collectively, these observations suggest that VGCCs might be integrated into a holistic understanding of plasticity as having primary effects on induction thresholds, post-synaptic excitation and dendritic processing (discussed below), along with various secondary impacts. A key research goal will therefore be determining the relative importance of each for plasticity at any given synapse.
Post-synaptic calcium is also directly manipulated by calcium-permeable AMPA receptors (CP-AMPARs) [20, 23]. Typically, AMPARs contain at least one (post transcriptionally "edited") GluA2 subunit, making them impermeable to calcium. CP-AMPARs, on the other hand, are typically GluA1 homomeric. They are often inserted into the post-synaptic density during plasticity induction as a result of trafficking from endosomes, only to be subsequently removed [98,99,100]. Some neurons also appear to express long lasting CP-AMPARs however, including cortical and hippocampal interneurons [101,102,103,104]. It is not well understood what effects either the transient or long-lasting changes in calcium transmission have, but the broader calcium theory suggests they should lower the thresholds for inducing both synaptic depression (given weak additional input) and further potentiation (given strong additional input). Complicating the matter, intracellular polyamides close CP-AMPARs at significant depolarizations, making them voltage gated [105, 106]. This voltage gating appears to allow CP-AMPARs expressed by hippocampal interneurons to mediate "anti-Hebbian" plasticity [101, 103, 104], and may be important in principal-interneuron plasticity generally [102, 103, 107, 108]. (In this case, excitatory synapses onto inhibitory neurons are potentiated when pre-synaptic firing occurs without post-synaptic firing.) A number of regions have also been shown to express CP-AMPARs under pathological conditions, but here too, little is known about their precise effects on plasticity [21, 23]. As with VGCCs, it may therefore be reasonable to consider CP-AMPARs according to a first-order effect on calcium-based threshold change and diverse second-order effects. Whether these "second order" effects are really secondary remains to be seen, but the idea that transiently expressed CP-AMPARs should facilitate plasticity induction (both LTP and LTD) in the short term appears plausible.
Finally, as noted above, a number of metabotropic, neuromodulatory receptors interact with the CaMKII, PKA, and PKC calcium signalling cascades, providing further levers for controlling plasticity [19, 62, 80, 81]. These G-protein coupled receptors (GPCRs) form a very large class of trans-membrane proteins. Much of the diversity of this class occurs in the extracellular components, whereas intracellularly, GPCRs are characterized by their bound hetero-trimeric α, β, and γ subunits [80, 81]. Each subunit has a variety of subtypes as well. Extracellular ligand binding dissociates the αβγ trimers from the transmembrane elements, and generally further subdivides them into free α and a free βγ dimer (often denoted Gα and Gβγ or similar). The Gα proteins can be clustered into families identified with Gαi (alpha types i, o, z, t), Gαs (types s, olf), Gαq (types q, 11, 14, 15) and Gα12 (types 12, 13). Of these, Gi inhibits adenylyl cyclase (AC), which lowers production of cAMP, and thereby reduces the activity of PKA [19, 62]. Gs does the opposite, upregulating AC, and hence cAMP, and PKA [19, 62]. And finally, Gq produces DAG and IP3, which activate PKC and Ca2+ release from endoplasmic reticula via IP3Rs [19, 62]. Metabotropic receptors for all of the major neurotransmitters bind these G-proteins (i.e. Gi, Gs, and Gq families), which mediate complex intracellular activity. As one would predict by their actions on PKA, Gs coupled receptors appear to generally promote LTP, whereas Gi coupled receptors often promote LTD [62]. Gq coupled receptors show mixed effects on plasticity, with mGluRs being generally associated with LTD, whereas M1 AChRs are demonstrably involved in both LTD and LTP (discussed below). For any given receptor and context, the exact relationship to plasticity likely varies according to the balance of different G-protein mediated effects and the conditions they’re exerted in. Nonetheless, because of their strong relationships with macroscopic theories of brain function, we now discuss several instances of DAR, mAChR, and β–AR modulated plasticity in further detail.
DA, ACh, and NE modulate both activity and plasticity
DA, ACh, and Ne receptors all modulate intracellular signals involved in plasticity, and they change cellular and network properties governing activity as well. These changes occur simultaneously in several systems, suggesting that they may generally act in concert, but how this occurs, and what it accomplishes, are not well understood. (Indeed this article examines potential syntheses.) We consider network integration primarily in a later section, and for now continue with some of the cellular and molecular relationships.
Empirically, DA dependence is well established at cortico-striatal synapses, for example [10, 62, 109,110,111]. The striatum is the input region of the basal ganglia and is broadly recognized as having roles in action planning and execution, working memory and attention, and reward-based learning. Dopamine is believed to signal reward-prediction errors (RPEs) and motivational variables, so DA dependence of plasticity is broadly in line with these theories [10, 62, 109,110,111,112,113]. This modulation acts differentially on the two primary types of neurons in the striatum, which express different dopamine receptors. The "direct pathway" medium spiny neurons (dMSNs) primarily express D1Rs, and these D1-MSNs are metabotropically rendered more excitable by DA. The "indirect pathway" projections (iMSNs) primarily express D2Rs, and these D2-MSNs are metabotropically inhibited by it. Furthermore, the plasticity of cortico-striatal synapses onto each type of neuron is unidirectional or bidirectional depending on local DA concentration [114], with positive RPEs and negative RPEs preferentially driving reinforcement of the D1 and D2 pathways. These effects occur in part because D1Rs and D2Rs impinge on the calcium-cAMP-PKA signalling pathway via the Gs and Gi family α subunits respectively [115]. DA-based plasticity in MSNs is further gated by ACh, which is signaled by tonically active neurons [116,117,118], and requires co-agonism by endocannabinoids and adenosine [114]. Because DA appears to multiplex various signals, these additional requirements may serve to specify exactly when and how plasticity should occur in response to only relevant dopaminergically communicated information. More broadly, computational accounts of these pathways, which we discuss below, draw on theories of modulated Hebbian plasticity to suggest normative roles for the opponency and modulation noted here.
DA-dependent plasticity has also been established in the PFC of mice, but is less well understood theoretically or empirically. Empirically, D1Rs are expressed in a bilaminar pattern across frontal cortex, with elevated density in layers I-III and V-VI, and low density in layer IV (in primates, with some results in rodents) [119,120,121,122,123]. These receptors appear to be predominantly located on dendrites of pyramidal neurons, but are also located pre-synaptically on principal cells targeting distal dendrites of other glutamatergic neurons, and on parvalbumin positive GABAergic interneurons [121, 124, 125]. Maximal frontal D1R concentrations appear in dlPFC, which hosts strong recurrent connectivity that is mediated, unusually, by NMDAR rather than AMPAR activity [126, 127]. Posterior regions are relatively devoid of both D1Rs and dopamine, with the significant exception of the lateral intraparietal area, which is also noted for its recurrent activity and its role in working memory [123, 126]. Occipital cortex, by contrast, hosts both an extremely low density of D1Rs and little to no dopaminergic innervation [123, 126]. D2Rs appear to be expressed relatively uniformly, in much smaller quantities, across layer V pyramidal neurons throughout cortex [119, 120]. In frontal regions, they have also been found on GABAergic parvalbumin positive interneurons [128,129,130,131]. The D2Rs in layer V pyramidal neurons were recently reported to be Gs coupled rather than Gi coupled, and hence to enhance, rather than reduce, cell excitability given dopamine application [132].
In terms of direct (rather than network) impacts, plasticity induction at excitatory L2/3 synapses onto L5 pyramidal neurons shows dopamine dependence in mice [130, 133]. DA application at several time-points after these authors’ spike-pairing protocol produced D1R dependent Hebbian LTP. The longer-delay application (30 ms after pairing) was found to depend on both D1R activity post-synaptically and D2R mediated reductions in GABAergic interneuron firing rates. The shorter-delay application (10 ms) required only the latter, demonstrating a clear case of activity dependence, disentangled from post-synaptic GPCR signalling. These findings were subsequently extended by showing that changing the spike pairing to a post-before-pre protocol, which classically would induce LTD, and concurrently applying dopamine, produced LTP instead [133]. This was dependent on post-synaptic D1R activity, but not pre-synaptic D2R activity.
In contrast with dopamine, acetylcholine and norepinephrine have been shown to modulate cortical plasticity in several sensory areas. NE and ACh dependence occur at V1 L4 to L2/3 cortical synapses, for example, and appear to be required for plasticity induction there in adult rodents [134,135,136]. The mAChRs present are Gq coupled (M1 family), and interact with the PLC-DAG-PKC signalling cascade to bias plasticity towards LTD [136,137,138]. Co-located β-adrenergic receptors are Gs coupled and interact with the AC-cAMP-PKA cascade to promote LTP [135, 136, 139, 140]. When both are activated, these aspects combine to specifically gate causal spike-time dependent plasticity, meaning LTP occurs when pre-synaptic input precedes post-synaptic depolarization, and LTD occurs when this order is reversed [136]. Notably, α1-adrenergic receptors are also present post-synaptically, which have higher affinity for norepinephrine than β-adrenergic receptors, are Gq coupled, and have often been reported to facilitate LTD as well as M1-AChRs [135,136,137, 139, 141, 142]. In line with these points, one study found that low NE concentrations in isolation produced LTD, whereas high NE concentrations activated both receptors and re-instantiated Hebbian STDP [135].
Finally, all three neuromodulators (DA, ACh, NE) modify hippocampal plasticity as well. DA dependence has been noted on Schaffer collateral synapses, which connect CA3 to CA1, and in the perforant pathway, which connects entorhinal cortex to the dentate gyrus [143,144,145,146,147,148], whereas NE and ACh modulation have been reported at Schaffer collaterals [149,150,151,152]. β-adrenergic modulation was also noted to extend the temporal window for time-dependent LTP, by making CA1 pyramidals more excitable [149]. More generally, NE has often been reported to facilitate hippocampal LTP via the preferential Gs coupling of β-adrenergic receptors, as with visual cortex [92, 134, 149, 153,154,155]. Reported ACh effects have been more varied, with some studies indicating facilitation of LTP [154, 156,157,158,159,160,161,162,163,164,165,166,167] and others showing LTD induction [168,169,170,171,172,173]. The latter have generally seen LTD under weak or non-existent post-synaptic stimulation however, whereas the former have tended to look at enhancement of LTP or conversion from LTD, a potentially critical difference.
Several recent studies have also investigated how spike timing effects these processes. In one, ACh converted bidirectional STDP at Schaffer synapses into unidirectional LTD, whereas retroactive application of dopamine transformed this into LTP [150], in line with earlier reports of hippocampal DA-ergic modulation [143, 146, 147]. This was at odds with another group’s report that inhibition of mAChRs converted causal LTP to LTD, and prevented anti-causal LTD [151, 152], but the induction protocol used in the latter appears to have been significantly stronger. This strength discrepancy might mirror the general difference in LTP vs LTD biasing actions noted above, or may be mediated by the complexity of Gq signalling, specifically by different contextual implications of IP3-Ca2+ and PLC-DAG-PKC cascades. For example, M1 activity can both enhance SK channel (calcium-dependent, voltage-independent, small conductance potassium channel) activity via IP3-based internal Ca2+ release [174], and inhibit it via PKC [166, 175]. LTP induced by theta-burst stimulation of Schaffer collaterals can be facilitated by ACh via the latter mechanism, because closing the SK channels diminishes shunting current and enhances NMDAR Ca2+ flux [166, 175]. In fact, M1-AChRs can have a number of other impacts on K+, VGCC, and nonspecific cation channels [176], which makes the diversity of ACh mediated plasticity results perhaps less surprising, and generally indicates we have much to learn about the matter.
Summary of cellular and molecular data
To summarize, a number of different fundamental processes modulate intracellular calcium signalling cascades, and thereby plasticity. Up-regulating calcium, CaMKII, and PKA pathways tends to facilitate LTP, whereas down-regulating calcium or PKA, or up-regulating PKC related pathways tends to facilitate LTD. This is compatible with a model of plasticity in which small, but non-negligible amounts of calcium facilitate LTD and large amounts facilitate LTP. Although we did not address it above, it is important to note that one open question in this regard is exactly which properties of calcium fluxes, such as their amplitude, duration, or location, govern this behavior [56]. Nonetheless, a number of results are clear. Voltage gated calcium channels contribute to these effects, at minimum, by modulating calcium directly, through their interactions with NMDARs, by changing cell excitability, and through complexes with β-adrenergic receptors. Calcium-permeable AMPA receptors also directly mediate calcium currents and are mostly expressed transiently, but may be long lasting in some synapses. The implications of their short-term facilitation of calcium currents is not well understood, but presumably interacts with the same signalling cascades just described. These interactions are likely complicated by the fact that CP-AMPARs are themselves targets of said cascades, because they are AMPARs, and by polyamide-based gating. Lastly, the major neuromodulatory systems all engage GPCR signalling. Gα subunits, which are categorized by family (Gi, Gs, Gq, and G12) engage the PKA, IP3, and PKC pathways (in addition to diverse effects we have not discussed). As a result, they are expected to directly modify plasticity induction, and indeed multiple areas including striatum, pre-frontal cortex, visual cortex, and hippocampus display neuromodulated plasticity. Often this occurs as "gating" of LTD or LTP, or by converting one to the other. Because these are Hebbian forms of plasticity, models of their function should build on research examining un-modulated Hebbian rules and knowledge of what modulating these can accomplish.
Controlling plasticity dictates key network properties
How can we understand the impacts of the diverse biological phenomena reviewed above? One answer is to interpret them in light of mathematical models of network function and learning. Many of the modulations of LTP or LTD discussed so far control specifically Hebbian synaptic change. By this we mean change at an existing synapse determined by a presynaptic factor and a postsynaptic factor. As such, formal theories of Hebbian plasticity are highly relevant, and we proceed to review them here. Then we consider how metaplasticity can be exerted by augmenting Hebbian change with so-called "third" factors. These can represent reward or attention, and can mathematically model the calcium based or PKA, PKC, and CaMKII impacts discussed above [62, 111, 177,178,179,180]. Mathematical analyses indicate that such modulation vastly expands the universe of resulting network functions. Collectively, these considerations indicate how important network properties can be related to metaplasticity, providing an interpretive framework for the calcium signalling observations discussed above and knitting local synapse changes into functional network ones.
Unconditional Hebbian rate theories
The primary mathematical formulations of Hebbian plasticity are rate-based and spike-based, whereby synapses change according to time-averaged activities or according to timing relations among peri-synaptic depolarizations. A diversity of rate-based theories exist, but the Bienenstock-Cooper-Munroe theory (BCM) is probably the most well known and well validated in neuroscience [53, 55]. The canonical spike-based theory is termed "Spike-time dependent plasticity" (STDP) [44, 45, 48, 49, 111, 180, 181]. Both theories are based on the idea that pre-synaptic activity producing post-synaptic activity should increase synaptic efficacy, i.e., on Hebb’s postulate.
Dependence on pre- and post-synaptic activity suggests Hebbian change should respond to covariances, an intuition which is appropriate across a range of models. In the most basic rate formulations, one set of neurons (principal cells in V1, say) receives feed-forward input from another (principals in thalamus). The simplest synaptic changes are products Δw = yx, where w denotes synaptic efficacy ("weight"), Δw denotes a change in efficacy, y denotes post-synaptic activity, and x denotes pre-synaptic activity. When one set of neurons drives a second set like this, covariance between pre- and post-synaptic activity primarily reflects covariation within the driving set. Since the main dimension in which a collection of data varies is termed its principal component (PC), this suggests that Hebbian synaptic plasticity transforms the weights (and hence receptive fields) to reflect principal components of the data, as illustrated in Fig. 2b [182,183,184,185]. When homeostatic elements such as floating plasticity thresholds are modelled, neurons develop more complex receptive fields like Gabor filters [53, 55]. Including inhibitory competition forces neurons’ receptive fields to specialize [186, 187], and heterosynaptic competition tends to modify the properties of those fields somewhat [188]. Top-down feed-back and recurrent inputs generalize these models further, and since Δw = yx has interchangeable x and y, one might expect recurrent weights to become reciprocal, which is indeed common in cortical networks [189]. Symmetric connectivity stabilizes persistent activity within recurrent sub-networks [30, 187], or recruits similarly tuned neurons to excite one another (see Fig. 2d-f). Models of visual processing based on these ideas predicted aspects of cortical stimulus selectivity and map-formation, such as visual tuning properties and stronger connections between similarly tuned neurons [190,191,192,193,194,195,196,197]. Hippocampal encoding of memories in recurrent activity also essentially relies on this logic, for example [35, 198].

Blue indicates less activity or reduction in efficacy, and red indicates the opposite throughout. a A feed-forward network with two stimulus presentations, "trial 1" and "trial 2". Neurons 1-4 are connected to 5 and 6, and connections to 5 are shown with arrows. Synapses are strengthened from neurons with higher than average activity (red) and weakened from those with weaker activity (blue). b The synapse updates arranged in an array, numbered according to neurons as (To,From). The principal component of the trial-by-trial variability in a is shown, also labelled by neuron. This vector is the same as the top row of the weight updates, illustrating how the update is a "matched filter" (parallel vector) for the PC. The response of unit 5 is the sum over all pairs of elements multiplied together, i.e. the sum of the weight number (5,1) times the activity in unit 1, the weight (5,2) times the activity in unit 2, etc. c RPE-modulated feature detectors extract features from subsets of the data, picking up the covariance of input-output transformations with reward, or generating matched filters for PCs that drive rewarding output activity. Such updates can perform reinforcement learning. d Recurrent network with two stimulus presentations, and weight changes under symmetric and asymmetric update rules (assuming neuron 2 fires before neuron 1, in asymmetric case). e Weight matrix updates, under the same convention as in (b). Grey diagonal elements indicate that neurons don't innervate themselves. Grey off diagonal elements show unchanged weights. Symmetric and antisymmetric updates are generated by rate-coded Hebbian, and causal STDP rules. Nilpotent updates can occur when an anti-symmetric update can't lower a weight any further. These are linked to so called "non-normal" dynamics, of which transient synfire chains are an example. f Activity propagation from unit 1 (with time represented as evolving downward) if weights were as shown in (e) (and neurons turn themselves off). In the symmetric case, 1 and 2 are mutually excitatory, passing spikes back and forth. In the latter cases, 1 excites 2, but is not reciprocally excited, so activity is transient.
Theoretical work on unconditional, rate-based Hebbian plasticity mainly proceeded from these foundations by examining Hebbian interactions with other important features of biology, especially homeostasis. Naive Hebbian dynamics are unstable, with weak synapses disappearing and strong synapses often increasing indefinitely. As noted above, the BCM rule posited a floating plasticity threshold partly to resolve this issue [53, 55], but other early approaches included modelling weights as bounded or conserved in aggregate, and plasticity as a diminishing function of strength [191, 199, 200]. Rough conservation in aggregate has largely been born out in the form of "synaptic scaling" [201, 202], and remarkably there is also evidence that local excitatory-inhibitory balance and synaptic strength are conserved on dendritic sub-domains as well [5, 11]. Plasticity of neural excitability, inhibitory plasticity, and autonomous spine fluctuation dynamics have also been identified as contributors to homeostasis, and likely also serve computational roles [14, 107, 108, 203,204,205,206,207]. Synaptic scaling appears to be crucial for maintaining correlative relations between neurons, whereas network firing rate homeostasis is more strongly impacted by excitability changes, for example [206]. Inhibitory plasticity, on the other hand, may be fundamental for controlling detailed forms of excitatory-inhibitory balance, which in turn impact a number of important network properties such as the desynchronized states associated with attention and alertness [107, 108]. Finally, unconditional Hebbian plasticity in networks can also regularize learning (i.e., make it less flexible but more targeted) or otherwise bias networks towards certain representations [196, 208,209,210,211,212]. One example of this is aiding the development of systematic representations that facilitate generalization [208]. Though it is beyond our scope to address these in detail, we suspect that biological multiplexing of different types of plasticity has signatures in calcium pathway dynamics, and that investigating these will be a key area for cross-talk between theoretical and empirical research. Ideally results from these areas would be rationalized along with other mechanisms by reverse engineering accounts, as has been done somewhat with stability and representation learning, for example (e.g., [210, 213, 214]).
Unconditional Hebbian spike theories
Spike-timing models inherit basic properties from rate-based models [200, 215]. The canonical formulation of STDP computes the time difference between every pair of spikes occurring in a connected pair of neurons, translates each into an increment or decrement of the synaptic efficacy, adds these all together, and updates the synapse. Post-synaptically, neurons can accomplish this online by maintaining an "eligibility trace" of the times at which they received input, comparing this with their own activity, and modifying their synapses accordingly [180]. Physiologically, the eligibility trace is interpreted as calcium or other intra-cellular products which are elevated by synaptic input and decay over time. Backpropagating action potentials or other retrograde signals are interpreted as communicating post-synaptic activity to the synapse. The eligibility-trace formulation immediately suggests that there may be cases in which plasticity is retroactively expressed, by converting a latent change into an expressed one, and this has been widely observed [13, 62, 147, 150, 216,217,218,219]. This retroactive expression is also important for computational models of metaplasticity and may relate considerably to empirical "consolidation" and "synaptic tag and capture" ideas, although they have been little integrated thus far (see [15, 36, 180, 220,221,222,223,224,225], for example.)
A more general mathematical description involves the use of Volterra expansions [200]. The net plasticity induced by a sequence of spikes in a pair of neurons can be described as application of a "functional" to the pre- and post-synaptic neurons’ spike trains. The STDP functional takes two spike-trains and returns a synaptic weight change. The weight change depends on a relation quantifying the temporal order and proximity of pairs of spikes, called a kernel. STDP is determined by convolving the kernel with one of the spike trains, then taking the inner product of the result with the other spike train. This procedure is illustrated in Fig. 3A, as are a number of different potential pairwise kernels for Volterra expansions. These descriptions are motivated by the fact that Volterra expansions express the simplest pairwise interactions, and can be naturally expanded to include higher or lower order interactions as well. For example, they can model spike-triplet effects [226,227,228,229] or the impacts of unpaired output spiking [200], and can also be expanded to account for other temporal variables, such as membrane voltage deviations [230, 231].

A STDP is established empirically by inducing pre- and post-synaptic spiking and observing synapse efficacy changes. This determines a "kernel" or window function, which can be used to model plasticity. (a-i) Kernel components can theoretically be mixed and matched, and in-vivo they appear to be. The vertical axis is change in synaptic efficacy, horizontal is time from presynaptic to post-synaptic spike. The canonical STDP kernel is in (c), but unipolar LTP (a), unipolar LTD (i) and anti-causal STDP (g) also appear under conditions of neuromodulation. The NE, ACh and DA-dependent plasticity discussed in text can often be regarded as specifying these kernels. Hippocampal DA appears to convert canonical STDP (c) to unipolar LTP (a), whereas hippocampal ACh appears to convert canonical STDP (c) to unipolar LTD (i) in some circumstances, for example. In some PFC synapses, DA gates unipolar potentiation (a). Cases (d), (b), (f), and (h) also seem to arise in other conditions. See [62] for a detailed review, and references in text. Note that any particular effect may be protocol dependent. B Illustration of connectivity effects. Organizing synaptic efficacies as a matrix, updates based on different kernels have different mathematical properties. Unipolar LTP or LTD are both symmetric, classic STDP and inverted STDP are both anti-symmetric, and the remaining possibilities, are called nilpotent. Symmetric matrices have real eigendecompositions, meaning roughly that networks with symmetric connections produce stable recurrent activity. Anti-symmetric network weights and nilpotent weights favor transient, "moving" activity, such as synfire chains. Because activities and weights in actual networks are rectified, asymmetric updates will often be rectified to nilpotent ones, and thereby push networks to have feed-forward sub-networks. Here, a network of three neurons with positive symmetric initial weights between neurons 2 and 3 undergoes asymmetric updates based on the classic asymmetric STDP kernel to produce unidirecitonal connectivity from neuron 2 to 3. C Example application of the REINFORCE algorithm, specifying a three-factor plasticity rule, to a two neuron network. On repeated trials, activity in unit 2 dictates network performance such that being closer than average to some hypothetical target T is rewarded, being further is punished. These outcomes generate reward prediction errors relative to average reward both of which serve to increase the weight from unit 1 to unit 2 under the equation for dW, the change in synapse strength.
The kernel description also provides flexibility in modeling timing and order dependence. In the standard case, kernels are constructed with spike-spike interactions that decay exponentially as a function of absolute time differences. These are also causal, so that pre-before-post spiking generates LTP, and post-before-pre spiking generates LTD, as noted above. This results in plasticity that recapitulates much (but far from all) empirical synaptic data [48, 49, 232]. The kernel approach suggests that these aspects might be mixed and matched however, for example to model forms of plasticity under which post-before-pre spiking does not generate change, or for which both temporal orders of spiking produce LTP. Indeed, beyond the canonical form of STDP, many such alternative forms have now been described [62]. The most well understood of these is probably anti-Hebbian STDP at climbing-fiber homologous synapses onto Purkinje cells in the cerebellum like structures of weakly electric fish. These are thought to play a role in signal cancellation [233,234,235], but debates about climbing-fiber plasticity more broadly appears unresolved [236]. Anti-Hebbian STDP has also been found in neuro- and inhibition-modulated preparations [62, 237], however, and in excitatory-inhibitory plasticity [101, 103, 104]. This may be related to network stability or competitive specialization, as theoretical work has suggested for anti-Hebbian plasticity broadly [213, 214], but a general theory is lacking. The impacts of different kernels or their associations with, for example, different network connections between cell classes, is in need of greater exploration (but see [238, 239]). Especially so, since neuromodulation can change these kernels, as discussed below.
At the network level, canonical STDP predicts structured timing relations among groups of neurons. In networks with significant feed-forward pathways and similar transmission delays, canonical STDP predicts the existence of "synfire chains", groups of neurons that propagate activity as volleys of synchronous action potentials [240,241,242, 242,243,244,245,246,247,248,249,250,251]. More general structured asynchronous activity generalizes this. Specifically, when transmission delays, refractory activity, and other elements of biology are incorporated into spiking neural network models, STDP tends to produce overlapping ensembles of co-active neurons with conserved timing. Izhikevich referred to this as "polychronization", and showed that these ensembles are essentially intermingled synfire chains with timing offsets [245]. An interesting consequence of these models is that random membrane fluctuations, which drive resting state activity, interact with STDP to randomly shrink and enlarge the pools of neurons participating in different chains [245].
Synfire and polychronization ideas have suggested a number of hypotheses about in-vivo network function. Reproducible, non-stationary activity has often been considered as a potential substrate for compositional representation and routing, for example [246, 249, 251,252,253,254]. Recurrently, these properties can also be used as a form of active maintenance, providing spike-based, asymmetric counterparts to the Hopfield dynamics initially proposed to model memory [241, 242, 247, 250, 255]. More generally, modelling suggests that networks subject to STDP would need to co-opt polychronizing dynamics to fulfill their functions, because they would otherwise be subject to constant plasticity-induced degradation. This is a spike-timing-based example of regularization, which we noted above for unconditional Hebbian theories, and is perhaps most well understood for cases of reward-modulated STDP [179, 256,257,258,259]. When, where, and how unconditional STDP interacts with more complex forms of learning remains a major open question however, as was the case with rate models. One straightforward question, for example, is when and where symmetric vs asymmetric network restructuring occur (see Fig. 2b, d–f) [230, 260].
More empirically, groups of neurons with conserved timing relations (as are generally predicted with STDP rules) have been identified in various circuits. For example, auditory cortex has long been recognized to encode stimuli with precise spike timing [261, 262], and several of the ideas discussed above seem to apply there. Primary auditory L5 neurons (excitatory and inhibitory, in rats) exhibit fluctuating spontaneous activity that is highly reminiscent of polychronization [263,264,265]. These transient sequences were most highly conserved at transitions from cortical down states to cortical up states, and became less conserved the longer the transient activity persisted (as predicted by a polychronization model). Furthermore, a subset of up-states propagated as travelling waves, and local groups of neurons with conserved timing relations were activated in stereotyped order, regardless of the direction of wave travel or whether their responses were initiated by the waves at all [263]. Follow up work found that these stereotyped synfire-like responses provided a "vocabulary" of neural activity that stimuli could elicit, with the same pattern of decaying spike-time precision over stimulus presentation time [264, 265]. The computational analysis of spiking involved in these types of investigations are statistically non-trivial however, hampering progress [266,267,268]. Complicating the matter further, more complex situations, such as spike-phase coupling to oscillatory local field potentials, are plausibly the more common contexts for conserved timing that actions of plasticity should be investigated in (see e.g. [269,270,271,272]). Nonetheless, the asymmetric activity generated by STDP is of significant interest in computational neuroscience for generating transients of population activity, such as sequences [273,274,275], and could reasonably be expected to either generate or adapt "hidden Markov model like" neural dynamics.
Metaplasticity makes Hebbian updates conditional
Metaplasticity co-opts the plasticity discussed above, generalizing Hebbian change by making it conditional. In the simplest case this means gating plasticity, such that it only happens under certain circumstances but is unchanged in form. A more complicated situation involves both gating plasticity and modulating its sign, i.e. converting LTP to LTD or vice versa. Finally, these can be combined with alterations in the magnitude of induced changes. This perspective is useful over and above thinking about different synapses as merely having different time-, history-, or neuromodulator-dependent forms of plasticity, because it leaves the Hebbian aspect intact and allows asking what controlling it in constrained ways can accomplish.
Mathematical analyses indicate that such control vastly expands the potential uses of plasticity [27, 28]. In computational theories of neural networks, there are several classes of algorithmic learning procedures, which are categorized by the informativeness of the feedback provided to the learning algorithm [27, 28]. Supervised learning entails providing maximally informative feedback, which instructs networks with the output that should have been produced, such as when a teacher corrects the way a student reads a word. Reinforcement learning requires ordinal feedback, indicating when network outputs are better or worse, making it a form of trial and error learning. Unsupervised learning occurs without feedback, and network connections come to reflect statistics of the training environment. Classical Hebbian learning, such as unmodulated spike-time dependent or BCM plasticity, are forms of unsupervised learning. Metaplasticity that flexibly controls Hebbian updates essentially converts this into reinforcement learning based on "goals" which are implicitly defined by the modulation.
In reinforcement learning, a neural network needs to determine the gradient of some function with respect to network parameters (e.g., weights) in order to change those parameters in a way that increases or decreases the value of the function [36, 177, 276,277,278]. This function is called the "error" or the "loss", and encodes the "goal" of the network. The gradient indicates what the best possible local change of weights is for improving performance. This situation is distinguished from supervised learning by the fact that the gradient must be estimated based on scalar evaluative feedback (e.g., reward outcomes rather than specific information about what should have been produced). The behavior being optimized is referred to as a policy, and in a neural network this policy is a function of synaptic strengths [36]. Mathematically, for a set of presynaptic neurons connected to a set of post-synaptic neurons the gradient is the average of a Hebbian three-factor update involving the pre-synaptic input, the post-synaptic output, and a reward-prediction error [177, 180]. The classical algorithm demonstrating this is known as the REINFORCE algorithm (Fig. 3C) [177], and it forms the basis for a number of works that have examined reinforcement learning via synaptic plasticity since (e.g. [110, 116, 212, 276,277,278,279,280,281,282,283,284,285,286,287,288,289]).
When a network’s policy gradient is the expected value of a three-factor rule, and in particular has a scalar modulatory term, it is natural to interpret as a prescription for metaplasticity. Modulation acts to turn Hebbian plasticity up or down, on or off, or to invert it depending on feedback from the environment. From the perspective of a local network, "the environment" can include neuromodulatory signals indicating arousal, attention, surprise, or reward, for example. This is a key connection, because the modulatory term in the RL formulation manipulates the "error" that the network is trying to minimize, or "reward" being maximized. (Mathematically speaking, we are informally considering the Helmholtz-Hodge decomposition of the flow generated by a plasticity rule, with modulation defining a transformation of the terms.) By implication, converging manipulation of intracellular calcium signalling pathways, which themselves control LTP and LTD, can be intuitively thought of as defining network level reinforcement learning problems. The "error" or "reward" in these problems are not necessarily error or reward from the standpoint of the organism, but are arbitrary function evaluations related to the implicitly defined "purpose" of the local network (although one expects consistency conditions to complicate this intuition). The minimization steps used to solve these problems are selective applications of the same Hebbian updates that would otherwise lead to feature detectors, associative memory, sequential dynamics, etc. As a result, such algorithms operate in much the same way, with the caveats that unrewarding situations are ignored, the "feature detector" or "associative memory" seeking updates occur conditional on positive reward, and negative reward moves networks away from such associations. This informal account has technical caveats, but describes basic REINFORCE algorithms and more recent work on surprise-modulated plasticity accurately [177, 290, 291]. (Important details and more information on this topic can also be found in [177, 259, 276, 278, 279, 292,293,294,295,296].) Before illustrating these functional ideas in a broader biological context, we pause to discuss one of the critical connections with calcium dynamics.
Considering metaplasticity as reinforcement learning hinges on the capacity for neurons to distinguish between inputs requiring different responses [36, 177, 297]. While many mechanisms might allow this, dendritic signal integration is well positioned to do so [297, 298]. Both active and passive properties of dendrites control plasticity, and dendrites support diverse electrical signals including the generation of Ca2+ spikes, Na+ spikes, and "plateau potentials" [299,300,301]. Furthermore, calcium signals can be segregated between dendrites and spines [302,303,304], GABAergic interneurons can selectively shape dendritic branch currents [305, 306], and back-propagating action potentials can be differentially attenuated on the basis of morphology or GABA-ergic input [4, 307]. These mechanisms collectively allow complex control of local calcium via VGCCs and NMDARs. In one highly relevant study, specifically addressing stimulus disambiguation, branch-specific Ca2+ spikes related to two different tasks were shown to be gated by somatostatin positive interneurons (SOMs) on the apical tufts of L5 pyramidals in mouse motor cortex [308]. Behavioral learning and synaptic plasticity were both found to be causally related to this segregated activity, with interference between tasks arising when SOM-based separation of signals was disrupted [308]. Moreover, reactivation during sleep specifically reinforced such branch-specific localization of plasticity, in support of learning [309]. Elsewhere, spatially controlled dendritic plasticity has been shown to support functional linking of memories, for example [310], supporting the hypothesis that similar mechanisms may be at play broadly. While we cannot expand these points in further detail here, they form a key connection between biology and theory.
Summary of metaplasticity and network computation
To recap, basic theories of unconditional Hebbian plasticity qualitatively account for various phenomena. They predict that feed-forward neural pathways, such as thalamo-cortical connections, should develop feature detectors. Local recurrence, as seen between L4 or L2/3 principal neurons, should enhance bidirectional connectivity among similarly tuned neurons, insofar as plasticity reflects symmetric Hebbian rules. To the extent asymmetric rules like classical STDP govern recurrent change, synapses are expected to become more asymmetric. In the former case, recurrent connectivity should produce stable dynamics, whereas in the latter, recurrence should produce sequence-like activity. These types of dynamics have been used to model memory, central pattern generation, and directional association. The implications of non-classical STDP rules, such as those using non-traditional Volterra kernels, are less well understood. When any of these processes are subject to metaplasticity, they are expected to retain some of their fundamental features, as well as gaining new ones.
Theoretical work on modulated plasticity indicates that it can accomplish several things. Making the unconditional forms of plasticity just discussed conditional in an all or none way should produce essentially unsupervised results (feature extractors, Hopfield dynamics, synfire chains), but based only on that subset of the data for which plasticity is applied. More complex modulation by third factors can convert Hebbian forms of plasticity into reinforcement learning algorithms, rather than unsupervised ones, which navigate down gradients of error functions that are implicitly defined by the modulatory inputs and the details of the plasticity rule in question. Lastly, fine grained dendritic calcium dynamics appear critical for generating targeted plasticity that can make use of these theoretical possibilities.
Architectural specialization and metaplasticity
To this point, we have reviewed mechanisms for controlling plasticity and the local network properties they should be related to. But the function of plasticity likely depends on the networks and brain regions in which it occurs. A straightforward hypothesis is then that network specialization and divisions of labor across brain regions determine the implicit functional objectives discussed above, and that plasticity is controlled (via Ca2+ etc) to attain these. For example, predictive coding theories hypothesize that the purpose of sensory systems is largely to transmit unexpected sensory information for further processing [311,312,313]. In this case "minimizing sensory prediction error" might define the relevant optimization problem that plasticity is used to solve. Acetylcholine and norepinephrine, which are related to attention, arousal, and orienting to external stimuli, widely modulate sensory plasticity and may tailor it to solving this problem. In regions more closely dedicated to controlling interactions with the world, behaving in reward-maximizing ways may be the main goal [314]. The cortico-basal-ganglia-thalamic system, which plays a prominent role in action selection and reinforcement learning, appears to support this, and cortico-striatal synaptic plasticity in particular is strongly modulated by dopaminergic reward prediction errors. As we discuss below, motor-associated cortex may operate similarly. Rather than merely being passive (if specialized) substrates for learning, however, large scale networks also route information. This changes the activity observed by plasticity mechanisms, implicitly conditioning them further. Moreover, routing is also an effect of learning, especially at "high leverage" connections, as between cortical and subcortical areas. We review examples of these ideas along with neuromodulation-as-optimization here.
Arousal, surprise, and attention modulate sensory plasticity
A well supported hypothesis regarding ACh and NE holds that they cooperatively tune sensory cortex to adaptively improve sensory processing [315,316,317]. Behaviorally, both acetylcholine and norepinephrine are associated with arousal, and norepinephrine is also closely associated with orienting behavior [318,319,320]. Acetylcholine, furthermore, is associated with directed attention [176, 321,322,323,324]. Cortically, the need for tuning may reflect configuration trade offs, such as between ideal cortical states for performing detection versus discrimination [325,326,327]. Of these, the ideal state for sensory detection appears to be characterized by a number of coordinated changes, including modest network depolarization, diminished endogenous LFP fluctuations, decorrelated activity across neurons, and middling cortical response amplitudes to sensory stimuli [328,329,330,331,332]. Behavioral correlates of this state include alertness, attentiveness, wakefulness, and dilated pupils, which generally co-occur with increased task accuracy, decreased response biases, lower response times and reduced response variability [328,329,330,331,332]. Correlative and causal evidence both suggest that rapid fluctuations in acetylcholine and norepinephrine drive the tuning process producing all of these outcomes, with some evidence suggesting that NE may relatively reliably precede ACh activity as well [328,329,330,331,332].
Both NE and ACh exert local network effects through direct neuromodulation as well as indirectly through modified network inputs, as noted above. Acetylcholine projections appear to be fairly targeted, in the sense that different basal forebrain nuclei preferentially innervate different areas, whereas norepinephrine projections appear more uniform [333, 334]. Locally, the direct effects of Ach can be further subdivided into ionotropic and metabotropic components, since nicotinic acetylcholine receptors (nAChRs) are excitatory ligand gated ion channels. These local AChR impacts are often coupled with attentional recruitment of feedback projections between cortical areas [335,336,337]. As for norepinephrine, ionotropic receptors have not been reported, but NE can exert ionotropic-like effects through β2-adrenergic coupling to Cav1.2 channels and modifications of K+, H-type, and A-type currents, for example [142]. Indirect effects also arise from the coupling of locus coeruleus activity (the NE projection nucleus) to basolateral amygdala neurons with widespread projections, particularly to frontal cortex and hippocampus [15, 338, 339].
Of the "optimal cortical state" effects noted above, acetylcholine is most closely linked to desynchronizing network activity. In sensory circuits, it acts locally to depolarize vasoactive intestinal peptide positive interneurons (VIPs) in L2/3 and L5a via nAChRs [340,341,342,343,344]. These inhibit somatostatin positive interneurons (SOMs) and a subset of parvalbumin positive interneurons (PVs), resulting in disinhibition of principal cells [340,341,342,343,344]. Simultaneously, mAChRs and nAChRs upregulate excitability and activity in subsets of L2/3 and L5a SOMs that are not inhibited by VIPs, as well as the majority of SOMs in L5 [345, 346]. These SOMs target PVs and PYs, and appear to have primarily disinhibitory effects on PYs [345, 346]. In concert with these actions, mAChRs appear to upregulate the excitability of local pyramidal cells and reduce their spike-frequency adaptation [347, 348]. Which pieces of this are necessary and which are sufficient for decorrelating local networks is not entirely clear; both VIP and SOM targeting studies have independently reported sufficiency, but joint engagement certainly appears adequate.
The functional roles of norepinephrine in modulating perceptual processing are less clear. A diverse array of excitatory, inhibitory, and modulatory effects have long been reported, reflecting a difficult to determine internal logic [320, 349, 350]. In local networks, the primary differential effects of LC activity appear to be modifying signal-to-noise ratios, neural gain, neural tuning profiles, and thalamocortical information transmission [350,351,352,353]. A major source of these changes, at least in rodent somatosensory cortex, appears to be modified interactions between the ventral posteromedial nucleus of the thalamus (VPm) and the local thalamo-reticular nucleus (TRN) [351]. Specifically, increases in LC activity were found to drive TRN-VPm interactions via α-adrenergic receptor and T-type calcium currents, biasing thalamic relay of trigeminal ganglion information towards tonic firing rather than bursting. This caused a significant increase in decodable stimulus information conserved by thalamic responses to trigeminal ones [351]. These findings are consistent with previous work showing similar shifts of thalamo-cortical activity away from bursting and towards spiking with noradrenergic activation [354, 355]. More broadly, they appear consistent with the idea of systematically modifying neural gains to improve sensory responsiveness [350]. As such, whereas direct NE impacts on cortical state (via local adrenergic receptors) continue to be somewhat murky, NE may significantly increase perceptual information transmission.
Why should sensory plasticity be modulated by ACh and NE, given these observations? One possibility is that the various changes discussed above maximize the fidelity of sensory responses at precisely the time when feature processing should be most plastic. Salient, arousing stimuli that animals attend to are also those they learn about most quickly [356], and this learning should probably be based on the most highly resolved percepts possible. This information-maximizing approach to plasticity, whereby an organism is trying to extract as much information as possible from important sensory inputs, can be implemented by neuromodulated Hebbian rules based on surprise, in fact [290, 291]. Such rules take the implicit function to be minimized by REINFORCE-like RL algorithms, as discussed above, to be (Bayesian) sensory surprise, which has also been considered in the context of cholinergic modulation by others [357]. These surprise-modulated plasticity rules push neurons to extract independent components of their inputs (per ICA), which have had a long history of consideration in theoretical work on sensory processing [358]. Isomura and Toyoizumi were the first to show how synapse-local information could be used to generate this plasticity however, and the results are of course directly interpretable as feature extractors, per our earlier discussion. Such an account would be compatible with the observation that both high concentrations of NE and moderate concentrations of NE, coupled with ACh, gated the sensory plasticity we discussed above, for example [135]. This could be interpreted as promoting general responsiveness (of plasticity) to highly arousing conditions on the one hand, and targeted responsiveness to alert directed attention on the other.
Dopamine based reinforcement learning adapts behavior
Reward prediction error based modulation plays an especially plausible role in instantiating reinforcement learning algorithms at synapses onto neurons controlling behavioral output [36, 177]. RL algorithms maximize reward attainment by using RPEs to improve action selection. Given the RPE theory of dopamine, this would suggest that plasticity in the striatum, the motor cortex, and the cerebellum, along with pyramidal tract neurons across cortex, should be especially likely to exhibit dopaminergic modulation, because each impacts motor behavior fairly directly. Striatum and frontal cortex, including motor cortex, express high densities of DARs, as noted above, and both cortico-striatal synapses and L5 pyramidal neurons in mPFC have been shown to exhibit dopamine dependent plasticity [13, 114, 130, 133]. DAR expression in the cerebellum is established in some areas, as is expression in L5 throughout cortex broadly, but these facts have received less attention.
The cortico-striatal dopamine modulation discussed above has been integrated into neural network models of reinforcement learning [110, 280]. A major feature of cortico-striatal learning that distinguishes it from the type of simple policy gradient algorithms discussed so far (such as REINFORCE) is that the D1-MSN pathway and the D2-MSN pathway both operate as reinforcement-learners. Furthermore, the extent of learning and the extent to which each pathway has an impact on behavior is also modulated by dopamine (Fig. 4c–e). The Opponent Actor Learning (OpAL) model [287], which summarizes the computations of such networks, is an "actor-critic" model, meaning it represents behavioral policies and assessments of how valuable states of the world are separately [36]. The ventral striatum (and associated cortical and subcortical areas) are hypothesized to track information about how rewarding states of the world are, and these are compared with the results of behavior to generate reward prediction errors (Fig. 4d). Cortico-striatal connections in the D1-MSN and D2-MSN pathways determine action selection. Notably, while these pathways are classically thought to have opposite effects on behavior, the incorporation of neuromodulated, activity-dependent plasticity rules renders them non-redundant. Indeed, plasticity rules that mimic those described above in vitro [114] give rise to specialized representations, whereby the D1 MSNs discriminate between rewarding outcomes, while the D2 MSNs specialize in developing a high resolution policy for avoiding poor ones [287]. Many studies confirm the necessity and sufficiency of these opponent pathways (and synaptic transmission therein) for learning from positive and negative reward outcomes respectively (e.g., [115, 359, 360]). Intracellularly, they implicate PKA, cAMP and DARPP-32 in reinforcement learning [361], and genetic variation in such signaling predicts behavioral learning (including in humans, reviewed in [362]).

a Striatal medium spiny neuron plasticity and excitability depend on DA, ACh, adenosine, and NMDARs. Balance between the two pathways is mediated by DA. Figure follows Shen et al. 2008. b Cortico-basal-ganglia loops connect the cortex to the striatum recurrently and hierarchically. Figure follows Obeso et al 2014. c Internal detail of a single loop, showing dual pathways from striatal D1 and D2-expressing neurons to different output structures along the direct and indirect pathways. D1 MSNs largely reside in the "Go" pathway, D2 MSNs in the "NoGo" pathway. GP denotes globus pallidus. d The Opponent Actor Learning model, which captures cortico-striatal contributions to reinforcement learning computationally. See text for details. e When dopamine is high, the biological details of the model recapitulate empirically observed enhancements in learning to pick the best among good options. When DA is low, the model recapitulates enhanced ability to avoid bad options. These effects arise from the Hebbian reward prediction error modulated strengthening of Go and No-Go pathway MSNs. ΔG = GE ΔN = NE E f Pathological feedback of low DA on enhanced No-Go learning.
Interestingly, DA-based plasticity in MSNs is further gated by ACh, which is transmitted by tonically active striatal interneurons (TANs) [117, 118]. In theoretical models, this TAN gating of RPEs allows striatal learning rates to be adapted online according to uncertainty and reward volatility, a form of metaplasticity that resembles Bayesian learning [116]. Moreover, the plasticity induced by RPEs onto a striatal medium spiny neuron is further tuned by the time delays between glutamatergic and dopaminergic activity at a given spine [13]. As such, spatiotemporal dynamics of dopamine signaling across the striatum in the form of traveling waves can support a form of credit assignment (meaning appropriately targeted plasticity) to the underlying striatal region most relevant for the current behavioral context [113]. Finally, aside from the evidence that opponent D1/D2 pathways mediate learning, recent theoretical work suggests that this metaplasticity scheme is adaptive and provides a lever by which online ("tonic") dopamine levels can rapidly re-weight the contributions of D1 or D2 MSNs to action selection, depending on which population is more well suited to the task environment [363].
Finally, outside of striatal circuits, the reward prediction error theory of dopamine may also accord with the hypothesis that frontal cortex is an evolutionary expansion (and generalization) of motor cortex [364, 365]. From this perspective, DARs could be expected to be widespread frontally (as they are) because learning to take rewarding actions is the typical goal of reward-based reinforcement learning. Cortical L5 generally contains both intratelencephalic (IT) and pyramidal tract (PT) neurons, with the former projecting to cortex and striatum, and the latter projecting to thalamic and lower motor regions [366]. Both classes of neuron are poorly understood in general, but the L5 pyramidal tract of motor cortex disynaptically controls voluntary muscle contraction [366]. A simple hypothesis is therefore that DA-ergic gating of L2/3 to L5 synapses potentiates cortical motor pathways involved in generating subsequent unpredicted reward. This may be in line with observations that dopaminergic innervation of forelimb associated cortex is substantially stronger than that related to hindlimbs in rodents, for example [367], and potentially with dopamine as a requirement for motor learning generally in M1 [368, 369]. The fact that DAR mRNA has been found in corticocortical, corticothalamic, and corticostriatal neurons, but not corticospinal or corticopontine ones complicates this idea however [370].
Metaplasticity via activity routing within and across networks
One of the major forms of specialization in brains is selective routing of information, which we take as our last example of an architectural context for metaplasticity. Routing can also be considered a form of metaplasticity itself, in fact, because it determines which neuronal populations are active and the information they receive, which conditions plasticity and determines what populations can learn about, when, and how. The hippocampus and amygdala route activity locally to this effect, through inhibitory competition and changes in intrinsic excitability over time. This appears to link memories in each, and in the hippocampus it arbitrates a trade-off between pattern separation and pattern completion. Corticostriatal circuits, by contrast, control inter-area routing. Using task context to gate activity to and from prefrontal populations, these circuits can support attention and can condition learning in motor areas. We address each of these points, in turn.
Amygdala and memory allocation
In the amygdala, intrinsic cellular excitability determines which neurons participate in memory encoding [371,372,373,374,375,376,377,378,379]. When fear related activity is initiated in the lateral amygdala, more excitable cells out-compete their neighbors via lateral inhibition, which gates plasticity encoding the relevant memory [373, 378, 380, 381]. Both microscopic and macroscopic processes determine which cells display increased excitability. Intracellularly, cAMP responsive element binding protein (CREB), a downstream target of calcium entry and phosphorlyation by PKA [382, 383], determines intrinsic excitability, which drifts over time [378], depends on recent activity [15, 374, 382, 384], and appears to be mediated by changes in voltage gated sodium and potassium channels [372, 373, 383, 385]. Evidence from several circuits suggests that increased CREB activity increases depolarizing sodium currents at rest, while also decreasing hyperpolarizing potassium currents [372] and reducing spiking adaptation and after-hyperpolarization [373, 385]. These changes collectively enhance neural responsiveness to ongoing inputs. Systemically, whether or not a given fear memory is activated and learned about also depends on input from medial prefrontal cortex, for example, which can support context-dependent "safety signals" mediating extinction via new learning [386, 387].
Hippocampus, pattern separation and pattern completion
In the hippocampus, a long history of research has assessed the question of how activity should be allocated to produce useful plasticity [35, 198]. Memories (or indices for them) are generally believed to be stored in recurrent connections that can be used to complete input patterns or recall associations on the basis of partial information, according to Hopfield like dynamics [15]. Intuitively, a trade-off occurs between storing representations with significant overlap, thereby encoding semantic content in the joint activity of many neurons, and having representations with little overlap and hence little interference. The former case, which can be achieved with relatively dense network interconnectivity, has been considered good for learning generally useful, abstracted forms of information, whereas the latter, which can be achieved with relatively sparse, strong connectivity, has been considered a good substrate for flashbulb or episodic memory [198]. This "complementary learning systems" perspective historically suggested a cortical substrate for the former and a hippocampal substrate for the latter, but more recent work has suggested an internal division of labor between the EC-CA1 "monosynaptic" pathway and the EC-DG-CA3-CA1 "trisynaptic" pathway could accomplish a similar goal [35]. Specifically, several studies found that the trisynaptic pathway is required for fast, single-exposure learning whereas the monosynaptic pathway was required for incremental learning [388,389,390,391]. Pattern separation was strongly dependent on EC-DG-CA3 interactions [388,389,390], and required NMDAR activation in DG in particular [389]. These mechanisms presumably interact with excitability based changes (e.g. [385, 392,393,394,395]) which, mirroring the amygdala work just discussed, have been found to mediate temporal linking of memories via ensemble overlap in CA1 as well [395, 396].
Corticostriatal circuits and task-rule gating
As a final example, we return again to cortico-basal-ganglia loops. Disinhibitory pathways from the striatum to the substantia nigra and thalamus are thought to support gating of information into and out of distinct sub-populations within frontal cortex [397,398,399]. These pathways are situated in PFC-BG loops and are organized hierarchically; anterior loops appear to gate abstract task information that contextualizes activity and attention in posterior regions, and to modulate response selection in motor loops [286]. This architecture allows for flexible action selection, inference about "latent states", and transfer of learning across tasks [400]. In particular, frontal and posterior regions can maintain information recurrently in distinct neural sub-populations, which can be "output gated" as needed [286, 397, 401, 402]. Such a scheme theoretically allows the PFC-BG-thalamus system to route information for specific uses, to learn and switch between tasks, to reuse old learning in new contexts, and to flexibly combine information across tasks [397, 403, 404]. By gating attractor dynamics within prefrontal cortex, PFC-BG loops also facilitate behavioral plasticity on faster time scales than synaptic plasticity may generally afford, as presumably does all ongoing activity.
From the standpoint of metaplasticity, each of these gating operations is a conditioning dependency. The hierarchical selection of which activity to propagate and when conditions reward-modulated reinforcement learning at cortico-striatal synapses. Higher level task and goal-relevant information gated into PFC can then influence the representations that are learned downstream, reducing interference that would otherwise arise between tasks [400]. Such gating processes can also control the degree to which prefrontal regions contribute to learning compositional or hierarchical rules, or to focusing on conjunctive coding of multiple features instead [286]. Humans can flexibly switch between these strategies according to task demands, and doing so recruits corticostriatal circuits [405,406,407]. Finally, to the extent that unsupervised plasticity is ongoing in any regions involved, routing will condition the activity statistics locally. These points suggest that the balance of these forms of metaplasticity - i.e. conditional unsupervised and conditional reinforcement learning - is likely to play an important, finely calibrated role in the proper function of such systems.
Summary of architectural relations to metaplasticity
In this section we’ve discussed several examples of circuit specialization and network architecture that we expect to interact with metaplasticity. As one set of examples, we considered how the general functional roles of neuromodulators, such as dopamine, acetylcholine, and norepinephrine, may align with their impacts on plasticity. Acetylcholine and norepinephrine, in their capacities to tune sensory cortex towards robust processing of the external world, suggest that they may be gating plasticity in sensory areas so that it occurs "when it most counts" and "when the circuit has maximal fidelity." Such a function would also align with information maximizing plasticity, facilitated by surprise-based neuromodulation. As a second example, we discussed dopaminergic RPE signalling and neuromodulated plasticity at cortico-striatal synapses, noting that DAergic modulation may play a similar role at L5 pyramidal tract neurons. In closing we discussed how activity routing, one of the most important architectural specializations in brains, can be considered a form of metaplasticity, which it also contextualizes. We illustrated this in three circuits. First, we noted that neurons recruited in lateral amygdala to encode memories are selected on the basis of intrinsic excitability and inhibitory competition, thereby making a seemingly unsupervised, associative form of learning conditional. Next, we noted that the monosynaptic and trisynaptic pathways in the hippocampus route information locally to control functional properties like pattern separation and pattern completion, and to control fast vs incremental aspects of learning. Finally, we discussed the capacity of cortico-striatal gating mechanisms, tuned by DAergic reinforcement learning, to mediate downstream forms of learning across fronto-posterior networks. This is accomplished by directing working memory and attention, and facilitates proper attribution of improved behavioral outcomes to specific internal processes.
Pathology from a scale-integrative metaplasticity perspective
We conclude our review by considering how the ideas discussed above may be related to several pathologies. Many pathologies perturb normal function across multiple scales, so scale-spanning aspects of metaplasticity may help reconcile their diverse results. This is inherently speculative, presently, because most psychiatric illnesses are poorly understood at any network scale, much less across them. Some diseases will be more amenable to metaplasticity-based analyses than others however. In this regard, autism spectrum disorders, with their noted impacts on learning, synaptic plasticity, and large-scale brain network integration may be relatively approachable. Likewise, because Parkinson’s disease involves well established changes in metaplasticity, which are recapitulated as side-effects of anti-psychotic treatments for Schizophrenia, this too should be a reasonable application area.
Negative symptoms in schizophrenia may relate to Parkinsonian metaplasticity
The proximal cause of Parkinsonian symptoms is loss of midbrain dopaminergic projection neurons in the substantia nigra [408, 409]. This chronically lowers DA concentrations in the striatum, impacting excitability and biasing the opponent learning processes instantiated across the D1 and D2 MSN populations as described above. Recall that functionally, the D1 pathway is preferentially recruited to facilitate responses producing rewarding outcomes, whereas the D2 pathway is recruited to inhibit responses resulting in aversive ones. When DA concentrations are lowered via drug effects or Parkinson’s, learning from negative RPEs is exaggerated, and even skills that have been learned under normal dopaminergic conditions become subject to degradation through practice [409, 410]. This is a result of enhanced, aberrant plasticity in the D2 pathway [410], which has direct relevance for understanding the emergence of motor symptoms induced by antipsychotics (D2 blockers) in schizophrenia [411]. Moreover, this aberrant plasticity can be disentangled from the proximal effects of DA depletion (or D2 blockade), on motor performance. Indeed, when Beeler et al administered an adenosine antagonist known to block plasticity in the D2 pathway [114], aberrant motor learning was prevented in dopamine depleted mice, despite the preservation of acutely impaired performance induced by D2 blockade. An analogue of motor symptom progression via the enhanced impacts of negative RPEs may also be responsible for progressing motivational avolition in schizophrenia [411]. For example, negative symptoms of schizophrenia are associated with reduced motivation to exert physical effort, an effect that is most prominent in those who have taken antipsychotics with high D2 affinity [412]. These effects accord with preclinical work showing causal effects of striatal D2 blockade on motivation to pursue effortful behaviors [413]. Thus, therapeutically, this account might also provide an avenue for preventing the progression of motor and motivational symptoms in both PD and as a result of antipsychotics in schizophrenia.
Autism and pathological LTD in Fragile X
Cognitively, it has been hypothesized that autism spectrum disorders (ASDs) may be characterized by an inability to extract and generalize information [414]. Various alterations in the control of plasticity we’ve reviewed above might produce these developmental learning impairments. For example, alterations in metaplasticity at the synaptic level via changes in calcium signalling would impact, among other functions, the dynamic ability to regulate floating thresholds in learning models. This would manifest in changes in the ability to extract statistical structure in independent, compositional ways (as with PCA, ICA, or BCM). Systemically, dysregulated excitatory-inhibitory balance might impact which populations learne about which inputs, and thus contextual use of memory [415]. Similarly, disrupted corticostriatal learning, which is occurs in ASD [416], could impair the ability to conditionally gate hierarchical behavioral rules, which, as reviewed earlier, could lead to failures in generalization, inference and compositionality that are also well documented in ASD [414, 417]. Empirical and computational work also suggests that healthy people can learn to flexibly represent both conjunctive and compositional rule structures and generalize based on either depending on the statistical structure of the environment [406, 418]. Such "meta generalization" would require not only adequate learning and metaplasticity within individual brain systems, but learning to adaptively switch between them. Because these points are fairly abstract, we now review some of the more concrete evidence for altered plasticity at the cellular level, then consider further systemic effects.
Mechanistically, ASDs appear to be diverse and multi-factorial [419, 420] (but see [421]). A number of autism risk genes are involved in regulating synaptic plasticity [422], but several, at minimum, cause well defined monogenic syndromes, with high phenotypic penetrance of autism. Of these, fragile X syndrome (FX) is the largest single such cause, and is one of the most well understood [423]. Individuals with FX frequently suffer from seizures, sensory hypersensitivity, and intellectual disability, all of which are also common among patients with autism more generally [424].
Fragile X results from a loss-of-function mutation in the FMR1 (Fragile X Mental Retardation 1) gene, which has a copy-number-variable sequence that blocks transcription when 200 or more repeats are present [425]. The transcript of FMR1, FMRP, itself regulates mRNA transcription in dendrites and spines, and is involved in the "consolidation" phase of synaptic plasticity. This occurs approximately 30 minutes after plasticity induction, and is regulated by protein synthesis from local mRNA [15]. FMRP transcription is, among other things, a direct consequence of postsynaptic mGluR5 activation (metabotropic glutamate receptor, a GPCR) [426], which mediates an NMDAR-dissociable form of LTD [427, 428]. This form of LTD requires calcium influx from T-type VGCCs and PKC activity [427]. When FMRP is absent, mGluR5 induced LTD is enhanced [429], which suggests an "mGluR theory of fragile X syndrome" [430]. In animal models, many of the physiological and behavioral effects of fragile X can, in fact, be reversed with treatments inhibiting mGluR5 activity [431] (also see [432, 433]), but translation to human patients has produced mixed effects. Perhaps unsurprisingly, ongoing work has shown that FMRP is involved in an enormous number of processes, many of which involve other plasticity related proteins [423, 434]. Fragile X, therefore, is itself diverse in its impacts, and significant heterogeneity likely occurs between individuals in the penetrance of any particular FMRP-related perturbation.
Nonetheless, a number of cellular and network changes have been observed which overlap with other autism phenotypes as well. These include altered excitatory-inhibitory balance (more excitable principal cells) [435] and widespread spine aberrations [436,437,438]. Regarding the latter, FX results in increased numbers of immature spines, decreased numbers of mature ones, and increased localization of spines to the distal dendrites of principal cells [439,440,441,442,443,444]. Various changes in regulation of spine morphology related proteins, such as PSD-95 (a critical regulator of post-synaptic AMPAR densities) have also been described [445, 446], as have delayed developmental reversal of GABAA post-synaptic potentials [447] and reduced GABAA receptor densities throughout the brain [448]. Many of these changes may in fact be homeostatic attempts to compensate for pathologically enhanced synaptic LTD [449], and treatment of FMR1 knockout mice with mGluR antagonists rescues many of the spine abnormalities just discussed [449, 450]. Treatments and their rescued processes are reviewed in [432].
If the FX phenotype was primarily a result of runaway LTD, what might our understanding of metaplasticity imply as a result? Presumably this would depend strongly on the balance between developmental structuring of connectivity and use-based refinement in any particular system, and on the degree to which the former of these is spared by FMRP loss. Nonetheless, excessive excitability might combine with surviving capacities for potentiation to degrade structured connectivity, especially in areas with persistently expressed unsupervised plasticity. This could lead to structurally exacerbated noisy network activity, and especially disordered processing in systems requiring finely calibrated spike timing, such as auditory cortex and potentially fine-motor coordination. Sensory systems may fail to self-organize (i.e., develop differentiated, cooperative feature extractors) in robust ways, further exaggerating network SNR problems.
Hyper-excitability and the need for LTP induction to overcome a persistent LTD bias might both suggest that plasticity across areas would shift towards processing highly arousing and aversive experiences, and towards generalizing them. Specifically, increased excitation mediated participation in amygdalar engrams and preferential hippocampal encoding of memories associated with high norepinephrine concentrations might result in preferential memory for negative experience. This could produce a feedback loop, shifting reinforcement learning systems on the basis of tonically reduced dopamine towards increased avoidance behavior, in concert with persistent negative reward prediction errors arising from a combination of poor predictive capacity and over-generalization of fear. Such an account could explain the increased aversiveness of sensory stimuli, given the apparently normal total evoked responses observed in FX, for example. Widespread noise increases could lead to reduced functional connectivity and inter-area signal fidelity, which could reduce the capacity of routing systems such as the cortico-basal-ganglia loops to coordinate changes across areas. This could thereby degrade the types of compositional, hierarchical, and generalizable learning these systems otherwise facilitate. While these hypotheses are highly speculative, they indicate how the ideas about metaplasticity we have reviewed above might be applied to ASDs generally.
Discussion
We conclude by briefly noting the connections discussed, in the service of a holistic perspective on metaplasticity. This review has examined research on plasticity at several scales, with an emphasis on the connections between them. It covered established cellular and molecular processes involved in regulating plasticity, network properties related to Hebbian change, theoretical features of modulated plasticity, and how these elements might be coordinated in specialized functional circuits. These indicated how metaplasticity can orchestrate scale-spanning functional adaptation. Specifically, combinations of calcium-pathway manipulations and activity routing (in which we include excitability changes) serve to specify which synapses, in which places, learn about which types of inputs. They do so by controlling when and where plasticity takes place, and to what extent. The statistics of these manipulations can implicitly define "error functions", which modulated forms of Hebbian plasticity can minimize. Such error functions should be empirically determined, but phenomena that define them can suggest rough hypotheses. Observations across scales can then be checked for consistency under the logic of any such synthesis.
The specific sections at each scale highlighted both connections that could be made presently and phenomena that appear relevant in expanding these connections. At the cellular-molecular scale, we discussed Ca2+ currents, closely related CaMKII, PKA, and PKC pathways, and some of the impacts that VGCCs, CP-AMPARs, and GPCRs, such as neuromodulatory receptors, have on these pathways. This demonstrated ways that diverse biological processes converge intracellularly to regulate Hebbian LTP and LTD induction. At the network level, we reviewed the basic functions of Hebbian plasticity to produce feature detectors and cell ensembles with different manifestations of recurrent activity. Then we reviewed work on how metaplasticity can co-opt these synaptic dynamics to fulfill functional roles. We discussed several ways that neuromodulated plasticity, in particular, can be framed as performing this co-optation to facilitate specialized circuit properties, like maximizing information transmission and supporting behavioral reinforcement learning. We noted that these types of plasticity critically require some amount of stimulus selectivity, enhancing or suppressing responsiveness to different inputs, and that complex dendritic processes appear more than sufficient for this. Macroscopically, we reviewed cellular and neuromodulatory aspects of basal-ganglia loops, sensory circuits, and memory encoding, with an emphasis on how reinforcement learning, surprise-minimization, and local activity routing might guide plasticity. This indicated again how coordination of cellular, network, and systemic changes can converge to turn Hebbian changes into functional ones. Finally, we addressed how metaplasticity considerations might prove fruitful for understanding pathology, by considering aberrant Parkinsonian metaplasticity in medicated schizophrenia, and disruptions of statistical information processing in autism. Together, these points have also indicated a number of important directions for future research integrating plasticity across scales. Understanding when and where different forms of plasticity operate in concert or antagonism, and how tuning them online changes their ultimate effects, are two important such goals.
References
Sales EC, Heckman EL, Warren TL, Doe CQ. Regulation of subcellular dendritic synapse specificity by axon guidance cues. eLife. 2021;8:e43478. https://doi.org/10.7554/eLife.43478.
Jamann N, Dannehl D, Lehmann N, Wagener R, Thielemann C, Schultz C. et al. Sensory input drives rapid homeostatic scaling of the axon initial segment in mouse barrel cortex. Nat Commun. 2021;12:23. https://doi.org/10.1038/s41467-020-20232-x.
Arikkath J. Molecular mechanisms of dendrite morphogenesis. Front Cell Neurosci. 2012;6:61. https://doi.org/10.3389/fncel.2012.00061.
Froemke R, Letzkus J, Kampa B, Hang G, Stuart G. Dendritic synapse location and neocortical spike-timing-dependent plasticity. Front Synaptic Neurosci. 2010;2:29. https://doi.org/10.3389/fnsyn.2010.00029.
Iascone DM, Li Y, Sümbül U, Doron M, Chen H, Andreu V. et al. Whole-neuron synaptic mapping reveals spatially precise excitatory/inhibitory balance limiting dendritic and somatic spiking. Neuron. 2020;106:566–78. https://doi.org/10.1016/j.neuron.2020.02.015.
Tripodi M, Evers JF, Mauss A, Bate M, Landgraf M. Structural homeostasis: Compensatory adjustments of dendritic arbor geometry in response to variations of synaptic input. PLoS Biol. 2008;6:e260. https://doi.org/10.1371/journal.pbio.0060260.
Knott GW, Quairiaux C, Genoud C, Welker E. Formation of dendritic spines with GABAergic synapses induced by whisker stimulation in adult mice. Neuron. 2002;34:265–73. https://doi.org/10.1016/S0896-6273(02)00663-3.
Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M. Experience leaves a lasting structural trace in cortical circuits. Nature. 2009;457:313–7. https://doi.org/10.1038/nature07487.
Kalantzis G, Shouval HZ. Structural plasticity can produce metaplasticity. PLoS ONE. 2009;4:e8062. https://doi.org/10.1371/journal.pone.0008062.
Wickens JR. Synaptic plasticity in the basal ganglia. Behavioural Brain Res. 2009;199:119–28. https://doi.org/10.1016/j.bbr.2008.10.030.
Bourne JN, Harris KM. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 2011;21:354–73. https://doi.org/10.1002/hipo.20768.
O’Donnell C, Nolan MF, van Rossum MCW. Dendritic spine dynamics regulate the long-term stability of synaptic plasticity. J Neurosci. 2011;31:16142–56. https://doi.org/10.1523/JNEUROSCI.2520-11.2011.
Yagishita S, Hayashi-Takagi A, Ellis Davies GCR, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science. 2014;345:1616–20. https://doi.org/10.1126/science.1255514.
Humble J, Hiratsuka K, Kasai H, Toyoizumi T. Intrinsic spine dynamics are critical for recurrent network learning in models with and without autism spectrum disorder. Front Computational Neurosci. 2019;13:38. https://doi.org/10.3389/fncom.2019.00038.
Rudy J W. The neurobiology of learning and memory. 2nd ed. Sinauer Associates; Sunderland, Massachusetts; 2021.
Lindskog M, Kim M, Wikström MA, Blackwell KT, Kotaleski JH. Transient calcium and dopamine increase PKA activity and DARPP-32 phosphorylation. PLOS Computational Biol. 2006;2:e119. https://doi.org/10.1371/journal.pcbi.0020119.
Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–82. https://doi.org/10.1038/nrn3192.
Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: The last 25 years. Neuron. 2013;80:704–17. https://doi.org/10.1016/j.neuron.2013.10.025.
Mäki-Marttunen T, Iannella N, Edwards AG, Einevoll GT, Blackwell KT. A unified computational model for cortical post-synaptic plasticity. eLife. 2020;9:e55714. https://doi.org/10.7554/eLife.55714.
Purkey AM, Dell’Acqua ML. Phosphorylation-dependent regulation of Ca2+-permeable AMPA receptors during hippocampal synaptic plasticity. Front Synaptic Neurosci. 2020;12:8. https://doi.org/10.3389/fnsyn.2020.00008.
Wolf M, Tseng K-Y. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci. 2012;5:72. https://doi.org/10.3389/fnmol.2012.00072.
Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. 2018;98:466–81. https://doi.org/10.1016/j.neuron.2018.03.017.
Cull-Candy SG, Farrant M. Ca2+-permeable AMPA receptors and their auxiliary subunits in synaptic plasticity and disease. J Physiol. 2021;599:2655–71. https://doi.org/10.1113/JP279029.
McCulloch WS, Pitts W. A logical calculus of the ideas immanent in nervous activity. Bull Math biophysics. 1943;5:115–33.
Rosenblatt F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychological Rev. 1958;65:386–408. https://doi.org/10.1037/h0042519.
Widrow B, Lehr M. 30 years of adaptive neural networks: perceptron, madaline, and backpropagation. Proc IEEE. 1990;78:1415–42. https://doi.org/10.1109/5.58323.
Bishop CM, Nasrabadi NM. Pattern recognition and machine learning. Vol. 4; Springer: New York; 2006.
Goodfellow I, Bengio Y, Courville A. Deep learning. MIT press: Cambridge, Massachusetts; 2016.
Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks. In Advances in Neural Information Processing Systems, vol. 25 (Curran Associates, Inc.; 2012). https://papers.nips.cc/paper/2012/hash/c399862d3b9d6b76c8436e924a68c45b-Abstract.html.
Hopfield JJ. Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci USA. 1982;79:2554–8.
Fahlman SE, Hinton, GE, Sejnowski TJ. Massively parallel architectures for AI: METL, THISTLE, and boltzmann machines. In Proceedings of National Conference on AI. AAAI Press: Menlo Park, California 1983.
Ackley DH, Hinton GE, Sejnowski TJ. A learning algorithm for boltzmann machines. Cogn Sci. 1985;9:147–69. https://doi.org/10.1207/s15516709cog0901_7.
O’Reilly RC, Norman KA. Hippocampal and neocortical contributions to memory: Advances in the complementary learning systems framework. Trends Cogn Sci. 2002;6:505–10.
O’Reilly RC, Bhattacharyya R, Howard MD, Ketz N. Complementary learning systems. Cogn Sci. 2014;38:1229–48. https://doi.org/10.1111/j.1551-6709.2011.01214.x.
Schapiro AC, Turk-Browne NB, Botvinick MM, Norman KA. Complementary learning systems within the hippocampus: a neural network modelling approach to reconciling episodic memory with statistical learning. Philos Trans R Soc B: Biol Sci. 2017;372:20160049. https://doi.org/10.1098/rstb.2016.0049.
Sutton RS, Barto AG. Reinforcement Learning: An Introduction (The MIT Press, 2018), second edn. http://incompleteideas.net/book/the-book-2nd.html.
Badre D, Frank MJ, Moore CI. Interactionist neuroscience. Neuron. 2015;88:855–60. https://doi.org/10.1016/j.neuron.2015.10.021.
Sehgal M, Song C, Ehlers VL, Moyer JR. Learning to learn-intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol Learn Mem. 2013;105:186–99. https://doi.org/10.1016/j.nlm.2013.07.008.
Müller-Dahlhaus F, Ziemann U. Metaplasticity in human cortex. Neuroscientist. 2015;21:185–202. https://doi.org/10.1177/1073858414526645.
Farashahi S, Donahue CH, Khorsand P, Seo H, Lee D, Soltani A. Metaplasticity as a neural substrate for adaptive learning and choice under uncertainty. Neuron. 2017;94:401–14. https://doi.org/10.1016/j.neuron.2017.03.044.
Bliss TVP, Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56. https://doi.org/10.1113/jphysiol.1973.sp010273.
Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–9. https://doi.org/10.1038/361031a0.
Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF. Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science. 1993;260:1518–21. https://doi.org/10.1126/science.8502997.
Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–5. https://doi.org/10.1126/science.275.5297.213.
Bi G-Q, Poo M-M. Synaptic modifications in cultured hippocampal neurons: Dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–72. https://doi.org/10.1523/JNEUROSCI.18-24-10464.1998.
Thomas MJ, Watabe AM, Moody TD, Makhinson M, O’Dell TJ. Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci. 1998;18:7118–26. https://doi.org/10.1523/JNEUROSCI.18-18-07118.1998.
Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574:283–90. https://doi.org/10.1113/jphysiol.2006.111062.
Markram HA. history of spike-timing-dependent plasticity. Front Synap Neurosci. 2011;3. https://doi.org/10.3389/fnsyn.2011.00004.
Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–71. https://doi.org/10.1016/j.neuron.2012.08.001.
Edelmann E, Cepeda-Prado E, Franck M, Lichtenecker P, Brigadski T, Leßmann V. Theta burst firing recruits BDNF release and signaling in postsynaptic CA1 neurons in spike-timing-dependent LTP. Neuron. 2015;86:1041–54. https://doi.org/10.1016/j.neuron.2015.04.007.
Athalye VR, Santos FJ, Carmena JM, Costa RM. Evidence for a neural law of effect. Science. 2018;359:1024–9. https://doi.org/10.1126/science.aao6058.
Citri A, Malenka RC. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. https://doi.org/10.1038/sj.npp.1301559.
Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. https://doi.org/10.1523/JNEUROSCI.02-01-00032.1982.
Bear MF, Cooper LN, Ebner FF. A physiological basis for a theory of synapse modification. Science. 1987;237:42–8.
Cooper LN, Bear MF. The BCM theory of synapse modification at 30: interaction of theory with experiment. Nat Rev Neurosci. 2012;13:798–810. https://doi.org/10.1038/nrn3353.
Evans RC, Blackwell KT. Calcium: Amplitude, duration, or location? Biol Bull. 2015;228:75–83. https://doi.org/10.1086/BBLv228n1p75.
Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. https://doi.org/10.1113/jphysiol.1983.sp014478.
Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–21. https://doi.org/10.1038/305719a0.
MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–22. https://doi.org/10.1038/321519a0.
Ascher P, Nowak L. The role of divalent cations in the n-methyl-d-aspartate responses of mouse central neurones in culture. J Physiol. 1988;399:247–66. https://doi.org/10.1113/jphysiol.1988.sp017078.
Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. https://doi.org/10.1186/1756-6606-5-14.
Foncelle A, Mendes A, Jedrzejewska-Szmek J, Valtcheva S, Berry H, Blackwell KT, et al. Modulation of spike-timing dependent plasticity: Towards the inclusion of a third factor in computational models. Front Comput Neurosci. 2018;12. https://doi.org/10.3389/fncom.2018.00049.
Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M. et al. Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc Natl Acad Sci. 1998;95:7097–102. https://doi.org/10.1073/pnas.95.12.7097.
Mangiavacchi S, Wolf ME. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase a. J Neurochemistry. 2004;88:1261–71. https://doi.org/10.1046/j.1471-4159.2003.02248.x.
Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X. et al. Phosphorylation of AMPA receptors: Mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32:237–50. https://doi.org/10.1385/MN:32:3:237.
Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–73. https://doi.org/10.1038/nrn2867.
Greger IH, Watson JF, Cull-Candy SG. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron. 2017;94:713–30. https://doi.org/10.1016/j.neuron.2017.04.009.
Malenka RC, Bear MF. LTP and LTD: An embarrassment of riches. Neuron. 2004;44:5–21. https://doi.org/10.1016/j.neuron.2004.09.012.
Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Science’s STKE 2004;2004. https://doi.org/10.1126/stke.2552004re16.
Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M. et al. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–4. https://doi.org/10.1126/science.1096615.
Massey PV. Differential roles of NR2a and NR2b-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–8. https://doi.org/10.1523/JNEUROSCI.1697-04.2004.
Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. https://doi.org/10.1016/j.neuron.2005.08.034.
Philpot BD, Cho KK, Bear MF. Obligatory role of NR2a for metaplasticity in visual cortex. Neuron. 2007;53:495–502. https://doi.org/10.1016/j.neuron.2007.01.027.
Cho KKA, Khibnik L, Philpot BD, Bear MF. The ratio of NR2a/b NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc Natl Acad Sci. 2009;106:5377–82. https://doi.org/10.1073/pnas.0808104106.
Smith GB, Heynen AJ, Bear MF. Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex. Philos Trans R Soc B: Biol Sci. 2009;364:357–67. https://doi.org/10.1098/rstb.2008.0198.
Xu Z, Chen RQ, Gu QH, Yan JZ, Wang SH, Liu SY. et al. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2a/NR2b ratio. J Neurosci. 2009;29:8764–73. https://doi.org/10.1523/JNEUROSCI.1014-09.2009.
Lee M-C, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66:859–70. https://doi.org/10.1016/j.neuron.2010.05.015.
Evans RC, Morera-Herreras T, Cui Y, Du K, Sheehan T, Kotaleski JH. et al. The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLOS Computational Biol. 2012;8:e1002493. https://doi.org/10.1371/journal.pcbi.1002493.
Fong MF, Finnie PS, Kim T, Thomazeau A, Kaplan ES, Cooke SF. et al. Distinct laminar requirements for NMDA receptors in experience-dependent visual cortical plasticity. Cereb Cortex. 2020;30:2555–72. https://doi.org/10.1093/cercor/bhz260.
Strader CD, Fong TM, Tota MR, Underwood D, Dixon RA. Structure and function of g protein-coupled receptors. Annu Rev Biochem. 1994;63:101–32.
Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of g-protein-coupled receptors. Nature. 2009;459:356–63. https://doi.org/10.1038/nature08144.
Hell JW, Westenbroek RE, Warner C, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class c and class d l-type calcium channel txl subunits. J Cell Biol. 1993;123:14.
Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-methyl-d-aspartate receptor-induced proteolytic conversion of postsynaptic class c l-type calcium channels in hippocampal neurons. Proc Natl Acad Sci. 1996;93:3362–7. https://doi.org/10.1073/pnas.93.8.3362.
Berger SM, Bartsch D. The role of l-type voltage-gated calcium channels cav1.2 and cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014;357:463–76. https://doi.org/10.1007/s00441-014-1936-3.
Parajuli LK, Nakajima C, Kulik A, Matsui K, Schneider T, Shigemoto R. et al. Quantitative regional and ultrastructural localization of the cav2.3 subunit of r-type calcium channel in mouse brain. J Neurosci. 2012;32:13555–67. https://doi.org/10.1523/JNEUROSCI.1142-12.2012.
Feng Z, Glebov OO. Synaptic NMDA receptor signalling controls r-type calcium channel recruitment. Eur J Neurosci. 2021;54:4133–40. https://doi.org/10.1111/ejn.15250.
Huber KM, Mauk MD, Kelly PT. Distinct LTP induction mechanisms: contribution of NMDA receptors and voltage-dependent calcium channels. J Neurophysiol. 1995;73:270–9. https://doi.org/10.1152/jn.1995.73.1.270.
Freir DB, Herron CE. Inhibition of l-type voltage dependent calcium channels causes impairment of long-term potentiation in the hippocampal CA1 region in vivo. Brain Res. 2003;967:27–36. https://doi.org/10.1016/S0006-8993(02)04190-2.
Moosmang S. Role of hippocampal cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–92. https://doi.org/10.1523/JNEUROSCI.1531-05.2005.
Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves-Cintrón M. et al. Phosphorylation of ser1928 mediates the enhanced activity of the l-type Ca2+ channel Cav 1.2 by the β2 -adrenergic receptor in neurons. Sci Signal. 2017;10:eaaf9659. https://doi.org/10.1126/scisignal.aaf9659.
Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ. et al. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav 1.2. Science. 2001;293:98–101. https://doi.org/10.1126/science.293.5527.98.
Qian H, Matt L, Zhang M, Nguyen M, Patriarchi T, Koval OM. et al. β2-adrenergic receptor supports prolonged theta tetanus-induced LTP. J Neurophysiol. 2012;107:2703–12. https://doi.org/10.1152/jn.00374.2011.
Theis A-K, Rózsa B, Katona G, Schmitz D, Johenning FW. Voltage gated calcium channel activation by backpropagating action potentials downregulates NMDAR function. Front Cell Neurosci. 2018;12:109. https://doi.org/10.3389/fncel.2018.00109.
Shah M, Haylett DG. Ca2+ channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2554–61. https://doi.org/10.1152/jn.2000.83.5.2554.
Lima PA, Marrion NV. Mechanisms underlying activation of the slow AHP in rat hippocampal neurons. Brain Res. 2007;1150. https://doi.org/10.1016/j.brainres.2007.02.067.
McKinney BC, Sze W, Lee B, Murphy GG. Impaired long-term potentiation and enhanced neuronal excitability in the amygdala of CaV1.3 knockout mice. Neurobiol Learn Mem. 2009;92:519–28. https://doi.org/10.1016/j.nlm.2009.06.012.
Szucs A, Rátkai A, Schlett K, Huerta R. Frequency-dependent regulation of intrinsic excitability by voltage-activated membrane conductances, computational modeling and dynamic clamp. Eur J Neurosci. 2017;46:2429–44. https://doi.org/10.1111/ejn.13708.
Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ. et al. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–4. https://doi.org/10.1038/nn1678.
Yang Y, Wang X-b, Zhou Q. Perisynaptic GluR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc Natl Acad Sci. 2010;107:11999–2004. https://doi.org/10.1073/pnas.0913004107.
Jaafari N, Henley JM, Hanley JG. PICK1 mediates transient synaptic expression of GluA2-lacking AMPA receptors during glycine-induced AMPA receptor trafficking. J Neurosci. 2012;32:11618–30. https://doi.org/10.1523/JNEUROSCI.5068-11.2012.
Lamsa KP, Heeroma JH, Somogyi P, Rusakov DA, Kullmann DM. Anti-hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science. 2007;315:1262–6. https://doi.org/10.1126/science.1137450.
Camire O, Topolnik L. Dendritic calcium nonlinearities switch the direction of synaptic plasticity in fast-spiking interneurons. J Neurosci. 2014;34:3864–77. https://doi.org/10.1523/JNEUROSCI.2253-13.2014.
Lalanne T, Oyrer J, Farrant M, Sjöström PJ. Synapse-specific expression of calcium-permeable AMPA receptors in neocortical layer 5. J Physiol. 2018;594:837–61. https://doi.org/10.1113/JP271394.
Lalanne T, Oyrer J, Farrant M, Sjöström PJ. Synapse type-dependent expression of calcium-permeable AMPA receptors. Front Synaptic Neurosci. 2018;10:34. https://doi.org/10.3389/fnsyn.2018.00034.
Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–62. https://doi.org/10.1016/0896-6273(95)90049-7.
Donevan SD, Rogawski MA. Intracellular polyamines mediate inward rectification of ca(2+)-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc Natl Acad Sci USA. 1995;92:9298–302.
Vogels TP, Sprekeler H, Zenke F, Clopath C, Gerstner W. Inhibitory plasticity balances excitation and inhibition in sensory pathways and memory networks. Science. 2011;334:1569–73.
Hennequin G, Agnes EJ, Vogels TP. Inhibitory plasticity: Balance, control, and codependence. Annu Rev Neurosci. 2017;40:557–79. https://doi.org/10.1146/annurev-neuro-072116-031005.
Balleine BW, Liljeholm M, Ostlund SB. The integrative function of the basal ganglia in instrumental conditioning. Behav Brain Res. 2009;199:43–52. https://doi.org/10.1016/j.bbr.2008.10.034.
Gurney KN, Humphries MD, Redgrave P. A new framework for cortico-striatal plasticity: Behavioural theory meets in vitro data at the reinforcement-action interface. PLOS Biol. 2015;13:e1002034. https://doi.org/10.1371/journal.pbio.1002034.
Brzosko Z, Mierau SB, Paulsen O. Neuromodulation of spike-timing-dependent plasticity: Past, present, and future. Neuron. 2019;103:563–81. https://doi.org/10.1016/j.neuron.2019.05.041.
Schultz W, Dickinson A. Neuronal coding of prediction errors. Annu Rev Neurosci. 2000;23:473–500. https://doi.org/10.1146/annurev.neuro.23.1.473.
Hamid AA, Frank MJ, Moore CI. Wave-like dopamine dynamics as a mechanism for spatiotemporal credit assignment. Cell. 2021;184:2733–49. https://doi.org/10.1016/j.cell.2021.03.046.
Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–51. https://doi.org/10.1126/science.1160575.
Lee SJ, Lodder B, Chen Y, Patriarchi T, Tian L, Sabatini BL. Cell-type-specific asynchronous modulation of PKA by dopamine in learning. Nature. 2021;590:451–6. https://doi.org/10.1038/s41586-020-03050-5.
Franklin NT, Frank MJ. A cholinergic feedback circuit to regulate striatal population uncertainty and optimize reinforcement learning. eLife. 2015;4:e12029. https://doi.org/10.7554/eLife.12029.
Cragg SJ. Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci. 2006;29:125–31. https://doi.org/10.1016/j.tins.2006.01.003.
Reynolds JNJ, Avvisati R, Dodson PD, Fisher SD, Oswald MJ, Wickens JR. et al. Coincidence of cholinergic pauses, dopaminergic activation and depolarisation of spiny projection neurons drives synaptic plasticity in the striatum. Nat Commun. 2022;13:1296. https://doi.org/10.1038/s41467-022-28950-0.
Lidow MS, Goldman-Rakic PS, Gallager DW, Rakic P. Distribution of dopaminergic receptors in the primate cerebral cortex: Quantitative autoradiographic analysis using [3h]raclopride, [3h]spiperone and [3h]SCH23390. Neuroscience. 1991;40:657–71. https://doi.org/10.1016/0306-4522(91)90003-7.
Lidow MS, Wang F, Cao Y, Goldman-Rakic PS. Layer v neurons bear the majority of mRNAs encoding the five distinct dopamine receptor subtypes in the primate prefrontal cortex. Synapse. 1998;28:10–20.
Paspalas CD. Presynaptic d1 dopamine receptors in primate prefrontal cortex: Target-specific expression in the glutamatergic synapse. J Neurosci. 2005;25:1260–7. https://doi.org/10.1523/JNEUROSCI.3436-04.2005.
Zilles K, Palomero-Gallagher N. Multiple transmitter receptors in regions and layers of the human cerebral cortex. Front Neuroanatom. 2017;11:78.
Jacob SN, Nienborg H. Monoaminergic neuromodulation of sensory processing. Front Neural Circuits 2018;12:51.
Muly EC, Szigeti K, Goldman-Rakic PS. D 1 receptor in interneurons of macaque prefrontal cortex: Distribution and subcellular localization. J Neurosci. 1998;18:10553–65. https://doi.org/10.1523/JNEUROSCI.18-24-10553.1998.
Gorelova N, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol. 2002;88. https://doi.org/10.1152/jn.00335.2002.
Froudist-Walsh S, Bliss DP, Ding X, Rapan L, Niu M, Knoblauch K. et al. A dopamine gradient controls access to distributed working memory in the large-scale monkey cortex. Neuron. 2021;109:3500–20. https://doi.org/10.1016/j.neuron.2021.08.024.
Cools R, Arnsten AFT. Neuromodulation of prefrontal cortex cognitive function in primates: the powerful roles of monoamines and acetylcholine. Neuropsychopharmacology. 2021;1–20. https://doi.org/10.1038/s41386-021-01100-8. Bandiera_abtest: a Cc_license_type: cc_by Cg_type: Nature Research Journals Primary_atype: Reviews Publisher: Nature Publishing Group Subject_term: Cognitive control;Excitability Subject_term_id: cognitive-control;excitability.
Mrzljak L, Bergson C, Pappy M, Huff R, Levenson R, Goldman-Rakic PS. Localization of dopamine d4 receptors in GABAergic neurons of the primate brain. Nature. 1996;381:245–8. https://doi.org/10.1038/381245a0.
Chiu CQ, Puente N, Grandes P, Castillo PE. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J Neurosci. 2010;30:7236–48. https://doi.org/10.1523/JNEUROSCI.0736-10.2010.
Xu T-X, Yao W-D. D1 and d2 dopamine receptors in separate circuits cooperate to drive associative long-term potentiation in the prefrontal cortex. Proc Natl Acad Sci. 2010;107:16366–71. https://doi.org/10.1073/pnas.1004108107.
Cousineau J, Lescouzères L, Taupignon A, Delgado-Zabalza L, Valjent E, Baufreton J. et al. Dopamine d2-like receptors modulate intrinsic properties and synaptic transmission of parvalbumin interneurons in the mouse primary motor cortex. eneuro. 2020;7:ENEURO.0081–20.2020. https://doi.org/10.1523/ENEURO.0081-20.2020.
Robinson SE, Sohal VS. Dopamine d2 receptors modulate pyramidal neurons in mouse medial prefrontal cortex through a stimulatory g-protein pathway. J Neurosci. 2017;37:10063–73. https://doi.org/10.1523/JNEUROSCI.1893-17.2017.
Ruan H, Saur T, Yao W-D. Dopamine-enabled anti-hebbian timing-dependent plasticity in prefrontal circuitry. Front Neural Circuits 2014;8. https://doi.org/10.3389/fncir.2014.00038.
Jedrzejewska-Szmek J, Luczak V, Abel T, Blackwell KT. beta-adrenergic signaling broadly contributes to LTP induction. PLOS Computational Biol. 2017;13:e1005657. https://doi.org/10.1371/journal.pcbi.1005657.
Salgado H, Köhr G, Treviño M. Noradrenergic ’tone’ determines dichotomous control of cortical spike-timing-dependent plasticity. Sci Rep. 2012;2:417. https://doi.org/10.1038/srep00417.
Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K. et al. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–29. https://doi.org/10.1016/j.neuron.2007.08.013.
Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–609. https://doi.org/10.1523/JNEUROSCI.19-05-01599.1999.
Choi S-Y. Multiple receptors coupled to phospholipase c gate long-term depression in visual cortex. J Neurosci. 2005;25:11433–43. https://doi.org/10.1523/JNEUROSCI.4084-05.2005.
Huang S, Treviño M, He K, Ardiles A, de Pasquale R, Guo Y. et al. Pull-push neuromodulation of LTP and LTD enables bidirectional experience-induced synaptic scaling in visual cortex. Neuron. 2012;73:497–510. https://doi.org/10.1016/j.neuron.2011.11.023.
Hong SZ, Mesik L, Grossman CD, Cohen JY, Lee B, Lee HK. et al. Norepinephrine potentiates and serotonin depresses visual cortical responses by transforming eligibility traces. Nat Commun. 2022;13:3202. https://doi.org/10.1038/s41467-022-30827-1.
Trevino M, Frey S, Kohr G. Alpha-1 adrenergic receptors gate rapid orientation-specific reduction in visual discrimination. Cereb Cortex. 2012;22:2529–41. https://doi.org/10.1093/cercor/bhr333.
Salgado H, Treviño M, Atzori M. Layer- and area-specific actions of norepinephrine on cortical synaptic transmission. Brain Res. 2016;1641:163–76. https://doi.org/10.1016/j.brainres.2016.01.033.
Zhang J-C, Lau P-M, Bi G-Q. Gain in sensitivity and loss in temporal contrast of STDP by dopaminergic modulation at hippocampal synapses. Proc Natl Acad Sci. 2009;106:13028–33. https://doi.org/10.1073/pnas.0900546106.
Edelmann E, Lessmann V. Dopamine modulates spike timing-dependent plasticity and action potential properties in CA1 pyramidal neurons of acute rat hippocampal slices. Front Synap Neurosci. 2011;3:6.
Edelmann E, Lessmann V. Dopamine regulates intrinsic excitability thereby gating successful induction of spike timing-dependent plasticity in CA1 of the hippocampus. Front Neurosci. 2013;7.
Yang K, Dani JA. Dopamine d1 and d5 receptors modulate spike timing-dependent plasticity at medial perforant path to dentate granule cell synapses. J Neurosci. 2014;34:15888–97. https://doi.org/10.1523/JNEUROSCI.2400-14.2014.
Brzosko Z, Schultz W, Paulsen O. Retroactive modulation of spike timing-dependent plasticity by dopamine. eLife. 2015;4:e09685. https://doi.org/10.7554/eLife.09685.
Edelmann E, Cepeda-Prado E, Leßmann V. Coexistence of multiple types of synaptic plasticity in individual hippocampal CA1 pyramidal neurons. Front Synap Neurosci. 2017;9:7.
Lin Y-W, Min M-Y, Chiu T-H, Yang H-W. Enhancement of associative long-term potentiation by activation of β-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 2003;23:4173–81.
Brzosko Z, Zannone S, Schultz W, Clopath C, Paulsen O. Sequential neuromodulation of hebbian plasticity offers mechanism for effective reward-based navigation. eLife. 2017;6. https://doi.org/10.7554/eLife.27756.
Sugisaki E, Fukushima Y, Tsukada M, Aihara T. Cholinergic modulation on spike timing-dependent plasticity in hippocampal CA1 network. Neuroscience. 2011;192:91–101. https://doi.org/10.1016/j.neuroscience.2011.06.064.
Sugisaki E, Fukushima Y, Fujii S, Yamazaki Y, Aihara T. The effect of coactivation of muscarinic and nicotinic acetylcholine receptors on LTD in the hippocampal CA1 network. Brain Res. 2016;1649:44–52. https://doi.org/10.1016/j.brainres.2016.08.024.
Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent β-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–82.
Connor SA, Maity S, Roy B, Ali DW, Nguyen PV. Conversion of short-term potentiation to long-term potentiation in mouse CA1 by coactivation of β-adrenergic and muscarinic receptors. Learn Mem. 2012;19:535–42. https://doi.org/10.1101/lm.026898.112.
Maity S, Rah S, Sonenberg N, Gkogkas CG, Nguyen PV. Norepinephrine triggers metaplasticity of LTP by increasing translation of specific mRNAs. Learn Mem. 2015;22:499–508. https://doi.org/10.1101/lm.039222.115.
Ito T, Miura Y, Kadokawa T. Effects of physostigmine and scopolamine on long-term potentiation of hippocampal population spikes in rats. Can J Physiol Pharmacol. 1988;66:1010–6. https://doi.org/10.1139/y88-165.
Blitzer RD, Gil O, Landau EM. Cholinergic stimulation enhances long-term potentiation in the CA1 region of rat hippocampus. Neurosci Lett. 1990;119:207–10. https://doi.org/10.1016/0304-3940(90)90835-W.
Boddeke EW, Enz A, Shapiro G. SDZ ENS 163, a selective muscarinic m1 receptor agonist, facilitates the induction of long-term potentiation in rat hippocampal slices. Eur J Pharmacol. 1992;222:21–5. https://doi.org/10.1016/0014-2999(92)90457-F.
Huerta PT, Lisman JE. Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature. 1993;364:723–5.
Auerbach JM, Segal M. A novel cholinergic induction of long-term potentiation in rat hippocampus. J Neurophysiol. 1994;72:2034–40. https://doi.org/10.1152/jn.1994.72.4.2034.
Huerta PT, Lisman JE. Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron. 1995;15:1053–63. https://doi.org/10.1016/0896-6273(95)90094-2.
Natsume K, Kometani K. θ-activity-dependent and-independent muscarinic facilitation of long-term potentiation in guinea pig hippocampal slices. Neurosci Res. 1997;27:335–41.
Patil MM, Linster C, Lubenov E, Hasselmo ME. Cholinergic agonist carbachol enables associative long-term potentiation in piriform cortex slices. J Neurophysiol. 1998;80:2467–74. https://doi.org/10.1152/jn.1998.80.5.2467.
Ovsepian SV, Anwyl R, Rowan MJ. Endogenous acetylcholine lowers the threshold for long-term potentiation induction in the CA1 area through muscarinic receptor activation: in vivo study. Eur J Neurosci. 2004;20:1267–75. https://doi.org/10.1111/j.1460-9568.2004.03582.x.
Shinoe T. Modulation of synaptic plasticity by physiological activation of m1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci. 2005;25:11194–200. https://doi.org/10.1523/JNEUROSCI.2338-05.2005.
Buchanan KA, Petrovic MM, Chamberlain SE, Marrion NV, Mellor JR. Facilitation of long-term potentiation by muscarinic m1 receptors is mediated by inhibition of SK channels. Neuron. 2010;68:948–63. https://doi.org/10.1016/j.neuron.2010.11.018.
Dennis SH, Pasqui F, Colvin EM, Sanger H, Mogg AJ, Felder CC. et al. Activation of muscarinic m1 acetylcholine receptors induces long-term potentiation in the hippocampus. Cereb Cortex. 2016;26:414–26. https://doi.org/10.1093/cercor/bhv227.
Massey PV, Bhabra G, Cho K, Brown MW, Bashir ZI. Activation of muscarinic receptors induces protein synthesis-dependent long-lasting depression in the perirhinal cortex. Eur J Neurosci. 2001;14:145–52. https://doi.org/10.1046/j.0953-816x.2001.01631.x.
Jo J, Ball SM, Seok H, Oh SB, Massey PV, Molnar E. et al. Experience-dependent modification of mechanisms of long-term depression. Nat Neurosci. 2006;9:170–2. https://doi.org/10.1038/nn1637.
Volk LJ, Pfeiffer BE, Gibson JR, Huber KM. Multiple gq-coupled receptors converge on a common protein synthesis-dependent long-term depression that is affected in fragile x syndrome mental retardation. J Neurosci. 2007;27:11624–34. https://doi.org/10.1523/JNEUROSCI.2266-07.2007.
Dickinson BA, Jo J, Seok H, Son GH, Whitcomb DJ, Davies CH. et al. A novel mechanism of hippocampal LTD involving muscarinic receptor-triggered interactions between AMPARs, GRIP and liprin-α. Mol Brain. 2009;2:18. https://doi.org/10.1186/1756-6606-2-18.
Wu L-J, Wang Y-T, Zhuo M. Hook-up of GluA2, GRIP and liprin-α for cholinergic muscarinic receptor-dependent LTD in the hippocampus. Mol Brain. 2009;2:17. 1756–6606–2–17. https://doi.org/10.1186/1756-6606-2-17.
Xiong CH, Liu MG, Zhao LX, Chen MW, Tang L, Yan YH. et al. M1 muscarinic receptors facilitate hippocampus-dependent cognitive flexibility via modulating GluA2 subunit of AMPA receptors. Neuropharmacology. 2019;146:242–51. https://doi.org/10.1016/j.neuropharm.2018.12.005.
Gulledge AT. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–20. https://doi.org/10.1523/JNEUROSCI.2697-05.2005.
Giessel AJ, Sabatini BL. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron. 2010;68:936–47. https://doi.org/10.1016/j.neuron.2010.09.004.
Thiele A. Muscarinic signaling in the brain. Annu Rev Neurosci. 2013;36:271–94. https://doi.org/10.1146/annurev-neuro-062012-170433.
Williams RJ. Simple statistical gradient-following algorithms for connectionist reinforcement learning. Mach Learn. 1992;8:229–56.
Pawlak V, Wickens J, Kirkwood A, Kerr J.Timing is not everything: Neuromodulation opens the STDP gate. Front Synap Neurosci. 2010;2. https://doi.org/10.3389/fnsyn.2010.00146.
Frémaux N, Gerstner W. Neuromodulated spike-timing-dependent plasticity, and theory of three-factor learning rules. Front Neural Circuits 2016;9. https://doi.org/10.3389/fncir.2015.00085.
Gerstner W, Lehmann M, Liakoni V, Corneil D, Brea J. Eligibility traces and plasticity on behavioral time scales: Experimental support of NeoHebbian three-factor learning rules. Front Neural Circuits. 2018;12:53. https://doi.org/10.3389/fncir.2018.00053.
Caporale N, Dan Y. Spike timing-dependent plasticity: A hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. https://doi.org/10.1146/annurev.neuro.31.060407.125639.
Sanger TD. Optimal unsupervised learning in a single-layer linear feedforward neural network. Neural Netw. 1989;2:459–73. https://doi.org/10.1016/0893-6080(89)90044-0.
Oja E. Simplified neuron model as a principal component analyzer. J Math Biol. 1982;15:267–73. https://doi.org/10.1007/BF00275687.
Karhunen J. Adaptive algorithms for estimating eigenvectors of correlation type matrices. ICASSP ’84 IEEE Int Conf Acoust, Speech, Signal Process. 1984;9:592–5. https://doi.org/10.1109/ICASSP.1984.1172323.
Hyvärinen A, Oja E. Independent component analysis: algorithms and applications. Neural Netw. 2000;13:411–30.
Grossberg S. Competitive learning: From interactive activation to adaptive resonance. Cogn Sci. 1987;11:23–63. https://doi.org/10.1111/j.1551-6708.1987.tb00862.x.
Hertz J, Krogh A, Palmer RG. Introduction to the theory of neural computation. CRC Press: Boca Raton, Florida; 1991.
Miller KD, MacKay DJC. The role of constraints in hebbian learning. Neural Comput. 1994;6:100–26. https://doi.org/10.1162/neco.1994.6.1.100.
Song S, Sjöström PJ, Reigl M, Nelson S, Chklovskii DB. Highly nonrandom features of synaptic connectivity in local cortical circuits. PLOS Biol. 2005;3:e68. https://doi.org/10.1371/journal.pbio.0030068.
Sirosh J, Miikkulainen R. Cooperative self-organization of afferent and lateral connections in cortical maps. Biol Cybern. 1994;71:65–78.
Miller KD. Synaptic economics: Competition and cooperation in synaptic plasticity. Neuron. 1996;17:371–4. https://doi.org/10.1016/S0896-6273(00)80169-5.
Buonomano DV, Merzenich MM. CORTICAL PLASTICITY: From synapses to maps. Annu Rev Neurosci. 1998;21:149–86. https://doi.org/10.1146/annurev.neuro.21.1.149.
Feldman DE, Brecht M. Map plasticity in somatosensory cortex. Science. 2005;310:810–5. https://doi.org/10.1126/science.1115807.
Cossell L, Iacaruso MF, Muir DR, Houlton R, Sader EN, Ko H. et al. Functional organization of excitatory synaptic strength in primary visual cortex. Nature. 2015;518:399–403. https://doi.org/10.1038/nature14182.
Lee WCA, Bonin V, Reed M, Graham BJ, Hood G, Glattfelder K. et al. Anatomy and function of an excitatory network in the visual cortex. Nature. 2016;532:370–4. https://doi.org/10.1038/nature17192.
Lindsay GW, Rigotti M, Warden MR, Miller EK, Fusi S. Hebbian learning in a random network captures selectivity properties of the prefrontal cortex. J Neurosci. 2017;37:11021–36. https://doi.org/10.1523/JNEUROSCI.1222-17.2017.
Peron S, Pancholi R, Voelcker B, Wittenbach JD, Ólafsdóttir HF, Freeman J. et al. Recurrent interactions in local cortical circuits. Nature. 2020;579:256–9. https://doi.org/10.1038/s41586-020-2062-x.
Mcclelland JL, Mcnaughton BL, O’Reilly RC. Why there are complementary learning systems in the hippocampus and neocortex: Insights from the successes and failures of connectionist models of learning and memory. Psychological Rev. 1995;102:419–57.
von der Malsburg C. The correlation theory of brain function. In: Domany E, van Hemmen JL, Schulten K, editors. Models of Neural Networks, 95-119, https://doi.org/10.1007/978-1-4612-4320-5_2 (Springer New York; 1994).
Gerstner W, Kistler WM. Mathematical formulations of hebbian learning. Biol Cybern. 2002;87:404–15. https://doi.org/10.1007/s00422-002-0353-y.
Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–6. https://doi.org/10.1038/36103.
Turrigiano G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736–a005736. https://doi.org/10.1101/cshperspect.a005736.
Toyoizumi T, Kaneko M, Stryker MP, Miller KD. Modeling the dynamic interaction of hebbian and homeostatic plasticity. Neuron. 2014;84:497–510. https://doi.org/10.1016/j.neuron.2014.09.036.
Keck T, Toyoizumi T, Chen L, Doiron B, Feldman DE, Fox K. et al. Integrating hebbian and homeostatic plasticity: the current state of the field and future research directions. Philos Trans R Soc B: Biol Sci. 2017;372:20160158. https://doi.org/10.1098/rstb.2016.0158.
Fox K, Stryker M. Integrating hebbian and homeostatic plasticity: introduction. Philos Trans R Soc B: Biol Sci. 2017;372:20160413. https://doi.org/10.1098/rstb.2016.0413.
Wu YK, Hengen KB, Turrigiano GG, Gjorgjieva J. Homeostatic mechanisms regulate distinct aspects of cortical circuit dynamics. Proc Natl Acad Sci. 2020;117:24514–25. https://doi.org/10.1073/pnas.1918368117.
Shimizu G, Yoshida K, Kasai H, Toyoizumi T. Computational roles of intrinsic synaptic dynamics. Curr Opin Neurobiol. 2021;70:34–42. https://doi.org/10.1016/j.conb.2021.06.002.
O’reilly RC. Generalization in interactive networks: The benefits of inhibitory competition and hebbian learning. Neural Comput. 2001;13:1199–241.
Krotov D, Hopfield JJ. Unsupervised learning by competing hidden units. Proc Natl Acad Sci. 2019;116:7723–31. https://doi.org/10.1073/pnas.1820458116.
Cao Y, Summerfield C, Saxe A. Characterizing emergent representations in a space of candidate learning rules for deep networks. In: Larochelle H, Ranzato M, Hadsell R, Balcan MF, Lin H, editors. Advances in Neural Information Processing Systems, vol. 33, 8660-70. Curran Associates, Inc: Red Hook, New York; 2020.
Flesch T, Juechems K, Dumbalska T, Saxe A, Summerfield C. Rich and lazy learning of task representations in brains and neural networks. bioRxiv. 2021;2021.04.23.441128. https://doi.org/10.1101/2021.04.23.441128.
Nassar MR, Scott D, Bhandari A. Noise correlations for faster and more robust learning. J Neurosci. 2021;41:6740–52. https://doi.org/10.1523/JNEUROSCI.3045-20.2021.
Pehlevan C, Sengupta A, Chklovskii DB. Why do similarity matching objectives lead to hebbian/anti-hebbian networks? Neural Comput. 2018;30:84–124. https://doi.org/10.1162/neco_a_01018.
Kozachkov L, Lundqvist M, Slotine J-J, Miller EK. Achieving stable dynamics in neural circuits. PLOS Computational Biol. 2020;16:e1007659. https://doi.org/10.1371/journal.pcbi.1007659.
Sjöström J, Gerstner W. Spike-timing dependent plasticity. Scholarpedia. 2010;5:1362. https://doi.org/10.4249/scholarpedia.1362.
He K, Huertas M, Hong SZ, Tie X, Hell JW, Shouval H. et al. Distinct eligibility traces for LTP and LTD in cortical synapses. Neuron. 2015;88:528–38. https://doi.org/10.1016/j.neuron.2015.09.037.
Bittner KC, Milstein AD, Grienberger C, Romani S, Magee JC. Behavioral time scale synaptic plasticity underlies CA1 place fields. Science. 2017;357:1033–6. https://doi.org/10.1126/science.aan3846.
Shindou T, Shindou M, Watanabe S, Wickens J. A silent eligibility trace enables dopamine-dependent synaptic plasticity for reinforcement learning in the mouse striatum. Eur J Neurosci. 2019;49:726–36. https://doi.org/10.1111/ejn.13921.
Yamaguchi K, Maeda Y, Sawada T, Iino Y, Tajiri M, Nakazato R. et al. A behavioural correlate of the synaptic eligibility trace in the nucleus accumbens. Sci Rep. 2022;12:1921. https://doi.org/10.1038/s41598-022-05637-6.
Redondo RL, Okuno H, Spooner PA, Frenguelli BG, Bito H, Morris RGM. Synaptic tagging and capture: Differential role of distinct calcium/calmodulin kinases in protein synthesis-dependent long-term potentiation. J Neurosci. 2010;30:4981–9. https://doi.org/10.1523/JNEUROSCI.3140-09.2010.
Redondo RL, Morris RGM. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci. 2011;12:17–30. https://doi.org/10.1038/nrn2963.
Lisman J, Grace AA, Duzel E. A neoHebbian framework for episodic memory; role of dopamine-dependent late LTP. Trends Neurosci. 2011;34:536–47. https://doi.org/10.1016/j.tins.2011.07.006.
Park P, Kang H, Sanderson TM, Bortolotto ZA, Georgiou J, Zhuo M. et al. On the role of calcium-permeable AMPARs in long-term potentiation and synaptic tagging in the rodent hippocampus. Front Synap Neurosci. 2019;11:4.
Luboeinski J, Tetzlaff C. Memory consolidation and improvement by synaptic tagging and capture in recurrent neural networks. Commun Biol. 2021;4:275. https://doi.org/10.1038/s42003-021-01778-y.
Okuda K, Højgaard K, Privitera L, Bayraktar G, Takeuchi T. Initial memory consolidation and the synaptic tagging and capture hypothesis. Eur J Neurosci. 2021;54:6826–49. https://doi.org/10.1111/ejn.14902.
Pfister J-P. Triplets of spikes in a model of spike timing-dependent plasticity. J Neurosci. 2006;26:9673–82. https://doi.org/10.1523/JNEUROSCI.1425-06.2006.
Gjorgjieva J, Clopath C, Audet J, Pfister J-P. A triplet spike-timing-dependent plasticity model generalizes the bienenstock-cooper-munro rule to higher-order spatiotemporal correlations. Proc Natl Acad Sci. 2011;108:19383–8. https://doi.org/10.1073/pnas.1105933108.
Babadi B, Abbott LF. Stability and competition in multi-spike models of spike-timing dependent plasticity. PLOS Computational Biol. 2016;12:e1004750. https://doi.org/10.1371/journal.pcbi.1004750.
Montangie L, Miehl C, Gjorgjieva J. Autonomous emergence of connectivity assemblies via spike triplet interactions. PLOS Computational Biol. 2020;16:e1007835. https://doi.org/10.1371/journal.pcbi.1007835.
Clopath C, Büsing L, Vasilaki E, Gerstner W. Connectivity reflects coding: a model of voltage-based STDP with homeostasis. Nat Neurosci. 2010;13:344–52. https://doi.org/10.1038/nn.2479.
Clopath C, Gerstner W. Voltage and spike timing interact in STDP—a unified model. Front Synap Neurosci. 2010;2. https://doi.org/10.3389/fnsyn.2010.00025.
Dan Y, Poo M-m. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. https://doi.org/10.1016/j.neuron.2004.09.007.
Bell, C. C., Caputi, A., Grant, K. & Serrier, J.Storage of a sensory pattern by anti-hebbian synaptic plasticity in an electric fish. Proc Natl Acad Sci. 1993;90. https://doi.org/10.1073/pnas.90.10.4650.
Han VZ, Grant K, Bell CC. Reversible associative depression and nonassociative potentiation at a parallel fiber synapse. Neuron. 2000;27:611–22. https://doi.org/10.1016/S0896-6273(00)00070-2.
Roberts P, Leen T. Anti-hebbian spike-timing-dependent plasticity and adaptive sensory processing. Front Comput Neurosci. 2010;4:156.
Lang EJ, Apps R, Bengtsson F, Cerminara NL, De Zeeuw CI, Ebner TJ. et al. The roles of the olivocerebellar pathway in motor learning and motor control. a consensus paper. Cerebellum. 2017;16:230–52. https://doi.org/10.1007/s12311-016-0787-8.
Paille V, Fino E, Du K, Morera-Herreras T, Perez S, Kotaleski JH. et al. GABAergic circuits control spike-timing-dependent plasticity. J Neurosci. 2013;33:9353–63. https://doi.org/10.1523/JNEUROSCI.5796-12.2013.
Kempter R, Gerstner W, Hemmen JLV. Intrinsic stabilization of output rates by spike-based hebbian learning. Neural Comput. 2001;13:2709–41. https://doi.org/10.1162/089976601317098501.
Sboev A, Vlasov D, Serenko A, Rybka R, Moloshnikov I. A comparison of learning abilities of spiking networks with different spike timing-dependent plasticity forms. J Phys: Conf Ser. 2016;681:012013. https://doi.org/10.1088/1742-6596/681/1/012013.
Abeles M. Local cortical circuits: an electrophysiological study. Springer Science & Business Media: New York; 1982.
Diesmann M, Gewaltig M-O, Aertsen A. Stable propagation of synchronous spiking in cortical neural networks. Nature. 1999;402:529–33. https://doi.org/10.1038/990101.
Aviel Y, Mehring C, Abeles M, Horn D. On embedding synfire chains in a balanced network. Neural Comput. 2003;15:1321–40. https://doi.org/10.1162/089976603321780290.
Yazdanbakhsh A, Babadi B, Rouhani S, Arabzadeh E, Abbassian A. New attractor states for synchronous activity in synfire chains with excitatory and inhibitory coupling. Biol Cybern. 2002;86:367–78. https://doi.org/10.1007/s00422-001-0293-y.
Mehring C, Hehl U, Kubo M, Diesmann M, Aertsen A. Activity dynamics and propagation of synchronous spiking in locally connected random networks. Biol Cybern. 2003;88:395–408. https://doi.org/10.1007/s00422-002-0384-4.
Izhikevich EM. Polychronization: Computation with spikes. Neural Comput. 2006;18:245–82. https://doi.org/10.1162/089976606775093882.
Hosaka R, Araki O, Ikeguchi T. STDP provides the substrate for igniting synfire chains by spatiotemporal input patterns. Neural Comput. 2008;20:415–35. https://doi.org/10.1162/neco.2007.11-05-043.
Kumar A, Rotter S, Aertsen A. Conditions for propagating synchronous spiking and asynchronous firing rates in a cortical network model. J Neurosci. 2008;28:5268–80. https://doi.org/10.1523/JNEUROSCI.2542-07.2008.
Kumar A, Rotter S, Aertsen A. Spiking activity propagation in neuronal networks: reconciling different perspectives on neural coding. Nat Rev Neurosci. 2010;11:615–27. https://doi.org/10.1038/nrn2886.
Trengove C, van Leeuwen C, Diesmann M. High-capacity embedding of synfire chains in a cortical network model. J Comput Neurosci. 2013;34:185–209. https://doi.org/10.1007/s10827-012-0413-9.
Zheng P, Triesch J. Robust development of synfire chains from multiple plasticity mechanisms. Front Comput Neurosci. 2014;8. https://doi.org/10.3389/fncom.2014.00066.
Wang Z, Sornborger AT, Tao L. Graded, dynamically routable information processing with synfire-gated synfire chains. PLOS Comput Biol. 2016;12:e1004979. https://doi.org/10.1371/journal.pcbi.1004979.
Bienenstock E. A model of neocortex. Netw: Comput neural Syst. 1995;6:179–224.
Abeles M, Hayon G, Lehmann D. Modeling compositionality by dynamic binding of synfire chains. J Compu Neurosci. 2004;17:179–201. https://doi.org/10.1023/B:JCNS.0000037682.18051.5f.
Hanuschkin A, Herrmann JM, Morrison A, Diesmann M. Compositionality of arm movements can be realized by propagating synchrony. J Comput Neurosci. 2011;30:675–97. https://doi.org/10.1007/s10827-010-0285-9.
Aviel Y, Pavlov E, Abeles M, Horn D. Synfire chain in a balanced network. Neurocomputing. 2002;44:285–92.
Izhikevich EM. Solving the distal reward problem through linkage of STDP and dopamine signaling. Cereb Cortex. 2007;17:2443–52. https://doi.org/10.1093/cercor/bhl152.
Farries MA, Fairhall AL. Reinforcement learning with modulated spike timing-dependent synaptic plasticity. J Neurophysiol. 2007;98:3648–65. https://doi.org/10.1152/jn.00364.2007.
Legenstein R, Pecevski D, Maass W. A learning theory for reward-modulated spike-timing-dependent plasticity with application to biofeedback. PLoS Computational Biol. 2008;4:e1000180. https://doi.org/10.1371/journal.pcbi.1000180.
Fremaux N, Sprekeler H, Gerstner W. Functional requirements for reward-modulated spike-timing-dependent plasticity. J Neurosci. 2010;30:13326–37. https://doi.org/10.1523/JNEUROSCI.6249-09.2010.
Babadi B. Network Structures Arising from Spike-Timing Dependent Plasticity. Ph.D. thesis, 2011. https://academiccommons.columbia.edu/doi/10.7916/D8DF6Z6S. Accessed 16 February 2022.
Kayser C, Logothetis NK, Panzeri S. Millisecond encoding precision of auditory cortex neurons. Proc Natl Acad Sci. 2010;107. https://doi.org/10.1073/pnas.1012656107.
Wang X. Cortical coding of auditory features. Annu Rev Neurosci. 2018;41:527–52. https://doi.org/10.1146/annurev-neuro-072116-031302.
Luczak A, Bartho P, Marguet SL, Buzsaki G, Harris KD. Sequential structure of neocortical spontaneous activity in vivo. Proc Natl Acad Sci. 2007;104:347–52. https://doi.org/10.1073/pnas.0605643104.
Luczak A, Barthó P, Harris KD. Spontaneous events outline the realm of possible sensory responses in neocortical populations. Neuron. 2009;62:413–25. https://doi.org/10.1016/j.neuron.2009.03.014.
Luczak A, Bartho P, Harris KD. Gating of sensory input by spontaneous cortical activity. J Neurosci. 2013;33:1684–95. https://doi.org/10.1523/JNEUROSCI.2928-12.2013.
Schrader S, Grün S, Diesmann M, Gerstein GL. Detecting synfire chain activity using massively parallel spike train recording. J Neurophysiol. 2008;100:2165–76. https://doi.org/10.1152/jn.01245.2007.
Gerstein GL, Williams ER, Diesmann M, Grün S, Trengove C. Detecting synfire chains in parallel spike data. J Neurosci Methods. 2012;206:54–64. https://doi.org/10.1016/j.jneumeth.2012.02.003.
Russo E, Durstewitz D. Cell assemblies at multiple time scales with arbitrary lag constellations. eLife. 2017;6:e19428. https://doi.org/10.7554/eLife.19428.
Gulati T, Ramanathan DS, Wong CC, Ganguly K. Reactivation of emergent task-related ensembles during slow-wave sleep after neuroprosthetic learning. Nat Neurosci. 2014;17:1107–13. https://doi.org/10.1038/nn.3759.
Hasenstaub A, Otte S, Callaway E. Cell type-specific control of spike timing by gamma-band oscillatory inhibition. Cerebral Cortex. 2015. https://doi.org/10.1093/cercor/bhv044.
Luz Y, Shamir M. Oscillations via spike-timing dependent plasticity in a feed-forward model. PLOS Comput Biol. 2016;12. https://doi.org/10.1371/journal.pcbi.1004878.
Silversmith DB, Lemke SM, Egert D, Berke JD, Ganguly K. The degree of nesting between spindles and slow oscillations modulates neural synchrony. J Neurosci. 2020;40:4673–84. https://doi.org/10.1523/JNEUROSCI.2682-19.2020.
Fiete IR, Senn W, Wang CZ, Hahnloser RH. Spike-time-dependent plasticity and heterosynaptic competition organize networks to produce long scale-free sequences of neural activity. Neuron. 2010;65:563–76. https://doi.org/10.1016/j.neuron.2010.02.003.
Mazzucato L, Fontanini A, Camera GL. Dynamics of multistable states during ongoing and evoked cortical activity. J Neurosci. 2015;35:8214–31. https://doi.org/10.1523/JNEUROSCI.4819-14.2015.
La Camera G, Fontanini A, Mazzucato L. Cortical computations via metastable activity. Curr Opin Neurobiol. 2019;58:37–45. https://doi.org/10.1016/j.conb.2019.06.007.
Vasilaki E, Frémaux N, Urbanczik R, Senn W, Gerstner W. Spike-based reinforcement learning in continuous state and action space: When policy gradient methods fail. PLoS Comput Biol. 2009;5. https://doi.org/10.1371/journal.pcbi.1000586.
Frémaux N, Sprekeler H, Gerstner W. Reinforcement learning using a continuous time actor-critic framework with spiking neurons. PLOS Computational Biol. 2013;9:e1003024. https://doi.org/10.1371/journal.pcbi.1003024.
Scott DN, Frank MJ. Beyond gradients: Noise correlations control hebbian plasticity to shape credit assignment. 2021. https://doi.org/10.1101/2021.11.19.466943.
Xie X, Seung HS. Learning in neural networks by reinforcement of irregular spiking. Phys Rev E. 2004;69:041909. https://doi.org/10.1103/PhysRevE.69.041909.
Frank MJ. Dynamic dopamine modulation in the basal ganglia: A neurocomputational account of cognitive deficits in medicated and nonmedicated parkinsonism. J Cogn Neurosci. 2005;17:51–72. https://doi.org/10.1162/0898929052880093.
Baras D, Meir R. Reinforcement learning, spike-time-dependent plasticity, and the BCM rule. Neural Comput. 2007;19:2245–79. https://doi.org/10.1162/neco.2007.19.8.2245.
Florian RV. Reinforcement learning through modulation of spike-timing-dependent synaptic plasticity. Neural Comput. 2007;19:1468–502. https://doi.org/10.1162/neco.2007.19.6.1468.
di Castro D, Volkinshtein D, Meir R. Temporal difference based actor critic learning—convergence and neural implementation. In: Advances in Neural Information Processing Systems, vol. 21. Curran Associates, Inc.: Red Hook, NY; 2008.
Urbanczik R, Senn W. Reinforcement learning in populations of spiking neurons. Nat Neurosci. 2009;12:250–2. https://doi.org/10.1038/nn.2264.
Law C-T, Gold JI. Reinforcement learning can account for associative and perceptual learning on a visual-decision task. Nat Neurosci. 2009;12:655–63. https://doi.org/10.1038/nn.2304.
Frank MJ, Badre D. Mechanisms of hierarchical reinforcement learning in corticostriatal circuits 1: Computational analysis. Cereb Cortex. 2012;22:509–26. https://doi.org/10.1093/cercor/bhr114.
Collins AGE, Frank MJ. Opponent actor learning (OpAL): Modeling interactive effects of striatal dopamine on reinforcement learning and choice incentive. Psychological Rev. 2014;121:337–66. https://doi.org/10.1037/a0037015.
Song HF, Yang GR, Wang X-J. Reward-based training of recurrent neural networks for cognitive and value-based tasks. eLife. 2017;6:e21492. https://doi.org/10.7554/eLife.21492.
Wang JX, Kurth-Nelson Z, Kumaran D, Tirumala D, Soyer H, Leibo JZ, et al. Prefrontal cortex as a meta-reinforcement learning system. Nat Neurosci. 2018;21:860–8. https://doi.org/10.1038/s41593-018-0147-8.
Isomura T, Toyoizumi T. A local learning rule for independent component analysis. Sci Rep. 2016;6:1–17. https://doi.org/10.1038/srep28073.
Isomura T, Toyoizumi T. Error-gated hebbian rule: A local learning rule for principal and independent component analysis. Sci Rep. 2018;8:1835. https://doi.org/10.1038/s41598-018-20082-0.
Seung H. Learning in spiking neural networks by reinforcement of stochastic synaptic transmission. Neuron. 2003;40:1063–1073. https://doi.org/10.1016/S0896-6273(03)00761-X.
Xie X, Seung HS. Equivalence of backpropagation and contrastive hebbian learning in a layered network. Neural Comput. 2003;15:441–54.
Fiete IR, Seung HS. Gradient learning in spiking neural networks by dynamic perturbation of conductances. Phys Rev Lett. 2006;97:048104. https://doi.org/10.1103/PhysRevLett.97.048104.
Loewenstein Y, Seung HS. Operant matching is a generic outcome of synaptic plasticity based on the covariance between reward and neural activity. Proc Natl Acad Sci. 2006;103:15224–9. https://doi.org/10.1073/pnas.0505220103.
Loewenstein Y. Robustness of learning that is based on covariance-driven synaptic plasticity. PLOS Comput Biol. 2008;4:e1000007. https://doi.org/10.1371/journal.pcbi.1000007.
Richards BA, Lillicrap TP. Dendritic solutions to the credit assignment problem. Curr Opin Neurobiol. 2019;54:28–36. https://doi.org/10.1016/j.conb.2018.08.003.
Mel BW. Why have dendrites? a computational perspective. In: Dendrites, 22. 2nd ed. Oxford University Press: Oxford, England; 2006.
Antic SD, Zhou W-L, Moore AR, Short SM, Ikonomu KD. The decade of the dendritic NMDA spike. J Neurosci Res. 2010;88:2991–3001. https://doi.org/10.1002/jnr.22444.
Major G, Larkum ME, Schiller J. Active properties of neocortical pyramidal neuron dendrites. Annu Rev Neurosci. 2013;36:1–24. https://doi.org/10.1146/annurev-neuro-062111-150343.
Gambino F, Pagès S, Kehayas V, Baptista D, Tatti R, Carleton A, et al. Sensory-evoked LTP driven by dendritic plateau potentials in vivo. Nature. 2014;515:116–9. https://doi.org/10.1038/nature13664.
Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–52. https://doi.org/10.1016/S0896-6273(02)00573-1.
Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313.
Bloodgood BL, Sabatini BL. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2007;17:345–51. https://doi.org/10.1016/j.conb.2007.04.003.
Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci. 2011;31:4221–32. https://doi.org/10.1523/JNEUROSCI.4561-10.2011.
Higley MJ. Localized GABAergic inhibition of dendritic Ca2+ signalling. Nat Rev Neurosci. 2014;15:567–72. https://doi.org/10.1038/nrn3803.
Tsubokawa H, Ross WN. IPSPs modulate spike backpropagation and associated [Ca2+]i changes in the dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:2896–2906. https://doi.org/10.1152/jn.1996.76.5.2896.
Cichon J, Gan W-B. Branch-specific dendritic Ca2+ spikes cause persistent synaptic plasticity. Nature. 2015;520:180–5. https://doi.org/10.1038/nature14251.
Yang G, Lai CSW, Cichon J, Ma L, Li W, Gan WB. Sleep promotes branch-specific formation of dendritic spines after learning. Science. 2014;344:1173–8. https://doi.org/10.1126/science.1249098.
Sehgal M, Filho DA, Kastellakis G, Kim S, Lee J, Martin S, et al. Co-allocation to overlapping dendritic branches in the retrosplenial cortex integrates memories across time. 2021. https://doi.org/10.1101/2021.10.28.466343.
Rao RPN, Ballard DH. Predictive coding in the visual cortex: a functional interpretation of some extra-classical receptive-field effects. Nat Neurosci. 1999;2:79–87. https://doi.org/10.1038/4580.
Bastos AM, Usrey WM, Adams RA, Mangun GR, Fries P, Friston KJ. Canonical microcircuits for predictive coding. Neuron. 2012;76:695–711. https://doi.org/10.1016/j.neuron.2012.10.038.
Millidge B, Seth A, Buckley CL. Predictive coding: a theoretical and experimental review. arXiv:2107.12979 [cs, q-bio]. (2021)
Dayan P, Daw ND. Decision theory, reinforcement learning, and the brain. Cogn, Affect, Behav Neurosci. 2008;8:429–53. https://doi.org/10.3758/CABN.8.4.429.
Harris KD, Thiele A. Cortical state and attention. Nat Rev Neurosci. 2011;12:509–23. https://doi.org/10.1038/nrn3084.
Sachidhanandam S, Sreenivasan V, Kyriakatos A, Kremer Y, Petersen CCH. Membrane potential correlates of sensory perception in mouse barrel cortex. Nat Neurosci. 2013;16:1671–7. https://doi.org/10.1038/nn.3532.
Neske GT, Nestvogel D, Steffan PJ, McCormick DA. Distinct waking states for strong evoked responses in primary visual cortex and optimal visual detection performance. J Neurosci. 2019;39:10044–59. https://doi.org/10.1523/JNEUROSCI.1226-18.2019.
Sara SJ, Bouret S. Orienting and reorienting: The locus coeruleus mediates cognition through arousal. Neuron. 2012;76:130–41. https://doi.org/10.1016/j.neuron.2012.09.011.
Eldar E, Cohen JD, Niv Y. The effects of neural gain on attention and learning. Nat Neurosci. 2013;16:1146–53. https://doi.org/10.1038/nn.3428.
Waterhouse BD, Navarra RL.The locus coeruleus-norepinephrine system and sensory signal processing: A historical review and current perspectives. Brain Res. 2019;1709. https://doi.org/10.1016/j.brainres.2018.08.032.
Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron. 2012;76:116–29. https://doi.org/10.1016/j.neuron.2012.08.036.
Polack P-O, Friedman J, Golshani P. Cellular mechanisms of brain state-dependent gain modulation in visual cortex. Nat Neurosci. 2013;16:1331–9. https://doi.org/10.1038/nn.3464.
Ljubojevic V, Luu P, Gill PR, Beckett LA, Takehara-Nishiuchi K, De Rosa E. Cholinergic modulation of frontoparietal cortical network dynamics supporting supramodal attention. J Neurosci. 2018;38:3988–4005. https://doi.org/10.1523/JNEUROSCI.2350-17.2018.
Sajedin A, Menhaj MB, Vahabie A-H, Panzeri S, Esteky H. Cholinergic modulation promotes attentional modulation in primary visual cortex- a modeling study. Sci Rep. 2019;9:20186. https://doi.org/10.1038/s41598-019-56608-3.
Ollerenshaw DR, Zheng HJ, Millard DC, Wang Q, Stanley GB. The adaptive trade-off between detection and discrimination in cortical representations and behavior. Neuron. 2014;81:1152–64. https://doi.org/10.1016/j.neuron.2014.01.025.
Zheng HJV, Wang Q, Stanley GB. Adaptive shaping of cortical response selectivity in the vibrissa pathway. J Neurophysiol. 2015;113:3850–65. https://doi.org/10.1152/jn.00978.2014.
Schriver BJ, Bagdasarov S, Wang Q. Pupil-linked arousal modulates behavior in rats performing a whisker deflection direction discrimination task. J Neurophysiol. 2018;120:1655–70. https://doi.org/10.1152/jn.00290.2018.
McGinley MJ, David SV, McCormick DA. Cortical membrane potential signature of optimal states for sensory signal detection. Neuron. 2015;87:179–92. https://doi.org/10.1016/j.neuron.2015.05.038.
McGinley MJ, Vinck M, Reimer J, Batista-Brito R, Zagha E, Cadwell CR. et al. Waking state: Rapid variations modulate neural and behavioral responses. Neuron. 2015;87:1143–61. https://doi.org/10.1016/j.neuron.2015.09.012.
Vinck M, Batista-Brito R, Knoblich U, Cardin JA. Arousal and locomotion make distinct contributions to cortical activity patterns and visual encoding. Neuron. 2015;86:740–54. https://doi.org/10.1016/j.neuron.2015.03.028.
Reimer J, McGinley MJ, Liu Y, Rodenkirch C, Wang Q, McCormick DA. et al. Pupil fluctuations track rapid changes in adrenergic and cholinergic activity in cortex. Nat Commun. 2016;7:13289. https://doi.org/10.1038/ncomms13289.
Lee CC, Kheradpezhouh E, Diamond ME, Arabzadeh E. State-dependent changes in perception and coding in the mouse somatosensory cortex. Cell Rep.2020;32:108197. https://doi.org/10.1016/j.celrep.2020.108197.
Muñoz W, Rudy B. Spatiotemporal specificity in cholinergic control of neocortical function. Curr Opin Neurobiol. 2014;26:149–60. https://doi.org/10.1016/j.conb.2014.02.015.
Kim JH, Jung AH, Jeong D, Choi I, Kim K, Shin S. et al. Selectivity of neuromodulatory projections from the basal forebrain and locus ceruleus to primary sensory cortices. J Neurosci. 2016;36:5314–27. https://doi.org/10.1523/JNEUROSCI.4333-15.2016.
Kanashiro T, Ocker GK, Cohen MR, Doiron B. Attentional modulation of neuronal variability in circuit models of cortex. eLife. 2017;6:e23978. https://doi.org/10.7554/eLife.23978.
Ruff DA, Cohen MR. Simultaneous multi-area recordings suggest that attention improves performance by reshaping stimulus representations. Nat Neurosci. 2019;22:1669–76. https://doi.org/10.1038/s41593-019-0477-1.
Ferro D, Kempen Jv, Boyd M, Panzeri S, Thiele A. Directed information exchange between cortical layers in macaque v1 and v4 and its modulation by selective attention. Proc Natl Acad Sci. 2021;118. https://doi.org/10.1073/pnas.2022097118.
McGaugh JL. THE AMYGDALA MODULATES THE CONSOLIDATION OF MEMORIES OF EMOTIONALLY AROUSING EXPERIENCES. Annu Rev Neurosci. 2004;27:1–28. https://doi.org/10.1146/annurev.neuro.27.070203.144157.
McIntyre CK, McGaugh JL, Williams CL. Interacting brain systems modulate memory consolidation. Neurosci Biobehav Rev. 2012;36:1750–62. https://doi.org/10.1016/j.neubiorev.2011.11.001.
Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 2013;16:1068–76.
Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–4. https://doi.org/10.1038/nature12676.
Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Darcy DP, Nicoll RA. et al. A cortical circuit for gain control by behavioral state. Cell. 2014;156:1139–52. https://doi.org/10.1016/j.cell.2014.01.050.
Eggermann E, Kremer Y, Crochet S, Petersen CC. Cholinergic signals in mouse barrel cortex during active whisker sensing. Cell Rep.2014;9:1654–60. https://doi.org/10.1016/j.celrep.2014.11.005.
Gasselin C, Hohl B, Vernet A, Crochet S, Petersen CC. Cell-type-specific nicotinic input disinhibits mouse barrel cortex during active sensing. Neuron. 2021;109:778–87. https://doi.org/10.1016/j.neuron.2020.12.018.
Chen N, Sugihara H, Sur M. An acetylcholine-activated microcircuit drives temporal dynamics of cortical activity. Nat Neurosci. 2015;18:892–902. https://doi.org/10.1038/nn.4002.
Muñoz W, Tremblay R, Levenstein D, Rudy B. Layer-specific modulation of neocortical dendritic inhibition during active wakefulness. Science. 2017;355:954–9. https://doi.org/10.1126/science.aag2599.
Hasselmo ME, Giocomo LM. Cholinergic modulation of cortical function. J Mol Neurosci. 2006;30:133–6. https://doi.org/10.1385/JMN:30:1:133.
Gulledge AT, Park SB, Kawaguchi Y, Stuart GJ. Heterogeneity of phasic cholinergic signaling in neocortical neurons. J Neurophysiol. 2007;97:2215–29. https://doi.org/10.1152/jn.00493.2006.
Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. 2003;42:33–84. https://doi.org/10.1016/S0165-0173(03)00143-7.
Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: Adaptive gain and optimal performance. Ann Rev Neurosci. 2005;28:403–50.
Rodenkirch C, Liu Y, Schriver BJ, Wang Q. Locus coeruleus activation enhances thalamic feature selectivity via norepinephrine regulation of intrathalamic circuit dynamics. Nat Neurosci. 2019;22:120–33. https://doi.org/10.1038/s41593-018-0283-1.
McBurney-Lin J, Lu J, Zuo Y, Yang H. Locus coeruleus-norepinephrine modulation of sensory processing and perception: A focused review. Neurosci Biobehav Rev. 2019;105:190–9. https://doi.org/10.1016/j.neubiorev.2019.06.009.
McBurney-Lin J, Lu J, Zuo Y, Yang H. Bidirectional pharmacological perturbations of the noradrenergic system differentially affect tactile detection. Neuropharmacology. 2020;174:108151. https://doi.org/10.1016/j.neuropharm.2020.108151.
McCormick DA, Prince DA. Noradrenergic modulation of firing pattern in guinea pig and cat thalamic neurons, in vitro. J Neurophysiol. 1988;59:978–96. https://doi.org/10.1152/jn.1988.59.3.978.
Pape H-C, McCormick DA. Noradrenaline and serotonin selectively modulate thalamic burst firing by enhancing a hyperpolarization-activated cation current. Nature. 1989;340:715–8. https://doi.org/10.1038/340715a0.
Nassar MR, Rumsey KM, Wilson RC, Parikh K, Heasly B, Gold JI. Rational regulation of learning dynamics by pupil-linked arousal systems. Nat Neurosci. 2012;15:1040–6. https://doi.org/10.1038/nn.3130.
Iglesias S, Mathys C, Brodersen KH, Kasper L, Piccirelli M, den Ouden HE, et al. Hierarchical prediction errors in midbrain and basal forebrain during sensory learning. Neuron. 2013;80:519–30. https://doi.org/10.1016/j.neuron.2013.09.009.
Lee T-W, Girolami M, Bell A, Sejnowski T. A unifying information-theoretic framework for independent component analysis. Computers Math Appl. 2000;39:1–21. https://doi.org/10.1016/S0898-1221(00)00101-2.
Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci. 2012;15:816–8. https://doi.org/10.1038/nn.3100.
Hikida T, Yawata S, Yamaguchi T, Danjo T, Sasaoka T, Wang Y. et al. Pathway-specific modulation of nucleus accumbens in reward and aversive behavior via selective transmitter receptors. Proc Natl Acad Sci. 2013;110:342–7. https://doi.org/10.1073/pnas.1220358110.
Stipanovich A, Valjent E, Matamales M, Nishi A, Ahn JH, Maroteaux M. et al. A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature. 2008;453:879–84. https://doi.org/10.1038/nature06994.
Frank MJ, Fossella JA. Neurogenetics and pharmacology of learning, motivation, and cognition. Neuropsychopharmacology. 2011;36:133–52. https://doi.org/10.1038/npp.2010.96.
Jaskir A, Frank MJ. On the normative advantages of dopamine and striatal opponency for learning and choice. bioRxiv. 2022. https://www.biorxiv.org/content/10.1101/2022.03.10.483879v1.abstract. Accessed March 13, 2022.
Ito T, Murray JD. Multi-task representations in human cortex transform along a sensory-to-motor hierarchy, https://doi.org/10.1101/2021.11.29.470432. Company: Cold Spring Harbor Laboratory Distributor: Cold Spring Harbor Laboratory Label: Cold Spring Harbor Laboratory Section: New Results Type: article. (2021)
Fine JM, Hayden BY. The whole prefrontal cortex is premotor cortex. Philos Trans R Soc B: Biol Sci. 2022;377:20200524. https://doi.org/10.1098/rstb.2020.0524.
Harris KD, Shepherd GMG. The neocortical circuit: themes and variations. Nat Neurosci. 2015;18:170–81. https://doi.org/10.1038/nn.3917.
Hosp JA, Nolan HE, Luft AR. Topography and collateralization of dopaminergic projections to primary motor cortex in rats. Exp Brain Res. 2015;233:1365–75. https://doi.org/10.1007/s00221-015-4211-2.
Molina-Luna K, Pekanovic A, Röhrich S, Hertler B, Schubring-Giese M, Rioult-Pedotti MS. et al. Dopamine in motor cortex is necessary for skill learning and synaptic plasticity. PLoS ONE. 2009;4:e7082. https://doi.org/10.1371/journal.pone.0007082.
Hosp JA, Pekanovic A, Rioult-Pedotti MS, Luft AR. Dopaminergic projections from midbrain to primary motor cortex mediate motor skill learning. J Neurosci. 2011;31:2481–7. https://doi.org/10.1523/JNEUROSCI.5411-10.2011.
Gaspar P, Bloch B, Le Moine C. D1 and d2 receptor gene expression in the rat frontal cortex: Cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–63. https://doi.org/10.1111/j.1460-9568.1995.tb01092.x.
Josselyn SA, Shi C, Carlezon WA, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–12. https://doi.org/10.1523/JNEUROSCI.21-07-02404.2001.
Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, et al. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–7. https://doi.org/10.1038/nn1661.
Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA. et al. Neuronal competition and selection during memory formation. Science. 2007;316:457–60. https://doi.org/10.1126/science.1139438.
McKay BM, Matthews EA, Oliveira FA, Disterhoft JF. Intrinsic neuronal excitability is reversibly altered by a single experience in fear conditioning. J Neurophysiol. 2009;102:2763–70. https://doi.org/10.1152/jn.00347.2009.
Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J. et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–43. https://doi.org/10.1038/nn.2405.
Yiu AP, Mercaldo V, Yan C, Richards B, Rashid AJ, Hsiang HLL. et al. Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron. 2014;83:722–35. https://doi.org/10.1016/j.neuron.2014.07.017.
Gouty-Colomer LA, Hosseini B, Marcelo IM, Schreiber J, Slump DE, Yamaguchi S. et al. Arc expression identifies the lateral amygdala fear memory trace. Mol Psychiatry. 2016;21:364–75. https://doi.org/10.1038/mp.2015.18.
Rashid AJ, Yan C, Mercaldo V, Hsiang HLL, Park S, Cole CJ. et al. Competition between engrams influences fear memory formation and recall. Science. 2016;353:383–7. https://doi.org/10.1126/science.aaf0594.
Josselyn SA, Frankland PW. Memory allocation: Mechanisms and function. Annu Rev Neurosci. 2018;41:389–413. https://doi.org/10.1146/annurev-neuro-080317-061956.
Kim D, Samarth P, Feng F, Pare D, Nair SS. Synaptic competition in the lateral amygdala and the stimulus specificity of conditioned fear: a biophysical modeling study. Brain Struct Funct. 2016;221:2163–82. https://doi.org/10.1007/s00429-015-1037-4.
Morrison DJ, Rashid AJ, Yiu AP, Yan C, Frankland PW, Josselyn SA. Parvalbumin interneurons constrain the size of the lateral amygdala engram. Neurobiol Learn Mem. 2016;135:91–9. https://doi.org/10.1016/j.nlm.2016.07.007.
Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–48.
Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–40. https://doi.org/10.1016/j.tins.2010.02.001.
Adams JP, Dudek SM. Late-phase long-term potentiation: getting to the nucleus. Nat Rev Neurosci. 2005;6:737–43. https://doi.org/10.1038/nrn1749.
Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, Barco A. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. https://doi.org/10.1523/JNEUROSCI.3850-07.2007.
Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72.
Gershman SJ, Jones CE, Norman KA, Monfils M-H, Niv Y. Gradual extinction prevents the return of fear: implications for the discovery of state. Front Behav Neurosci. 2013;7:64.
Leutgeb JK, Leutgeb S, Moser M-B, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–6. https://doi.org/10.1126/science.1135801.
McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK. et al. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–9. https://doi.org/10.1126/science.1140263.
Bakker A, Kirwan CB, Miller M, Stark CEL. Pattern separation in the human hippocampal CA3 and dentate gyrus. Science. 2008;319:1640–2. https://doi.org/10.1126/science.1152882.
Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–4. https://doi.org/10.1126/science.1151120.
Moyer Jr. JR, Thompson LT, Disterhoft JF. Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci. 1996;16:5536–46. https://doi.org/10.1523/JNEUROSCI.16-17-05536.1996.
Kaczorowski CC, Disterhoft JF. Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learn Mem. 2009;16:362–6.
Jancic D, Lopez de Armentia M, Valor LM, Olivares R, Barco A. Inhibition of cAMP response element-binding protein reduces neuronal excitability and plasticity, and triggers neurodegeneration. Cereb Cortex. 2009;19:2535–47. https://doi.org/10.1093/cercor/bhp004.
Cai DJ, Aharoni D, Shuman T, Shobe J, Biane J, Song W. et al. A shared neural ensemble links distinct contextual memories encoded close in time. Nature. 2016;534:115–8. https://doi.org/10.1038/nature17955.
Mankin EA, Sparks FT, Slayyeh B, Sutherland RJ, Leutgeb S, Leutgeb JK. Neuronal code for extended time in the hippocampus. Proc Natl Acad Sci. 2012;109:19462–7. https://doi.org/10.1073/pnas.1214107109.
O’Reilly RC, Frank MJ. Making working memory work: A computational model of learning in the prefrontal cortex and basal ganglia. Neural Comput. 2006;18:283–328. https://doi.org/10.1162/089976606775093909.
Stocco A, Lebiere C, Anderson JR. Conditional routing of information to the cortex: A model of the basal ganglia’s role in cognitive coordination. Psychological Rev. 2010;117:541–74. https://doi.org/10.1037/a0019077.
Dayan P. How to set the switches on this thing. Curr Opin Neurobiol. 2012;22:1068–74.
Collins AGE, Frank MJ. Cognitive control over learning: Creating, clustering, and generalizing task-set structure. Psychological Rev. 2013;120:190–229. https://doi.org/10.1037/a0030852.
Chatham CH, Frank MJ, Badre D. Corticostriatal output gating during selection from working memory. Neuron. 2014;81:930–42. https://doi.org/10.1016/j.neuron.2014.01.002.
Rac-Lubashevsky R, Frank MJ. Analogous computations in working memory input, output and motor gating: Electrophysiological and computational modeling evidence. PLOS Comput Biol. 2021;17:e1008971. https://doi.org/10.1371/journal.pcbi.1008971.
Calderon CB, Verguts T, Frank MJ. Thunderstruck: The ACDC model of flexible sequences and rhythms in recurrent neural circuits. PLOS Comput Biol. 2022;18:e1009854. https://doi.org/10.1371/journal.pcbi.1009854.
Kriete T, Noelle DC, Cohen JD, O’Reilly RC. Indirection and symbol-like processing in the prefrontal cortex and basal ganglia. Proc Natl Acad Sci. 2013;110:16390–5. https://doi.org/10.1073/pnas.1303547110.
Badre D, Frank MJ. Mechanisms of hierarchical reinforcement learning in cortico-striatal circuits 2: Evidence from fMRI. Cereb Cortex. 2012;22:527–36. https://doi.org/10.1093/cercor/bhr117.
Franklin NT, Frank MJ. Generalizing to generalize: Humans flexibly switch between compositional and conjunctive structures during reinforcement learning. PLoS Comput Biol. 2020;16:e1007720.
Leong YC, Radulescu A, Daniel R, DeWoskin V, Niv Y. Dynamic interaction between reinforcement learning and attention in multidimensional environments. Neuron. 2017;93:451–63. https://doi.org/10.1016/j.neuron.2016.12.040.
Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic parkinson’s disease. N. Engl J Med. 1988;318:876–80.
Frank MJ, Seeberger LC, O’Reilly RC. By carrot or by stick: Cognitive reinforcement learning in parkinsonism. Science. 2004;306:1940–3. https://doi.org/10.1126/science.1102941.
Beeler JA, Frank MJ, McDaid J, Alexander E, Turkson S, Sol Bernandez M. et al. A role for dopamine-mediated learning in the pathophysiology and treatment of parkinson’s disease. Cell Rep.2012;2:1747–61. https://doi.org/10.1016/j.celrep.2012.11.014.
Maia TV, Frank MJ. An integrative perspective on the role of dopamine in schizophrenia. Biol Psychiatry. 2017;81:52–66. https://doi.org/10.1016/j.biopsych.2016.05.021.
Gold JM, Waltz JA, Frank MJ. Effort cost computation in schizophrenia: A commentary on the recent literature. Biol Psychiatry. 2015;78:747–53. https://doi.org/10.1016/j.biopsych.2015.05.005.
Salamone JD, Correa M, Nunes EJ, Randall PA, Pardo M. The Behavioral Pharmacology of Effort-related Choice Behavior: Dopamine, Adenosine and Beyond. J Exp Anal Behav. 2012;97. https://doi.org/10.1901/jeab.2012.97-125.
Plaisted, KC. Reduced generalization in autism: An alternative to weak central coherence. In: Burack JA, Charman T, Yirmiya N, Zelazo PR, editors. The Development of Autism, 152–71 (Routledge; 2001), 0 edn.
Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain Behav. 2003;2:255–67. https://doi.org/10.1034/j.1601-183X.2003.00037.x.
Li W, Pozzo-Miller L. Dysfunction of the corticostriatal pathway in autism spectrum disorders. J Neurosci Res. 2020;98:2130–47. https://doi.org/10.1002/jnr.24560.
Solomon M, Frank MJ, Smith AC, Ly S, Carter CS. Transitive inference in adults with autism spectrum disorders. Cogn, Affect, Behav Neurosci. 2011;11:437–49. https://doi.org/10.3758/s13415-011-0040-3.
Liu RG, Frank MJ. Hierarchical clustering optimizes the tradeoff between compositionality and expressivity of task structures in reinforcement learning, https://doi.org/10.1101/2021.07.20.453122. Section: New Results Type: article. (2021)
Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu Rev Pathol: Mechanisms Dis. 2015;10:111–44. https://doi.org/10.1146/annurev-pathol-012414-040405.
de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med. 2016;22:345–61. https://doi.org/10.1038/nm.4071.
Mottron L, Bzdok D. Autism spectrum heterogeneity: fact or artifact?. Mol Psychiatry. 2020;25:3178–85. https://doi.org/10.1038/s41380-020-0748-y.
Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–63. https://doi.org/10.1038/nrn3992.
Richter JD, Zhao X. The molecular biology of FMRP: new insights into fragile x syndrome. Nat Rev Neurosci. 2021;22:209–22. https://doi.org/10.1038/s41583-021-00432-0.
Fernandez BA, Scherer SW. Syndromic autism spectrum disorders: moving from a clinically defined to a molecularly defined approach. Dialog Clin Neurosci. 2017;19:353–71.
Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF. et al. Fragile x syndrome. Nat Rev Dis Prim. 2017;3:17065. https://doi.org/10.1038/nrdp.2017.65.
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K. et al. Fragile x mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci. 1997;94:5395–400. https://doi.org/10.1073/pnas.94.10.5395.
Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–82. https://doi.org/10.1016/S0896-6273(00)80336-0.
Kemp N. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. https://doi.org/10.1016/S0028-3908(98)00222-6.
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile x mental retardation. Proc Natl Acad Sci. 2002;99:7746–50. https://doi.org/10.1073/pnas.122205699.
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile x mental retardation. Trends Neurosci. 2004;27:370–7. https://doi.org/10.1016/j.tins.2004.04.009.
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG. et al. Chronic pharmacological mGlu5 inhibition corrects fragile x in adult mice. Neuron. 2012;74:49–56. https://doi.org/10.1016/j.neuron.2012.03.009.
Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile x syndrome. Nat Rev Neurosci. 2015;16:595–605. https://doi.org/10.1038/nrn4001.
Asiminas A, Jackson AD, Louros SR, Till SM, Spano T, Dando O. et al. Sustained correction of associative learning deficits after brief, early treatment in a rat model of fragile x syndrome. Sci Transl Med. 2019;11:eaao0498. https://doi.org/10.1126/scitranslmed.aao0498.
Sidorov MS, Auerbach BD, Bear MF. Fragile x mental retardation protein and synaptic plasticity. Mol Brain. 2013;6:15. https://doi.org/10.1186/1756-6606-6-15.
Contractor A, Klyachko VA, Portera-Cailliau C. Altered neuronal and circuit excitability in fragile x syndrome. Neuron. 2015;87:699–715. https://doi.org/10.1016/j.neuron.2015.06.017.
He C, Portera-Cailliau C. The trouble with spines in fragile x syndrome: density, maturity and plasticity. Neuroscience. 2013;251:120–8. https://doi.org/10.1016/j.neuroscience.2012.03.049.
Martínez-Cerdeño V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Developmental Neurobiol. 2017;77:393–404. https://doi.org/10.1002/dneu.22417.
Bagni C, Zukin RS. A synaptic perspective of fragile x syndrome and autism spectrum disorders. Neuron. 2019;101:1070–88. https://doi.org/10.1016/j.neuron.2019.02.041.
Hinton V, Brown W, Wisniewski K, Rudelli R. Analysis of neocortex in three males with the fragile x syndrome. Am J Med Genet. 1991;41:289–94.
Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ. et al. Abnormal dendritic spines in fragile x knockout mice: Maturation and pruning deficits. Proc Natl Acad Sci. 1997;94:5401–4. https://doi.org/10.1073/pnas.94.10.5401.
Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ. et al. Dendritic spine and dendritic field characteristics of layer v pyramidal neurons in the visual cortex of fragile-x knockout mice. Am J Med Genet. 2002;111:140–6. https://doi.org/10.1002/ajmg.10500.
Galvez R, Gopal AR, Greenough WT. Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile x mental retardation syndrome. Brain Res. 2003;971:83–9.
Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile x mental retardation syndrome. Am J Med Genet Part A. 2005;135:155–60.
McKinney BC, Grossman AW, Elisseou NM, Greenough WT. Dendritic spine abnormalities in the occipital cortex of c57bl/6 fmr1 knockout mice. Am J Med Genet Part B: Neuropsychiatr Genet. 2005;136:98–102.
Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, et al. Reversible inhibition of psd-95 mrna translation by mir-125a, fmrp phosphorylation, and mglur signaling. Mol cell. 2011;42:673–88.
Ifrim MF, Williams KR, Bassell GJ. Single-molecule imaging of PSD-95 mRNA translation in dendrites and its dysregulation in a mouse model of fragile x syndrome. J Neurosci. 2015;35:7116–30. https://doi.org/10.1523/JNEUROSCI.2802-14.2015.
He Q, Nomura T, Xu J, Contractor A. The developmental switch in GABA polarity is delayed in fragile x mice. J Neurosci. 2014;34:446–50. https://doi.org/10.1523/JNEUROSCI.4447-13.2014.
D’Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA. et al. Decreased expression of the GABAA receptor in fragile x syndrome. Brain Res. 2006;1121:238–45. https://doi.org/10.1016/j.brainres.2006.08.115.
Kim SW, Cho KJ. Activity-dependent alterations in the sensitivity to BDNF-TrkB signaling may promote excessive dendritic arborization and spinogenesis in fragile x syndrome in order to compensate for compromised postsynaptic activity. Med Hypotheses. 2014;83:429–35. https://doi.org/10.1016/j.mehy.2014.07.007.
Su T, Fan HX, Jiang T, Sun WW, Den WY, Gao MM, et al. Early continuous inhibition of group 1 mglu signaling partially rescues dendritic spine abnormalities in the fmr1 knockout mouse model for fragile x syndrome. Psychopharmacology. 2011;215:291–300.
Acknowledgements
We would like to thank Peter Hitchcock, Alexander More, and Megha Sehgal for providing feedback on this manuscript and for helpful discussions.
Funding
This work was supported by NIMH training grant T32MH115895 (PI’s: Frank, Badre, Moore), as well as NIMH R01 MH084840-08A1. Computing was supported by NIH Office of the Director grant S10OD025181. We have no disclosures to make.
Author information
Authors and Affiliations
Contributions
DNS and MJF determined the content and perspective. DNS drafted the manuscript. DNS and MJF edited and redrafted the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Scott, D.N., Frank, M.J. Adaptive control of synaptic plasticity integrates micro- and macroscopic network function. Neuropsychopharmacol. 48, 121–144 (2023). https://doi.org/10.1038/s41386-022-01374-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01374-6
This article is cited by
-
Goal-directed learning in adolescence: neurocognitive development and contextual influences
Nature Reviews Neuroscience (2024)
-
Lack of effects of eight-week left dorsolateral prefrontal theta burst stimulation on white matter macro/microstructure and connection in autism
Brain Imaging and Behavior (2024)
