
Abstract
Altered striatal regulation of the GluN2B subunit of N-methyl-D-aspartate (NMDA) glutamate receptors by the Fyn/Src family of protein tyrosine kinases has been implicated in animal alcohol consumption. Previously, we have described differences between individuals positive (FHP) and negative (FHN) for familial alcohol use disorder (AUD) in the ventral striatal (VS) activation associated with monetary incentive delay task (MIDT) performance during functional magnetic resonance imaging (fMRI). Here, we used AZD0530 (saracatinib), a centrally active Fyn/Src inhibitor to probe the role of Fyn/Src regulation of NMDA receptors (NMDAR) in VS activation differences between FHP and FHN individuals during fMRI MIDT performance. We studied 21 FHN and 22 FHP individuals, all without AUD. In two sessions, spaced 1 week apart, we administered 125 mg of saracatinib or placebo in a double-blind manner, prior to measuring VS signal during fMRI MIDT performance. MIDT comprises reward prospect, anticipation, and outcome phases. During the initial (prospect of reward) task phase, there was a significant group-by-condition interaction such that, relative to placebo, saracatinib reduced VS BOLD signal in FHP and increased it in FHN individuals. This study provides the first human evidence that elevated signaling in striatal protein kinase A-dependent pathways may contribute to familial AUD risk via amplifying the neural response to the prospect of reward. As Fyn kinase is responsible for NMDAR upregulation, these data are consistent with previous evidence for upregulated NMDAR function within reward circuitry in AUD risk. These findings also suggest a possible therapeutic role for Src/Fyn kinase inhibitors in AUD risk.
Similar content being viewed by others

Introduction
Approximately half of the risk for alcohol use disorder (AUD) is inherited [1, 2]. Individuals with a strong family history of AUD (FHP), like those with AUD, tend to respond more to stimuli signaling high levels of immediate reward, contributing to their “sensation seeking” or “impulsivity” [3,4,5,6,7]. Also, while euphoric effects of ethanol are intact or increased in high-risk groups, negative alcohol effects that normally constrain heavy drinking [8, 9] are reduced in healthy FHP individuals, increasing their AUD risk [10]. Ethanol acts through many signaling mechanisms including blockade of N-methyl-D-aspartate glutamate receptor (NMDA-R). FHP individuals FHP for AUD, relative to FHNs, also show enhanced euphoria and reduced dysophoric, cognitive, and neural responses to a wide range of NMDA-R antagonists and partial agonists including ketamine, memantine, and nitrous oxide [11,12,13,14,15]. These studies have been interpreted as suggesting that the familial risk for AUD is associated with with upregulation of NMDA-R function, altering ethanol responses and perhaps other circuit mechanisms contributing to AUD risk (Krystal et al. 2003; Krystal et al. 2002) and that AUD might be treated by targeting glutamate signaling [16]. However the cause of NMDA-R upregulation associated with AUD risk is not yet clear.
The biology of the heritable risk for AUD is both complex and poorly understood. It appears to involve plasticity within cortico-striatal pathways, modulated by midbrain dopamine input [17,18,19]. The striatal indirect pathway, expressing dopamine D2 receptors (D2Rs), serves a protective role in addiction, while the striatal direct pathway, expressing dopamine D1 receptors (D1Rs) is implicated in both reward and addiction [20]. D1Rs promote striatal plasticity by activating protein kinase A (PKA) intracellular signaling pathways with steps that converge upon the phosphorylation of Fyn kinase, which phosphorylates GluN2B subunits and thereby promotes the synaptic presence of glutamate receptors, promoting striatal excitability and neuroplasticity [21,22,23]. In contast, activation of D2Rs has the opposite effect. In animal models, activation of D2R striatal signaling and the indirect pathway is protective against alcohol self-administration [24, 25]. In humans, higher levels of striatal D2Rs also appear to be protective against drinking in social drinkers [26], and individuals with AUD show reductions in striatal D2Rs [27, 28]. Furthermore, preclinical studies suggest that inhibitors of Fyn/Src kinases reduce alcohol reward and alcohol-seeking in mice [29, 30], and Fyn kinase is upregulated after repeated ethanol exposures, particularly in the dorso-medial striatum [31]. These findings suggest that relative deficits in D2R signaling relative to signaling via D1R, i.e., an imbalance of signaling via the direct and indirect pathways, might contribute to AUD risk in part via enhanced PKA signaling, activation of Fyn kinase, synaptic upregulation of striatal GluN2B-containing NMDA-Rs, and increased striatal activation and neuroplasticity associated with reward processing.
The VS has been particularly implicated in reward processing with FHP individuals, showing similar blunting of activation during reward anticipation processing as seen in individuals with AUD and other addictions [32,33,34,35]. The VS is important not only in connecting motivation to action [36], but also as a site where both abstract/learned rewards (such as money) and abused substances manifest their rewarding effects [19, 37]. Furthermore, VS activation can be assessed quantitatively in living volunteers using fMRI in studies employing the Monetary Incentive Delay Task (MIDT). The original task distinguished anticipatory and consummatory phases of reward and loss processing [38, 39], while a modified version can be used to evaluate the prospect of reward as well [32]. Our ability to separately model prospect and anticipation phases permitted identification in FHP of enhanced VS activation during the prospect phase of winning and blunted activation during the anticipation and outcome of winning phases [32].
To test the hypothesis that enhanced signaling via PKA-dependent pathways might contribute to enhanced VS activation during the prospect of reward in FHP, we compared the effects of the Fyn/Src kinase inhibitor, saracatinib (AZD0530) [40], to placebo during performance of the MIDT in family history negative (FHN) and FHP individuals without AUD. We chose to focus exclusively on VS activation during reward processing. We hypothesized that the altered VS signals in FHP individuals during prospect (relatively increased versus FHN) and anticipatory and outcome (relatively decreased versus FHN) phases of reward processing during performance of the MIDT would be replicated and normalized by saracatinib administration. The resulting findings should provide a better understanding of neural signaling mechanisms in persons at high familial risk of AUD.
Methods and materials
Participants
From N = 79 initial volunteers, 51 subjects met study eligibility criteria and were allocated to the intervention. Of these, 50 received both active drug and placebo, and 43 had analyzable data following image inspection for missing data, motion and artifacts. The latter consisted of 22 FHN subjects (age 23.09 yr ± 4.2 yr; 54.3% female) and 21 FHP subjects (age 25.10 yr ± 5.0 yr; 85.7% female). Although our intial intent was to complete 80 subjects in the trial, based on estimated power, the study was stopped when 51 subjects had been recruited into the study, due to expiration of the 125 mg study drug supply. Neither group contained subjects with past or current alcohol dependence. The FHP group had multiple affected first-degree relatives including at least 1 parent, and the FHN group had no family history in any first- or second-degree relative. See Table 1 for additional demographic information. Further study inclusion criteria included age 18–45 years and estimated full-scale IQ > 85 estimated using the WASI-II [41]. Participants were excluded for histories of brain trauma sufficient to cause loss of consciousness for >10 min, major medical condition (e.g., cancer), Axis-1 psychiatric disorders (assessed via the SCID-V-RV [42]), current pregnancy (by urine testing), positive urine drug or breath alcohol test at time of MRI scan visits, and having non MRI-safe metal implants or severe claustrophobia.
Subjects were recruited from 6/23/2015 to 4/26/2019 at the Olin Center, Institute of Living, Hartford CT, from community advertisements placed by the Institute of Living at Hartford Hospital and Yale University. Subjects meeting inclusion criteria were entered consecutively into the study. All participants signed informed consent approved by Hartford Hospital and Yale University Institutional Review Boards.
Experiment
Each subject was administered either placebo or 125 mg dose of saracatinib on 2 different days, one week apart. The identical-appearing doses were administered in a randomized, double-blind, counterbalanced manner with assignment supervised by the hospital pharmacy, using randomization tables. Doses were supplied by the pharmacy in sequentially numbered packets for each subject. The research assistants (AG, AD) were blind to drug placebo dosing; the study physician (GDP) and image analyst (KTP) were blind to both subject FH status and to drug/placebo allocation. Subjects were also blind to the drug they received. The drug dose was chosen based on prior reports in human trials [43]. Side effects likely due to the study medication were minimal and are reported in Supplementary Table 1. The study was originally designed and funded for two different saracatnib doses (50 mg and 125 mg) and placebo. AstraZeneca subsequently discontinued the 50 mg dose and recommended that we test the 125 mg dose exclusively. At the time this decision was made, only 15 total subjects had received an additional 50 mg dose; therefore, these lower-dose data were not analyzed. We only incuded the 125 mg dose and placebo scans in the current analyses. The CONSORT flowchart (Supplementary Fig. S1) includes details of the full study design. This study is registered on ClinicalTrial.gov (Title: “A Phase One Study Investigating the Tolerability and Effects of AZD0530 on Functional Neuroimaging Responses in Individuals With or Without a Family History of Alcoholism”; Registration: NCT02262026; URL: https://clinicaltrials.gov/ct2/show/NCT02262026).
FMRI data were acquired on a Siemens Skyra 3 T MRI scanner (Siemens, Malvern PA). Functional MIDT data were acquired using a multiband gradient-echo sequence (Axial, TR = 720 ms, TE = 30 ms, FOV = 240 mm, flip angle = 60°, acquisition matrix = 80 × 80, voxel size = 3 mm3, number of slices × 48, multi band factor = 8, iPAT = 1).
fMRI task
Prior to entering the scanner, ~5 h post drug administration, (a time representing optimal trade-off between the drug’s median tmax range of 2–7 h and feasibility within the study day [40]), subjects practiced the MIDT for 15 min to ensure that they understood task instructions and to assess individual reaction times (RTs). The in-scanner MIDT was adapted to each individual subject’s RT to ensure winning/failing to lose on ~66% of trials. Briefly, the task included three major phases: Prospect, Anticipation and Outcome of reward and loss. Prospect phase: In a single 12-minute session, each subject was shown 6 possible word cues (duration 1000msec) indicating possible monetary rewards ($0, $1, $5) or losses ($0, $1, $5), following which they prepared to respond with a button press on a box cue that appeared on the display screen for a variable length (RT ± SD) of time. Anticipation phase: participants were asked to press the button as quickly as possible; if they did so within the individually pre-determined time span, they won/did not lose money. If they did not press the button sufficiently quickly, then they did not win/lost money. Outcome phase: Finally, feedback on outcome was provided on the screen by notifying whether the participant had won/did not win (receipt/non-receipt of reward) or had not lost/lost (avoidance/receipt of loss) during each trial. Cumulative winnings were displayed on the feedback screen. Participants were paid one-third of their overall cumulative winnings to maximize performance incentive, as detailed previously [32, 44]. See Supplementary Fig. S2 for a flow chart of the MIDT design.
Subjects were also administered psychological and behavioral self-report batteries, incuding the Alcohol Use Disorder Test (AUDIT) [45], Fagerstrom Test for Nicotine Dependence (FTND) [46], Short Inventory of Problems (SIP-2R) [47], Barratt Impulsivity Scale-11 (BIS11) [48] and Behavioral Inhibition System/ Behavioral Acivation System Scale (BIS-BAS) [49]. Of subjects who were included in the study, 8 had missing data from one or more of the above-mentioned psychological test batteries. See Table 1 for group statistics.
Analysis
Our analyses included only subjects with complete data for both placebo and active (125 mg) drug scans. Six subjects were excluded from analyses due to missing MIDT behavioral data (N = 2), in-scanner motion greater than 1.5 voxel (N = 2), technically compromised scan quality (N = 1), different scan TR (N = 1) or pre-visit screening compliance issues (N = 1). Some subjects had >1 disqualifier. We assessed fMRI BOLD response during reward/loss processing phases. MIDT data were pre-processed using Statistical Parametric Mapping (SPM v12) software (https://www.fil.ion.ucl.ac.uk/spm/software/spm12/). fMRI data were realigned and normalized to the SPM default tissue probability map image (TPM.nii). Data were smoothed using a Gaussian smoothing kernel of 8 mm full-width half-maximum in SPM 12. The MIDT was modeled using SPM12 for reward and loss phases including $0, $1, $5 trials. Based on our previous findings in FHP [32], we investigated VS activity during reward phases. Subjects were evaluated for number of trials per phase to eliminate noise due to very few trials. All subjects had >4 trials for all three reward phases.
We created an in-house left and right binary VS mask as previously reported [44]. This region is largely identical to VS definitions from other atlases (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/Atlases). We used the MarsBaR [50] region of interest tool in SPM12 to extract raw mean values for these pre-selected left and right VS ROIs for reward-phase-related task contrasts including (1) Prospect of Win$1 and Win$5 vs. implicit baseline, (2) Anticipation of Win$1 and Win$5 vs. implicit baseline, and (3) Outcome of Win$1 and Win$5 vs. implicit baseline. Extracted mean values were then imported and analyzed in the R software lmer4 package [51] using a linear mixed model fit with restricted maximum likelihood (ReML), including group (FHN/FHP) and drug conditions (Placebo/125 mg Saracatinib dose) as fixed effects and subjects as random effects. We included group and condition main effects as well as group x condition interaction effects in a mixed model linear regression. We ensured that our sample followed normal distribution patterns using Q-Q plot (Supplementary Fig. S3) and density plot (Supplementary Fig. S4) using the ggplot2 [52] package in R. We used False Discovery Rate (FDR) correction for multiple comparisons and corrected for for 18 tests conducted using p.adjust function in R. We also adjusted for gender effects in the analysis as the groups were unbalanced. We calculated the likelihood ratio using the ANOVA function in R to compare models and to determine whether models were significantly different. To calculate likelihood ratios, we compared regression models a. with and without interation term b with and without group main effect and c with and without condition main effect while keeping remaining terms constant in each test.
Results
Uncorrected activation maps for Prospect, Anticipation and Outcome of Win trials are illustrated in Supplementary Fig. S5. None of the analyses results were significantly different when adjusted for gender due to unbalanced male/female ratio in groups. We report results that not gender adjusted and are p < 0.05 FDR-corrected
Prospect of win
-
a
Right VS: There were no significant group main effects (Table 2). We found significant condition main effects (t value = −2.843; Puncorrected = 0.007; PFDR = 0.042) and GroupxCondition interation effects (t value = 2.885, Puncorrected = 0.006 PFDR = 0.042; Table 2) (Table 3; Fig. 1). For interaction effects, the likelihood ratio was significant (Pr | ChiSq | = 0.0037). The likelihood of the model without an interaction term (LogLik = −229.92) was a poorer fit compared to the model with an interaction term (LogLik = −225.70). We also detected a relatively large effect size (Cohen’s d = 0.90, Table 2) for the interaction model during this phase. During the Prospect Of Win phase, FHP individuals showed significantly more right VS BOLD activation than FHN individuals following placebo administration (Fig. 2). With active drug administration, right VS BOLD signal decreased in FHP and increased in FHN individuals. Detailed statistics of the interaction model fit are described in Table 3.
Table 2 Table illustrates main effects of Group (family history positive, family history negative) and Condition (drug: saracatnib/placebo) as well as interaction of Group x Condition for right and left ventral striatum during MID prospect, anticipation and outcome of win conditions. Table 3 Interaction model fit for Prospect of Win phase of the Monetary Incentive Delay task. Fig. 1: Boxplot for MIDT ventral striatum activity in FHN and FHP groups during drug and placebo conditions. 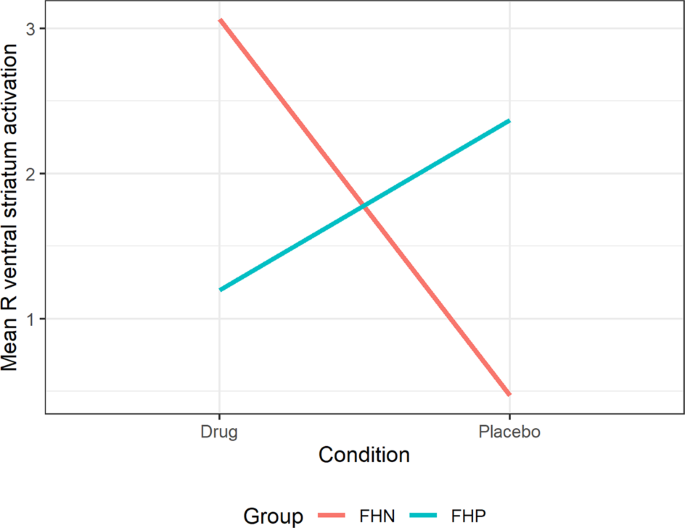
Illustrates boxplot of right VS (ventral striatum) activity during prospect of winning (Prospect of Win) phase during performance of the monetary incentive delay task for each group (FHN = family history negative; FHP = family history positive) and each drug condition (saracatinib/placebo) with mean and standard deviation.
Fig. 2: Group by dose interaction for ventral striatum activity during MIDT. 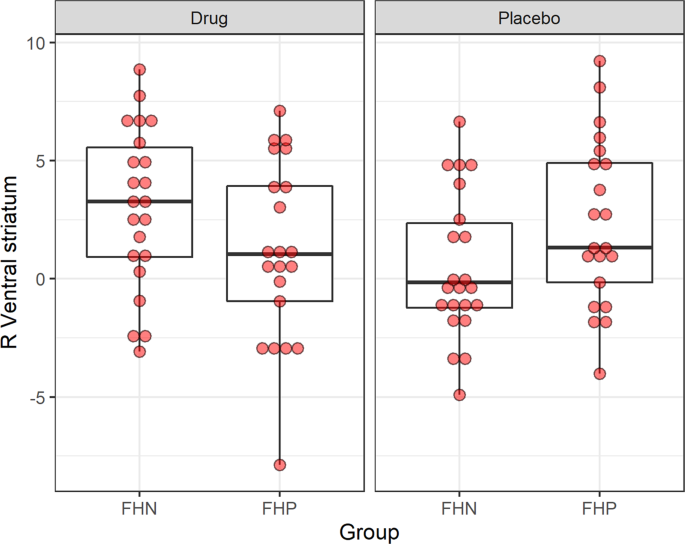
Figure illustrates group (FHN/FHP) x dose (saracatinib 125 mg/ Placebo) interaction during the MIDT prospect of winning phase (Prospect of Win) in the right VS (ventral striatum). FHN Family history negative, FHP Family history positive, MIDT Monetary Incentive Delay Task.
-
b
Left VS: There were no significant group or condition main effects. GroupxCondition interaction effects were only significant at uncorrected p values (Table 2).


Anticipation of Win
Right and Left VS: There were no significant group or condition main effects or GroupxCondition interaction effects.
Outcome of Win
-
a
Right VS: Group main effect was not significant during Outcome Of Win. We found a condition main effect (t value = −2.927; p uncorrected = 0.005; p FDR = 0.042). A GroupxCondition interaction was significant only at uncorrected levels (Table 2).
-
b
Left VS: There were no significant group or condition main effects or GroupxCondition interaction effects.
Discussion
The principal finding of this study was an interactive impact of family history and saracatinib on ventral striatum activation associated with the prospect of reward, but not the anticipation of a delayed and uncertain reward or the receipt of the reward phases of MIDT. This interactive effect reflected, under the placebo condition, a replication of the increased VS activation during the prospect of reward phase in FHP relative to FHN reported by Andrews et al. [32]. Also, saracatinib reduced VS activation in FHP but increased it in FHN. These findings were most robust in the right VS, and no significant group differences or interactions were observed for the Anticipation of Win and Outcome of Win phases. One might say that saractinib “normalized” the FHP response in that it returned it to a level similar to the FHN, but it may have “pathologized” the FHN response. These data suggest that there may be an optimal level of signaling via Fyn/Src and that hyperactivity, as seen in FHP, and deficits, as seen in FHN administered saractinib, augment VS activity during the prospect of reward, biasing individuals toward impulsive reward-related decision making (Andrews et al. 2011). In other words, there may be an “inverted U curve” describing the relationship between Fyn kinase activation and VS activation when processing cues associated with the prospect of reward.
This study provides the first human evidence that accentuated signaling via Fyn/Src kinase may contribute to the familial risk for AUD by enhancing activation of reward circuitry. As Fyn/Src are downstream from PKA in ventral striatal signaling pathways, it is tempting to place the current findings within the context of the drivers of PKA activity in the striatum. In particular, the possibility that increased signaling via Fyn/Src in FHP is a consequence of an alteration in the balance between D1R signaling in the direct pathway relative to D2R signaling in the indirect pathway. Both accentuation of D1R signaling and deficits in D2R signaling could explain the current findings. As noted earlier, higher D2R levels were found to be protective against drinking (Thanos et al,. 2008), leading one to hypothesize reductions in D2R in FHP. This might be consistent with increased DNA methylation of the DRD2 gene in white blood cells from FHP [53]. However, reductions in D2R/D3R were not found in FHP relative to FHN [54] under resting conditions. This finding does not rule out state-dependent dopaminergic alterations in FHP. In fact, in FHP, increased dopamine release has been observed in association with anticipation of alcohol or exposure to alcohol cues [55, 56]. If this were the case, it would suggest that the dopaminergic dysregulation originates from another primary risk mechanism.
The current findings also converge with studies reviewed earlier suggesting that upregulation of NMDA-Rs constitute an AUD risk mechanism. One possible interpretation of the current findings would be that upregulation of ventral striatal GluN2B receptors in the synapse in FHP, perhaps as a consequence of enhanced PKA-Fyn activation, was ameliorated by Fyn kinase inhibition, which would be expected to downregulate NMDA-Rs without blocking them. This hypothesis might be consistent with the evidence of NMDA-R upregulation in FHP (Petrakis et al. 2004; Krystal et al. 2003) and the reductions in alcohol craving produced by memantine in heavy drinking FHP [57] and AUD [58]. However, this study also found evidence of increased VS activation in FHN and this might be expected to increase impulsive decision making and to undermine anti-drinking effects of memantine, as has been reported previously in both a laboratory study (Krishnan-Sarin) and a clinical trial [59].
There are strengths and weaknesses to the current study. Saracatinib inhibits Src as well as Fyn, so that the mechanism of the drug’s primary mode of action on NMDA-R that we are hypothesizing may not be entirely specific. We selected FHP individuals without AUD to avoid confounding alcohol effects with risk, but acknowledge that by doing so we may have selected a group that was at lower alcoholism risk than those FHP individuals who have already gone on to develop AUD. Also, individuals with chronic AUD may differ from those at risk without that status, and the current results may or may not generalize to them. Randomization of the study was executed successfully, as subjects could not identify accurately whether they had received drug or placebo when queried following drug admistration. This study was orginally designed for two doses of saracatnib, but we needed to stop administering the smaller dose early in the study, precluding analysis of its effects. Thus, we did not compare multiple doses of saracatnib, as intended. We also adopted an acute challenge study using a single drug dose administration rather than building up to steady state with repeated dosing. This latter approach would have required a longer, more complex study with likely increased cost, subject burden and dropout. Future studies should examine other doses and durations. Researchers should also attempt to replicate our findings in a larger dataset and in treatment-seeking AUD individuals before clinical application, including in a 2 × 2 design (FHP with and without AUD and FHN with and without AUD). While a strength of our approach was to focus on the VS in a hypothesis-driven manner, we may have missed other regional brain activation differences. A Go/No-Go fMRI paradigm was administered to all subjects immediately after the MIDT; results from that portion of the study will be reported separately elsewhere.
Given saracatinib’s functional effects on the VS, it might seem reasonable to test it as a possible therapeutic agent in individuals with AUD, although brain effects do not necessarily equate to behavioral alterations. In that regard, although various behavioral measures such as impulsivity assessments were quantified out of scanner at the baseline visit, prior to important for administration of drug/placebo, these assessments were not repeated at subsequent drug/placebo challenge visits to measure whether there were altered by the study drug. This is a limitation of the study that should be addressed in future attempts at replication and extension of the current data. Another future direction might be to examine the MIDT findings in younger FHN and FHP individuals (e.g., in the ABCD cohort, which is using the MIDT). An earlier report using a MIDT version that did not separate the Prospect and Anticipation phases [60] found diminished VS response to anticipated rewards and punishments in at-risk adolescents. However, young individuals and adolescents in general show diminished VS responses to rewards on the MIDT [61]. Another limitation of the study is that ethnicity, race and SIP total scores differed significantly between FHN and FHP groups.
To summarize, we replicate in a new sample the prior finding of increased VS activation during the MIDT Prospect of Win phase in FHP compared to FHN healthy subjects, and show that the Src/Fyn kinase inhibitor, saracatinib, a non-selective regulator of NMDA-R, attenuates this VS over-activation in FHP, eliminating the group VS activation difference between FHP and FHN individuals, albeit also altering FHN response in addition. This study provides an initial test of the hypothesis that upregulation of NMDA-R function associated with FHP may be dependent on signaling mechanisms downstream from dopamine actions at dopamine receptors, (i.e., PKA pathway signaling proteins STEP and Fyn). Although Src/Fyn inhibitors reduce alcohol reward and alcohol-seeking in mice [29, 30], these data may not necessarily translate into equivalent human behaviors. Nonetheless, the current findings suggest possible novel therapeutic approaches to target reward/motivation disturbances that contribute to AUD risk.
References
Kendler KS, Ohlsson H, Sundquist J, Sundquist K. Prediction of onset of substance-induced psychotic disorder and its progression to Schizophrenia in a Swedish national sample. Am J Psychiatry. 2019;176:711–9.
Richmond-Rakerd LS, Slutske WS, Lynskey MT, Agrawal A, Madden PA, Bucholz KK, et al. Age at first use and later substance use disorder: shared genetic and environmental pathways for nicotine, alcohol, and cannabis. J Abnorm Psychol. 2016;125:946–59.
Whipple SC, Berman SM, Noble EP. Event-related potentials in alcoholic fathers and their sons. Alcohol. 1991;8:321–7.
Finn PR, Earleywine M, Pihl RO. Sensation seeking, stress reactivity, and alcohol dampening discriminate the density of a family history of alcoholism. Alcohol Clin Exp Res. 1992;16:585–90.
Hesselbrock MN, Hesselbrock VM. Relationship of family history, antisocial personality disorder and personality traits in young men at risk for alcoholism. J Stud Alcohol. 1992;53:619–25.
Petry NM, Kirby KN, Kranzler HR. Effects of gender and family history of alcohol dependence on a behavioral task of impulsivity in healthy subjects. J Stud Alcohol. 2002;63:83–90.
Saunders B, Farag N, Vincent AS, Collins FL Jr, Sorocco KH, Lovallo WR. Impulsive errors on a Go-NoGo reaction time task: disinhibitory traits in relation to a family history of alcoholism. Alcohol Clin Exp Res. 2008;32:888–94.
Mello NK, Mendelson JH. Behavioral studies of sleep patterns in alcoholics during intoxication and withdrawal. J Pharm Exp Ther. 1970;175:94–112.
Mello NK, Mendelson JH. Experimentally induced intoxication in alcoholics: a comparison between programed and spontaneous drinking. J Pharm Exp Ther. 1970;173:101–16.
Schuckit MA. A 10-year follow-up of sons of alcoholics: preliminary results. Alcohol Alcohol Suppl. 1991;1:147–9.
Narayanan B, Stevens MC, Jiantonio RE, Krystal JH, Pearlson GD. Effects of memantine on event-related potential, oscillations, and complexity in individuals with and without family histories of alcoholism. J Stud Alcohol Drugs. 2013;74:245–57.
Dickerson D, Pittman B, Ralevski E, Perrino A, Limoncelli D, Edgecombe J, et al. Ethanol-like effects of thiopental and ketamine in healthy humans. J Psychopharmacol. 2010;24:203–11.
Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, et al. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–82.
Walsh K, Das RK, Kamboj SK. The subjective response to nitrous oxide is a potential pharmaco-endophenotype for alcohol use disorder: a preliminary study with heavy drinkers. Int J Neuropsychopharmacol. 2017;20:346–50.
Jamadar S, DeVito EE, Jiantonio RE, Meda SA, Stevens MC, Potenza MN, et al. Memantine, an NMDA receptor antagonist, differentially influences Go/No-Go performance and fMRI activity in individuals with and without a family history of alcoholism. Psychopharmacol (Berl). 2012;222:129–40.
Holmes A, Spanagel R, Krystal JH. Glutamatergic targets for new alcohol medications. Psychopharmacol (Berl). 2013;229:539–54.
Robbins C. Sex differences in psychosocial consequences of alcohol and drug abuse. J Health Soc Behav. 1989;30:117–30.
Jentsch JD, Roth RH, Taylor JR. Role for dopamine in the behavioral functions of the prefrontal corticostriatal system: implications for mental disorders and psychotropic drug action. Prog Brain Res. 2000;126:433–53.
Wise RA, Koob GF. The development and maintenance of drug addiction. Neuropsychopharmacology. 2014;39:254–62.
Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, et al. Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat Neurosci. 2013;16:632–8.
Mao LM, Wang JQ. Dopamine D2 receptors are involved in the regulation of Fyn and metabotropic glutamate receptor 5 phosphorylation in the rat striatum in vivo. J Neurosci Res. 2016;94:329–38.
Trepanier CH, Jackson MF, MacDonald JF. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012;279:12–9.
Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharm. 2004;65:121–9.
Thanos PK, Taintor NB, Rivera SN, Umegaki H, Ikari H, Roth G, et al. DRD2 gene transfer into the nucleus accumbens core of the alcohol preferring and nonpreferring rats attenuates alcohol drinking. Alcohol Clin Exp Res. 2004;28:720–8.
Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13:635–41.
Thanos PK, Wang GJ, Volkow ND. Positron emission tomography as a tool for studying alcohol abuse. Alcohol Res Health. 2008;31:233–7.
Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemann R, Ding YS, et al. Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcohol Clin Exp Res. 1996;20:1594–8.
Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A, et al. Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychiatry. 2005;58:779–86.
Morisot N, Berger AL, Phamluong K, Cross A, Ron D. The Fyn kinase inhibitor, AZD0530, suppresses mouse alcohol self-administration and seeking. Addict Biol. 2019;24:1227–34.
Yaka R, Tang KC, Camarini R, Janak PH, Ron D. Fyn kinase and NR2B-containing NMDA receptors regulate acute ethanol sensitivity but not ethanol intake or conditioned reward. Alcohol Clin Exp Res. 2003;27:1736–42.
Wang J, Lanfranco MF, Gibb SL, Yowell QV, Carnicella S, Ron D. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–98.
Andrews MM, Meda SA, Thomas AD, Potenza MN, Krystal JH, Worhunsky P, et al. Individuals family history positive for alcoholism show functional magnetic resonance imaging differences in reward sensitivity that are related to impulsivity factors. Biol Psychiatry. 2011;69:675–83.
Luijten M, Schellekens AF, Kuhn S, Machielse MW, Sescousse G. Disruption of reward processing in addiction: an image-based meta-analysis of functional magnetic resonance imaging studies. JAMA psychiatry. 2017;74:387–98.
Beck A, Schlagenhauf F, Wustenberg T, Hein J, Kienast T, Kahnt T, et al. Ventral striatal activation during reward anticipation correlates with impulsivity in alcoholics. Biol Psychiatry. 2009;66:734–42.
Wrase J, Schlagenhauf F, Kienast T, Wustenberg T, Bermpohl F, Kahnt T, et al. Dysfunction of reward processing correlates with alcohol craving in detoxified alcoholics. Neuroimage 2007;35:787–94.
Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–76.
Robinson TE, Berridge KC. The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 2000;95:S91–117. Suppl 2.
Knutson B, Fong GW, Adams CM, Varner JL, Hommer D. Dissociation of reward anticipation and outcome with event-related fMRI. Neuroreport 2001;12:3683–7.
Knutson B, Momenan R, Rawlings RR, Fong GW, Hommer D. Negative association of neuroticism with brain volume ratio in healthy humans. Biol Psychiatry. 2001;50:685–90.
Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP. et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem.2006;49:6465–88.
Wechsler D (1999): Wechsler Abbreviated Scale of Intelligence. San Antonio, TX: The Psychological Corporation.
First MB, Williams, JB, Karg, RS, Spitzer, RL (2015): User’s Guide for the Structured Clinical Interview for DSM-5 Disorders, Research Version (SCID-5-RV). Arlington, VA: American Psychiatric Association.
Nygaard HB, Wagner AF, Bowen GS, Good SP, MacAvoy MG, Strittmatter KA, et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimers Res Ther. 2015;7:35.
Patel KT, Stevens MC, Meda SA, Muska C, Thomas AD, Potenza MN, et al. Robust changes in reward circuitry during reward loss in current and former cocaine users during performance of a monetary incentive delay task. Biol Psychiatry. 2013;74:529–37.
World Health O (2001): AUDIT: the Alcohol Use Disorders Identification Test: guidelines for use in primary health care/Thomas F. Babor, [et al.]. 2nd ed ed. Geneva: World Health Organization.
Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO. The Fagerstrom Test for Nicotine Dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict. 1991;86:1119–27.
Miller WR, Tonigan, JS and Longbaugh, R (1995): The Drinker Inventory of Consequences (DrInC): An instrument for assessing adverse consequences of alcohol abuse (Test Manual). Bethesda, MD: Department of Health and Human Services, pp 95–3911.
Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt impulsiveness scale. J Clin Psychol. 1995;51:768–74.
Carver CS, White TL. Behavioral inhibition, behavioral activation, and affective responses to impending reward and punishment: the BIS/BAS scales. J Pers Soc Psychol. 1994;67:319–33.
Brett M, Anton JL, Valabregue R, Poline JB (2002): Region of interest analysis using an SPM toolbox. 8th International Conference on Functional Mapping of the Human Brain. Sendai, Japan: Neuroimage.
Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:48.
Wickham H (2016): ggplot2: Elegant graphics for data analysis. New York: Springer-Verlang.
Hill SY, Sharma VK. DRD2 methylation and regional grey matter volumes in young adult offspring from families at ultra-high risk for alcohol dependence. Psychiatry Res Neuroimaging. 2019;286:31–8.
Alvanzo AA, Wand GS, Kuwabara H, Wong DF, Xu X, McCaul ME. Family history of alcoholism is related to increased D2 /D3 receptor binding potential: a marker of resilience or risk? Addict Biol. 2017;22:218–28.
Kegeles LS, Horga G, Ghazzaoui R, Rosengard R, Ojeil N, Xu X, et al. Enhanced striatal dopamine release to expectation of alcohol: a potential risk factor for alcohol use disorder. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:591–8.
Oberlin BG, Dzemidzic M, Tran SM, Soeurt CM, Albrecht DS, Yoder KK, et al. Beer flavor provokes striatal dopamine release in male drinkers: mediation by family history of alcoholism. Neuropsychopharmacology. 2013;38:1617–24.
Krishnan-Sarin S, O’Malley SS, Franco N, Cavallo DA, Morean M, Shi J, et al. N-methyl-D-aspartate receptor antagonism has differential effects on alcohol craving and drinking in heavy drinkers. Alcohol Clin Exp Res. 2015;39:300–7.
Krupitsky EM, Neznanova O, Masalov D, Burakov AM, Didenko T, Romanova T, et al. Effect of memantine on cue-induced alcohol craving in recovering alcohol-dependent patients. Am J Psychiatry. 2007;164:519–23.
Evans SM, Levin FR, Brooks DJ, Garawi F. A pilot double-blind treatment trial of memantine for alcohol dependence. Alcohol Clin Exp Res. 2007;31:775–82.
Yau WY, Zubieta JK, Weiland BJ, Samudra PG, Zucker RA, Heitzeg MM. Nucleus accumbens response to incentive stimuli anticipation in children of alcoholics: relationships with precursive behavioral risk and lifetime alcohol use. J Neurosci. 2012;32:2544–51.
Dhingra I, Zhang S, Zhornitsky S, Le TM, Wang W, Chao HH, et al. The effects of age on reward magnitude processing in the monetary incentive delay task. Neuroimage. 2020;207:116368.
Funding information
This study was funded by 2P50 AA012870 to JK. Study drug and placebo were provided by Astra-Zeneca. Dr. Potenza has consulted for and advised Game Day Data, the Addiction Policy Forum, AXA, Idorsia, and Opiant Therapeutics; has received research support from the Connecticut Council on Problem Gambling, Mohegan Sun Casino, and the National Center for Responsible Gaming (now the International Center for Responsible Gambling); has participated in surveys, mailings, or telephone consultations related to addictions, impulse-control disorders or other health topics; has consulted for law offices and the federal public defender’s office in issues related to impulse-control and addictive disorders; has provided clinical care in the Connecticut Department of Mental Health and Addiction Services Problem Gambling Services Program; has performed grant reviews for the National Institutes of Health and other agencies; has edited journals and journal sections; has given academic lectures in grand rounds, CME events and other clinical/scientific venues; and has generated books or chapters for publishers of mental health texts. All other authors report no biomedical financial interests or potential conflicts of interest.
Author information
Authors and Affiliations
Contributions
KTP, GDP, MCS, SOM MNP & JHK all contributed to manuscript authorship. KTP performed statistical analyses. AD and AG were responsible for subject recruitment and data collection. KDM was responsible for administrative aspects of the study. All authors reviewed & commented on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Patel, K.T., Stevens, M.C., Dunlap, A. et al. Effects of the Fyn kinase inhibitor saracatinib on ventral striatal activity during performance of an fMRI monetary incentive delay task in individuals family history positive or negative for alcohol use disorder. A pilot randomised trial. Neuropsychopharmacol. 47, 840–846 (2022). https://doi.org/10.1038/s41386-021-01157-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01157-5
