
Abstract
The hypothalamic–pituitary–adrenal (HPA) axis, a neuroendocrine network that controls hormonal responses to internal and external challenges in an organism’s environment, exhibits strikingly sex-biased activity. In adult female rodents, acute HPA function following a stressor is markedly greater than it is in males, and this difference has largely been attributed to modulation by the gonadal hormones testosterone and estradiol. These gonadal hormones are produced by the hypothalamic–pituitary–gonadal (HPG) axis and have been shown to determine sex differences in adult HPA function after acute stress via their activational and organizational effects. Although these actions of gonadal hormones are well supported, the possibility that sex chromosomes similarly influence HPA activity is unexplored. Moreover, questions remain regarding sex differences in the activity of the HPA axis following chronic stress and the underlying contributions of gonadal hormones and sex chromosomes. The present review examines what is currently known about sex differences in the neuroendocrine response to stress, as well as outstanding questions regarding this sex bias. Although it primarily focuses on the rodent literature, a brief discussion of sex differences in the human HPA axis is also included.
Similar content being viewed by others


Introduction
The ability of all mammals to cope with any environmental or homeostatic challenge (i.e., stressor), or with perceptual threats to homeostasis, relies upon activation of a neuroendocrine signaling cascade called the hypothalamic–pituitary–adrenal (HPA) axis. The HPA axis is activated in response to real or perceived stressors and culminates in the production and secretion of glucocorticoids by the adrenal glands. These act upon virtually all tissues to facilitate a body-wide stress response. When acutely elevated by stressors, glucocorticoids induce physiological and behavioral changes that are beneficial and indispensable for survival [1, 2]. However, persistent rises in glucocorticoids due to chronic stress or disease states are detrimental and increase risk for stress-related pathology [2,3,4]. Strikingly, women are at twice the risk of men for developing many of these diseases, likely due to sex differences in the function and regulation of the HPA axis [5]. Thus, understanding the nature and causes of such sex differences in the HPA axis following stress has important implications for understanding sex-biased risk for disease.
Much of what is known about sex differences in the stress-induced activity of the HPA axis and their underlying mechanisms comes from studies done in rodents. Such studies have demonstrated that sex differences in the HPA axis can arise from the influence of gonadal hormones during adulthood or during key developmental periods [6,7,8,9]. Additionally, they support the possibility that sex biases result from sex chromosomal effects, although supporting evidence is currently limited [10]. The present review accordingly focuses on advances revealed from rodent studies in our understanding of sex differences in the stress-induced activity of the HPA axis in adulthood. We primarily outline what is known about how gonadal hormones and sex chromosomes modulate HPA axis activity following acute stress, and then focus on sex-biased HPA axis activity post-chronic stress, which is far less well understood. We conclude with a brief discussion of sex differences in the human HPA axis with the caveat that they are less pronounced than in rodents and are largely dependent on the stress modality.
Overview of the HPA axis
Activation of the HPA axis
Activation of the neuroendocrine response to stressors occurs at the level of the hypothalamic paraventricular nucleus (PVN), which receives varying inputs depending on the nature of the stressor. Some stressors involve an immediate threat to physiological homeostasis and require the rapid relay of peripheral signals to PVN neurons via direct serotonergic or catecholaminergic projections from brainstem nuclei [9, 11]. Other stressors, alternatively, have psychological components that can activate neuroendocrine responses in the absence of a direct threat to survival [12]. These responses involve anticipation of a homeostatic challenge that requires interpretation by higher brain structures in order to assign significance to an external event based on instinctual fears and/or prior experiences with homeostatic challenges [12]. Because these stressors likely involve preparing for a threat to survival rather than immediately coping with the threat itself, there is time for information to first be processed in limbic forebrain regions before it reaches the PVN [12].
The amygdala is a limbic structure that plays a notable role in activating the neuroendocrine response to various stressors [13]. It is divided into subnuclei, including the central (CeA), medial (MeA), and basolateral amygdaloid (BLA) nuclei, that all have this excitatory effect [13]. However, each subnucleus is preferentially responsive to specific types of stressors (see ref. [13] for review) and may have sex-dependent function [14,15,16,17]. Notably, the amygdala sends few direct projections to the PVN [12, 18]. Thus, it largely modulates PVN neuronal activity by first innervating limbic or brainstem relay centers [19]. For instance, both the MeA and CeA send GABAergic projections to discrete regions of the bed nucleus of the stria terminalis (BNST) that ultimately function to decrease its GABAergic input to the PVN [12].
Ultimately, regardless of the nature of the stressor and corresponding neural circuitry, all pathways converge on PVN neurons that initiate the HPA axis and eventual hormonal response to stress [11] (Fig. 1).

Stress-related neuronal inputs to the PVN are candidates for gonadal hormone influence on the HPA activity. A diagrammatic representation of the major limbic and brainstem structures that send projections to the PVN to either enhance or inhibit the activity of the HPA axis in response to stressors is shown. Projections either directly contact PVN neurosecretory neurons or are indirect in nature and first synapse in the peri-PVN or limbic relay nuclei, such as the BNST. Excitatory and inhibitory projections are indicated by blue plus and red minus signs, respectively. Because their limbic and brainstem origins express androgen and/or estrogen receptors, these projections are potential targets for gonadal hormone modulation of HPA activity. PVN paraventricular nucleus, PFC prefrontal cortex, LS lateral septum, BNST bed nucleus of the stria terminalis, MPOA medial preoptic area, AMY amygdala, LC locus coeruleus, NTS nucleus of the solitary tract, Raphe represents both dorsal and medial raphe nuclei
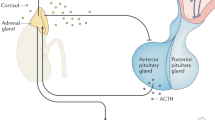
The HPA axis is activated by a distinct population of hypophysiotropic neurons in the medial parvocellular division of the PVN [12, 20]. These neurons synthesize a cocktail of neuropeptides, including corticotropin-releasing hormone (CRH), vasopressin (AVP), and oxytocin (OT), which are carried through the hypothalamo-hypophyseal portal vasculature where they can stimulate production of adrenocorticotropin (ACTH) by corticotrophs in the anterior pituitary gland [21,22,23]. The subsequent increase in the release of ACTH into the systemic circulation can then act in the adrenal cortex to induce the synthesis and release of glucocorticoids (i.e., cortisol in humans, and corticosterone (CORT) in rats and mice). Glucocorticoids act on virtually all tissues to mobilize energy stores, enhance cognition, and increase cardiovascular activity, while suppressing immune, reproductive, and digestive functions [2]. In the short term, stress-related elevations in glucocorticoids permit the beneficial physiological and behavioral changes necessary for acute stress responses. Unfortunately, prolonged elevations in glucocorticoids induced by chronic stress can have detrimental health consequences. Thus, numerous mechanisms exist to enable the tight regulatory control of the HPA axis [2,3,4].
Inhibition of the HPA axis
Glucocorticoid production by the HPA axis is partially maintained within homeostatic limits via neural inhibitory pathways (Fig. 1). Local brain areas, including the BNST, numerous hypothalamic nuclei, and the region immediately surrounding the PVN (i.e., peri-PVN), project to and directly inhibit PVN neuronal activity [9, 11]. These relay centers contain dense populations of GABAergic neurons. Thus, glutamatergic projections from limbic structures, such as the prefrontal cortex and hippocampus, can inhibit PVN neuronal activation by increasing tone of the inhibitory relay center [12, 24]. Notably, the involvement of limbic structures in constraining the activity of the HPA axis is largely restricted to stressors with a psychological component [11]. The neural inhibition of HPA activity induced by physical stress is far less well understood, but may involve inhibition of excitatory glucagon-like peptide 1-expressing neurons [25, 26]. Nevertheless, inhibitory neural pathways cannot act alone to inhibit the HPA axis and synthesis of glucocorticoids.
The actions of such inhibitory neural pathways are complemented by a glucocorticoid-mediated negative feedback system [11]. In this system, glucocorticoids alter the gain of key HPA regulatory regions, including limbic structures (i.e., hippocampus and prefrontal cortex), the PVN, and the anterior pituitary, to ultimately reduce adrenal glucocorticoid production [26]. Glucocorticoids act through either the mineralocorticoid receptor (MR) or the glucocorticoid receptor (GR) to exert these effects [27]. However, the differential expression and affinity for CORT of the MR and GR give them functionally distinct feedback roles. The MR is mostly found in limbic structures and has a high affinity for CORT [28, 29]. Consequently, MRs are thought to be occupied when CORT is at basal levels and are important for maintaining low basal glucocorticoid secretion [27, 28, 30]. GR, alternatively, is expressed in limbic areas, the PVN, and the anterior pituitary, and it has relatively lower affinity for CORT [27, 28]. It is selectively occupied by stress-induced elevations in CORT levels and, therefore, plays a significant role in reducing HPA axis activity following a stressor [28, 30]. Accordingly, GR mediates the majority of glucocorticoid negative feedback processes [31]. In support of this, GRs regulate two well-established phases of glucocorticoid feedback on the HPA axis: fast and delayed. Fast feedback involves membrane-bound GRs that induce endocannabinoid suppression of the HPA axis [32, 33], whereas delayed feedback involves a classical nuclear receptor-mediated mechanism [34, 35]. In this classical mechanism, unoccupied GR is found in the cell cytoplasm and translocates to the cell nucleus upon ligand binding, where it can directly influence gene transcription by binding glucocorticoid response elements and/or by interacting with other transcriptional regulators [36]. It ultimately adjusts HPA axis activity by altering expression of HPA-related genes, such as Crh and Avp [37, 38]. A thorough discussion of the mechanisms of GR action can be found in ref. [39].
Sex differences in HPA axis activity
In rodents, prominent sex differences exist in the HPA axis response to stress. Females typically have a more robust neuroendocrine response to acute stress, as evidenced by their increased CORT and ACTH levels compared to those of males following exposure to a number of stressors with different modalities [40,41,42,43,44]. Sex differences at each level of the HPA axis, as well as in regulatory limbic structures, may underlie this sex-biased HPA output. In the PVN, neuronal activation following numerous stressors, as measured by the expression of immediate early genes such as c-Fos, is greater in females [6, 40, 45]. Additionally, expression of HPA-related genes is sexually dimorphic. Female rats have been shown to have greater expression of AVP and CRH mRNA in the PVN and greater expression of the ACTH precursor, proopiomelanocortin (POMC) mRNA, in the anterior pituitary following acute stressors than males [40, 42, 43, 46].
In addition to their greater hormonal response to stress, female rats have been shown to have a delayed return to baseline ACTH and CORT levels after acute stress, indicating sex differences in the negative feedback regulation of the HPA axis [40,41,42,43]. Sex differences are present in neural pathways that may explain this sex-biased inhibition. In limbic structures known to activate inhibitory inputs to the HPA axis, including the frontal cortex, cingulate cortex, piriform cortex, and hippocampus, neuronal activation following acute restraint stress is reduced in females compared to males [47].
Sex differences also exist in glucocorticoid feedback mechanisms that may similarly contribute to the less robust negative feedback on the HPA axis in females. Glucocorticoid binding is lower in the hypothalamus of female versus male rats, suggesting that females have fewer hypothalamic corticosteroid receptors [48]. In the pituitary, female rats also have a reduced density of MRs and GRs compared to males [49]. Furthermore, regulation of corticosteroid receptors in response to acute stressors varies between males and females in structures regulating HPA activity, including the hippocampus, hypothalamus, and pituitary [50, 51]. For example, acute stressors upregulate GR and MR mRNA in the hypothalamus of male, but not female rats [51].
GR knockout studies have further highlighted sex differences in the feedback regulation of the HPA axis by glucocorticoids. In mice with GR selectively depleted in forebrain regions, including the hippocampus, medial prefrontal cortex, and basolateral amygdala, sex differences have been identified in the necessity of GR in these regions for HPA axis regulation [52]. The loss of forebrain GR results in HPA axis dysregulation in males but not females, suggesting that forebrain GR is largely inconsequential for normal HPA regulation in females [52]. The same group also examined the effects of selectively deleting GR in the PVN in male versus female mice [53]. Interestingly, males show increases in ACTH and CORT responses to acute stress above those of wild-type animals, but this response is absent in females [53]. Accordingly, PVN GR may be necessary for feedback inhibition of stress-induced ACTH and CORT in males, whereas GR in other, unexamined areas may be more important in females.
The availability of corticosteroids in brain regions where they can modulate HPA axis function may also influence their sex-dependent actions. Following their release from the adrenal gland, most corticosteroids are bound by corticosteroid-binding globulin (CBG), a glycoprotein produced by the liver [4]. The role of CBG is to protect corticosteroids from degradation as they are transported in the plasma to their target tissues [4]. Upon reaching the target, CBG releases corticosteroids by a number of mechanisms (see ref. [54] for review), as corticosteroids can only bind their intracellular receptors if they are not bound by CBG [55]. Thus, CBGs modulate the amount of plasma corticosteroids available to act on their targets, and it is important to make the distinction between total plasma CORT levels and bioavailable “free” CORT when considering sex differences in the activity of the HPA axis [54]. One study examining sex differences in basal CBG and free CORT levels found that females have approximately twice the binding activity of males, but only slightly higher (not statistically significant) levels of free CORT [56]. Consequently, the higher basal levels of total CORT in females may be partly buffered by their higher levels of CBG [56, 57]. It is also possible that the increased CBG levels contribute to the enhanced basal and stress-induce activity of the HPA axis observed in females, because CBG makes CORT less available for the negative feedback mechanisms that shut down the HPA axis. In males, CBG is negatively regulated by acute stress, which makes CORT more available for negative feedback [58]. Whether or not this is true for females as well remains to be determined. Thus, higher CBG levels may make show increases vulnerable to elevated HPA axis activity following acute stress but further research is necessary.
Activational effects of gonadal hormones
Adult sex differences in the neuroendocrine response to acute stress are due in part to interactions between the HPA axis and a parallel neuroendocrine network that controls reproduction, the hypothalamic–pituitary–gonadal (HPG) axis [59]. The HPG axis culminates in the production of testosterone and synthesis of estrogens from aromatizable androgens in the testis and ovary, respectively. Both testosterone and estradiol can in turn modulate the HPA axis in adulthood (i.e., have activational effects) and contribute to its sex-dependent function, as will be explored in this section. Notably, gonadal steroids also play a role in altering HPA axis function in aging rodents, although this topic fall beyond the scope of this review [60].
Gonadal hormone receptors and their localization
Estrogens and androgens influence the activity of the HPA axis in adulthood by acting through estrogen receptors (ERs) and androgen receptors (ARs), respectively [9]. The androgens, testosterone and its metabolite, dihydrotestosterone (DHT), both bind the androgen receptor, whereas the potent endogenous estrogen, estradiol, can activate either of two major ER subtypes: estrogen receptor alpha (ERα) or beta (ERβ). AR, ERα, and ERβ all belong to the same family of receptors, nuclear receptor subfamily 3, which also includes GR, MR, and the progesterone receptor (PR) [61]. All receptors in this subfamily function classically as ligand-activated transcription factors [62]. They reside as multiprotein complexes in the nucleus or cytoplasm until ligand binding triggers their shuttling to chromatin DNA, where they can influence transcription [62]. To alter transcription, these receptors (1) bind directly to hormone response elements on DNA and/or (2) interact with other transcriptional regulators [62,63,64]. As is the case for other members of nuclear receptor subfamily 3, ERs and ARs can also be found as membrane receptors that have faster (non-classical) influences on neuronal function and/or transcriptional activity by modulating second messenger pathways and ion channels [65, 66]. Moreover, a membrane estrogen receptor, G-protein-coupled estrogen receptor (GPER), has been identified that has high affinity for estradiol and can also mediate its rapid effects [67]. Thus, through classical and non-classical roles, gonadal hormone receptors enable gonadal hormones to influence HPA axis activity in a rapid and delayed manner.
The widespread expression of gonadal steroid hormone receptors in key parts of the neural circuitry controlling the HPA axis enables gonadal steroids to modify the neuroendocrine response to stress in concert with changes in reproductive function (Fig. 1). AR, ERα, and ERβ expression accordingly has been found in cortex, hippocampus, MeA, BNST, and numerous hypothalamic areas, including the medial preoptic area (MPOA) [68]. GPER is also expressed in the hippocampus and hypothalamic areas [67]. In neuroendocrine PVN neurons that direct the activity of the HPA axis; however, ERβs and GPERs are robustly expressed, whereas ARs are limited, suggesting that gonadal steroids modulate HPA activity by distinct mechanisms of action [41, 69]. Estradiol can directly alter HPA function by acting in neuroendocrine PVN neurons or indirectly by acting through upstream brain regions that project to the PVN; androgens, alternatively, have been shown to have predominantly indirect effects [41]. The MPOA and BNST, for example, are particularly important mediators of androgens’ actions, as they have abundant AR expression and project to the PVN and peri-PVN [9, 70, 71].
The estrous cycle and the HPA axis
Sex differences in the activity of the HPA axis may largely be influenced by HPG-mediated fluctuations in the levels of estradiol that occur across a 4-day estrous cycle in female rodents. Early investigations revealed that, as estradiol concentrations increase throughout the estrous cycle in female rats, so do the basal and stress-induced activity of the HPA axis. Accordingly, female rodents in diestrus (low estradiol) are similar to males, in that they exhibit low resting glucocorticoid secretion and a relatively quick on–off response to stressors [72, 73]. Females in proestrus (high estradiol, high progesterone) and estrus (recent exposure to peak estradiol), alternatively, have elevated basal and stress-induced ACTH and CORT [72,73,74]. Notably, elevations in HPA output are greatest on proestrous morning, when estradiol levels are peaking, but elevations in progesterone have not yet occurred. Progesterone appears to reduce estradiol’s effects on HPA output. Accordingly, in OVX’d female rats replaced with physiological levels of estradiol with or without progesterone to mimic the estrous cycle, estradiol treatment enhances HPA activity more than estradiol and progesterone treatments combined [72]. Thus, the extent of estradiol’s ability to increase HPA output on proestrus depends on whether there is a high or low background level of progesterone [72, 75]. This is well aligned with the findings of rodent studies demonstrating that progesterone treatment alone suppresses HPA axis stress reactivity [76, 77].
Female rats in proestrus and estrus also have a delayed return to baseline glucocorticoid secretion following stress [72, 73]. This may be due to less robust mechanisms of glucocorticoid negative feedback on the HPA axis and/or lesser input from limbic structures that are known to inhibit the HPA axis [47, 73]. Indeed, neuronal activation in cortical and hippocampal areas is lower in proestrous and estrous females than diestrous females [47]. Overall, studies of HPA axis function across the reproductive cycle have highlighted an important role for estradiol in controlling the neuroendocrine response to stress.
Estradiol and the HPA axis
In rodents, estradiol often enhances the activity of the HPA axis. Numerous studies correspondingly report that the removal of most endogenous estrogens by ovariectomy decreases stress-induced ACTH and CORT levels, while estradiol treatment has the opposite effects [6, 72, 78]. However, there are exceptions to this pattern, as some studies demonstrate that estradiol treatment can inhibit the HPA axis [79, 80]. Such exceptions will be discussed shortly. Nonetheless, support for the excitatory effects of estradiol has been found at all levels of the HPA axis. In the PVN, estradiol has been shown to increase stress-induced neuronal activation [45], as well as Crh and Avp gene expression [6, 81]. Additionally, estradiol increases POMC mRNA [6] and ACTH content [82] in the pituitary; and it increases sensitivity to ACTH in the adrenal gland [78]. Lastly, estradiol disrupts GR-mediated negative feedback on the HPA axis, as it reduces the repression of PVN crh by glucocorticoids [83] and interferes with GR expression and binding in the pituitary and hippocampus [84]. Estradiol also upregulates CBG levels [85], but this is not necessarily responsible for estradiol effects on the acute neuroendocrine stress response, since estradiol upregulates the HPA axis even in the absence of CBG changes [86].
Other studies show that estradiol inhibits neuroendocrine responses to stress or has no effect [42, 79, 80]. Accordingly, under certain physiological conditions, estradiol has been shown to decrease PVN neuronal activation [78] and Crh gene expression [79, 87]. Reports of such apparently opposing effects of estradiol on HPA activity may be due to varying experimental conditions, such as the dose or duration of estradiol treatment. Interestingly, a recent study measuring the effects of estradiol on stress-related, anxiety-like behaviors showed that the response to estradiol is opposite if females are ovariectomized and fed a standard chow versus a phytoestrogen-free diet [88]. HPA axis activity was not measured in this study, unfortunately, but its findings suggest that food composition can greatly affect the way that animals respond to estradiol.
Additionally, it is likely that estradiol will have contrasting actions based on whether ERα or ERβ-mediated signaling is invoked [9]. Whereas selective activation of ERα with propylpyrazoletriol (PPT) increases glucocorticoid secretion following stressors, selective activation of ERβ with diarylpropionitrile (DPN) decreases it in ovariectomized female rats [9, 89,90,91]. ERα and ERβ also modulate HPA activity via distinct mechanisms of action. ERβ can directly modify PVN action and downstream HPA output, as it is co-expressed by PVN neurons that express CRH, AVP, and OT [90, 92, 93]. Notably, ERβ co-expression with these neuropeptides varies based on the species and neuropeptide examined. For example, ERβ is found in 2/3 of AVP neurons in the rat PVN, whereas it is expressed in <20% of these neurons in the mouse PVN [92, 94]. A total of 60–80% of OT neurons, alternatively, co-express ERβ in both rats and mice [92, 94]. In contrast to the presumably direct actions of ERβ, ERα may influence HPA activity more indirectly. Although some ERα expression has recently been reported in mouse PVN neurons, very limited expression has been found in the rat PVN [92, 94]. More substantial expression of ERα has been found in brain regions that project to the PVN, such as the peri-PVN, BNST, and hippocampus [89, 93], supporting its indirect effects on HPA activity. Accordingly, studies indicate that ERα can inhibit glucocorticoid negative feedback on the HPA axis by disinhibiting the inhibitory signal from the peri-PVN to the PVN [89].
Moreover, estradiol may decrease excitatory inputs to the PVN by binding GPERs. The PVN receives serotonergic projections from the median and dorsal raphe nuclei of the brainstem that generally induce activation of the HPA axis (Fig. 1) [95], and estradiol has been shown to partially decrease this effect by desensitizing serotonin receptor signaling in the PVN [96]. Studies in ovariectomized rats have correspondingly shown that treatment with a serotonin receptor agonist increases plasma ACTH levels and that estradiol administration limits this effect [97]. Interestingly, GPER, but not ERβ, has been shown to be necessary for the desensitizing effects of estradiol on serotonin signaling in the PVN [97, 98].
Androgens and the HPA axis
Androgens generally inhibit the activity of the HPA axis. Thus, removal of most endogenous androgens by gonadectomy increases stress-induced ACTH and CORT secretion, whereas testosterone treatment has the opposite effects [6, 46, 99]. Notably, the reduction of testosterone to the more potent androgen, DHT, is necessary for its suppression of glucocorticoid secretion after stress [100]. Accordingly, inhibition of the enzyme that converts testosterone to DHT, 5α-reductase, with central infusion of finasteride increases stress-enhanced glucocorticoid secretion in male rats [100]. This effect is reversed by treatment with DHT, but not testosterone, indicating that testosterone’s inhibition of HPA activity depends on 5α-reductase activity within the central nervous system in males [100]. Moreover, estradiol does not potentiate the effects of DHT on the HPA axis in adulthood as may occur during development [101] and reproductive behaviors [102]. DHT placed near the PVN in adult, gonadectomized male rats reduces, whereas estradiol and ERα− selective agonists increase the ACTH and CORT responses to acute stress [90].
As illustrated for estrogens, the overall effect of androgens on the neuroendocrine stress response is partially mediated by androgen effects on the hypothalamus. In the PVN, androgens decrease neuronal activation, as well as elevations in CRH and AVP mRNA brought on by varying types of stressors [6, 103, 104]. These effects of androgens on PVN function and neuropeptide expression are likely mediated by ARs in limbic structures upstream of the PVN, because neuroendocrine PVN neurons do not express ARs [69, 105]. The MPOA and BNST, alternatively, are rich in AR expression and send AR immunoreactivity expressing projections to the PVN [70, 71]. Thus, they are favorably positioned to facilitate regulation of the HPA axis by androgens. Accordingly, androgen treatment administered directly to the MPOA decreases neuronal activation and Avp gene expression in the PVN, as well as glucocorticoid secretion following acute stress [70, 106]. Stereotaxic placement of DHT in the BNST, on the other hand, increases PVN AVP mRNA and stress-induced neuronal activation [107]. While further examination is necessary to determine if this unexpected role of androgens in the BNST depends on the type or duration of stress exposure, it supports the idea that androgens can act on brain regions upstream of the PVN to modulate the function of the HPA axis [107].
Androgens also act downstream of the PVN to influence HPA axis activity. At the level of the pituitary, POMC mRNA is downregulated by androgens [6]. Androgens also decrease pituitary ACTH content [82] and adrenal CORT content [108]. Moreover, androgens enhance GR-mediated negative feedback on the HPA axis. This is indirectly supported by evidence that testosterone increases GR binding in the MPOA, an area that mediates some of the inhibitory effects of glucocorticoids on the HPA axis [106]. It is also supported by evidence that testosterone decreases plasma CBG levels, which makes CORT more accessible to its receptors that facilitate glucocorticoid negative feedback [99].
Importantly, most studies examining androgen effects on the HPA axis employ DHT, which has high affinity for ARs, but cannot be aromatized to estradiol [105]. However, DHT can be metabolized to 5α-androstane-3β, 17β-diol (3β-diol), which has been shown to bind and activate ERβ [105]. Like DHT and ERβ agonists, 3β-diol inhibits the HPA axis response to stress [90, 105]. Accordingly, administration of either DHT or 3β-diol just above the PVN decreases ACTH and CORT responses to an acute stressor [90]. It has been hypothesized that 3β-diol and DHT, via its metabolism to 3β-diol, act through ERβs in PVN neurons to exert these effects, as rat PVN neuroendocrine neurons do not express ARs [105]. In support of this hypothesis, an ER but not AR antagonist blocks the local actions of DHT and 3β-diol in the PVN [90]. Additionally, 3β-diol has been shown to directly regulate the OT and AVP promoters through binding to ERβ [109, 110]. Together, these findings suggest that the inhibitory effects of DHT on HPA axis activity may be partially mediated by 3β-diol signaling through ERβ. They also emphasize the importance of further considering the roles ERβ may play in facilitating androgen regulation of the HPA axis.
Organizational effects of gonadal hormones
Perinatal gonadal hormones and the HPA axis
Sex differences in HPA function appear to also be organized by exposure to gonadal steroids during key developmental periods that program lasting differences in the HPA axis (i.e., organizational effects) [8] (Fig. 2). During perinatal development, male rats and mice encounter two surges in testosterone release that can presumably masculinize/defeminize the brain prior to puberty. One occurs late in gestation (G18 in the rat) and the other occurs shortly after birth [111, 112]. Accordingly, if neonatal testosterone is removed by gonadectomy, or if ARs are antagonized with flutamide, during the prenatal or postnatal periods in male rats, increased basal and stress-induced concentrations of CORT are found in adulthood [7, 113,114,115]. Male rats treated perinatally with flutamide also have altered patterns of basal CORT secretion as adults [7]. Normally, both male and female adult rodents secrete basal glucocorticoids in a pulsatile fashion throughout the day; but females have higher frequency and amplitude pulses than do males [6]. Perinatal AR antagonism in males increases their frequency and amplitude of CORT pulses in adulthood such that they resemble those of females. Notably, the effects of perinatal gonadectomy on basal and stress-enhanced glucocorticoid secretion in male rats are reversed by a single neonatal dose or neonatal doses of testosterone, but not prolonged testosterone treatment in adulthood [113, 114]. In females, a single neonatal dose of testosterone also dampens their HPA activity as adults [116]. Together, these findings indicate that neonatal testosterone acts through AR to produce lasting masculinization of glucocorticoid secretion by the HPA axis.

Organizational actions of gonadal hormones program lasting changes in the adult male HPA axis response to stress. Testosterone surges that occur during the perinatal period and puberty play important roles in masculinizing the HPA axis response to stress in adult male rodents. During the perinatal period, testosterone, largely via its conversion to estradiol by the aromatase enzyme, is involved in establishing a blueprint for a male typical pattern of HPA axis activity in adulthood. Pubertal testosterone then acts on this blueprint to complete the development of the masculinized HPA axis. Thus, in adulthood, males have decreased HPA axis responses to acute stressors characterized by decreased PVN neuronal activation and gene expression, decreased pituitary expression of POMC, and decreased ACTH and CORT responses to acute stressors. Adult males also have enhanced negative feedback resulting from their relatively reduced CBG levels, their increased PVN and pituitary GR gene expression, and their increased neuronal activation in limbic regions that inhibit the HPA axis. Organizational effects of testosterone, therefore, are important contributors to sex differences in the adult HPA axis in which females have relatively enhanced activity. Triangles indicate neuronal activation
In rodents, the conversion of testosterone to estradiol by the aromatase enzyme, and estradiol’s subsequent actions in organizing brain function, set the stage for functional sex differences in the HPA axis in adulthood (Fig. 2). Support for such estradiol-dependent sexual differentiation of the HPA axis comes from the identification of high levels of estrogen receptors [117] and aromatase enzymes [118] in the prenatal and neonatal rat brain. Additionally, treatment with an aromatase inhibitor or estradiol during the perinatal period increases and decreases basal and stress-induced HPA output in adult male rats, respectively [7, 114, 119]. Similarly, chronic estradiol treatment (estrogenization) of female neonates results in an evening corticosterone rise in adulthood that resembles that of male rather than diestrous female rats [120]. Thus, both early androgen and estrogen actions through ARs or ERs can establish sex differences in glucocorticoid secretion in adulthood, which are supported by sexually dimorphic activity at all levels of the HPA axis.
Current evidence supports a role for neonatal exposure to gonadal hormones in altering PVN neuroendocrine function during adulthood. Gonadectomy of male rats as neonates increases their basal and restraint-induced c-Fos expression (a measure of neuronal activation) in the medial parvocellular part of the PVN in adulthood [113]. These effects are reversed by neonatal, but not adult, treatment with testosterone, suggesting that neonatal testosterone contributes to male typical stress-induced PVN neuronal activation [113]. Furthermore, adult male rats treated with an aromatase inhibitor (1,4,6-androstatriene-3,17-dione (ATD)) as neonates have increased restraint-stimulated c-Fos mRNA in the PVN [119]. Collectively, these findings suggest that neonatal testosterone treatment, at least partially via its conversion to estradiol, contributes to male typical PVN neuronal activation following stress.
Early exposure to gonadal hormones also alters the expression of genes associated with HPA activation in adulthood. A single injection of testosterone in neonatal females is sufficient to decrease their stress-generated levels of PVN CRH, PVN AVP, and anterior pituitary POMC mRNAs as adults [116]. Additionally, estradiol treatment of female neonatal rats reduces basal PVN Crh gene expression in adulthood such that it resembles that of males rather than that of control diestrous females [120]. Such estrogenized females also do not exhibit increases in PVN CRH mRNA following adult ovariectomy and estradiol replacement as do control females [120]. In males, on the other hand, perinatal exposure to either an AR antagonist (flutamide) or ATD markedly increases PVN CRH and anterior pituitary POMC mRNA following stress in adulthood [7]. Consequently, both testosterone and its aromatization to estradiol are important for the development of masculinized HPA gene expression, which is reduced in adult males compared to females post stress.
Limbic regulation of the HPA axis is also subject to organizational actions of gonadal steroids. Neonatal treatment with an aromatase inhibitor, ATD, increases neural activation in limbic brain regions known to regulate the HPA axis in adult males, such as the PFC, lateral septum, and MeA [119]. Moreover, neonatal gonadectomy modifies expression of AR in the MeA and BNST, two areas that are important for regulating PVN neuronal activation and glucocorticoid secretion in response to stress [113]. Both stressed and unstressed adult male rats that were gonadectomized as neonates have decreased numbers of AR-expressing cells in both brain regions, and this effect is reversed by neonatal testosterone treatment [113]. Consequently, early testosterone exposure and aromatization enable the normal development of limbic structures that regulate the HPA axis in adult males.
Gonadal hormones similarly influence glucocorticoid negative feedback on the HPA axis, which is generally enhanced in males relative to females in adulthood. Accordingly, GR expression is often greater in males than females in structures important for driving the HPA axis, such as the hippocampus, PVN, and pituitary [48, 50, 51]. However, this sex difference in adulthood likely depends on the estrous stage of the females examined, as one study found that adult diestrous females have greater hippocampal and PVN GR expression than males [120]. Nonetheless, early gonadal hormone exposure has been shown to modulate GR expression and contribute to its sex-biased expression in adulthood. One study found that neonatal estradiol treatment decreases adult levels of hippocampal GR mRNA such that they are indistinguishable from those in adult males, suggesting that early estradiol exposure has “defeminizing” effects [120]. Furthermore, PVN GR mRNA is decreased, or “feminized”, in adult male rats treated neonatally with either an AR antagonist or an aromatase inhibitor, whereas it is increased (defeminized) in adult females treated neonatally with testosterone or estradiol [7, 116, 120]. In the anterior pituitary, adult males also have decreased GR binding if they were neonatally gonadectomized [114]. Lastly, perinatal gonadectomy elevates CBG levels in adult males [114]. Collectively, these findings suggest that the increased HPA negative feedback observed in adult males may be due, at least in part, to androgen-mediated effects via ARs and ERs on the epigenetic control of corticosteroid receptor expression and ligand availability.
Studies indicating sex differences in the necessity of forebrain and PVN GRs for HPA axis suppression may further support an organizational hypothesis of gonadal hormone action. These studies revealed that the loss of forebrain and PVN GRs results in dysregulation of HPA axis activity in males but is largely inconsequential in females [52, 53]. The use of randomly cycling females in these studies leaves open the possibility that activational effects of gonadal hormones influence GR involvement in HPA regulation. However, the substantial differences in HPA function between control females and those containing selective GR depletion—all of which were randomly cycling—would suggest that the effects of GR knockdown are not modified by estrous cycle stage. Rather, organizational effects of gonadal hormones may be at play and suggest that females use a different mechanism or neurocircuitry to inhibit HPA axis function.
Pubertal gonadal hormones and the HPA axis
Rises in gonadal steroids that occur at puberty provide another signal that can drive permanent changes in brain function. Although limited, some evidence indicates that a second critical period for the organizational actions of gonadal hormones on the HPA axis may occur near puberty [121]. The increases in gonadal steroids during this period drive numerous neural and behavioral changes to produce adult-like characteristics [121]. These include changes in neuroendocrine function, such as the stress-induced activity of the HPA axis. Accordingly, rats of both sexes have greater glucocorticoid responses to acute stressors before puberty than they do in adulthood [122, 123]. A thorough review of such age-dependent changes in the HPA axis can be found in ref. [124].
Notably, neuroendocrine sex differences that begin to develop in the perinatal period are completed during puberty [121]. As is the case for testosterone exposure during the perinatal period, exposure during puberty is important for the masculinization of neuroendocrine responses to stress in adulthood [125]. Adult female mice given testosterone during puberty have decreased CORT levels following acute restraint stress in adulthood, which are similar to those observed in males [125]. Moreover, pubertal testosterone exposure is important for organizing the adult male typical sensitivity of the HPA axis to androgen regulation [126]. Unlike males gonadectomized in adulthood, adult male rats gonadectomized before puberty do not respond to testosterone administration with decreases in basal or stress-induced PVN gene expression or glucocorticoid secretion [126]. Thus, puberty is potentially a critical organizational period during which rising levels of androgens play a vital role in sculpting the correct development of a male typical, androgen-sensitive adult HPA phenotype.
Pubertal rises in estradiol may also play a role, although less pronounced, in the organization of the adult HPA axis. Prior to puberty, estradiol decreases the activity of the HPA axis in female rats, whereas in adulthood it increases HPA activity [127]. Interestingly, this change in the HPA response to estrogens does not appear to depend on ovarian steroids [127]. In adult rats, estradiol treatment increases basal and stress-induced glucocorticoid secretion regardless of whether they were ovariectomized before or after puberty [127]. Thus, pubertal estrogen exposure is not essential for establishing the stimulatory effects of estradiol on the HPA axis as in adult females.
Adolescence and the HPA axis
Adolescence marks a transitional period between weaning and adulthood characterized by significant brain development, including changes in the HPA axis [128]. Although adolescence includes puberty and the associated effects of gonadal steroids on the HPA that occur during this period (discussed above), it can be more specifically divided into three stages: (1) prepubescence/early adolescence, (2) mid-adolescence, which includes pubertal onset, and (3) late adolescence during which puberty terminates [128]. Sex differences in HPA activity have been reported to emerge throughout adolescence. For example, adrenal volume increases in the late adolescence period more so in females than in males [129]. Sex biases also exist in the process whereby GRs translocate from the cytosol to the nucleus to influence gene transcription. One study demonstrated that GR translocation increases following acute stress in female, but not male, adolescent rats [130]. However, when adolescent rats have a history of chronic stress exposure, co-chaperones that inhibit GR translocation are upregulated and glucocorticoid negative feedback is impaired following an acute stressor only in females [130]. Additionally, gonadal hormones have been shown to play a role in regulating HPA function during adolescence, which can have lasting consequences. One study demonstrated that exposure to androgens irreversibly masculinizes CBG concentrations in adulthood. Accordingly, adult males have increased levels of CBG (resembling those of females) if gonadectomized during early adolescence [131]. A thorough review of sex differences in the HPA axis during adolescence and the influence of gonadal hormones can be found in refs. [8] and [132].
Remaining questions regarding sex differences in the HPA axis
Do sex chromosomes affect the HPA axis?
Although activational and organizational actions of gonadal hormones are essential mediators of sex differences in neural and behavioral phenotypes, more recent evidence suggests that sex chromosomes can have direct effects in non-gonadal tissues that act alongside or even independent of gonadal hormones secretions [10]. Such chromosome effects partially involve expression of male- and female-specific genes by the Y and X chromosomes, respectively (see ref. [133] for review). For example, expression of the gene Sry by the Y chromosome in males not only causes differentiation of the testis, but can also act in the brain to alter its function [133, 134]. Thus, differences intrinsic to XX and XY cells may be key mediators of sex differences in diverse aspects of physiology and behavior, including the neuroendocrine response to stress [133].
Unfortunately, studies examining the effects of XX versus XY on sex differences in the adult HPA axis are lacking. Nevertheless, rodent models now exist that would greatly aid in such investigations. The four core genotypes model has been used most frequently, and involves the uncoupling of gonadal (ovaries versus testes) and chromosomal (XX versus XY) sex. Subjects accordingly include: (1) XX gonadal females, (2) XY gonadal females, (3) XX gonadal males, and (4) XY gonadal males (see ref. [135] for review). Thus, the four core genotypes allow for testing of the organizational/activational effects of hormones resulting from gonadal sex independent of the intrinsic differences in XX versus XY cells [10]. This model has previously been used to examine sex differences in the morphology of AVP fibers in the lateral septum, a region known to modulate activity of the HPA axis. Results indicatethat sexually dimorphic septal AVP not only arises due to organizational/activational effects of gonadal steroids, but also due to chromosomal effects [136, 137]. Thus, it is highly likely that such sex chromosomal effects alter other aspects of HPA axis regulation, but this remains to be determined.
Are there sex differences in HPA axis following chronic stress?
Acute activation of the HPA axis by stress is considered an adaptive response to the increased metabolic demand required to deal with the stressor. Yet, in the face of chronic stress, this same response can be considered maladaptive and lead to greater risk for disease states [4]. A particularly well-established association exists between hyperactivity of the HPA axis and the presentation of affective disorders, such as depression and anxiety [5, 138]. Such disorders are of notable interest since they exhibit a striking sex bias in prevalence. Women are at least twice as likely as men to present with these conditions, and this bias is likely driven (at least in part) by sex differences in the physiological response to chronic stress [5]. Thus, some studies have begun to examine sex differences in the activity of the HPA axis under chronic stress conditions in the interest of understanding why women have increased vulnerability to affective disorders.
Models frequently used to examine HPA dysregulation that may predispose or trigger stress-related diseases are the repeated (homotypic) stress and the chronic mild stress (CMS)/chronic variable stress (CVS) models. The repeated stress model involves presentation of the same stimulus continually until a reduction in the physiological response, or “habituation”, to that stressor occurs [139]. The CMS/CVS model, alternatively, prevents habituation of the stress responses by exposing animals to randomly presented stressors over a prolonged period [140]. Both models are thought to be etiologically relevant for human disease risk and have been used to examine sex differences in the HPA axis response to chronic stress as discussed below.
Sex differences following repeated stress
After a number of exposures to the same stressor, sex differences are present in the basal activity of the HPA axis in which female rats have higher plasma CORT and CBG levels than males [141, 142]. Additionally, sex differences may be found in the habituation of HPA activity in repeated stress scenarios. In both sexes, this habituation is characterized by decreased ACTH and CORT responses to a stressor, as well as reduced stress-induced neuronal activation in the medial parvocellular part of the PVN, as the number of exposures increases [143]. However, the degree to which the habituation of HPA activity occurs in male and female rodents may vary. Some studies report similar habituation between the sexes. Accordingly, CORT responses to restraint or loud noise stressors incrementally decrease by the same amount in male and female rats [144]. The number of activated, c-Fos-expressing neurons that co-express AVP following restraint stress also similarly decreases in males and female rats [143]. Other studies, alternatively, suggest that habituation in a repeated restraint stress paradigm is reduced or slower in female versus male rats [142, 145, 146], Notably, this sex difference has also been shown to be accompanied by a decrease in plasma CBG levels only in female rats [142].
Morphological changes may also accompany chronic stressors and show sex biases. Whereas repeatedly restrained males exhibit apical dendritic atrophy of CA3c pyramidal neurons in the hippocampus, females show a decrease in basal dendritic branching [142]. Ultimately, whether or not habituation is sex-biased likely depends on the type of stressor and/or experimental paradigm. Nonetheless, findings to date largely support the possibility that females have enhanced HPA axis responses during repeated stress, which may contribute to their increased susceptibility to stress-related diseases.
Some studies have also examined sex differences in facilitation, a process that occurs when repeatedly stressed subjects have an equal or greater response to a novel stressor than control subjects [147]. When exposed to a novel acute restraint stress, chronically cold-stressed male mice exhibit similar ACTH and CORT responses to control males [148]. Cold-stressed females, on the other hand, have lower ACTH responses than, but similar CORT responses to, unstressed control females following a novel stressor [148]. These findings suggest that both sexes show facilitation, but that it may be more prominent in males. Conversely, another study found no sex differences in the HPA axis response to a novel environment following repeated exposure to either audiogenic or restraint stress [144]. Together, these studies suggest that sex differences in facilitation are stressor dependent.
The influence of gonadal hormones on HPA responses to repeated stressors has also been investigated. During repeated restraint stress, estradiol has been shown to limit habituation. Thus, in OVX female rats treated with estradiol, the decrease in plasma CORT responses following repeated stress exposure is lesser than it is in vehicle-treated controls [81]. In the same estradiol-treated OVX females, PVN CRF and AVP mRNA levels are also enhanced by chronic stress, and the habituation of AVP mRNA is impaired [81]. These findings suggest that, like in acute stress studies, estradiol increases HPA responses to repeated stress in female rats. Conversely, estradiol has been shown to dampen the effects of repeated footshock stress on activity in the PVN [149]. Accordingly, in OVX female rats given estradiol, c-Fos expression is lesser in the PVN following repeated stress than in vehicle-treated controls [149]. The differential effects of estradiol on the repeated stress-induced activity of the HPA axis may be due to varying experimental parameters, such as the type of stressor or the treatment duration. However, it is also possible that estradiol has varying effects depending on whether ERα or ERβ-mediated mechanisms are involved, as discussed above for acute stress situations. In support of this possibility, the study in ref. [149] demonstrated that estradiol treatment increases the number of ERβ-expressing cells in the PVN observed during chronic footshock stress, supporting ERβ-mediated mechanisms of PVN neuronal inhibition. Regardless of the direction of influence, the studies presented here demonstrate an important role for estradiol in determining how females adapt to chronic stress, which may influence sex differences in stress-related pathology.
The effect of gonadal hormones on chronic stress-induced HPA activity has also been investigated in male subjects. In male rats exposed to repeated restraint stress, gonadectomy enhances the habituation of the plasma CORT response to the restraint. However, habituation of the neuronal c-Fos response to repeated restraint stress in the PVN, MeA, and locus coeruleus are not affected by gonadectomy [148]. Therefore, androgens may minimally decrease HPA activity during repeated stress, but further study is necessary to fully determine this. Another study demonstrated that testosterone has organizational rather than activational effects that influence the habituation of the HPA axis during repeated psychogenic stress in male rats [101]. Accordingly, males treated with an androgen receptor antagonist (flutamide) or an aromatase inhibitor (ATD) during the perinatal period have a reduced decline in ACTH and CORT responses to repeated stress in adulthood. When adult males are gonadectomized, on the other hand, no changes in the habituation of the CORT response to stress are evident [101]. Thus, both the activation of androgen receptors and the conversion of testosterone to estradiol during the perinatal period—but not androgen exposure during adulthood—are essential for males to exhibit habituation during adulthood [101].
Sex differences following chronic mild or variable stress
Studies using the CMS and CVS models in male rodents have revealed that chronic stress generally enhances HPA axis activity, as indicated by increased basal CORT secretion, increased PVN CRH and AVP mRNA, and decreased GR expression in inhibitory brain regions [150, 151]. Although some studies have begun to compare HPA activity following CMS/CVS in male and female rodents, these are relatively few in number.
HPA axis dysregulation has been shown to be greater following CMS and CVS in female versus male rats, as females have larger elevations in basal CORT secretion than males post CMS and CVS [141, 152, 153]. Yet, chronically stressed males do exhibit adrenal hypertrophy and have elevated CORT levels when faced with a novel stressor, whereas chronically stressed females respond similarly to control, non-CMS females [141, 153]. This suggests that in males, but not females, a chronic stress history results in a potentiated HPA axis response to a novel stressor. Thus, females may be more vulnerable than males to chronic stress in the first place, but they utilize different or better coping skills when faced with a novel stressor. Whether these results indicate that one sex is better or worse off cannot be determined without more information. Females may adapt better in a CMS scenario, which may be important for stress resilience. However, if an acute response to a novel stressor following CMS is beneficial, females may be relatively impaired.
Sex differences in PVN neuronal activity following CMS have also been examined. In CMS-exposed rats, c-Fos expression is increased in the PVN of socially housed males, but not socially housed females [154]. Yet, both males and females have increased PVN c-Fos expression following CMS when singly housed [154]. These findings suggest that social support for females enables them to better cope with stress and is beneficial, whereas social housing in males is not beneficial and may even exacerbate their vulnerability to stress [154]. They support the possibility that social environment modifies the activity of the HPA axis [155]. Additionally, sex differences in PVN gene expression changes have been observed following CMS [156]. Constitutive PVN Crh gene expression is increased in males, but not females, post CMS which may be due to the females’ increased basal PVN CRH mRNA levels relative to those of males [156]. Accordingly, males may require greater increases in CRH from their lower basal levels to meet the demands of CMS. It is also possible that females have greater increases in Avp or Ot gene expression to make up for their lower CRH levels post CMS; or females could have CBG and/or MR levels that result in decreased glucocorticoid negative feedback. Although these possibilities require further investigation, the findings of this study and that showing sex differences in PVN neuronal activation support the possibility that HPA axis activity post CMS is increased in males and socially isolated, but not group housed, females.
Future directions for chronic stress studies
Further examination of the HPA axis response to chronic stress in both sexes is necessary to better assess sex differences. Although studies have begun to examine sex differences in PVN gene expression and neuronal activation following repeated stress and CMS, questions remain regarding sex differences at other levels of HPA axis regulation. For instance, are there sex differences in the limbic and brainstem structures that control the activity of the HPA axis following chronic stress? One study suggests that female, but not male rats, have deceased serotonergic activity in the hippocampus after CMS [153]. This difference may underlie the increased vulnerability of females to CMS, but it remains unclear how other regulatory brain regions may be affected to further contribute to this sensitivity. Similarly, following repeated stress, sex-biased changes in the morphology of hippocampal neurons have been observed; however, investigation of changes in other regulatory brain regions is lacking [142]. Additionally, whether or not sex differences exist in Ot and Avp gene expression in the PVN and/or in POMC expression in the anterior pituitary remains to be better determined using both chronic stress models. Sex differences in adrenal sensitivity to ACTH and glucocorticoid feedback regulation are similarly under-investigated. One study did examine the role of the PVN GR in the regulation of chronic stress reactivity, but found that selective deletion of GR in the PVN has no effect on the CORT response to CMS in either male or female mice [53]. This finding suggests that glucocorticoid control of the HPA axis after CMS likely does not involve the PVN and raises questions about whether other limbic and brainstem regions and/or the anterior pituitary serve as compensatory targets for glucocorticoid negative feedback in a sex-dependent manner. In a repeated stress model, alternatively, females show greater numbers of c-Fos and GR-positive PVN cells than males, suggesting that females may be more sensitive to glucocorticoid negative feedback in this case, but further investigation is necessary [143]. Furthermore, in both chronic stress models, sex differences in the mechanisms used by the GR and MR to negatively regulate the activity of the HPA axis have not been examined. Sex differences in receptor activation, assembly, or transactivation, for instance, may be present. Ultimately, it will be important to investigate sex differences at all levels of HPA axis regulation in response to repeated and variable chronic stressors moving forward. Such investigations will provide a more complete picture of the neurobiological mechanisms that coordinate to produce sex differences in vulnerability and resilience to chronic stress.
Moreover, few, if any, studies have examined the influence of gonadal hormones on sex-dependent HPA activity following CMS/CVS. The corresponding studies following repeated stress are greater in number, but still limited. This leaves questions regarding whether changes at each level of HPA axis regulation are influenced by the organizational and/or activational actions of gonadal hormones as has been demonstrated in acute stress studies. There are also especially large gaps in our understanding of how gonadal steroids may come to program lasting sex differences in the HPA axis response to stress in females [157]. Although further investigation is necessary, the rodent literature certainly supports the possibility that gonadal hormones alter the chronic stress-induced activity of the HPA axis in both sexes. For one, chronic stress is well associated with a dysregulation of the HPG axis, which involves inhibition of estrogen and testosterone secretion, as well as desynchronization of estrous cycling in females [153] (see refs. [54] and [158] for review). Thus, it is likely that chronic stress disrupts the HPG axis to contribute to the dysregulation of the HPA axis classically found following chronic stress [159]. Additionally, endogenous gonadal hormone levels may have a profound influence on HPA activity when altered by chronic stress exposure during critical periods. Early life stress, for instance, can disrupt key organizing actions of gonadal hormones that masculinize the HPA axis and produce lasting effects on rodents’ vulnerability to chronic stress in the future [60]. Similarly, the activational action of gonadal hormones can be disturbed by chronic stress in adulthood [60]. Ultimately, understanding how gonadal hormones modulate the HPA axis response to chronic stress has important implications for understanding the pathology of affective disorders, which are associated with major changes in gonadal hormone levels (see ref. [160] for review).
Sex differences in the HPA axis and disease in humans
Understanding sex differences in the human HPA axis has been a topic of much interest, as dysregulation of the HPA axis (hypo- or hyper-reactivity) is a hallmark of many stress-related diseases, which are known to differentially present in men versus women [5]. In humans, as in rodents, sex differences exist in the HPA axis response to stress and are largely age- and modality-dependent, although findings are not always consistent between the species. Before puberty, male and female rodents exhibit limited differences in their physiological stress response, as do boys and girls [161, 162]. However, during adolescence and into adulthood, male rodents begin to show a blunted HPA axis response to stress relative to females and human findings are much less consistent [5, 163]. While some studies suggest that adult women have enhanced HPA responses to acute stressors compared to males, others indicate greater responses in males or no significant difference [5, 162, 164,165,166]. These contradicting findings could be due to a number of factors, with the type of stressor being important, as well as the age, overall health, and menstrual cycle stage of the female participants [5, 164, 166]. For a more thorough review of sex differences in the human HPA axis, please see refs. [5] and [162].
Organizational/activational effects of gonadal hormones also influence sex differences in the human HPA axis as they do in rodents. Studies examining females in varying stages of the menstrual cycle, compared to males, have accordingly found that sex differences in the salivary cortisol response to psychosocial stress are present when women are in the low-estrogen state of the follicular phase but not the lueteal phase [167]. This suggests a role for estrogen and progesterone in regulating stress responsiveness in women across the menstrual cycle. Moreover, support for a role of gonadal hormones comes from the sex-biased prevalence of stress-related diseases, such as major depressive disorder, which develops after after puberty [60]. During early gestation, exposure to maternal and/or environmental stress produces more adverse outcomes in male than female offspring [60, 168]. Accordingly, neurodevelopmental disorders, such as autism spectrum disorder and attention-deficit hyperactivity disorder, are more common in boys than girls [169, 170]. This discrepancy potentially reflects a failure of neonatal testosterone to prepare the brain in early life for adult levels of testosterone, which complete the masculinization of the brain during puberty (see ref. [60] for review). During adolescence and throughout adulthood, however, females have increased risk for affective disorders, such as depression and anxiety, especially if they have experienced early life adversity [5, 171]. Such findings are also consistent with the greater risk for comorbidity of major depressive disorder with cardiometabolic diseases in women, which may be related to sex-biased HPA activity as well as sex-biased activity in the autonomic nervous system in which females have enhanced parasympathetic tone [172, 173]. Collectively, these clinical findings indicate that the timing of developmental perturbations can have long-term sex-biased outcomes; and they support a role for adult levels of female gonadal hormones in increasing risk for stress-related diseases (see ref. [60] for review). Ultimately, prior to starting any therapy for stress-related diseases, clinicians should be aware of sex differences in HPA axis activity and their underlying sources.
Conclusions
Striking sex differences exist in the neuroendocrine response to acute stress and its underlying circuitry in rodents. Generally, females exhibit enhanced glucocorticoid secretion in response to various acute stressors, which is supported by evidence of sex differences at all levels of HPA axis regulation [9]. Abundant evidence also supports organizational and activational roles for gonadal hormones in driving sex differences in acute HPA axis activity [8]. However, further study is necessary to rule out the possibility of sex chromosome effects that act with or without gonadal steroids to modulate HPA activity. Moreover, sex differences in the neuroendocrine stress response after chronic stress remain largely under-investigated. Chronic rather than acute stress results in the dysregulation of the HPA axis that may increase risk for stress-related diseases in humans [4]. Thus, investigations of sex differences in chronic stress-induced HPA dysregulation and the underlying influences of gonadal hormones and sex chromosomes may have important therapeutic consequences in the future.
References
Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44.
Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89.
Holsboer F. Stress, hypercortisolism and corticosteroid receptors in depression: implicatons for therapy. J Affect Disord. 2001;62:77–91.
de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–75.
Bangasser DA, Valentino RJ. Sex differences in stress-related psychiatric disorders: neurobiological perspectives. Front Neuroendocrinol. 2014;35:303–19.
Seale JV, Wood SA, Atkinson HC, Harbuz MS, Lightman SL. Gonadal steroid replacement reverses gonadectomy-induced changes in the corticosterone pulse profile and stress-induced hypothalamic-pituitary-adrenal axis activity of male and female rats. J Neuroendocrinol. 2004;16:989–98.
Seale JV, Wood SA, Atkinson HC, Lightman SL, Harbuz MS. Organizational role for testosterone and estrogen on adult hypothalamic-pituitary-adrenal axis activity in the male rat. Endocrinology. 2005;146:1973–82.
Green MR, McCormick CM. Sex and stress steroids in adolescence: gonadal regulation of the hypothalamic–pituitary–adrenal axis in the rat. Gen Comp Endocrinol. 2016;234:110–6.
Handa RJ, Weiser MJ. Gonadal steroid hormones and the hypothalamo-pituitary-adrenal axis. Front Neuroendocrinol. 2014;35:197–220.
Arnold AP. The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Horm Behav. 2009;55:570–8.
Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84.
Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, et al. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–80.
Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008;1148:64–73.
Mitsushima D, Yamada K, Takase K, Funabashi T, Kimura F. Sex differences in the basolateral amygdala: the extracellular levels of serotonin and dopamine, and their responses to restraint stress in rats. Eur J Neurosci. 2006;24:3245–54.
Mizukami S, Nishizuka M, Arai Y. Sexual difference in nuclear volume and its ontogeny in the rat amygdala. Exp Neurol. 1983;79:569–75.
Toufexis D. Region- and sex-specific modulation of anxiety behaviours in the rat. J Neuroendocrinol. 2007;19:461–73.
Brunton PJ, Donadio MV, Russell JA. Sex differences in prenatally programmed anxiety behaviour in rats: differential corticotropin-releasing hormone receptor mRNA expression in the amygdaloid complex. Stress. 2011;14:634–43.
Prewitt CMF, Herman JP. Anatomical interactions between the central amygdaloid nucleus and the hypothalamic paraventricular nucleus of the rat: a dual tract-tracing analysis. J Chem Neuroanat. 1998;15:173–85.
Myers B, Mark Dolgas C, Kasckow J, Cullinan WE, Herman JP. Central stress-integrative circuits: forebrain glutamatergic and GABAergic projections to the dorsomedial hypothalamus, medial preoptic area, and bed nucleus of the stria terminalis. Brain Struct Funct. 2014;219:1287–303.
Whitnall MH. Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog Neurobiol. 1993;40:573–629.
Rivier C, Vale W. Interaction of corticotropin-releasing factor and arginine vasopressin on adrenocorticotropin secretion in vivo. Endocrinology. 1983;113:939–42.
Schlosser S, Almeida O, Patchev VK, Yassouridis A, Elands J. Oxytocin-stimulated release of adrenocorticotropin from the rat pituitary is mediated by arginine vasopressin receptors of the V1b type. Endocrinology. 1994;135:2058–63.
Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–7.
Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13:3839–47.
Zhang R, Packard BA, Tauchi M, D’Alessio DA, Herman JP. Glucocorticoid regulation of preproglucagon transcription and RNA stability during stress. Proc Natl Acad Sci USA. 2009;106:5913–8.
Herman JP, Mcklveen JM, Solomon MB, Carvalho-Netto E, Myers B. Neural regulation of the stress response: glucocorticoid feedback mechanisms. Braz J Med Biol Res. 2012;45:292–8.
Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–11.
Reul JM, van den Bosch FR, de Kloet ER. Relative occupation of type-I and type-II corticosteroid receptors in rat brain following stress and dexamethasone treatment: functional implications. J Endocrinol. 1987;115:459–67.
Ahima R, Krozowski Z, Harlan RE. Type I corticosteroid receptor‐like immunoreactivity in the rat CNS: distribution and regulation by corticosteroids. J Comp Neurol. 1991;313:522–38.
Spencer RL, Kim PJ, Kalman BA, Cole MA. Evidence for mineralocorticoid receptor facilitation of glucocorticoid receptor-dependent regulation of hypothalamic-pituitary-adrenal axis activity. Endocrinology. 1998;139:2718–26.
de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–7.
Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151:4811–9.
Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24.
Levin N, Shinsako J, Dallman MF. Corticosterone acts on the brain to inhibit adrenalectomy-induced adrenocorticotropin secretion. Endocrinology. 1988;122:694–701.
Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132:1033–44.
Aguilera G, Liu Y. The molecular physiology of CRH neurons. Front Neuroendocrinol. 2012;33:67–84.
Ferrini MG, Grillo CA, Piroli G, De Kloet ER, De Nicola AF. Sex difference in glucocorticoid regulation of vasopressin mRNA in the paraventricular hypothalamic nucleus. Cell Mol Neurobiol. 1997;17:671–86.
Keller-Wood M. Hypothalamic-pituitary-adrenal axis-feedback control. Compr Physiol. 2015;5:1161–82.
Viau V, Bingham B, Davis J, Lee P, Wong M. Gender and puberty interact on the stress-induced activation of parvocellular neurosecretory neurons and corticotropin-releasing hormone messenger ribonucleic acid expression in the rat. Endocrinology. 2005;146:137–46.
Handa RJ, Burgess LH, Kerr JE, O’Keefe JA. Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm Behav. 1994;28:464–76.
Babb JA, Masini CV, Day HE, Campeau S. Sex differences in activated corticotropin-releasing factor neurons within stress-related neurocircuitry and hypothalamic-pituitary-adrenocortical axis hormones following restraint in rats. Neuroscience. 2013;234:40–52.
Iwasaki-Sekino A, Mano-Otagiri A, Ohata H, Yamauchi N, Shibasaki T. Gender differences in corticotropin and corticosterone secretion and corticotropin-releasing factor mRNA expression in the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala in response to footshock stress or psychological. Psychoneuroendocrinology. 2009;34:226–37.
MacLusky NJ, Yuan H, Elliott J, Brown TJ. Sex differences in corticosteroid binding in the rat brain: an in vitro autoradiographic study. Brain Res. 1996;708:71–81.
Larkin JW, Binks SL, Li Y, Selvage D. The role of oestradiol in sexually dimorphic hypothalamic-pituitary-adrena axis responses to intracerebroventricular ethanol administration in the rat. J Neuroendocrinol. 2010;22:24–32.
Seale JV, Wood SA, Atkinson HC, Bate E, Lightman SL, Ingram CD, et al. Gonadectomy reverses the sexually diergic patterns of circadian and stress-induced hypothalamic-pituitary-adrenal axis activity in male and female rats. J Neuroendocrinol. 2004;16:516–24.
Figueiredo HF, Dolgas CM, Herman JP. Stress activation of cortex and hippocampus is modulated by sex and stage of estrus. Endocrinology. 2002;143:2534–40.
Turner BB, Weaver DA. Sexual dimorphism of glucocorticoid binding in rat brain. Brain Res. 1985;343:16–23.
Turner BB. Sex difference in glucocorticoid binding in rat pituitary is estrogen dependent. Life Sci. 1990;46:1399–406.
Karandrea D, Kittas C, Kitraki E. Contribution of sex and cellular context in the regulation of brain corticosteroid receptors following restraint stress. Neuroendocrinology. 2000;71:343–53.
Karandrea D, Kittas C, Kitraki E. Forced swimming differentially affects male and female brain corticosteroid receptors. Neuroendocrinology. 2002;75:217–26.
Solomon MB, Furay AR, Jones K, Packard AE, Packard BA, Wulsin AC, et al. Deletion of forebrain glucocorticoid receptors impairs neuroendocrine stress responses and induces depression-like behavior in males but not females. Neuroscience. 2012;203:135–43.
Solomon MB, Loftspring M, de Kloet AD, Ghosal S, Jankord R, Flak JN, et al. Neuroendocrine function after hypothalamic depletion of glucocorticoid receptors in male and female mice. Endocrinology. 2015;156:2843–53.
Panagiotakopoulos L, Neigh GN. Development of the HPA axis: where and when do sex differences manifest? Front Neuroendocrinol. 2014;35:285–302.
Henley DE, Lightman SL. New insights into corticosteroid-binding globulin and glucocorticoid delivery. Neuroscience. 2011;180:1–8.
Gala RR, Westphal U. Corticosteroid-binding globulin in the rat: studies on the sex difference. Endocrinology. 1965;77:841–51.
McCormick CM, Linkroum W, Sallinen BJ, Miller NW. Peripheral and central sex steroids have differential effects on the HPA axis of male and female rats. Stress. 2002;5:235–47.
Tannenbaum B, Rowe W, Sharma S, Diorio J, Steverman A, Walker M, et al. Dynamic variations in plasma corticosteroid-binding globulin and basal HPA activity following acute stress in adult rats. J Neuroendocrinol. 1997;9:163–8.
Viau V. Functional cross-talk between the hypothalamic-pituitary-gonadal and -adrenal axes. J Neuroendocrinol. 2002;14:506–13.
Bale TL, Epperson CN. Sex differences and stress across the lifespan. Nat Neurosci. 2015;18:1413–20.
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–9.
Pawlak M, Lefebvre P, Staels B. General molecular biology and architecture of nuclear receptors. Curr Top Med Chem. 2012;12:486–504.
Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010;42:813–27.
Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, et al. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science. 1997;277:1508–10.
Vasudevan N, Pfaff DW. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front Neuroendocrinol. 2008;29:238–57.
Foradori CD, Weiser MJ, Handa RJ. Non-genomic actions of androgens. Front Neuroendocrinol. 2008;29:169–81.
Brailoiu E, Dun SL, Brailoiu GC, Mizuo K, Sklar LA, Oprea TI, et al. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J Endocrinol. 2007;193:311–21.
Simerly RB, Chang C, Muramatsu M, Swanson LW. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J Comp Neurol. 1990;294:76–95.
Bingham B, Williamson M, Viau V. Androgen and estrogen receptor-β distribution within spinal-projecting and neurosecretory neurons in the paraventricular nucleus of the male rat. J Comp Neurol. 2006;499:911–23.
Williamson M, Bingham B, Gray M, Innala L, Viau V. The medial preoptic nucleus integrates the central influences of testosterone on the paraventricular nucleus of the hypothalamus and its extended circuitries. J Neurosci. 2010;30:11762–70.
Williamson M, Viau V. Androgen receptor expressing neurons that project to the paraventricular nucleus of the hypothalamus in the male rat. J Comp Neurol. 2007;503:717–40.
Viau V, Meaney MJ. Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology. 1991;129:2503–11.
Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol. 2016;6:603–21. https://doi.org/10.1002/cphy.c150015.
Carey MP, Deterd CH, de Koning J, Helmerhorst F, de Kloet ER. The influence of ovarian steroids on hypothalamic-pituitary-adrenal regulation in the female rat. J Endocrinol. 1995;144:311–21.
Patchev VK, Shoaib M, Holsboer F, Almeida OFX. The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience. 1994;62:265–71.
Owens MJ, Ritchie JC, Nemeroff CB. 5a-Pregnane-3a,21-diol-20-one (THDOC) attenuates mild stress-induced increases in plasma corticosterone via a non-glucocorticoid mechanism: comparison with alprazolam. Brain Res. 1992;573:353–5.
Patchev VK, Hassan AH, Holsboer DF, Almeida OF. The neurosteroid tetrahydroprogesterone attenuates the endocrine response to stress and exerts glucocorticoid-like effects on vasopressin gene transcription in the rat hypothalamus. Neuropsychopharmacology. 1996;15:533–40.
Figueiredo HF, Ulrich-Lai YM, Choi DC, Herman JP. Estrogen potentiates adrenocortical responses to stress in female rats. AJP Endocrinol Metab. 2006;292:E1173–82.
Ochedalski T, Subburaju S, Wynn PC, Aguilera G. Interaction between oestrogen and oxytocin on hypothalamic-pituitary-adrenal axis activity. J Neuroendocrinol. 2007;19:189–97.
Young EA, Altemus M, Parkison V, Shastry S. Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology. 2001;25:881–91.
Lunga P, Herbert J. 17β-oestradiol modulates glucocorticoid, neural and behavioural adaptations to repeated restraint stress in female rats. J Neuroendocrinol. 2004;16:776–85.
Kitay JI. Pituitary-adrenal function in the rat after gonadectomy and gonadal hormone replacement. Endocrinology. 1963;73:253–60.
Patchev VK, Almeida OF. Gonadal steroids exert facilitating and “buffering” effects on glucocorticoid-mediated transcriptional regulation of corticotropin-releasing hormone and corticosteroid receptor genes in rat brain. J Neurosci. 1996;16:7077–84.
Burgess LH, Handa RJ. Estrogen-induced alterations in the regulation of mineralocorticoid and glucocorticoid receptor messenger RNA expression in the female rat anterior pituitary gland and brain. Mol Cell Neurosci. 1993;4:191–8.
Gala R, Westphal U. Further studies on the corticosteroid-binding globulin in the rat: proposed endocrine control. Endocrinology. 1966;79:67–76.
Lund TD, Munson DJ, Haldy ME, Handa RJ. Dihydrotestosterone may inhibit hypothalamo-pituitary-adrenal activity by acting through estrogen receptor in the male mouse. Neurosci Lett. 2004;365:43–47.
Paulmyer-Lacroix O, Hery M, Pugeat M, Grino M. The modulatory role of estrogens on corticotropin-releasing factor gene expression in the hypothalamic paraventricular nucleus of ovariectomized rats: role of the adrenal gland. J Neuroendocr. 1996;8:515–9.
Russell AL, Grimes JM, Larco DO, Cruthirds DF, Westerfield J, Wooten L, et al. The interaction of dietary isoflavones and estradiol replacement on behavior and brain-derived neurotrophic factor in the ovariectomized rat. Neurosci Lett. 2017;640:53–9.
Weiser MJ, Handa RJ. Estrogen impairs glucocorticoid dependent negative feedback on the hypothalamic-pituitary-adrenal axis via estrogen receptor alpha within the hypothalamus. Neuroscience. 2009;159:883–95.
Lund TD, Hinds LR, Handa RJ. The androgen 5α-dihydrotestosterone and its metabolite 5α-androstan-3β, 17β-diol inhibit the hypothalamo–pituitary–adrenal response to stress by acting through estrogen receptor β-expressing neurons in the hypothalamus. J Neurosci. 2006;26:1448–56.
Lund TD, Rovis T, Chung WCJ, Handa RJ. Novel actions of estrogen receptor-beta on anxiety-related behaviors. Endocrinology. 2005;146:797–807.
Hrabovszky E, Kalló I, Steinhauser A, Merchenthaler I, Coen CW, Petersen SL, et al. Estrogen receptor-beta in oxytocin and vasopressin neurons of the rat and human hypothalamus: immunocytochemical and in situ hybridization studies. J Comp Neurol. 2004;473:315–33.
Laflamme N, Nappi RE, Drolet G, Labrie C, Rivest S. Expression and neuropeptidergic characterization of estrogen receptors (ERα and ERβ) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J Neurobiol. 1998;36:357–78.
Oyola MG, Thompson MK, Handa AZ, Handa RJ. Distribution and chemical composition of estrogen receptor β neurons in the paraventricular nucleus of the female and male mouse hypothalamus. J Comp Neurol. 2017;525:3666–82.
Van de Kar LD, Blair ML. Forebrain pathways mediating stress-induced hormone secretion. Front Neuroendocrinol. 1999;20:1–48.
Raap DK, Doncarlos L, Garcia F, Muma NA, Wolf WA, Battaglia G, et al. Estrogen desensitizes 5-HT(1A) receptors and reduces levels of G(z), G(i1) and G(i3) proteins in the hypothalamus. Neuropharmacology. 2000;39:1823–32.
McAllister CE, Creech RD, Kimball PA, Muma NA, Li Q. GPR30 is necessary for estradiol-induced desensitization of 5-HT1A receptor signaling in the paraventricular nucleus of the rat hypothalamus. Psychoneuroendocrinology. 2012;37:1248–60.
Rossi DV, Dai Y, Thomas P, Carrasco GA, DonCarlos LL, Muma NA, et al. Estradiol-induced desensitization of 5-HT1A receptor signaling in the paraventricular nucleus of the hypothalamus is independent of estrogen receptor-beta. Psychoneuroendocrinology. 2010;35:1023–33.
Viau V, Meaney MJ. Testosterone-dependent variations in plasma and intrapituitary corticosteroid binding globulin and stress hypothalamic-pituitary-adrenal activity in the male rat. J Endocrinol. 2004;181:223–31.
Handa RJ, Kudwa AE, Donner NC, McGivern RF, Brown R. Central 5-alpha reduction of testosterone is required for testosterone’s inhibition of the hypothalamo-pituitary-adrenal axis response to restraint stress in adult male rats. Brain Res. 2013;1529:74–82.
Bingham B, Gray M, Sun T, Viau V. Postnatal blockade of androgen receptors or aromatase impair the expression of stress hypothalamic-pituitary-adrenal axis habituation in adult male rats. Psychoneuroendocrinology. 2011;36:249–57.
D’Occhio MJ, Brooks DE. Effects of androgenic and oestrogenic hormones on mating behaviour in rams castrated before or after puberty. J Endocrinol. 1980;86:403–11.
Viau V, Lee P, Sampson J, Wu J. A testicular influence on restraint-induced activation of medial parvocellular neurons in the paraventricular nucleus in the male rat. Endocrinology. 2003;144:3067–75.
Lund TD, Munson DJ, Haldy ME, Handa RJ. Androgen inhibits, while oestrogen enhances, restraint-induced activation of neuropeptide neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol. 2004;16:272–8.
Handa RJ, Weiser MJ, Zuloaga DG. A role for the androgen metabolite, 5α-androstane-3β,17β-diol, in modulating oestrogen receptor β-mediated regulation of hormonal stress reactivity. J Neuroendocrinol. 2009;21:351–8.
Viau V, Meaney MJ. The inhibitory effect of testosterone on hypothalamic-pituitary-adrenal responses to stress is mediated by the medial preoptic area. J Neurosci. 1996;16:1866–76.
Bingham B, Myung C, Innala L, Gray M, Anonuevo A, Viau V. Androgen receptors in the posterior bed nucleus of the stria terminalis increase neuropeptide expression and the stress-induced activation of the paraventricular nucleus of the hypothalamus. Neuropsychopharmacology. 2011;36:1433–43.
Malendowicz LK, Młynarczyk W. Sex differences in adrenocortical structure and function. X. Lipid and corticosterone in the rat adrenal as affected by gonadectomy and testosterone or estradiol replacement. Endokrinologie. 1982;79:292–300.
Pak TR, Chung WC, Hinds LR, Handa RJ. Arginine vasopressin regulation in pre- and postpubertal male rats by the androgen metabolite 3beta-diol. Am J Endocrinol Metab. 2009;296:E1409–13.
Hiroi R, Lacagnina AF, Hinds LR, Carbone DG, Uht RM, Handa RJ. The androgen metabolite, 5alpha-androstane-3beta,17beta-diol (3beta-diol), activates the oxytocin promoter through an estrogen receptor-beta pathway. Endocrinology. 2013;154:1802–12.
Weisz J, Ward IL. Plasma testosterone and progesterone titers of pregnant rats, their male and female fetuses, and neonatal offspring. Endocrinology. 1980;106:306–16.
Corbier P, Edwards DA, Roffi J. The neonatal testosterone surge: a comparative study. Arch Physiol Biochem. 1992;100:127–31.
Bingham B, Viau V. Neonatal gonadectomy and adult testosterone replacement suggest an involvement of limbic arginine vasopressin and androgen receptors in the organization of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2008;149:3581–91.
McCormick CM, Furey BF, Child M, Sawyer MJ, Donohue SM. Neonatal sex hormones have “organizational” effects on the hypothalamic-pituitary-adrenal axis of male rats. Dev Brain Res. 1998;105:295–307.
McCormick CM, Mahoney E. Persistent effects of prenatal, neonatal, or adult treatment with flutamide on the hypothalamic-pituitary-adrenal stress response of adult male rats. Horm Behav. 1999;35:90–101.
Seale JV, Wood SA, Atkinson HC, Harbuz MS, Lightman SL. Postnatal masculinization alters the HPA axis phenotype in the adult female rat. J Physiol. 2005;563:265–74.
MacLusky NJ, Lieberburg I, McEwen BS. The development of estrogen receptor systems in the rat brain: perinatal development. Brain Res. 1979;178:129–42.
Shinoda K, Nagano M, Osawa Y. Neuronal aromatase expression in preoptic, strial, and amygdaloid regions during late prenatal and early postnatal development in the rat. J Comp Neurol. 1994;343:113–29.
Bingham B, Wang NX, Innala L, Viau V. Postnatal aromatase blockade increases c-fos mRNA responses to acute restraint stress in adult male rats. Endocrinology. 2012;153:1603–8.
Patchev VK, Hayashi S, Orikasa C, Almeida OF. Implications of estrogen-dependent brain organization for gender differences in hypothalamo-pituitary-adrenal regulation. FASEB J. 1995;9:419–23.
Romeo RD. Puberty: a period of both organizational and activational effects of steroid hormones on neurobehavioural development. J Neuroendocrinol. 2003;15:1185–92.
Romeo RD, Lee SJ, Chhua N, McPherson CR, McEwen BS. Testosterone cannot activate an adult-like stress response in prepubertal male rats. Neuroendocrinology. 2004;79:125–32.
Romeo RD, Lee SJ, McEwen BS. Differential stress reactivity in intact and ovariectomized prepubertal and adult female rats. Neuroendocrinology. 2004;80:387–93.
Romeo RD. The metamorphosis of adolescent hormonal stress reactivity: a focus on animal models. Front Neuroendocrinol. 2017;49:43–51.
Goel N, Bale TL. Organizational and activational effects of testosterone on masculinization of female physiological and behavioral stress responses. Endocrinology. 2008;149:6399–405.
Evuarherhe OM, Leggett JD, Waite EJ, Kershaw YM, Atkinson HC, Lightman SL. Organizational role for pubertal androgens on adult hypothalamic-pituitary-adrenal sensitivity to testosterone in the male rat. J Physiol. 2009;587:2977–85.
Evuarherhe O, Leggett J, Waite E, Kershaw Y, Lightman S. Reversal of the hypothalamo-pituitary-adrenal response to oestrogens around puberty. J Endocrinol. 2009;202:279–85.
McCormick CM, Green MR. From the stressed adolescent to the anxious and depressed adult: investigations in rodent models. Neuroscience. 2013;249:242–57.
Pignatelli D, Xiao F, Gouveia AM, Ferreira JG, Vinson GP. Adrenarche in the rat. J Endocrinol. 2006;191:301–8.
Bourke CH, Raees MQ, Malviya S, Bradburn CA, Binder EB, Neigh GN. Glucocorticoid sensitizers Bag1 and Ppid are regulated by adolescent stress in a sex-dependent manner. Psychoneuroendocrinology. 2013;38:84–93.
Mataradze GD, Kurabekova RM, Rozen VB. The role of sex steroids in the formation of sex-differentiated concentrations of corticosteroid-binding globulin in rats. J Endocrinol. 1992;132:235–40.
McCormick CM, Mathews IZ. HPA function in adolescence: Role of sex hormones in its regulation and the enduring consequences of exposure to stressors. Pharmacol Biochem Behav. 2007;86:220–33.
Arnold AP. A general theory of sexual differentiation. J Neurosci Res. 2017;95:291–300.
Dewing P, Chiang CWK, Sinchak K, Sim H, Fernagut PO, Kelly S, et al. Direct regulation of adult brain function by the male-specific factor SRY. Curr Biol. 2006;16:415–20.
Arnold AP, Chen X. What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Front Neuroendocrinol. 2009;30:1–9.
De Vries GJ, Rissman EF, Simerly RB, Yang LY, Scordalakes EM, Auger CJ, et al. A model system for study of sex chromosome effects on sexually dimorphic neural and behavioral traits. J Neurosci. 2002;22:9005–14.
Gatewood JD. Sex chromosome complement and gonadal sex influence aggressive and parental behaviors in mice. J Neurosci. 2006;26:2335–42.
Martin EI, Ressler KJ, Binder E, Nemeroff CB. The neurobiology of anxiety disorders: brain imaging, genetics, and psychoneuroendocrinology. Clin Lab Med. 2010;30:865–91.
Dhabhar FS, McEwen BS, Spencer RL. Adaptation to prolonged or repeated stress – comparison between rat strains showing intrinsic differences in reactivity to acute stress. Neuroendocrinology. 1997;65:360–8.
Willner P. The chronic mild stress (CMS) model of depression: history, evaluation and usage. Neurobiol Stress. 2017;6:78–93.
Vieira JO, Duarte JO, Costa-Ferreira W, Morais-Silva G, Marin MT, Crestani CC. Sex differences in cardiovascular, neuroendocrine and behavioral changes evoked by chronic stressors in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2018;81:426–37.
Galea LA, McEwen B, Tanapat P, Deak T, Spencer R, Dhabhar F. Sex differences in dendritic atrophy of CA3 pyramidal neurons in response to chronic restraint stress. Neuroscience. 1997;81:689–97.
Zavala JK, Fernandez AA, Gosselink KL. Female responses to acute and repeated restraint stress differ from those in males. Physiol Behav. 2011;104:215–21.
Babb JA, Masini CV, Day HE, Campeau S. Habituation of hypothalamic-pituitary-adrenocortical axis hormones to repeated homotypic stress and subsequent heterotypic stressor exposure in male and female rats. Stress. 2014;17:224–34.
Bhatnagar S, Lee TM, Vining C. Prenatal stress differentially affects habituation of corticosterone responses to repeated stress in adult male and female rats. Horm Behav. 2005;47:430–8.
Chadda R, Devaud LL. Differential effects of mild repeated restraint stress on behaviors and GABAA receptors in male and female rats. Pharmacol Biochem Behav. 2005;81:854–63.
Dallman MF, Jones MT. Corticosteroid feedback control of acth secretion: effect of stress-induced corticosterone secretion on subsequent stress responses in the rat. Endocrinology. 1973;92:1367–75.
Chen X, Herbert J. The effect of long‐term castration on the neuronal and physiological responses to acute or repeated restraint stress: interactions with opioids and prostaglandins. J Neuroendocrinol. 1995;7:137–44.
Gerrits M, Grootkarijn A, Bekkering BF, Bruinsma M, Den Boer JA, Ter Horst GJ. Cyclic estradiol replacement attenuates stress-induced c-Fos expression in the PVN of ovariectomized rats. Brain Res Bull. 2005;67:147–55.
Dallman MF. Stress update. Adaptation of the hypothalamic-pituitary-adrenal axis to chronic stress. Trends Endocrinol Metab. 1993;4:62–69.
Herman JP. Neural control of chronic stress adaptation. Front Behav Neurosci. 2013;7.
Xing Y, He J, Hou J, Lin F, Tian J, Kurihara H. Gender differences in CMS and the effects of antidepressant venlafaxine in rats. Neurochem Int. 2013;63:570–5.
Dalla C, Antoniou K, Drossopoulou G, Xagoraris M, Kokras N, Sfikakis A, et al. Chronic mild stress impact: are females more vulnerable? Neuroscience. 2005;135:703–14.
Westenbroek C, Den Boer JA, Ter Horst GJ. Gender-specific effects of social housing on chronic stress-induced limbic FOS expression. Neuroscience. 2003;121:189–99.
Kirschbaum C, Klauer T, Filipp SH, Hellhammer DH. Sex-specific effects of social support on cortisol and subjective responses to acute psychological stress. Psychosom Med. 1995;57:23–31.
Duncko R, Kiss A, Skultétyová I, Rusnák M, Jezová D, Ivana S, et al. Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology. 2001;26:77–89.
Viau V, Innala L. Organizational influences of the gonadal steroid hormones: lessons learned through the hypothalamic-pituitary-adrenal (HPA) axis. Sex Differ Physiol. 2016;193–203.
Joseph D, Whirledge S. Stress and the HPA axis: balancing homeostasis and fertility. Int J Mol Sci. 2017;18:2224.
Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. 2005;4:141–94.
Mueller SC, Grissom EM, Dohanich GP. Assessing gonadal hormone contributions to affective psychopathologies across humans and animal models. Psychoneuroendocrinology. 2014;46:114–28.
Romeo RD. Pubertal maturation and programming of hypothalamic-pituitary-adrenal reactivity. Front Neuroendocrinol. 2010;31:232–40.
Kudielka BM, Kirschbaum C. Sex differences in HPA axis responses to stress: a review. Biol Psychol. 2005;69:113–32.
Gomez F, Manalo S, Dallman MF. Androgen-sensitive changes in regulation of restraint-induced adrenocorticotropin secretion between early and late puberty in male rats. Endocrinology. 2004;145:59–70.
Seeman TE, Singer B, Wilkinson CW, Bruce McEwen. Gender differences in age-related changes in HPA axis reactivity. Psychoneuroendocrinology. 2001;26:225–40.
Uhart M, Chong RY, Oswald L, Lin PI, Wand GS. Gender differences in hypothalamic-pituitary-adrenal (HPA) axis reactivity. Psychoneuroendocrinology. 2006;31:642–52.
Kirschbaum C, Kudielka BM, Gaab J, Schommer NC, Hellhammer DH. Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamus-pituitary-adrenal axis. Psychosom Med. 1999;61:154–62.
Rohleder N, Schommer NC, Hellhammer DH, Engel R, Kirschbaum C. Sex differences in glucocorticoid sensitivity of proinflammatory cytokine production after psychosocial stress. Psychosom Med. 2001;63:966–72.
Sandman CA, Glynn LM, Davis EP. Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J Psychosom Res. 2013;75:327–35.
Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health. 2007;28:235–58.
Erksine HE, Ferrari AJ, Nelson P, Polanczyk GV, Flaxman AD, Vos T, et al. Epidemiological modelling of attention-deficit/hyperactivity disorder and conduct disorder for the Global Burden of Disease Study 2010. J Child Psychol Psychiatry. 2013;54:1263–74.
Heim C, Nemeroff CB. The impact of early adverse experiences on brain systems involved in the pathophysiology of anxiety and affective disorders. Biol Psychiatry. 1999;46:1509–22.
Goldstein JM, Cherkerzian S, Buka SL, Fitzmaurice G, Hornig M, Gillman M, et al. Sex-specific impact of maternal-fetal risk factors on depression and cardiovascular risk 40 years later. J Dev Orig Health Dis. 2011;2:353–64.
Goldstein JM, Handa RJ, Tobet SA. Disruption of fetal hormonal programming (prenatal stress) implicates shared risk for sex differences in depression and cardiovascular disease. Front Neuroendocrinol. 2014;35:140–58.
Funding
Funding is provided by NIH R01 DK105826 and Arizona Biomedical Research Commission ADHS14-082990.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Heck, A.L., Handa, R.J. Sex differences in the hypothalamic–pituitary–adrenal axis’ response to stress: an important role for gonadal hormones. Neuropsychopharmacol 44, 45–58 (2019). https://doi.org/10.1038/s41386-018-0167-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0167-9
This article is cited by
-
Sex differences of post-Covid patients undergoing outpatient pulmonary rehabilitation
Biology of Sex Differences (2024)
-
Sex-specific interactions between stress axis and redox balance are associated with internalizing symptoms and brain white matter microstructure in adolescents
Translational Psychiatry (2024)
-
Psychological Health and Ischemic Heart Disease in Women: A Review of Current Evidence and Clinical Considerations across the Healthspan
Current Atherosclerosis Reports (2024)
-
Pharmacokinetic Analysis of [18F]FES PET in the Human Brain and Pituitary Gland
Molecular Imaging and Biology (2024)
-
Die zentrale Steuerung des Menstruationszyklus und funktionelle Störungen – Neues zur Pathophysiologie und Therapieansätzen
Die Gynäkologie (2024)
