
Abstract
Endometrial serous carcinoma (ESC) is an aggressive type of endometrial carcinoma with a poor prognosis. Immune checkpoint blockade has evolved as a novel treatment option for endometrial cancers; however, data on expression of immune checkpoints that may be potential targets for immunotherapy in ESC are limited. We analyzed the prevalence and prognostic significance of PD-L1, TIM-3 and B7-H3 immune checkpoints in 99 ESC and evaluated their correlation with CD8 + tumor infiltrating lymphocytes. Applying the tumor proportion score (TPS) with a cutoff of 1%, PD-L1, TIM-3 and B7-H3 expression was present in 17%, 10% and 93% of cases, respectively. Applying the combined positive score (CPS) with a cutoff of 1, PD-L1, TIM-3 and B7-H3 expression was present in 63%, 67% and 94% of cases, respectively. Expression of these markers was largely independent of one another. PD-L1 correlated with higher CD8 + T-cell density when evaluated by either TPS (p = 0.02) or CPS (p < 0.0001). TIM-3 correlated with CD8 + T-cell density when evaluated by CPS (p < 0.0001). PD-L1 positivity was associated with improved overall survival (p = 0.038) when applying CPS. No association between PD-L1 expression and survival was found using TPS, and there was no association between TIM-3 or B7-H3 positivity and survival by either TPS or CPS. Using TPS, PD-L1 correlated with a higher tumor stage but not with survival, whereas the converse was true when PD-L1 was evaluated by CPS, suggesting that PD-L1 expression in immune cells correlates with prognosis and is independent of tumor stage. In conclusion, PD-L1, TIM-3 and B7-H3 may be potential therapeutic targets in selected patients with ESC. Further investigation of their roles as predictive biomarkers is needed.
Similar content being viewed by others


Introduction
Endometrial serous carcinoma (ESC) is a high-grade type of endometrial cancer with an aggressive behavior. Although it represents only approximately 10% of endometrial malignancies, it accounts for almost 40% of endometrial cancer-related deaths.1 Despite advances in treatment, the overall 5-year survival rate of patients with ESC is still less than 50%2, underscoring the need for novel treatment options in these tumors. As many cancers develop a mechanism of escaping immune surveillance through modulation of T-lymphocyte activity via upregulation of expression of immune checkpoint signaling molecules3,4, targeting these checkpoints with immune checkpoint blockade has emerged as a breakthrough therapeutic option in oncology in recent years. Success of the anti-PD-1 and anti-PD-L1 inhibitors in clinical trials has led to the approval by the Food and Drug Administration (FDA) of several immune checkpoint inhibitors (ICI) for treatment of advanced tumors of selected types,5 including endometrial cancer.6 A recent clinical trial which included a large cohort of advanced stage endometrial cancer patients, 25% of whom had ESC, demonstrated a significantly longer progression-free survival (PFS) and overall survival (OS) in patients receiving the multi-kinase inhibitor lenvatinib (LENVIMA®) in combination with the PD-1 inhibitor pembrolizumab (KEYTRUDA®) as compared with those in the chemotherapy group. Survival benefits were observed across all evaluated subgroups, including both mismatch repair (MMR)-proficient and MMR-deficient tumors and less-common but aggressive histologic subtypes.7
Although a durable response is achieved in some patients treated with immune checkpoint inhibitors, the fact that many patients fail to respond to these therapies indicates the need for elucidation of predictive biomarkers that could be used to individualize targeted treatment. Currently, knowledge regarding the complex immune tumor microenvironment in endometrial carcinoma in general, and ESC in particular, is limited.
The PD-1/PD-L1 axis, the most clearly illuminated immune checkpoint pathway to date, involves interaction between T-cell PD-1 receptors and PD-L1 expressed on various cell types including tumor and immune cells. In clinical trials, PD-L1 has emerged as a promising biomarker for some tumors, with efficacy of ICI contingent on PD-L1 expression. In other tumors, however, therapeutic success of ICI appears to be independent of PD-L1 expression.5 Of import, endometrial carcinoma comprises various histologic types with different molecular alterations and biological behavior. Although the prevalence of PD-L1 expression in endometrial cancers has been previously documented in the literature8,9,10,11,12,13,14,15,16,17, many studies have not stratified endometrial tumors by histologic type, or have studied cohorts skewed toward the (most common) endometrioid type while underrepresenting less common histologic types including ESC. In addition to PD-L1, several alternative T-cell checkpoints and associated predictive biomarkers are currently under investigation due to the need to identify novel immunotherapeutic targets.18,19,20 One of these checkpoints is T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), a molecule expressed by various immune cells including activated effector T cells and T regulatory cells (Tregs). TIM-3 has multiple ligands, is upregulated in various carcinomas21 and is generally considered a negative regulator of immune responses.22,23,24 B7-H3 is another immune checkpoint of interest, expression of which can be induced on T-cells and other immune cells.25 B7-H3 is also upregulated in many cancers and plays a complex role in tumor immunity with both stimulatory and inhibitory functions and synergetic effects with other immune checkpoints.26 Therapies targeting cells expressing these molecules have been developed in the last decade and tested in clinical trials, alone or in combination with anti-PD-1/PD-L1 drugs, demonstrating good efficacy and an acceptable safety profile.27,28 Identification of these and other potential immunotherapy targets in ESC could inform future studies aimed at improving patient outcomes.
In addition to the predictive role of immune checkpoint biomarkers, their prognostic significance has also lately gained attention. For solid tumors, published studies on the prognostic significance of PD-L1 expression have shown conflicting results,29 while TIM-3 and B7-H3 expression has been reported to be associated with predominantly worse outcomes.30,31,32 For endometrial carcinomas in particular, reports on the prognostic significance of PD-L1 expression have demonstrated inconsistent results,8,9,10,11,12,13,14,15,16,17 while data on the prognostic significance of TIM-3 and B7-H3 is largely lacking. In this study, we delved into selected aspects of the immune landscape of ESC by analyzing the prevalence and prognostic significance of PD-L1, TIM-3 and B7-H3 and their correlation with tumor-associated CD8 + T-lymphocytes in a large patient cohort. We found expression of the three immune checkpoints to be variable and largely independent from one another. While both PD-L1 and TIM-3 positivity by CPS was associated with higher CD8 + T cells, only PD-L1 expression correlated with survival.
Materials and methods
Case selection
After approval from the institutional review board of the University of Texas Southwestern Medical Center, 103 cases with a diagnosis of ESC accessioned between January 2008 and December 2020 were identified from Parkland Health and Hospital System and Clements University Hospital. Clinicopathological characteristics including patient age, tumor stage, tumor characteristics, treatment history, and clinical outcome data were extracted from the electronic medical records. Cases originally diagnosed as ESC but for which subsequent MMR or MSI testing was performed as part of clinical care (n = 37) and demonstrated an abnormal result (n = 3, loss of MMR expression by IHC) were classified as MMR-deficient and excluded from the study. Cases with unknown MMR/MSI status (n = 63) were tested for PMS2 and MSH6 and, in cases with an abnormal result, reflexed for testing for MLH1 or MSH2, respectively. Cases with loss of expression of any of these markers (n = 1, loss of MLH1/PMS) were excluded from the study.
Immunohistochemistry
Formalin-fixed paraffin embedded (FFPE) tumor tissue was used for immunohistochemistry.
Whole tissue sections from 99 tumors were tested for PD-L1 and CD8, and from 98 tumors for TIM-3 and B7-H3. Sixty-three cases with unknown MMR status were tested for PMS2 and MSH6. One case with an abnormal PMS2 result was tested for MLH1. Positively charged slides with tissue sections cut at 4 μm thickness were dried in an oven for 1 h at 56 °C to 60 °C. Deparaffinization, rehydration, and target retrieval were performed in a three-in-one procedure, per the manufacturer’s instructions, by fully submerging the slides in preheated (65 °C) EnVision™ FLEX Target Retrieval Solution at low pH (6.0) for PD-L1 and high pH (9.0) for TIM-3, B7-H3 and CD8, and incubating slides at 97 °C for 20 min. After incubation, when the temperature cooled to 70 °C, the slides were removed from the solution and immediately submerged in room temperature wash buffer diluted 1:20 for 5 min. After this, the slides were placed in an Autostainer Link 48 platform (Dako, Agilent), subjected to FLEX peroxidase block for 5 min, and then incubated with the respective primary antibody. The following primary antibodies and dilutions were used: monoclonal mouse PD-L1 (clone 22C3, 1:50 dilution, Dako/Agilent), monoclonal rabbit B7-H3 (clone D9M2L, 1:150 dilution, Cell Signaling Technology), monoclonal rabbit TIM-3 (clone D5D5R, 1:100 dilution, Cell Signaling Technology), monoclonal mouse CD8 (clone C8/144B, RTU, Dako/Agilent), human PMS2 (clone EP51, RTU, Dako/Agilent), MSH6 (clone EP49, RTU, Dako/Agilent) and MLH1 (clone ES05, RTU, Dako/Agilent). Incubation time with primary antibody was 60 min for PD-L1, TIM-3 and B7-H3, 20 min for CD8, 30 min for MSH6, and 40 min for MLH1. After incubation with the PD-L1 primary antibody, incubation with the EnVision™ FLEX + Mouse LINKER anti-mouse linker antibody was performed for 30 min. Subsequently, all slides were rinsed in wash buffer for 5 min, and then incubated with the EnVision™ FLEX HRP visualization reagent for 30 min for PD-L1, 20 min for MSH6, B7-H3, TIM-3 and CD8, and 40 min for PMS2 and MLH1 at room temperature. After rinsing in wash buffer for 5 min, enzymatic conversion of the subsequently added 3,3’-diaminobenzidine (DAB) tetrahydrochloride chromogen was performed for 10 min at room temperature. For PD-L1, this step was followed by addition of DAB enhancer for 5 min at room temperature. The instrument monitored the incubation time and rinsing of slides between reagents. The reagent times were preprogrammed in the Dako Link software. Slides were subsequently counterstained for 5 min with hematoxylin (Link) using a preprogrammed hematoxylin incubation time. Mounting was performed using non-aqueous permanent mounting media. Control tissues included a section of normal tonsil and a tissue microarray (TMA) with a known range of expression for PD-L1, a section of normal tonsil for TIM-3 and CD8, and sections of placenta and prostate for B7-H3.
Analysis of PD-L1, TIM-3 and B7-H3 expression
Two scoring methods, tumor proportion score (TPS) and combined positive score (CPS), were used for evaluation of PD-L1, TIM-3 and B7-H3 based on FDA-approval of these methods for assessment of PD-L1 expression in other solid tumors.5 TPS was calculated as the percentage of tumor cells with membranous PD-L1 expression. CPS was calculated as the number of PD-L1-staining cells (tumor cells, lymphocytes, and macrophages) divided by the total number of viable tumor cells, multiplied by 100. Both scores ranged from 0 to 100. For all three immune checkpoint markers, cutoff scores of ≥1% for TPS and ≥1 for CPS were used to define positivity, and additional cutoffs (10, 20, and 50) were used to further analyze the significance of immune checkpoint expression. In the CPS system, for scoring of mononuclear immune cells (lymphocytes and macrophages), only intratumoral and peritumoral (within one 20x field from the tumor nest edge) immune cells were counted. Stromal immune cells distant from the tumor were excluded. Due to the lack of established criteria for assessment of TIM-3 and B7-H3, the same scoring systems and positivity thresholds used for PD-L1 assessment were applied.
Evaluation of MMR status
Intact MMR protein expression was defined as nuclear expression in tumor cells, and loss of expression was defined as the absence of staining in tumor cells in the presence of positive reaction in internal control cells. All MMR-deficient cases were excluded from the study.
Image analysis and calculation of CD8 + TIL density
Whole slide images of CD8 immunostained slides were generated for image analysis by Aperio ScanScope AT Turbo scanner (Leica Biosystems, Heidelberg, Germany). Quantification of total tumor area and percentage of positive staining cells was performed by Aperio Analysis software. Areas for quantification were annotated using Aperio analysis tools. The percentage of weak, medium, and strong staining was determined using the color-deconvolution tool. CD8 + TIL density was calculated as total CD8 + cells (including weak, medium, and strong signals) divided by the area comprising the tumor and areas in direct contact with the tumor periphery (mm2).
Statistical analysis
Two-tailed Chi square test was used to compare the expression of immune checkpoints among various clinical stage groups. Unpaired student’s t-test was used to compare CD8 + TIL density among different immune checkpoint expression groups. Statistical analyses were performed using JMP 11.2.0 (SAS Institute Inc., Cary, NC). OS was calculated from the date of the pathologic diagnosis to the date of death or last follow-up. The progression interval was censored for patients for whom there was no recorded date of progression in the data field “months to new tumor event after initial diagnosis”. Log-rank test and Kaplan-Meier survival curves were used to compare OS and PFS between the respective immune checkpoint marker positive and -negative expression groups. A p-value <0.05 was considered to be statistically significant. GraphPad Prism 7.04 software (San Diego, CA) was used for survival analysis. Cohen’s Kappa (κ), an index that considers observed agreement with respect to agreement by chance, was used to measure the co-occurrence/agreement between individual markers. It has a range from −1 to 1, where 1 indicates perfect agreement, 0 random, and −1 perfect disagreement.
Results
Patient characteristics
Of 103 cases with an original diagnosis of ESC, four cases were found to be MMR-deficient and were excluded from the final study cohort. Ninety-nine cases were included in the analysis of immune checkpoint markers. The morphologic diagnosis of ESC cases included in the study was confirmed in each case by one or more gynecologic pathologists (E.L. and/or H.C.).
Patient demographics, clinical stage, and outcomes are summarized in Table 1. All patients were staged using the International Federation of Gynecology and Obstetrics (FIGO) system. Patient age ranged from 49 to 83 years (median 64 years). Complete follow-up information was available for 93 (93%) of 99 patients and was included in survival analysis. Median follow-up was 28 months (range, 3–151 months).
PD-L1, TIM-3, and B7-H3 are differentially expressed in ESC
The distribution of PD-L1, TIM-3, and B7-H3 expression varied among the tumors. In the majority of PD-L1 positive tumors, PD-L1 expression was heterogeneous with positive cells arranged in patchy, focal or multifocal distributions. In a few tumors, both tumor and immune PD-L1 positive cells were present predominantly at the advancing edge of the tumor, forming a cordon of varying intensity with accentuation at the tumor-stroma interface. Rare tumors demonstrated diffuse positivity in tumor cells. The distribution of TIM-3 positive cells within the tumor was predominantly patchy, focal, or multifocal with rare tumors showing accentuation at the tumor edge and tumor-stroma interface. B7-H3 expression was present in tumor cells in the majority of cases, and showed either diffuse or patchy distribution. An example of a case with expression of three immune checkpoint markers is illustrated in Fig. 1A.

A Examples of PD-L1, TIM-3, B7-H3 expression in endometrial serous carcinoma by immunohistochemistry. B, C PD-L1, TIM-3, and B7-H3 expression in individual cases according to a raw score group arranged in a case matrix. Results evaluated using (B) tumor proportion score (TPS) and (C) combined positive score (CPS). Color coding scheme is depicted on the bottom. NP indicates not performed; Neg Negative. Values in raw score groups are expressed as percentages for TPS and score number for CPS.
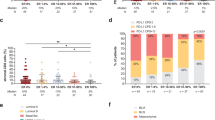
Applying TPS (with 1% positivity cutoff), PD-L1 expression was present in 17% (17/99) of cases, TIM-3 expression was present in 10% (10/98) of cases, and B7-H3 was present in 93% (91/98) of cases. Applying CPS (with 1 positivity cutoff), PD-L1 expression was present in 63% (62/99) of cases, TIM-3 expression was present in 67% (66/98) of cases, and B7-H3 was present in 94% (92/98) of cases (Fig. 1B, C and Table 2). Using TPS, 69% of cases were positive for one marker, 23% of cases were positive for two, and 2% were positive for all three markers (Fig. 2A). In contrast, using CPS, 16% of cases were positive for one, 35% for two and 46% for all three markers. (Fig. 2B) Using TPS, none to slight agreement was found between the expression of three immune checkpoints with κ = −0.01–0.03. (Fig. 2C) Using CPS, fair agreement was found between PD-L1 and TIM-3 (κ = 0.26), and none to slight agreement between PD-L1 and B7-H3 (κ = 0.09) and between TIM-3 and B7-H3 (κ = 0.06) (Fig. 2D). Overall, expression of the three biomarkers was largely independent from one another.

Individual bars reflect the proportion of cases expressing 0, 1, 2 or 3 markers by (A) tumor proportion score (TPS) and (B) by combined positive score (CPS). Cohen’s kappa agreement/co-occurrence between individual markers by (C) TPS and (D) CPS.
To investigate the correlation between the level of immune expression and prognosis, we further subdivided positive raw CPS scores into group 1 (score 1–9), group 2 (score 10–19), group 3 (score 20–49) and group 4 (score ≥ 50). For TPS, we grouped cases according to the same numerical values as for CPS but expressed as percentages. The majority of cases that were positive for PD-L1 or for TIM-3 demonstrated a relatively low level of positivity by both TPS and CPS and belonged to group 1 (score 1–9), while most B7-H3 positive cases showed a high positivity (score ≥ 50) by both TPS and CPS. The results of PD-L1, TIM-3, and B7-H3 expression including the distribution according to the raw score groups are summarized in Table 2.
Correlation between immune checkpoint expression and clinical stage
PD-L1 positivity by TPS was significantly lower in stage IA tumors as compared to other stages (p = 0.04) while no statistically significant difference was observed between more advanced stages (p = 0.39). There was no significant difference in PD-L1 expression by CPS between tumors of different stages (p = 0.80). (Fig. 3A, B) No statistically significant difference was found in TIM-3 or B7-H3 expression by either TPS or CPS between tumors of different stages (TIM-3: TPS, p = 0.67; CPS, p = 0.12; B7-H3: TPS, p = 0.35; CPS, p = 0.43). (Fig. 3C–F).

PD-L1 expression by (A) TPS and (B) CPS. TIM-3 expression by (C) TPS and (D) CPS. B7-H3 expression by (E) TPS and (F) CPS. *p < 0.05.
Correlation between immune checkpoint expression and CD8 + tumor infiltrating lymphocyte (TIL) density
Using CPS, ESC with positive PD-L1 expression demonstrated significantly higher CD8 + TIL density than PD-L1 negative tumors (1171 ± 120 cells/mm2 in PD-L1 positive tumors vs. 488 ± 65 cells/mm2 in PD-L1 negative tumors, p < 0.0001). Furthermore, we investigated the correlation between the level of PD-L1 expression and CD8 + TIL density in PD-L1 positive tumors, which we divided into “low” and “high” expression groups using CPS 10 or 20 as cutoffs. There was no statistically significant difference between CD8 + TIL density in these two groups (CPS cutoff 10: p = 0.07; CPS cutoff 20: p = 0.06). Using TPS, higher CD8 + TIL density was observed in PD-L1 positive than in PD-L1 negative tumors (1348 ± 226 cells/mm2 in PD-L1 positive tumor vs. 830 ± 75 cells/mm2in PD-L1 negative tumors, p = 0.02). ESC with positive TIM-3 expression demonstrated significantly higher CD8 + TIL density compared to TIM-3 negative tumors (1170 ± 112 cells/mm2 in TIM-3 positive tumors vs. 379 ± 38 cells/mm2 in TIM-3 negative tumor, p < 0.0001). Similar to PD-L1 positive tumors, no statistically significant difference was found between TIM-3 “low” or “high” groups when CPS 10 cutoff was applied p = 0.21). Due to limited power for TIM-3 CPS ≥ 20 group, we were not able to perform statistical analysis using CPS 20 as cutoff. Similar to PD-L1, no statistically significant difference was found between TIM-3 positive and TIM-3 negative tumors (1176 ± 232 cells/mm2 in TIM-3 positive tumor vs. 881 ± 74 cells/mm2 in TIM-3 negative tumors, p = 0.29). The limited number of cases within the PD-L1 positive (n = 17) and TIM-3 positive (n = 10) groups may have contributed to the failure to reach statistical significance. Due to the limited power of the B7-H3 negative tumor group (n = 7 by TPS, and n = 6 by CPS), we were not able to perform statistical analysis between B7-H3 positive and negative tumors. Due to the predominantly high expression of B7-H3 in ESC, we chose cutoffs of 20 or 50 (TPS or CPS) to subclassify tumors into “low” and “high” expression groups. There was no statistically significant difference between CD8 + TIL density in these groups (CPS cutoff 20: p = 0.74; CPS cutoff 50: p = 0.44; TPS cutoff 20%: p = 0.98; TPS cutoff 50%: p = 0.71). These results are illustrated in Fig. 4A–M.

A CD8 IHC and (B) corresponding image annotation of CD8 + TIL using Aperio image analysis tool: CD8 + lymphocytes are annotated and color-coded based on staining intensity as weak (yellow), medium (orange), and strong (red); blue color highlights nuclei. C–F Correlation of CD8 + TIL density with PD-L1 expression using : (C) CPS with positivity defined as ≥ 1; (D) CPS, low versus high score with cutoff point 10; (E) CPS, low vs. high score with cutoff point 20; (F) TPS with positivity defined as ≥1%. G–I, Correlation of CD8 + TIL density with TIM-3 expression using: (G) CPS with positivity defined as ≥ 1; (H) CPS, low vs. high score with cutoff point 10; (I) TPS with positivity defined as ≥ 1%. J–M Correlation of CD8 + TIL density with B7-H3 expression using: (J) CPS, low vs. high score with cutoff point 20; (K) CPS, low vs. high score with cutoff point 50; (L) TPS, low vs. high score with cutoff point 20; (M) TPS, low vs. high score with cutoff point 50. *p < 0.05.
Correlation between PD-L1 expression and survival
No statistically significant difference was observed in PFS between PD-L1 positive and PD-L1 negative tumors by CPS (p = 0.2, log-rank test; Fig. 5A), with 38-months median survival in the PD-L1-positive group compared with a 17-month median survival in PD-L1-negative group. The adjusted HR for PFS was 0.70 (CI: 0.39–1.26) for the PD-L1-positive group and 1.42 (95% CI: 0.79–2.54) for the PD-L1-negative group. Survival analysis demonstrated significantly better OS in the patient group with PD-L1 positive tumors by CPS compared to the PD-L1 negative group (p = 0.038; log-rank test, Fig. 5B), with an undefined median survival interval in the PD-L1 positive group compared with 31 months median survival in the PD-L1 negative group. The adjusted HR for OS was 0.53 (CI: 0.26–1.04) for the PD-L1 positive group and 1.90 (95% CI: 0.97–3.75) for the PD-L1 negative group. We further divided PD-L1 positive tumors into “low” and “high” expression groups using CPS 10 or 20 as cutoff points and found no significant difference between these groups (PFS, cutoff 10: p = 0.34 and cutoff 20: p = 0.38; OS, cutoff 10: p = 0.17 and cutoff 20: p = 0.83) (Fig. 5C–F). No significant difference was observed in PFS or OS between PD-L1 positive versus negative groups by TPS (PFS: p = 0.73; OS: p = 0.84) (Fig. 5G–H). Due to the limited power of PD-L1 positive cases by TPS, we were not able to perform statistical analysis between “low” and “high” expression groups by TPS.

A, B Kaplan–Meier curves of progression-free survival (PFS) and overall survival (OS) for patients in PD-L1-positive (red) and PD-L1-negative (blue) groups using CPS (cutoff 1): (A) PFS; (B) OS. C–F, Kaplan–Meier curves for patients in PD-L1-low (blue) and high (red) groups using 10 and 20 CPS cutoff points: (C) PFS, CPS cutoff 10; (D) OS, CPS cutoff 10; (E) PFS, CPS cutoff 20; (D) OS, CPS cutoff 20. G, H Kaplan–Meier curves for patients in PD-L1-positive (red) and PD-L1-negative (blue) groups using TPS (cutoff 1%): (G) PFS; (H) OS. *p < 0.05.
TIM-3 and B7-H3 expression do not correlate with clinical outcomes
Using CPS, with a threshold for positivity of 1, no significant difference was observed in PFS or OS between the TIM-3 positive and negative groups (PFS, p = 0.86; OS, p = 0.75). Furthermore, no significant difference was found between TIM-3 “low” and “high” groups using a CPS cutoff of 10 (PFS, p = 0.90; OS, p = 0.57) (Fig. 6A–D). Using TPS with a threshold for positivity of 1%, the power of TIM-3 negative group was too small (n = 10) and did not allow for comparison of survival between TIM-3 positive and negative groups. Similarly, due to the limited power of B7-H3 negative tumors (n = 6 by CPS, n = 7 by TPS), we were not able to compare survival between B7-H3 positive and negative groups. We further performed survival analysis for patients with B7-H3 positive ESC using different cutoffs. Due to the high level of B7-H3 expression, we chose 20 or 50 as cutoff for both TPS and CPS. Using CPS, no significant difference in survival was observed between B7-H3 “low” and “high” groups (PFS, cutoff 20: p = 0.78 and cutoff 50: p = 0.74; OS, cutoff 20: p = 0.31 and cutoff 50: p = 0.96). Similarly, no significant difference in survival was observed between B7-H3 “low” and “high” groups by TPS (PFS, cutoff 20: p = 0.39 and cutoff 50: p = 0.80; OS, cutoff 20: p = 0.78 and cutoff 50: p = 0.91) (Fig. 6E–L).

A, B Survival in TIM-3 positive (red) vs. TIM-3 negative (blue) groups using CPS (cutoff 1): (A) PFS; (B) OS. C, D Survival in TIM-3 low (blue) vs. high (red) groups using cutoff point 10: (C) PFS; (D) OS. E–L Survival in B7-H3 high (red) vs. B7-H3 low (blue) groups using CPS with cutoff point 20: (E) PFS; (F) OS; CPS with cutoff point 50: (G) PFS; (H) OS; TPS with cutoff point 20: (I) PFS; (J) OS; TPS with cutoff point 50: (K) PFS; (L) OS.
Discussion
In this study, we analyzed expression of three different immune checkpoints in a large cohort of ESC and correlated their positivity with CD8 + TIL and patient outcomes. Applying TPS, we found expression of PD-L1, TIM-3 and B7-H3 in tumor cells in 17%, 10%, and 93% of cases, respectively. Applying CPS (which takes both tumor and immune cells into account), the rate of positivity was 63%, 67% and 94% for PD-L1, TIM-3 and B7-H3, respectively. Expression of these markers was largely independent of one another. We found that PD-L1 positivity expressed as either TPS or CPS and TIM-3 positivity expressed as CPS were associated with higher CD8 + TIL density. Furthermore, PD-L1 expression scored using CPS was associated with improved OS. In our study, PD-L1 positivity in tumor cells (TPS) correlated with a higher tumor stage but not with survival, whereas the converse was true when PD-L1 was evaluated in both tumor and immune cells (CPS). These findings suggest that PD-L1 expression mainly in immune cells correlates with prognosis, and appears to be independent of tumor stage.
A number of studies have investigated the expression of PD-L1 in endometrial carcinomas,8,9,11,16,17,33,34,35,36,37,38,39, some of which reported it separately in ESC, with inconsistent results. In a recent study by Zhang et al. with a design similar to ours, the authors examined 79 cases of ESC and found PD-L1 expression by TPS (cut-off 1%) to be present in 43% and by CPS (cutoff 1) in 73.4% of cases. They found PD-L1 positivity by TPS but not CPS to positively correlate with stage II–IV tumor (p = 0.015).17 The rate of PD-L1 positivity by CPS and correlation of TPS with tumor stage in that study are in keeping with our findings. In a study of endometrial carcinomas utilizing a TMA that included 29 ESC, Pasanen et al. found that in ESC, 3.7% of cases expressed PD-L1 in tumor cells and 38.2% of tumors demonstrated PD-L1 positivity by CPS (cut-off 1). They also found that patients with advanced-stage (III to IV) tumors were more likely to show PD-L1 positivity in both tumor cells and by CPS compared to patients with stage I or II disease.14 Their observation that PD-L1 expression by TPS associated with advanced tumor stage is similar to our findings, although, in our study, only stage IA tumors demonstrated a lower level of positivity compared to more advanced stages. Bregar et al. examined PD-L1 expression in various types of endometrial carcinoma and found that tumors with high grade histology exhibited significant PD-L1 expression (in either tumor or immune cells) including a 30% (3/10) positivity rate in serous carcinomas.36 Kucukgoz Gulec et al. assessed PD-L1 in tumor cells and the local “microenvironment” (employing a threshold of > 5% positivity) in 53 type II endometrial carcinomas that included 17 serous carcinomas and found PD-L1 positivity in 15% of cases for tumor and 28% of cases for microenvironment. They found PD-L1 expression in the tumor cells to be associated with worse OS.12 In the study by Talhouk et al. performed on a TMA, the proportion of PD-L1 positive cases (>5% positivity threshold) in “p53 abnormal” tumors was 32%, lower than in polymerase epsilon (POLE)-mutated and MMR-deficient tumors but significantly higher than in p53-wild type tumors. These authors found PD-L1 positivity to lack prognostic significance.13 Zong at al. examined 113 ESC within a TMA and demonstrated the PD-L1 positivity rate by TPS (cut-off 1%) to be 17.7% and by CPS (cut-off 1) 54%.15 These results are similar to our observations. These authors found PD-L1 positivity in tumor cells to be associated with a favorable prognosis in patients with advanced-stage disease, non-endometrioid endometrial carcinoma, and high-risk endometrial cancer.15 We could not directly compare these results to ours because prognostic significance for ESC was not separately analyzed in that study. A study by Engerud et al. found a PD-L1 positivity rate in ESC (assessed in a TMA as “only glandular expression” with a positivity threshold of <10%) of 63% (41/65), and no association between PD-L1 positivity and disease-specific survival in either the endometrioid or non-endometrioid subgroup.38 Kir et al. investigated a group of 59 endometrial carcinomas including 5 serous carcinomas and 7 p53-mutant carcinomas and found p53 mutant-type tumors to demonstrate PD-L1 positivity in 14.3% (1/7) of cases by TPS employing a cutoff of ≥1%, in 0% (0/7) of cases by TPS employing a cutoff of ≥5%, and in 28.6% (2/7) of cases by CPS employing a cutoff of 1.39 In a study of 231 endometrial carcinomas that included 33 serous carcinomas, Amarin et al. found 27% of ESC to be positive for PD-L1 by TPS. In their study, in an unselected group of endometrial carcinomas, PD-L1 expression was not associated with overall survival.16 Direct comparison between results of various studies is limited due to such factors as different study designs, different immunostaining protocols, and variability in antibody clones used. Studies performed on a TMA are especially difficult to compare to studies utilizing whole tissue sections due to frequent and significant heterogeneity in PD-L1 expression within individual tumors. Because of the small size of tissue cores included in TMAs, there is a higher probability of sampling bias. Of note, we observed PD-L1 expression to be especially prominent at the advancing edge of the tumor, although with frequent heterogeneity of expression within different areas at this edge. The hope is that increasing understanding of patterns of immune checkpoint marker expression, further standardization of testing, and refinement of evaluation methods for specific tumor types will allow for greater data interchangeability and improved confidence in comparison of results across studies.
Our results demonstrated association of higher CD8 + TIL density with expression of PD-L1 by either TPS or CPS and TIM-3 by CPS. Similar findings on PD-L1 positivity in endometrial carcinomas with higher CD8 + TIL have previously been reported in other studies.15,40 Increased CD8 + T cell infiltrate is generally considered a good prognostic feature indicating a robust anti-tumor immune response in solid tumors in general and in endometrial carcinoma in particular.41,42 PD-L1 signaling within tumors is affected by co-activation of other specific immune regulators, and this co-activation requires presence of TILs and IFN-γ expression. Rather than being a constitutively active, oncogene-driven immunosuppressive function, PD-L1 upregulation likely represents, in at least some tumors, an adaptive immune resistance mechanism adopted by tumor cells in an attempt to avert destruction by TIL.43,44 Our observations support the hypothesis that in many ESC, immunosuppressive pathways are triggered by activated CD8 + T cells via negative feedback within the tumor microenvironment. Targeting such negative regulatory immune checkpoints might be beneficial for patients with a preexisting T cell-inflamed tumor microenvironment.44,45
Although the expression of TIM-3 has been evaluated in various tumors, far less is known about this immune checkpoint as compared to PD-L1. Prevalence and significance of TIM-3 expression in endometrial carcinomas are currently underexplored. In many solid tumors, increased TIM-3 expression has been shown to have an association with adverse outcomes.21 Moore et al. found tumoral and tumor-associated immune cell expression of TIM-3 by IHC (scored using TPS and CPS) to be common in endometrial carcinoma with particularly high levels of expression in MLH1-hypermethylated, MSH6-deficient, and intermediate- to high histologic grade tumors.46 Ramos et al. investigated the TIM-3 RNA expression signature in endometrial carcinomas including 10 ESC and found its levels to be significantly higher in endometrioid carcinoma compared to carcinosarcoma and serous carcinoma, and increased in microsatellite instability (MSI)-high versus MSI-low tumors.47 Although published results on B7-H3 prevalence and significance in solid tumors are not uniform, like TIM-3, it is overexpressed in many cancers and, in the majority of published studies, is associated with a worse prognosis.26,32 In their study of 107 endometrial carcinomas including 15 serous carcinomas, Brunner et al., using a semiquantitative immunoreactive score, demonstrated that high grade and type II tumors expressed significantly more B7-H3 than low grade and endometrioid carcinomas. They also found a correlation between B7-H3 expression on tumor cells and tumor T-cell infiltration.48 Nishimura et al. evaluated PD-L1 and B7-H3 expression (defined as staining “in > 10% area”) in 21 ESC and found no correlation with tumor characteristics, stage or recurrence, although B7-H3 expression showed a trend for correlation with nodal metastasis.49 Direct comparison of these results to our findings is precluded by significant differences in study design.
Correlation between clinical tumor stage or outcome and TIM-3 or B7-H3 expression was not found in our study. In addition, we found little concordance between PD-L1, TIM-3 and B7-H3 expression within individual tumors and did not detect a tendency for synergistic interaction of these molecules, although our data provide only a narrow view of a highly complex tumor microenvironment with an incompletely elucidated interplay of myriad factors. Independent expression of these markers could indicate their complementary roles in tumor immune evasion and suggest a potential benefit from targeted combination immunotherapy in these tumors. In some solid tumors including lung and head and neck carcinomas, upregulation of TIM-3 has been shown to be associated with the development of resistance to PD-1 blockade18,50, and dual blockade of the PD-1/PD-L1 axis with addition of anti-TIM-3 monoclonal antibodies showed encouraging results in clinical trials.51 As the B7-H3 receptor and the downstream events of its binding to recipient cells are still incompletely characterized, no ICI has yet been developed. A study by Yonesaka et al. demonstrated, however, that B7-H3 expression in lung cancer directly correlated with nonresponsiveness to anti-PD-1 immunotherapy. These authors also showed in a murine model improved antitumor immunity with increased number of CD8 + T cells and their recovered effector function after dual anti-B7-H3 and anti-PD-L1 antibody treatment.52 Other innovative B7-H3-based immunotherapy strategies are currently underway and show great promise.28,31
Many previous studies in endometrial carcinomas focused on correlation of PD-L1 expression in MSI high/MMR-deficient tumors, comparing it to microsatellite stable/MMR-intact tumors. Results of these investigations repeatedly confirmed significantly higher PD-L1 and CD8 + TIL levels in the former group.13,33,34,53 The postulated mechanism of such disparate results is the high number of somatic mutations and, thus, an increased tumor-specific neoantigen load in the former group, which would be expected to induce an enhanced antitumor immune response with a highly inflamed tumor phenotype. Another group of tumors associated with a high number of mutations and neoantigens is POLE mutated/ultramutated tumors, which also tend to be enriched with intratumoral infiltration of CD8 + T cells and increased PD-L1 expression.13,33,34,53 In our study we examined ESC, a histotype corresponding to a copy number high group by The Cancer Genome Atlas (TCGA) molecular classification.54 Tumors from this group characteristically contain the lowest number of mutations compared to tumors from other groups and thus are not expected to show a high number of neoantigens; nevertheless, a good number of these tumors demonstrated a robust immune response and an inflamed tumor phenotype manifesting as both immune checkpoint positivity and high numbers of CD8 + T cells.
These observations correlate with results reported by other investigators.13,34 It has been previously hypothesized that such an immune response may be aimed at self-antigens or cancer/testis (CT) antigens instead of neoepitopes34; however, the precise molecular interactions triggering immune reaction in these tumors remain to be elucidated. Irrespective of the underlying mechanisms, these observations suggest the existence of specific subsets of hypomutated endometrial carcinomas that are immunogenic and may be potential candidates for targeted immunotherapy. In such tumors, specifically in serous carcinoma where MMR status and other surrogate markers of potentially enhanced antitumor immune response are less relevant, immune checkpoint biomarkers such may have a particular value.
Our study has several strengths. First, to minimize sampling bias, we utilized whole tissue sections. Second, we studied a relatively homogenous group of tumors of identical histologic type. Finally, we analyzed our results using two different scoring methods (both recommended by the FDA in companion diagnostics for other solid tumors), as well as different positivity thresholds for each marker. A potential limitation of our study is a modest degree of correlation between PD-L1 expression and overall survival. Additional larger studies are needed to validate our results. Future investigations of the ESC immune landscape should also include evaluation of lesser investigated biomarkers (e.g., lymphocyte-activation gene 3 (LAG-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain immunoglobulin (Ig)-containing suppressor of T-cell activation (VISTA), and B7-H4), with correlation of their expression with tumor pathologic and molecular characteristics and clinical outcome. Exploring correlation of spatial distribution of immune checkpoints within tumors with outcomes may provide a more nuanced understanding of the significance of various expression patterns (e.g., diffuse expression versus predominantly at tumor advancing edge and stromal interface). Studying potential somatic and germline sources of interpatient heterogeneity to answer the question as to why some tumors generate a spontaneous antigen-specific T cell response while others do not may provide further insight into disparities in antitumor immune responses. Finally, studies correlating immune checkpoint expression with response to ICI therapies will contribute to a better understanding of their predictive value and inform design of future clinical trials and individualized therapeutic strategies.
In summary, results of our study contribute to the growing body of knowledge concerning prevalence, distribution and significance of PD-L1, TIM-3 and B7-H3 expression in ESC, and highlight the complexity of the immune landscape in these tumors. Our findings also suggest that these molecules may potentially be viable targets for individualized immunotherapy in ESC.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Hamilton CA, Cheung MK, Osann K, Chen L, Teng NN, Longacre TA et al. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br J Cancer 94, 642–646 (2006).
Han Y & Liu C. Clinicopathological characteristics and prognosis of uterine serous carcinoma: A SEER program analysis of 1016 cases. J Obstet Gynaecol Res 47, 2460–2472 (2021).
Keir ME, Butte MJ, Freeman GJ & Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26, 677–704 (2008).
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264 (2012).
Davis AA & Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer 7, 278 (2019).
Arora S, Balasubramaniam S, Zhang W, Zhang L, Sridhara R, Spillman D et al. FDA Approval Summary: Pembrolizumab plus Lenvatinib for Endometrial Carcinoma, a Collaborative International Review under Project Orbis. Clin Cancer Res 26, 5062–5067 (2020).
Makker V, Colombo N, Casado Herráez A, Santin AD, Colomba E, Miller DS et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N Engl J Med 386, 437–448. https://doi.org/10.1056/NEJMoa2108330 (2022).
Sloan EA, Ring KL, Willis BC, Modesitt SC & Mills AM. PD-L1 Expression in Mismatch Repair-deficient Endometrial Carcinomas, Including Lynch Syndrome-associated and MLH1 Promoter Hypermethylated Tumors. Am J Surg Pathol 41, 326–333 (2017).
Yamashita H, Nakayama K, Ishikawa M, Nakamura K, Ishibashi T, Sanuki K et al. Microsatellite instability is a biomarker for immune checkpoint inhibitors in endometrial cancer. Oncotarget 9, 5652–5664 (2018).
Li Z, Joehlin-Price AS, Rhoades J, Ayoola-Adeola M, Miller K, Parwani AV et al. Programmed Death Ligand 1 Expression Among 700 Consecutive Endometrial Cancers: Strong Association With Mismatch Repair Protein Deficiency. Int J Gynecol Cancer 28, 59–68 (2018).
Kim J, Kim S, Lee HS, Yang W, Cho H, Chay DB et al. Prognostic implication of programmed cell death 1 protein and its ligand expressions in endometrial cancer. Gynecol Oncol 149, 381–387 (2018).
Kucukgoz Gulec U, Kilic Bagir E, Paydas S, Guzel AB, Gumurdulu D & Vardar MA. Programmed death-1 (PD-1) and programmed death-ligand 1 (PD-L1) expressions in type 2 endometrial cancer. Arch Gynecol Obstet 300, 377–382 (2019).
Talhouk A, Derocher H, Schmidt P, Leung S, Milne K, Gilks CB et al. Molecular Subtype Not Immune Response Drives Outcomes in Endometrial Carcinoma. Clin Cancer Res 25, 2537–2548 (2019).
Pasanen A, Ahvenainen T, Pellinen T, Vahteristo P, Loukovaara M & Bützow R. PD-L1 Expression in Endometrial Carcinoma Cells and Intratumoral Immune Cells: Differences Across Histologic and TCGA-based Molecular Subgroups. Am J Surg Pathol 44, 174–181 (2020).
Zong L, Sun Z, Mo S, Lu Z, Yu S, Xiang Y et al. PD-L1 expression in tumor cells is associated with a favorable prognosis in patients with high-risk endometrial cancer. Gynecol Oncol 162, 631–637 (2021).
Amarin JZ, Mansour R, Al-Ghnimat S & Al-Hussaini M. Differential Characteristics and Prognosis of PD-L1-Positive Endometrial Carcinomas: A Retrospective Chart Review. Life (Basel) 11 (2021).
Zhang T, Liu Q, Zhu Y, Zhang S, Peng Q, Strickland AL et al. PD-L1 Expression in Endometrial Serous Carcinoma and Its Prognostic Significance. Cancer Manag Res 13, 9157–9165 (2021).
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7, 10501 (2016).
Hellmann MD, Friedman CF & Wolchok JD. Combinatorial Cancer Immunotherapies. Adv Immunol 130, 251–277 (2016).
Dempke WCM, Fenchel K, Uciechowski P & Dale SP. Second- and third-generation drugs for immuno-oncology treatment-The more the better? Eur J Cancer 74, 55–72 (2017).
Qin S, Dong B, Yi M, Chu Q & Wu K. Prognostic Values of TIM-3 Expression in Patients With Solid Tumors: A Meta-Analysis and Database Evaluation. Front Oncol 10, 1288 (2020).
Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143 (2007).
Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol 39, 2492–2501 (2009).
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol 2, 269–274 (2001).
Liu S, Liang J, Liu Z, Zhang C, Wang Y, Watson AH et al. The Role of CD276 in Cancers. Front Oncol 11, 654684 (2021).
Tian T & Li Z. Targeting Tim-3 in Cancer With Resistance to PD-1/PD-L1 Blockade. Front Oncol 11, 731175 (2021).
Zhou WT & Jin WL. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front Immunol 12, 701006 (2021).
Wang Q, Liu F & Liu L. Prognostic significance of PD-L1 in solid tumor: An updated meta-analysis. Medicine (Baltimore) 96, e6369 (2017).
Zang K, Hui L, Wang M, Huang Y, Zhu X & Yao B. TIM-3 as a Prognostic Marker and a Potential Immunotherapy Target in Human Malignant Tumors: A Meta-Analysis and Bioinformatics Validation. Front Oncol 11, 579351 (2021).
Liu C, Zhang G, Xiang K, Kim Y, Lavoie RR, Lucien F et al. Targeting the immune checkpoint B7-H3 for next-generation cancer immunotherapy. Cancer Immunol Immunother 71, 1549–1567. https://doi.org/10.1007/s00262-021-03097-x (2022).
Loos M, Hedderich DM, Friess H & Kleeff J. B7-h3 and its role in antitumor immunity. Clin Dev Immunol 2010, 683875 (2010).
Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol 1, 1319–1323 (2015).
Eggink FA, Van Gool IC, Leary A, Pollock PM, Crosbie EJ, Mileshkin L et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology 6, e1264565 (2017).
Mo Z, Liu J, Zhang Q, Chen Z, Mei J, Liu L et al. Expression of PD-1, PD-L1 and PD-L2 is associated with differentiation status and histological type of endometrial cancer. Oncol Lett 12, 944–950 (2016).
Bregar A, Deshpande A, Grange C, Zi T, Stall J, Hirsch H et al. Characterization of immune regulatory molecules B7-H4 and PD-L1 in low and high grade endometrial tumors. Gynecol Oncol 145, 446–452 (2017).
Asaka S, Yen TT, Wang TL, Shih IM & Gaillard S. T cell-inflamed phenotype and increased Foxp3 expression in infiltrating T-cells of mismatch-repair deficient endometrial cancers. Mod Pathol 32, 576–584 (2019).
Engerud H, Berg HF, Myrvold M, Halle MK, Bjorge L, Haldorsen IS et al. High degree of heterogeneity of PD-L1 and PD-1 from primary to metastatic endometrial cancer. Gynecol Oncol 157, 260–267 (2020).
Kir G, Soylemez T, Olgun ZC, Aydin A & McCluggage WG. Correlation of PD-L1 expression with immunohistochemically determined molecular profile in endometrial carcinomas. Virchows Arch 477, 845–856 (2020).
Vagios S, Yiannou P, Giannikaki E, Doulgeraki T, Papadimitriou C, Rodolakis A et al. The impact of programmed cell death-ligand 1 (PD-L1) and CD8 expression in grade 3 endometrial carcinomas. Int J Clin Oncol 24, 1419–1428 (2019).
Kondratiev S, Sabo E, Yakirevich E, Lavie O & Resnick MB. Intratumoral CD8+ T lymphocytes as a prognostic factor of survival in endometrial carcinoma. Clin Cancer Res 10, 4450–4456 (2004).
de Jong RA, Leffers N, Boezen HM, ten Hoor KA, van der Zee AG, Hollema H et al. Presence of tumor-infiltrating lymphocytes is an independent prognostic factor in type I and II endometrial cancer. Gynecol Oncol 114, 105–110 (2009).
Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 4, 127ra137 (2012).
Teng MW, Ngiow SF, Ribas A & Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res 75, 2139–2145 (2015).
Gajewski TF, Schreiber H & Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14, 1014–1022 (2013).
Moore M, Ring KL & Mills AM. TIM-3 in endometrial carcinomas: an immunotherapeutic target expressed by mismatch repair-deficient and intact cancers. Mod Pathol 32, 1168–1179 (2019).
Ramos A FS, Melchert V, Jenkins D, Borger DR, Growdon WB. Checkpoint inhibitor signatures across endometrial carcinoma histologic subtypes. Gynecol Oncol 149, 621–621 (2018).
Brunner A, Hinterholzer S, Riss P, Heinze G & Brustmann H. Immunoexpression of B7-H3 in endometrial cancer: relation to tumor T-cell infiltration and prognosis. Gynecol Oncol 124, 105–111 (2012).
Nishimura M MA, Abe A, Irahara M. PD-L1 and B7-H3 expression in serous carcinoma of endometrium. Cancer Research (2018).
Shayan G, Srivastava R, Li J, Schmitt N, Kane LP & Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 6, e1261779–e1261779 (2016).
Acharya N, Sabatos-Peyton C & Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. Journal for immunotherapy of cancer 8, e000911 (2020).
Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N et al. B7-H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clin Cancer Res 24, 2653–2664 (2018).
Victoor J, Borght SV, Spans L, Lehnert S, Brems H, Laenen A et al. Comprehensive immunomolecular profiling of endometrial carcinoma: A tertiary retrospective study. Gynecol Oncol 162, 694–701 (2021).
Murali R, Soslow RA & Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol 15, e268–278 (2014).
Acknowledgements
We acknowledge the assistance of the University of Texas Southwestern Tissue Resource, a shared resource at the Simmons Comprehensive Cancer Center, which is supported in part by the National Cancer Institute under award number 5P30CA142543. The authors thank staff of the Parkland Hospital Anatomic Pathology Laboratory for technical assistance.
Funding
The project was partially funded by the UTSW Department of Pathology; intramural research fund of the Department of Pathology, UTSW to H.C., intramural research fund of the Department of Pathology, UTSW to E.L., and Philip O’Bryan Montgomery, Jr., MD endowment fund, UTSW to K.M.
Author information
Authors and Affiliations
Contributions
E.L., H.C.: study conception and design, data acquisition, analysis, paper writing and editing; H.C.: statistical analyses; C.L.: performance of IHC and discussion; E.L., H.C., D.H.C., K.M., K.C, S.N., G. R.C., K.G., W.Z.: data interpretation and discussion; K.C., D.H.C., K.M., K.G.: paper editing; All authors: final editing and approval of the present version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the Institutional Review Board of University of Texas Southwestern Medical Center. This study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, H., Molberg, K., Carrick, K. et al. Prevalence and prognostic significance of PD-L1, TIM-3 and B7-H3 expression in endometrial serous carcinoma. Mod Pathol 35, 1955–1965 (2022). https://doi.org/10.1038/s41379-022-01131-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01131-6
This article is cited by
-
Overall survival prediction models for gynecological endometrioid adenocarcinoma with squamous differentiation (GE-ASqD) using machine-learning algorithms
Scientific Reports (2023)
-
Tumour-infiltrating lymphocytes: from prognosis to treatment selection
British Journal of Cancer (2023)
-
Bispecific antibody targeting both B7-H3 and PD-L1 exhibits superior antitumor activities
Acta Pharmacologica Sinica (2023)
