
Abstract
This editorial focuses on common issues that surround the diagnosis of usual interstitial pneumonia (UIP), a clinically significant pathologic diagnosis. Most of these issues stem from conflation of the pathologically defined entity UIP with the clinically defined entity IPF. A pathologic or radiologic diagnosis of UIP is required for the clinical/multidisciplinary diagnosis of idiopathic pulmonary fibrosis (IPF) but it has also been described in several other clinical settings. I offer my viewpoint on 5 important questions. 1. Is UIP a diagnosis or a “pattern”? Answer: UIP is a pathologic diagnosis and is better conceptualized as a “pattern” than as a specific clinical entity. Since all cases of UIP require pattern recognition, adding the word “pattern” to UIP is redundant. 2. Is pathology the gold standard for UIP? Answer: Yes. 3. How do you “prove” etiology of a given case of UIP? Answer: “Soft” histologic features can raise the possibility of certain etiologies but the final determination of etiology comes from the multidisciplinary team. With few exceptions, there are no findings pathognomonic for any etiology in UIP. 4. Does UIP imply IPF? Answer: No. 5. What should we do when pathology and HRCT are discordant? Answer: This depends on the specifics of the discrepancy. When HRCT suggests a non-UIP diagnosis such as NSIP and histology shows UIP, histology has been shown to predict prognosis in multiple studies. In other settings, the radiologic impression based on HRCT is often proven to be incorrect by the histologic findings.
Similar content being viewed by others

This editorial represents one of two “point-counterpoint” articles in the “Controversies in Pathology” special issue that examine an area of controversy in this evolving field; for the corresponding counterpoint article, see the accompanying editorial by B. Larsen published in this issue1.
Usual interstitial pneumonia (UIP) is best known as the pathologic correlate of the clinically defined entity idiopathic pulmonary fibrosis (IPF)2,3,4,5,6,7,8,9,10,11,12. Per current guidelines issued by the American Thoracic Society (ATS), European Respiratory Society (ERS), Japanese Respiratory Society (JRS) and the Latin American Thoracic Society (LATS), a pathologic or radiologic diagnosis of UIP is required for a clinical diagnosis of IPF12. In the ideal world envisioned by these guidelines, the clinical diagnosis of IPF is made after a multidisciplinary discussion between pulmonologists, radiologists and pathologists.
We have known for several years that the pathologic features of UIP can occur in a wide variety of clinical settings, including various forms of connective tissue disease (CTD)13,14,15,16, familial interstitial lung disease17, asbestos exposure18, fibrotic hypersensitivity pneumonitis19, etc. Most cases of UIP in lung biopsies are diagnosed in patients without an obvious underlying cause (IPF). In each of these settings, a pathologic diagnosis of UIP connotes a poor prognosis and a lack of response to immunomodulators when compared to other histologic tissue reactions such as non-specific interstitial pneumonia (NSIP) or organizing pneumonia5,13,14,15,16,20. Thus, UIP is a clinically significant pathologic diagnosis.
The histologic features of UIP are well known2,3,4,12. The features that help to separate UIP from other forms of fibrosing interstitial lung disease have been described in detail by Katzenstein et al.2,3. The main pathologic features of UIP are as follows (Figs. 1–4):
-
1.
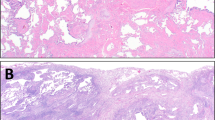
Interstitial fibrosis in a patchwork pattern (Fig. 1). This means that scarred lung with significant architectural distortion and/or honeycombing is abruptly juxtaposed to non-fibrotic lung. In UIP, fibrosis is always present in subpleural and paraseptal lung parenchyma, but also commonly extends deeper into the lobule and frequently involves peribronchiolar parenchyma. In advanced cases, the lobules are entirely obliterated. Note that most surgical lung biopsies do not sample more than a few centimeters of subpleural lung tissue. Therefore, truly central/perihilar lung parenchyma is never sampled in a surgical lung biopsy. Those with experience in lung explants will vouch that fibrosis in UIP can and often does extend very deep into the central lung, albeit to a lesser degree.
-
2.
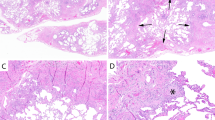
Scarring. This is fibrosis that distorts lung architecture (“architectural distortion”), and is the most essential feature of the diagnosis (Fig. 2). The difference between scarring (fibrosis with distortion of lung architecture) and fibrosis without architectural distortion is illustrated in Fig. 2B.
-
3.
Honeycomb change. This process also distorts lung architecture. It is characterized by clusters of dilated, mucin-filled epithelium-lined airspaces in a fibrotic background (Fig. 3). Honeycomb change may be visible grossly and on high-resolution chest tomography (HRCT). Honeycomb change consisting of smaller spaces that are not visible grossly and can only be seen microscopically is called “microscopic honeycomb change” (Fig. 4).
-
4.
Fibroblast foci. These are dome-shaped (convex on one side) collections of fibroblasts within the interstitium. They are common in UIP but are not specific (Fig. 5).

A Pre-transplant chest CT was read as “fibrotic interstitial lung disease with UIP imaging pattern”, with a comment that “the presence of significant air trapping makes the CT inconsistent with UIP per the ATS criteria…”. Note honeycomb change in the left lower lobe (arrow). Surgical lung biopsy 9 years prior to this CT was consistent with UIP. The patient was treated with Pirfenidone. B Honeycomb change in the left upper lobe (arrow). C Explanted lung shows UIP with interstitial fibrosis in a patchwork pattern. Fibrosis with architectural distortion (scarring, short arrows) is juxtaposed to normal lung (arrowheads). Note extensive fibrosis around airways causing traction bronchiolectasis (long arrow).

A Scarring distorts lung architecture (arrows). B This image shows the difference between interstitial fibrosis without architectural distortion (arrows) and scarring (arrowheads). Scarring is the most essential feature for the diagnosis of UIP.

Explanted lung from a 70-year-old man with a clinical diagnosis of IPF. A This section shows architectural distortion with extensive honeycomb change, manifesting as clusters of dilated, mucin-filled airspaces (short arrows). No normal lung architecture is identified. Compare honeycomb change (short arrows) with dilated airways (traction bronchiolectasis, long arrows). Note that honeycombing surrounds several airways. B Honeycomb change at high magnification. Dilated bronchus is at top. The honeycombed areas are filled with mucin (short arrows) and are lined by respiratory-type epithelium (arrowheads).

Explanted lung from a patient with a clinical diagnosis of IPF. The macroscopic image shown in (A) corresponds to (B) (microscopic); (C) (gross) corresponds to (D) (microscopic). A Subpleural lung (purple box indicates exact area shown in (B)). Short black arrow: pleura. Long black arrow: honeycombing visible grossly. Black arrowhead: fibrosis without grossly visible honeycombing. B Microscopy provides greater detail than appreciable grossly. Short black arrow: pleura. Long black arrow: honeycomb change. Black arrowhead: microscopic honeycombing. C Another area from same lung, more central (closer to hilum; purple box indicates exact section shown in (D)). Arrowhead: pulmonary artery. Long arrow: Bronchial cartilage. Short arrows: This area appears fibrotic grossly but does not show honeycombing. (D) Microscopic correlate of (C). Arrowhead: pulmonary artery. Long arrow: Bronchial cartilage. Short arrows: Extensive microscopic honeycombing is present. Images courtesy of Dr. Frido Bruehl.
Is UIP a diagnosis or a pattern?
UIP is a pathologic diagnosis2,3. I hope most pathologists would agree that “pathologic diagnosis” is the appropriate designation for a diagnosis that goes on the top-line of a pathology report. I have used UIP as a top-line diagnosis in pathology reports for the last 14 years and have seen many reports from experienced pulmonary pathologists who used this term as the top-line diagnosis in their pathology reports.

Note that these collections of fibroblasts (A–C) are located within the interstitium, with a layer of epithelial cells covering their luminal surface.
Now let’s address the word “pattern”. An enduring source of debate and disagreement among pulmonary pathologists is how to diagnose cases with histologic features of UIP that occur in clinical settings suggesting a diagnosis other than IPF, and cases of UIP with histologic features that stray from the norm. The word “pattern”, used in the 2002 and 2018 ATS/ERS guidelines4,12, has been appended to some cases of UIP, but the usage and implied meaning of this word depends on who is using it. Some clinicians use the phrase “UIP pattern” for cases that have radiologic or histologic features of UIP but are given a final clinical/multidisciplinary label other than IPF, such as CTD-related interstitial lung disease (CTD-ILD) or fibrotic hypersensitivity pneumonitis. These clinicians will say, after multidisciplinary discussion, “this is CTD-ILD with a UIP pattern”.
The way some pathologists use this word is quite different, and is based on the erroneous notion that a pathologic diagnosis of UIP must always line up with a clinical diagnosis of IPF. If these pathologists do not have access to all pertinent clinical information at the time that the pathologic diagnosis is issued, they use the phrase “UIP pattern” for cases that look like UIP on pathologic grounds but that they feel might not end up being labeled as IPF by the clinical team. Even when all pertinent clinical and radiologic information is known to the pathologist, there is no guarantee that the pathologist’s interpretation of the final clinical/multidisciplinary diagnosis will be the same as the clinician’s. Thus, using the term “UIP” for cases that a pathologist thinks will turn out to be IPF and “UIP pattern” for cases that they think will turn out to be something else is misguided.
Other pathologists use the phrase “UIP pattern” in a different way; they use this term for cases that look like UIP on histologic grounds but have some features (such as granulomas or lymphoid hyperplasia) that the pathologist feels might indicate an alternative etiology. What do these pathologists do if a case they diagnosed as UIP is adjudged to be a CTD-ILD after multidisciplinary discussion? Do they amend their pathologic diagnosis to “UIP pattern”? Alternatively, what if a case diagnosed as “UIP pattern” by histology is adjudged as IPF by multidisciplinary discussion? Does the pathologic diagnosis need to be amended to “UIP”?
The inconsistent use of the word “pattern” only adds more confusion to the endless alphabet soup of interstitial lung disease terminology. Some pathologists use UIP as their top-line diagnosis, others use “UIP pattern” for all cases of UIP, and yet others use UIP for cases that they think will turn out to be IPF clinically and “UIP pattern” for cases that look like UIP histologically but do not fit with IPF clinically. Some prefer to use a top-line diagnosis of UIP for histologically classic cases, preferring a descriptive diagnosis for cases with histologic features that they feel suggest an alternative etiology. Others do not like to ever make a top-line diagnosis of UIP, instead preferring to use descriptive diagnoses for all cases. It is clear that differing views exist on how to word the pathologic diagnosis in this area. Given the complexity of the issue and the vast variety of differing opinions, I do not think it is feasible to compel every pathologist to word every case in exactly the same way. My approach is to use the term UIP as the top-line pathologic diagnosis for all cases in which histologic features of UIP are present. I do mention “soft features” as potential clues to an etiology in a comment, depending on the type and extent of the histologic abnormality, but I recognize that most of these features can be overridden by compelling clinical information. For cases where the histology shows especially compelling features suggesting an alternative diagnosis, I prefer to make a descriptive diagnosis such as “chronic fibrosing and cellular interstitial pneumonia with granulomas” and describe my concerns regarding the possibility of hypersensitivity pneumonitis in a comment; I do not use UIP as the top-line diagnosis in such cases. Since IPF is a multidisciplinary diagnosis, I do not attempt to make a diagnosis of IPF based on histology alone.
Conceptually, every case of UIP in a lung biopsy is a “pattern” that denotes a poor prognosis within the context of diffuse parenchymal lung disease. To varying extents, many pathologic diagnoses are “patterns” that can be refined by the addition of clinical information, and imaging and laboratory findings. Since UIP is always a pattern, my view is that the phrase “UIP pattern” is redundant. Other expert pulmonary pathologists have made similar arguments previously21,22. In 2006, Dr. Anna-Luise Katzenstein wrote: “It seems superfluous to add “pattern” to what is already a discrete histologic finding. Moreover a similar approach using pattern is not utilized in other analogous situations, either in the lung (for example, one does not diagnose “non-necrotizing granuloma pattern” in sarcoidosis, or “eosinophilic pneumonia pattern” in chronic eosinophilic pneumonia), or in other organ systems (one does not diagnose “chronic dermatitis pattern” or “chronic hepatitis pattern”)”21. It has also been pointed out that the use of the word “pattern” implies that sorting patients with pathologic UIP into different clinical groups impacts therapy and prognosis, even though the bulk of the evidence suggests that patients with a biopsy diagnosis of UIP—regardless of the final clinical/multidisciplinary label—are relatively insensitive to immunomodulators and are likely to have a progressive clinical course. The same is true of patients with an exposure history suggesting chronic hypersensitivity pneumonitis, where a biopsy diagnosis of UIP predicts a natural history indistinguishable from IPF22,23.
Some pathologists argue “if I diagnose UIP, my clinician will think it’s IPF”. For clinicians that labor under this misunderstanding, the role of pathologist should be to educate them about the various settings in which UIP can be encountered, not to use neologisms such as “UIP of IPF”, which serve only to create further confusion and blur the line between a pathologic diagnosis and a clinical entity. This approach also leads to undesirable obfuscation of the pathologic findings, in that pathologists who wish to avoid the term UIP feel obliged to re-brand fibroblast foci as “organizing acute lung injury” or honeycomb change as “remodeling”. It is important to communicate honestly with clinicians about the limitations of pathology and not suggest that pathology by itself can clearly distinguish between “UIP of IPF”, “UIP of connective tissue disease”, “UIP of familial IPF”, “UIP of IPAF”, or myriad other etiologies that might be applied to a given case based on the vagaries of multidisciplinary discussion. The fact that soft histologic features may be helpful in individual cases of these entities, or certain findings may be more common in one entity than another, does not detract from the fact that the pathologic findings are almost never pathognomonic for a specific etiology in any of these conditions.
Is pathology the gold standard for UIP?
Yes. The entity of UIP was created and defined by pathologists based on histologic features24,25. Subsequently, the radiologic correlates of this diagnosis were described and refined over the years using pathology as the gold standard7,11,26,27. Eventually - based partially on concerns regarding risk of complications related to surgical lung biopsies - this led to the decision that UIP could be diagnosed by HRCT without biopsy confirmation in the subset of cases in which honeycombing is visible on CT scans28. At every step, the role of imaging has been refined and redefined using pathology as the gold standard. More recent techniques such as molecular studies and optical coherence tomography have also used pathology as the gold standard for the diagnosis of UIP29,30. Surgical lung biopsies continue to be used as a gold standard to make the diagnosis of UIP in cases where the HRCT findings are equivocal, unclear or even inconsistent with UIP7,11,27. The fact that radiologists are allowed to make a diagnosis of UIP on the basis of HRCT findings in a subset of cases is frequently cited to mislead readers into thinking that pathology is no longer the gold standard for the diagnosis of UIP. Would we ask a liver pathologist if pathology is the gold standard for the diagnosis of cirrhosis, and cite the fact that the diagnosis can now be made by vibration-controlled transient elastography (VCTE)? Liver pathologists might respond that the diagnosis of cirrhosis can be made by VCTE but there are well-known pitfalls associated with its use, and that in fact VCTE was validated using histology in liver biopsies as the gold standard. The same is true of pathology in UIP. The notion that HRCT is now an acceptable means of making the diagnosis of UIP in a subset of cases was only possible after evaluating its performance using lung biopsies as a gold standard. After 20 years of numerous studies that have used pathology as the gold standard for the diagnosis of UIP, we must not diminish the value of pathology.
The issue of whether multidisciplinary discussion is the gold standard in the diagnosis of idiopathic pulmonary fibrosis (IPF) is a separate question. For an entity such as IPF that is defined clinically based on determination of etiology in a multidisciplinary fashion (which is often a subjective judgment call based on parameters outside the control of pathologists), pathology cannot be the gold standard. For this reason, multidisciplinary discussion is currently considered the gold standard for the diagnosis of IPF.
Does UIP imply IPF?
No. UIP is a pathologic diagnosis, while IPF is a clinical (multidisciplinary) diagnosis. While a multidisciplinary diagnosis of IPF does require a pathologic (or radiologic) diagnosis of UIP, not all cases of pathologic UIP will be acceptable to a multidisciplinary group as IPF. This is analogous to DAD, which is a pathologic diagnosis, while ARDS is a clinical diagnosis. Many cases of pathologic DAD meet clinical criteria for ARDS, but some do not31,32,33. Similarly, some cases with a pathologic diagnosis of organizing pneumonia are eventually deemed idiopathic by the clinical team and given the clinical label of “cryptogenic organizing pneumonia” (COP). However, some cases of pathologic organizing pneumonia are attributed to specific etiologies by multidisciplinary teams, including infection, vaping, drug toxicity, CTD or aspiration32,34. Should we start labeling some cases as “organizing pneumonia of COP” based on histologic findings because some clinicians feel that a pathologic diagnosis of organizing pneumonia represents COP?
There are situations in which the treating physician may decide that - despite a pathologic diagnosis of UIP - the clinical setting does not fit with IPF. The clinical judgment as to whether a given case of UIP represents IPF or not takes into account many factors including the patient’s demographics, history, exposures, tempo of disease, radiologic findings, serologies, extrapulmonary clinical manifestations and other factors. For example, a pathologic diagnosis of UIP in a 64-year-old woman with rheumatoid arthritis may be interpreted by the clinician and the multidisciplinary team as being attributable to rheumatoid arthritis. The final clinical label would then be rheumatoid arthritis-related interstitial lung disease (RA-ILD) or rheumatoid arthritis-related UIP (RA-UIP), which is a form of CTD-ILD. A pathologic diagnosis of UIP in a 45-year-old man with a family history of interstitial lung disease and a telomerase reverse transcriptase (TERT) gene mutation is likely to be interpreted by the clinical team as being familial. The final clinical/multidisciplinary label would be familial IPF. Regardless of one’s views on the utility of such splitting, the practical consequence of the modern multidisciplinary paradigm for the diagnosis of IPF is that the pathologist has no control over the final clinical label or presumed etiology. We can only control what we say about the biopsy findings.
The key question where opinions differ is: is the histologic appearance of “UIP of IPF” specific for an idiopathic process and histologically distinct from the “UIP of rheumatoid arthritis” or the “UIP of IPAF” or the “UIP of hypersensitivity pneumonitis”? My answer to this question is that in a system where the final clinical/etiologic label is determined by a multidisciplinary team, the histologic findings can never be pathognomonic for a specific etiology. If the ATS/ERS diagnostic criteria allowed pathology to be the gold standard for determining etiology, the situation would be different and the role of pathology would be greatly enhanced, but this is not the case in practice, where etiology is often assigned based on history, serology, radiology, etc. Even in the occasional case of UIP in which the histologic findings are highly suggestive of a specific etiology, pathologists have no control over the final multidisciplinary diagnosis, which often relies heavily on clinical and radiologic findings.
How do you “prove” etiology of a given case of UIP?
In the vast majority of cases, a pathologic diagnosis of UIP implies that no clear-cut etiology is identifiable histologically. In my experience, most such cases also lack compelling clinical evidence of a specific etiology and in my opinion should be labeled as IPF. However, the issue is complicated by the fact that the ATS/ERS guidelines give clinicians considerable leeway to assign an etiology based on a wide array of features other than pathology, including exposures, tempo of disease, serologic studies, imaging findings, medications, history of radiation or chemotherapy, family history, genetic tests, symptoms and signs with an “autoimmune flavor”, and so on. Some clinicians are reluctant to label a case as IPF if the patient is young or has exposures or if the fibrosis is upper lobe predominant. Therefore, the pathologist has no control over whether a case of pathologic UIP - even a classic case without any histologic features suggesting a specific etiology - will end up being labeled as IPF by a multidisciplinary team. This arbitrary manner in which etiology is assigned is a major flaw in the current etiology-based definition of IPF because it creates a process whereby the diagnosis of IPF can be jettisoned for a wide variety of reasons subject to individual bias.
Pathologists who claim that certain histologic features in UIP (such as a few granulomas or lymphoid follicles) denote an alternative diagnosis must consider the power of the clinical setting in the final multidisciplinary diagnosis. As an example, consider the case of an 80-year-old male smoker with a 2-year history of progressive debilitating fibrosing interstitial lung disease with worsening pulmonary function tests, no history of connective tissue disease, negative serologic work-up for connective tissue disease, a lack of exposures or drugs associated with interstitial lung disease, and bilateral lower lobe predominant, peripheral-predominant interstitial lung disease interpreted as “possible UIP” on HRCT. In this scenario, a pathologic diagnosis of UIP on a surgical lung biopsy would correspond to IPF. What if this patient underwent a lung transplant, the lung was heavily sampled histologically and the pathologist found 3 giant cells in one of 20 slides? Would that override the entire clinical story and firmly establish a diagnosis of “chronic/fibrotic hypersensitivity pneumonitis”? What if the same patient underwent a surgical lung biopsy, showing UIP in all slides and several lymphoid follicles in 7 of 20 slides? Would this prove a diagnosis of CTD-ILD based on histology alone? I hope the reader would agree that these non-specific findings cannot be considered pathognomonic given the clinical setting.
Now let’s consider an alternative scenario. In an asbestos exposed 68-year-old man with evidence of pleural plaques and a pathologic diagnosis of UIP with several asbestos bodies, is the final diagnosis asbestosis with a “UIP pattern” or IPF in an asbestos-exposed individual? Asbestos bodies attest to higher-than-background exposure to asbestos but they do not prove that the asbestos caused the lung fibrosis. In fact, Attanoos et al. have elegantly demonstrated that UIP in asbestos-exposed individuals lacks a dose-response relationship to asbestos fibers on mineral analysis and thus likely represents IPF, not asbestosis18. Yet, many clinicians who consider IPF a diagnosis of exclusion would argue that the history of asbestos exposure and the finding of asbestos bodies “proves” an etiology and thus excludes IPF.
The alternative diagnoses/etiologies most commonly invoked for pathologic UIP are CTD and hypersensitivity pneumonitis. We will address these separately in the following sections.
First, it is important to emphasize that the vast majority of cases of CTD-ILD are diagnosed in patients with a known CTD who subsequently develop lung abnormalities13,14,15,16. Most of these cases are not biopsied, and if a lung biopsy is performed, the etiology is seldom in question; because of the way IPF is defined, a patient with a known CTD is essentially “immune” from IPF, not because CTD protects against IPF but because of the way the ATS/ERS criteria are framed. Consequently, regardless of what the lung biopsy shows (UIP, NSIP, organizing pneumonia, etc.), the default assumption is that the process is driven by the underlying CTD. In such cases, there is no need for the pathologist to “determine an etiology” histologically. If the pathologic diagnosis in a patient with a known CTD is UIP, the final multidisciplinary diagnosis is virtually always CTD-ILD.
This leaves the small subset of cases in which interstitial lung disease is the first clinical manifestation of a CTD. The key question is: are there any histologic findings that accurately predict which patients will subsequently develop CTD vs. those who will not? My answer to this question is a categorical “no”. It has never been proven that lymphoid follicles, germinal centers, plasma cells, pleuritis or any combination of these findings predicts with 100% certainty that a CTD will develop at some future date. This paucity of data does not preclude strong opinions on this issue. Some pathologists consider such findings as definitive evidence of CTD-ILD regardless of whether or not a CTD ever develops. According to this school of thought, just because the clinical and serologic work-up for CTD is negative, it does not mean the patient cannot develop a CTD-ILD at some unspecified date in the future. In fact, the entity interstitial pneumonia with autoimmune features (IPAF) was created by pulmonologists to bypass the traditional view of rheumatologists that a diagnosis of CTD requires a minimum set of well-defined criteria, not just a positive antinuclear antibody (ANA) and a mix of non-specific clinical features with “autoimmune flavor”. My view is that it is reasonable to use soft histologic features such as lymphoid follicles, germinal centers and plasma cells to prompt a work-up for CTD in a patient with pathologic UIP, but if no CTD is found clinically, a diagnosis of IPF is appropriate.
Now let us consider the assertion that granulomas in a case of pathologic UIP exclude IPF and should suggest “chronic/fibrotic hypersensitivity pneumonitis”. Hypersensitivity pneumonitis is not the only cause of granulomas in the lung35,36. In fact, many of the same pathologists who argue that granulomas constitute evidence of hypersensitivity pneumonitis in UIP do not issue a top-line diagnosis of hypersensitivity pneumonitis, even when the full constellation of findings of hypersensitivity pneumonitis including classic granulomas are present in a cellular (non-fibrotic) context. Why then should we consider these findings pathognomonic in the setting of UIP? Surely we need to consider if the type and distribution of granulomas is within the spectrum of what one encounters in cellular non-fibrotic examples of hypersensitivity pneumonitis. In fact, there are several reasons one could encounter granulomas or giant cells in a case of UIP that have nothing to do with hypersensitivity pneumonitis. These include pulmonary interstitial emphysema (which features giant cells and fibrosis)37, particulate matter aspiration (which features bronchiolocentric giant cells)34, infection, and even chronic stasis of mucus and other debris in areas of honeycomb change, which frequently results in multinucleated giant cells with cholesterol clefts that float within the mucinous debris. If granulomas with the appropriate morphology and distribution are present, I agree that the possibility of hypersensitivity pneumonitis should be raised in a case of UIP. In my experience, this situation is uncommon, and even when it occurs, it is up to the discretion of the multidisciplinary team to accept the final diagnosis of hypersensitivity pneumonitis.
The most common scenario that creates difficulties in practice is when the HRCT findings are thought to be suggestive of chronic/fibrotic hypersensitivity pneumonitis but a lung biopsy shows only UIP without granulomas. Some authors claim that even histologically classic UIP without granulomas may represent chronic/fibrotic hypersensitivity pneumonitis. This notion is based entirely on the belief that some HRCT findings are specific for hypersensitivity pneumonitis, a notion that has been shown to be incorrect by studies that have noted identical features in a subset of multidisciplinary discussion-confirmed IPF cases27. Moreover, it has been shown that cases of so-called chronic/fibrotic hypersensitivity pneumonitis with histologically classic UIP and no granulomas have a clinical course, unresponsiveness to corticosteroids, and poor prognosis indistinguishable from IPF. Thus, objective metrics do not support the notion that this radiologically defined “entity” is distinct in any meaningful way from IPF. In summary, my view is that if classic features of non-fibrotic hypersensitivity pneumonitis19 (giant cells, poorly formed granulomas, bronchiolitis, lymphocytic interstitial inflammation with peribronchiolar accentuation) are present in non-fibrotic lung in a case of UIP, the possibility of fibrotic hypersensitivity pneumonitis should be raised. If classic histologic features of hypersensitivity pneumonitis are absent, the case should be diagnosed as UIP.
What should we do in ILD when pathology and HRCT are discordant?
Discordance between HRCT and pathologic findings is the crux of difficulties faced by multidisciplinary groups in current ILD practice. When clinical findings, HRCT findings and pathology are in agreement, there is a high likelihood of consensus in multidisciplinary teams and diagnosis. However, in indeterminate situations where the radiology does not match the pathologic findings, it is much harder to determine which modality should trump the other.
There are many situations where HRCT must trump pathology. These include cases where the biopsy did not sample abnormal tissue or sampled tissue that shows only non-specific or non-diagnostic findings. Unfortunately, this scenario is common, especially with transbronchial lung biopsies. There are also scenarios where the combination of clinical history and radiologic findings may be so compelling that pathologic findings—especially when non-specific—are unlikely to change the final multidisciplinary diagnosis. For example if a 56-year-old female never-smoker with 4 pet parakeets develops cough and dyspnea with bilateral ground-glass opacities and the head-cheese/3 density sign on HRCT, a transbronchial biopsy showing only mild non-specific chronic inflammation does not argue against the diagnosis of hypersensitivity pneumonitis.
However, there are pathologic findings that are so specific or pathognomonic that they can and should override the radiologic impression. Examples of this include the finding of lymphangitic carcinoma or lymphoma when the radiologic findings suggest interstitial lung disease38, the pathologic diagnosis of particulate matter aspiration when the HRCT suggests sarcoidosis or organizing pneumonia34, the pathologic finding of sarcoidosis when the HRCT suggests NSIP, the finding of smoking-related interstitial fibrosis (SRIF) when the radiologic findings suggest hypersensitivity pneumonitis or NSIP, or the pathologic diagnosis of pulmonary Langerhans cell histiocytosis when HRCT features are suspicious for disseminated malignancy or infection39.
Now let us consider the scenario in which HRCT findings are inconsistent with UIP but the pathologic diagnosis on a surgical lung biopsy is UIP. Three well-designed studies have addressed this issue. In a 2016 study by Yagihashi et al. co-authored by expert lung pathologists (Drs. Thomas Colby and Henry Tazelaar), 241 HRCTs from patients who had also undergone surgical lung biopsies in 3 studies sponsored by the IPF Clinical Research Network (IPFnet) were reviewed by 2 thoracic radiologists blinded to the histologic diagnosis and the final multidisciplinary diagnosis of IPF27. Of the 241 HRCTs, 71 were interpreted as “inconsistent with UIP” by the radiologists based on ATS/ERS/JRS/ALAT guidelines. The basis of the “inconsistent with UIP” label was most often diffuse mosaic attenuation/air trapping (51/71, 71.8%), followed by upper/mid zone predominance (17/71, 23.9%), extensive ground-glass opacities (16/71, 22.5%), and peribronchovascular predominance (9/71, 12.7%). Surprisingly, of the 71 cases in which the HRCT was interpreted as inconsistent with UIP on the basis of such features, all had UIP on biopsy (55 definite UIP, 16 probable UIP). The reader is reminded that all of these cases had a multidisciplinary diagnosis of IPF, which is the current gold standard. The survival was uniformly poor and in line with IPF regardless of the radiologic interpretation. The authors concluded that the term “inconsistent with UIP” was misleading. My experience is very much in line with this study. The practical consequence of this type of radiologic-pathologic discrepancy is that many pathologists are unduly swayed by the knowledge that a case has been labeled “inconsistent with UIP” by radiology, leading to many cases of UIP being misdiagnosed as NSIP, fibrotic hypersensitivity pneumonitis, and other entities.
In a 2013 study co-authored by expert lung pathologists (Drs. Colby, Tazelaar and Leslie), 2 expert thoracic radiologists (Drs. Gruden and Panse) reviewed 44 biopsy-proven UIP cases that were diagnosed at Mayo Clinic Scottsdale over 9 years7. On HRCT, 5 had honeycombing, 21 had fibrosis but no honeycombing, 10 had minimal fibrosis, and 5 had ground-glass opacities. Nearly all patients died or progressed regardless of radiologic findings. The authors concluded: “histologic UIP predicts clinical course”. The main implication of this study cannot be overstated: not all cases of UIP have classic radiologic findings. In fact, some have an HRCT appearance that would be considered highly suggestive of an alternative diagnosis. Since histologic UIP predicts the clinical course, it is imperative that the pathologist should not be biased by the radiologic findings.
A 2010 study by Sverzellati et al. examined this issue from the perspective of expert thoracic radiologists (the expert lung pathologist was Dr. Andrew Nicholson)11. In this study, 55 HRCTs from biopsy-proven UIP cases with multidisciplinary discussion-confirmed IPF were presented to expert radiologists. The radiologists misdiagnosed 34/55 (62%) of these cases as NSIP, chronic hypersensitivity pneumonitis, organizing pneumonia and even sarcoidosis, often making these incorrect diagnoses with a “high degree of probability”. Several examples of such radiologic misdiagnosis are illustrated in the paper. In one case, UIP was misdiagnosed as chronic hypersensitivity pneumonitis by all three observers (radiologists) based on ground-glass opacities and air trapping. In another, biopsy-proven UIP in a 47-year-old man was misinterpreted as NSIP (with high probability) by all three observers. NSIP was by far the most commonly misdiagnosed entity.
Similar findings were reported in a recent study of 101 ILD cases by Shih et al. from Massachusetts General Hospital40. In this study, of the 63 cases that were reported as “alternative diagnosis” (i.e., a diagnosis other than UIP) by CT, 13 showed UIP by histopathology. The authors illustrated the CT and pathologic findings in two of these cases. In the first, a histologically proven case of UIP was interpreted as chronic hypersensitivity pneumonitis on imaging due to upper and mid-zone predominant reticulation, central bronchiectasis and mosaic attenuation with lobular lucency. In the second, a thin-slice CT with asymmetric, non-zonal fibrosis and extensive ground-glass opacity was classified as “alternative diagnosis” by the 2018 ATS and Fleischner guidelines. On histopathology, this case showed UIP with superimposed organizing pneumonia.
What these studies consistently demonstrate is that some cases of UIP cannot be recognized by HRCT and as a consequence are erroneously labeled as NSIP, chronic hypersensitivity pneumonitis, or other entities7,11,27,28. The pathologic explanation for this phenomenon is quite straightforward. Since HRCT criteria for UIP require honeycombing, radiologists do not diagnose UIP in cases where they cannot see honeycombing. Instead, they often reach for another label, which may or may not be correct. If a surgical lung biopsy is performed, many of these cases show microscopic honeycombing (Fig. 4) and other classic features of UIP that can be easily identified by pathology but fall below the resolution of HRCT. Such cases can and should be diagnosed as UIP by pathologists and behave as IPF clinically. In this specific scenario, it is clear that pathology trumps HRCT because it predicts clinical behavior, and because there is a clear-cut rationale for why imaging findings might be inaccurate in this setting.
Concluding remarks
I readily concede that many pathologists and clinicians are unhappy with the term UIP, as it does not appropriately convey the most significant histologic finding in this entity, which is interstitial fibrosis. Many also point out that this term was coined in the 1960s, which is true24. Dr. Averill Liebow was a pioneer who laid the foundation of the pathologic classification of interstitial lung disease during a time when interstitial lung disease consisted of “fibrosing alveolitis”, “Hamman-Rich syndrome”, farmer’s lung, sarcoidosis and occupational lung disease. It is not surprising that in the 60 years since then we have learned more about interstitial lung disease and many concepts have been added or modified. Some entities (such as desquamative interstitial pneumonia, or DIP) coined by Dr. Averill Liebow in that pre-immunohistochemistry, pre-HRCT era persist even though they are misleading and outdated misnomers41,42, others (such as lymphoid interstitial pneumonia, or LIP) have decreased dramatically in frequency as they have been gradually replaced by better-defined pathologic entities, and yet others (such as pulmonary alveolar proteinosis, or PAP) remain remarkably accurate to this day. It is no surprise, therefore, that UIP—as described by Dr. Liebow - has undergone considerable revision over the years. The modern description of UIP dates back to Katzenstein and Myers’ seminal review in 1998, which described the key pathologic features of UIP and outlined the relevance of histologic subtyping of what was then known as IPF into UIP and NSIP43. Four years later, in 2002, the ATS/ERS consensus classification decided to limit the diagnosis of IPF to cases with a pathologic diagnosis of UIP4.
Perhaps it is time for better nomenclature, and I would be happy to accept a better term to replace UIP in pathology reports. Unfortunately, 20 years after the ATS/ERS guidelines, there is still no better alternative term for a chronic fibrosing interstitial process with a patchwork pattern, scarring, honeycomb change and fibroblast foci. Instead, what we have is a confusing potpourri of strong opinions from pathologists, radiologists and clinicians on how things ought to be. Needless to say, these opinions vary by years of experience, pulmonary pathology subspecialty training, the presence of a multidisciplinary conference at one’s institution, who one was trained by, volume of interstitial lung disease seen, whether one is a lung subspecialist or a generalist, and exposure to lung explants (especially lungs removed in the course of transplantation for IPF and other forms of advanced fibrosis).
In practice, surgical lung biopsies continue to be performed in selected circumstances in patients with interstitial lung disease. Practitioners with experience in this field recognize the limitations of imaging and realize that histopathology provides important information regarding the “ground truth” that impacts therapy and prognosis. Increasingly, cases with atypical clinical and radiologic findings are the ones most likely to be biopsied, and it is in these scenarios that surgical lung biopsies are of greatest value.
In summary, UIP is and always has been a clinically significant pathologic diagnosis that connotes a poor prognosis, irreversibility, and poor response to corticosteroids and other immunomodulators. In contrast, IPF, as defined by the ATS/ERS guidelines, is a clinical diagnosis that requires multidisciplinary input and assimilation of all available clinical, imaging and laboratory data. By ATS/ERS definition, the diagnosis of IPF requires a pathologic or radiologic diagnosis of UIP, just as a clinical diagnosis of Barrett’s esophagus requires a pathologic diagnosis of intestinal metaplasia; however, it is important to remember that IPF is not a pathologic diagnosis. Disagreements between expert pulmonary pathologists regarding UIP revolve around the specificity of histologic findings for indicating an etiology of UIP, and in how pathologic findings should be worded in pathology reports.
References
Larsen, B. T. Usual interstitial pneumonia: a clinically significant pattern, but not the final word. Mod Pathol https://doi.org/10.1038/s41379-022-01054-2 (2022).
Katzenstein, A. L., Mukhopadhyay, S. & Myers, J. L. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum Pathol 39, 1275–1294 (2008).
Katzenstein, A. L., Mukhopadhyay, S. & Myers, J. L. “Erratum to Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases”. Hum Pathol 39, 1562–1581 (2008).
American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 165, 277–304 (2002)
Flaherty, K. R. et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur. Respir. J. 19, 275–283 (2002).
Flaherty, K. R. et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am. J. Respir. Crit. Care Med. 164, 1722–1727 (2001).
Gruden, J. F., Panse, P. M., Leslie, K. O., Tazelaar, H. D. & Colby, T. V. UIP diagnosed at surgical lung biopsy, 2000–2009: HRCT patterns and proposed classification system. AJR Am. J. Roentgenol. 200, W458–W467 (2013).
Katzenstein, A. A., Zisman, D. A., Litzky, L. A., Nguyen, B. T. & Kotloff, R. M. Usual interstitial pneumonia: Histologic study of biopsy and explant specimens. Am. J. Surg. Pathol. 261, 1567–1577 (2002).
Nadrous, H. F., Myers, J. L., Decker, P. A. & Ryu, J. H. Idiopathic pulmonary fibrosis in patients younger than 50 years. Mayo Clin. Proc. 80, 37–40 (2005).
Park, J. H. et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am. J. Respir. Crit Care Med. 175, 705–711 (2007).
Sverzellati, N. et al. Biopsy-proved idiopathic pulmonary fibrosis: spectrum of non-diagnostic thin-section CT diagnoses. Radiology 254, 957–964 (2010).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit Care Med. 198, e44–e68 (2018).
Lee, H. K. et al. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest 127, 2019–2027 (2005).
Fischer, A. et al. Clinically significant interstitial lung disease in limited scleroderma: histopathology, clinical features, and survival. Chest 134, 601–605 (2008).
Parambil, J. G., Myers, J. L., Lindell, R., Matteson, E. L. & Ryu, J. H. Interstitial lung disease in primary Sjögren syndrome. Chest 130, 1489–1495 (2006).
Sawal, N. et al. Interstitial lung disease in antisynthetase syndrome: a clinical approach. J. Thorac. Dis. 13, 5556–5571 (2021).
Diaz de Leon, A. et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One 5, e10680 (2010).
Attanoos, R. L., Alchami, F. S., Pooley, F. D. & Gibbs, A. R. Usual interstitial pneumonia in asbestos-exposed cohorts—concurrent idiopathic pulmonary fibrosis or atypical asbestosis? Histopathology 69, 492–498 (2016).
Raghu, G. et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 202, e36–e69 (2020).
Bjoraker, J. A. et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 157, 199–203 (1998).
Katzenstein, A. L. Idiopathic interstitial pneumonia. In: Katzenstein and Askin’s Surgical pathology of non-neoplastic lung disease. 4th ed. Saunders Elsevier. pp 75 (2006).
Myers, J. L. & Katzenstein, A. L. Beyond a consensus classification for idiopathic interstitial pneumonia: progress and controversies. Histopathology 54, 90–103 (2009).
Pérez-Padilla, R. et al. Mortality in Mexican patients with chronic pigeon breeder’s lung compared with those with usual interstitial pneumonia. Am. Rev. Respir. Dis. 148, 49–53 (1993).
Liebow, A. A. Definition and classification of interstitial pneumonias in human pathology. Prog. Respir. Res. 8, 1–31 (1975).
Katzenstein, A. A., Myers, J. L., Prophet, W. D., Corley, L. S. III & Shin, M. S. Bronchiolitis obliterans organizing pneumonia and usual interstitial pneumonia. A comparative clinicopathologic study. Am. J. Surg. Pathol. 10, 373–381 (1986).
Hunninghake, G. W. et al. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest 124, 1215–1223 (2003).
Yagihashi, K. et al. Radiologic-pathologic discordance in biopsy-proven usual interstitial pneumonia. Eur Respir. J. 47, 1189–1197 (2016).
Raghu, G. et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 (2011).
Nandy, S. et al. Diagnostic accuracy of endobronchial optical coherence tomography for the microscopic diagnosis of usual interstitial pneumonia. Am. J. Respir. Crit. Care Med. 204, 1164–1179 (2021).
Kim, S. Y. et al. Classification of usual interstitial pneumonia in patients with interstitial lung disease: assessment of a machine learning approach using high-dimensional transcriptional data. Lancet Respir. Med. 3, 473–482 (2015).
Mukhopadhyay, S. & Parambil, J. G. Acute interstitial pneumonia (AIP): relationship to Hamman-Rich syndrome, diffuse alveolar damage (DAD) and acute respiratory distress syndrome (ARDS). Semin. Respir. Crit. Care Med. 33, 476–485 (2012).
Mukhopadhyay, S. et al. Lung biopsy findings in severe pulmonary illness associated with e-cigarette use (vaping): a report of eight cases. Am. J. Clin. Pathol. 153, 30–39 (2020).
Barton, L. M., Duval, E., Stroberg, E., Ghosh, S. & Mukhopadhyay, S. COVID-19 autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 153, 725–733 (2020).
Mukhopadhyay, S. & Katzenstein, A. L. Pulmonary disease due to aspiration of food and other particulate matter: a clinicopathologic study of 59 cases diagnosed on biopsy or resection specimens. Am. J. Surg. Pathol. 31, 752–759 (2007).
Mukhopadhyay, S. et al. Causes of pulmonary granulomas: a retrospective study of 500 cases from seven countries. J. Clin. Pathol. 65, 51–57 (2012).
Mukhopadhyay, S. & Gal, A. A. Granulomatous lung disease. An approach to the differential diagnosis. Arch. Pathol. Lab Med. 134, 667–690 (2010).
Barcia, S. M., Kukreja, J. & Jones, K. D. Pulmonary interstitial emphysema in adults. A clinicopathologic study of 53 lung explants. Am. J. Surg. Pathol. 38, 339–345 (2014).
Inaty, H. et al. Diffuse large B-cell lymphoma presenting as diffuse bilateral ground-glass opacities and diagnosed on transbronchial lung biopsy. Ann. Am. Thorac. Soc. 14, 605–607 (2017).
Mukhopadhyay, S., Eckardt, S. & Scalzetti, E. M. Diagnosis of pulmonary Langerhans cell histiocytosis by CT-guided core biopsy of lung: a report of 3 cases. Thorax 65, 833–835 (2010).
Shih, A. R., Nitiwarangkul, C., Little, B. P., Roop, B. W., Nandy, S. & Szabari, M. V. et al. Practical application and validation of the 2018 ATS/ERS/JRS/ALAT and Fleischner Society guidelines for the diagnosis of idiopathic pulmonary fibrosis. Respir. Res. 22, 124 (2021).
Katzenstein, A. L., Mukhopadhyay, S., Zanardi, C. & Dexter, E. E. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum. Pathol. 41, 316–325 (2010).
Mukhopadhyay, S., Aesif, S. W. & Sansano, I. 5 simple reasons to discard DIP, or why we should stop calling dolphins big fish. J. Clin. Pathol. 73, 762–768 (2020).
Katzenstein, A. L. & Myers, J. L. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am. J. Respir. Crit. Care Med. 157, 1301–1315 (1998).
Author information
Authors and Affiliations
Contributions
SM drafted this manuscript, and takes full responsibility for its contents.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mukhopadhyay, S. Usual interstitial pneumonia (UIP): a clinically significant pathologic diagnosis. Mod Pathol 35, 580–588 (2022). https://doi.org/10.1038/s41379-022-01053-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01053-3
