
Abstract
Usual interstitial pneumonia (UIP) is a concept that is deeply entrenched in clinical practice and the prognostic significance of UIP is well established, but the field continues to suffer from the lack of a true gold standard for diagnosing fibrotic interstitial lung disease (ILD). The meaning and usage of UIP have shifted over time and this term is prone to misinterpretation and poor diagnostic agreement. For pathologists, it is worth reflecting on the limitations of UIP and our true role in the care of patients with ILD, a controversial topic explored in two point-counterpoint editorials published simultaneously in this journal. Current diagnostic guidelines are ambiguous and difficult to apply in clinical practice. Further complicating matters for the pathologist is the paradigm shift that occurred with the advent of anti-fibrotic agents, necessitating increased focus on the most likely etiology of fibrosis rather than simply the pattern of fibrosis when pulmonologists select appropriate therapy. Despite the wealth of information locked in tissue samples that could provide novel insights into fibrotic ILDs, pulmonologists increasingly shy away from obtaining biopsies, likely because pathologists no longer provide sufficient value to offset the risks of a biopsy procedure, and pathologic assessment is insufficiently reliable to meaningfully inform therapeutic decisionmaking. To increase the value of biopsies, pathologists must first recognize the problems with UIP as a diagnostic term. Second, pathologists must realize that the primary goal of a biopsy is to determine the most likely etiology to target with therapy, requiring a shift in diagnostic focus. Third, pathologists must devise and validate new classifications and criteria that are evidence-based, biologically relevant, easy to use, and predictive of outcome and treatment response. Only after the limitations of UIP are understood will pathologists provide maximum diagnostic value from biopsies to clinicians today and advance the field forward.
Similar content being viewed by others

What’s in a name? When applied to roses, this Shakespearean philosophical question is a poignant reminder that the essence of the subject itself is far more important than the name applied. Like any field of scientific inquiry, medicine strives for precision and accuracy in its terminology yet often comes up short when this terminology is applied to complex natural processes. Usual interstitial pneumonia (UIP) is a prime example of a woefully inadequate moniker for the complex and diverse disease processes to which it has been applied. The meaning and usage of UIP as a diagnostic term have shifted over time, and consequently, it is not surprising that this term means different things to different people and is prone to misinterpretation, interobserver variation, and poor diagnostic agreement1,2,3,4. Though this term is deeply entrenched in clinical practice and the clinical and prognostic significance of UIP is well established, the field continues to suffer from the lack of a true gold standard for diagnosing fibrotic interstitial lung disease (ILD) and the limitations of UIP as a diagnostic term are well recognized3,4,5. To fill this void, four major international societies in pulmonary medicine have adopted “multidisciplinary discussion” (MDD) as a substitute or silver standard for primary diagnosis of ILD that supersedes a diagnosis rendered by any one specialty in isolation, though this diagnostic approach has its own well-recognized limitations6,7. For pathologists who interpret lung biopsies, it is worth reflecting on the problems with UIP as a diagnostic term and our true role in the multidisciplinary diagnosis and care of patients with ILD. This editorial represents one of two “point-counterpoint” articles that examine an area of controversy in pulmonary pathology; for the corresponding counterpoint article, see the accompanying editorial by S. Mukhopadhyay published in this same issue.
Problems with the term UIP
Since the term UIP was first coined by Averil Liebow and Charles Carrington in 1969, the meaning and usage of this term have shifted considerably. A careful review of their original description of UIP reveals a broad spectrum of clinical presentations and pathologic findings ranging from acute disorders with diffuse alveolar damage to chronic disorders with well-established fibrosis, including those with end-stage fibrosis and honeycombing8. This broad spectrum is quite different than the UIP of today and its meaning has narrowed considerably as classification of ILDs has evolved. UIP is now reserved for chronic fibrosing interstitial pneumonias9 and is characteristic of the clinical syndrome “idiopathic pulmonary fibrosis” (IPF), though this general pattern or very similar patterns of fibrosis can also occur in the setting of connective tissue disease (CTD)-associated ILD, fibrotic hypersensitivity pneumonitis (HP), familial ILDs, and other disorders.
As defined by current international consensus criteria10, the UIP pattern is characterized histopathologically by (1) dense fibrosis with architectural distortion (i.e., destructive scarring and/or honeycombing), (2) predominant subpleural and/or paraseptal distribution of fibrosis, (3) patchy involvement of lung parenchyma by fibrosis, (4) presence of foci of active fibroblastic proliferation (i.e., “fibroblast foci”), and (5) absence of features suggesting an alternate diagnosis. The latter may include granulomas, hyaline membranes, prominent airway-centered changes, areas of interstitial inflammation lacking associated fibrosis, prominent lymphoid hyperplasia with secondary germinal centers, chronic fibrous pleuritis, and/or organizing pneumonia; when one or more of these features is present, an alternate diagnosis should be entertained, though it should also be remembered that organizing pneumonia or hyaline membranes may occur with acute exacerbation of IPF and their presence does not exclude a diagnosis of UIP. Recognizing that diagnostic uncertainty is the norm rather than the exception, the authors of the guidelines also provided histologic categories of diagnostic confidence (“UIP”, “Probable UIP”, “Indeterminate for UIP”, and “Alternative diagnosis”) to aid the pathologist in communicating the degree of certainty or uncertainty that a UIP pattern is seen. This pragmatic terminology was intended to provide a framework whereby imperfect or conflicting clinical, radiologic, and pathologic information can be integrated together to better determine the likelihood of IPF through MDD.
Though these criteria appear straightforward at first glance, their application is often difficult in practice. Per international consensus guidelines10, UIP is merely a pattern of fibrosis and is not synonymous with the clinicopathologic entity termed IPF, yet these terms are often used interchangeably by clinicians and not in a fashion intended by the guidelines. Does a diagnosis of UIP imply a diagnosis of IPF? A careful reading of the guidelines leaves the impression that the answer is “no”, yet the very language used in the guidelines is ambiguous in this regard, defining categories of diagnostic confidence that refer to UIP (and not to IPF), even though these guidelines are meant to identify patients with or without IPF. The diagnostic categories proposed in 2018 by the Fleischner Society (“Definite UIP-IPF”, “Probable UIP-IPF”, “Indeterminate for UIP-IPF”, and “Features most consistent with an alternative diagnosis”) more accurately reflect the intended use of the guidelines—to identify patients with or without IPF—yet the same inherent problems with UIP and other ambiguities remain11.
This confusing and inconsistent use of UIP has persisted in guidelines more recently proposed for fibrotic HP as well. In 2020, international consensus criteria were established for diagnosing fibrotic HP. In these criteria, the authors indirectly acknowledged the problem with UIP by choosing to omit the terms “UIP” and “UIP-like” from histopathologic criteria supporting a diagnosis of HP, yet they provide a histopathologic description for fibrotic HP that is remarkably similar to UIP, and UIP was retained in the radiologic criteria in this same document12. It is well recognized that UIP or a UIP-like pattern of fibrosis can occur in fibrotic HP that may be indistinguishable from UIP of IPF, representing one of the most challenging diagnostic problems in ILD practice13,14,15, yet these guidelines fail to provide specific criteria enabling their distinction and suggest MDD as a surrogate “silver standard” in the absence of a suitable diagnostic gold standard, similar to the guidelines for IPF. In contrast, a separate set of diagnostic guidelines for HP proposed in 2021 by the American College of Chest Physicians retained UIP as a histologic pattern associated with HP16, maintaining consistency with historical concepts yet adding to diagnostic confusion given the differences from other recent guidelines. Though intended to standardize the diagnosis of IPF and HP, the ambiguous criteria and terminology provided in these four overlapping but nonidentical consensus documents on IPF and HP have failed to resolve the confusion that continues to plague the field, even among experts, and perpetuate the field’s overreliance on the imprecise and troublesome term UIP.
Compounding the confusion surrounding UIP are well-recognized problems with accuracy and reproducibility in the histopathologic diagnosis thereof. Significant interobserver variability and fair to poor agreement among pathologists in the diagnosis of UIP are well recognized problems1,2 and the exact pathologic features that define UIP are not universally agreed upon, even among expert pulmonary pathologists3,4, an inconvenient fact that may surprise many pathologists and clinicians. While features suggesting an alternate non-UIP diagnosis (i.e. indicating a non-IPF clinicopathologic diagnosis) have been provided by consensus guidelines10,11,12, the manner in which these should be used in practice and thresholds or cutoffs for various histopathologic features have not been defined. This challenge is amplified in the transbronchial forceps biopsy or cryobiopsy setting where smaller samples increase the likelihood of sampling error and hamper efforts to fully characterize pulmonary architectural changes, a reality illustrated by conflicting results from the recent Cryo-PID and COLDICE trials14,17,18. There is also a dearth of guidance on the way that clinical, radiologic, and serologic information should be incorporated into or allowed to influence a histopathologic interpretation. Several other essential questions also remain unanswered or incompletely answered; a few examples are listed in Table 1.
The shifting landscape in ILD—forward progress or a step backward?
Further complicating matters for the pathologist, a paradigm shift has also occurred in the clinical practice of pulmonary medicine with the advent of anti-fibrotic agents for progressive fibrosing ILDs19,20 and clinical trial data indicating that multiagent immunosuppressive therapy is detrimental to patients with IPF21. With this evolution has come an increased emphasis on the most likely etiology of fibrosis rather than simply the pattern of fibrosis present, and pharmacologic therapies are now chosen to target the presumed primary driver of the disease process (e.g. anti-fibrotic agents targeting aberrant fibroblastic proliferation, or immunosuppression for chronic inflammatory activity)22,23. Curiously, an opposite trend has also arisen in recent years, with less emphasis being placed on the fibrotic pattern and presumed etiology in favor of the general clinical phenotype. With this trend, the various fibrotic disorders are being lumped into a broader category of “progressive fibrosing ILDs”24,25. Indeed, data from the recent INBUILD trial indicates that anti-fibrotic therapy may provide benefit to patients with progressive fibrosing ILDs regardless of presumed etiology26,27, and these agents have also shown benefit in patients with Sjögren syndrome-associated ILD28,29. This approach favoring lumping over splitting is not without merit30, given the need for a pragmatic solution to the ongoing diagnostic challenges with ILD patients using the limited set of therapeutic tools available to the pulmonologist. Yet, as the late psychologist Abraham Maslow wisely observed, “if the only tool you have is a hammer, it is tempting to treat everything as if it were a nail”31. This narrowminded approach in pulmonary medicine may help in the short term but may ultimately impede progress, as it ignores fundamental differences among these patients that may be biologically and clinically relevant.
While recent clinical trials of anti-fibrotic agents are providing new hope to patients with progressive fibrosing ILDs, the decreased focus on underlying pathobiology of these diverse diseases may represent a step backward. Thankfully, the potential problems with this trend and continued relevance of a more refined disease-specific approach have also been acknowledged by at least some leaders in the field32. As in any area of clinical medicine, it is naïve to expect that all patients with fibrotic ILDs will respond similarly to general therapies that ignore underlying disease mechanisms; one only needs to look at the field of oncology to see the benefit of understanding disease-specific pathobiology and the direct impact this has on development of new therapies that target disease-defining or disease-driving mechanisms. It is also worth remembering that many advancements in oncology initially stemmed from detailed morphology-based studies by pathologists that subsequently informed creation of biologically relevant classification schemes and deeper investigations into tumor biology and prognostic and therapeutic biomarker discovery.
Despite the wealth of information locked in tissue samples that could provide novel insights in patients with fibrotic ILDs, the field of pulmonary medicine continues to shift away from obtaining biopsies from these patients. Some of this shift is not surprising, given continued improvements in imaging technology and concerns about risks to patients from a biopsy procedure, combined with consensus guidelines that enable a definitive diagnosis of IPF without a biopsy for patients meeting certain clinical and radiologic criteria10. Indications, contraindications, risks, and alternatives to the surgical lung biopsy for patients with fibrotic ILDs are well recognized and have been reviewed recently33. Yet it remains worth asking a more fundamental question: Why do clinicians continue to shy away from obtaining biopsies, instead favoring presumptive diagnoses for their patients without input from pathology? It is also worth asking why some pulmonologists are eager to adopt a molecular surrogate for a histopathologic diagnosis using a proprietary genomic classifier test that stratifies patients crudely into two simple categories (“UIP” and “non-UIP”)34,35,36. This test still requires bronchoscopy and a transbronchial tissue biopsy with the same inherent risks to the patient, yet it completely bypasses histologic assessment by a surgical pathologist! These realities lead to only one logical conclusion: Pathologists no longer provide sufficient value to clinicians to justify histologic assessment in every case and this assessment is insufficiently reliable to meaningfully inform the multidisciplinary diagnosis and care of at least some patients with fibrotic ILDs.
How can pathologists provide more value?
Throughout all fields of clinical medicine, clinicopathologic correlation is an essential component to the diagnostic process. Without exception, tissue samples are obtained to inform the next step in a patient’s clinical management, yet it is easy for a busy surgical pathologist to overlook this fact in day-to-day practice. Pathologists play a critical role in medicine and are uniquely positioned to integrate clinical, radiologic, and laboratory findings in the pathologic context, thereby functioning as a proverbial Rosetta stone for clinical colleagues to facilitate the best diagnosis and optimum treatment. The value of the pathologist in the contemporary practice of medicine cannot be overstated but directly depends on the degree to which a pathologist understands this role and seeks to fulfill it. As pathologists, it is worth reminding ourselves that we are first and foremost physicians who care for patients and not simply technicians in the hospital basement; when faced with a lung biopsy from a patient with fibrotic ILD, it is far more important for us to consult with clinical colleagues and ensure that the best clinicopathologic diagnosis is reached so the patient can be appropriately treated than it is to render a “correct” pathologic diagnosis in a vacuum.
How then do we provide more value to our clinical colleagues from biopsies despite the imperfect tools we have at our disposal, and avoid becoming obsolete? A few suggestions are offered in Table 2. First, we must recognize the problems with and limitations of UIP as a diagnostic term. These problems and limitations have been reviewed in detail recently3,4,37 and will not be reiterated here, but they are numerous and well known to those who regularly interpret lung biopsies from patients with ILD. These problems with our current classification scheme continue to undermine our role in the diagnostic process and increasingly remove us from the process of caring for patients with fibrotic ILDs.
Second, we must realize that the primary goal of a biopsy in contemporary practice is to determine the most likely etiology of a disease and, by logical extension, the best treatment approach. This will necessitate a shift in our diagnostic approach. For many decades, when a case of fibrotic lung disease was encountered, it was generally taught that the primary goal of the pathologist was to identify UIP and distinguish it from all other patterns of ILD. This focus on UIP required the pathologist to ask the question “Why is this UIP, or why is this not UIP?”, with the diagnostic process often ending there. While this exercise remains important to this day, it is no longer sufficient. Recent shifts in pulmonary medicine necessitate a corresponding evolution in pathology practice to enhance our value and improve the care of ILD patients. With the advent of targeted therapy, it has become increasingly important for the pathologist to not only ask if a biopsy shows UIP, but also to ask “Why is this case likely to represent IPF? Or why is this case unlikely to represent IPF, and if so, what alternative etiology is most likely, and how should the patient be treated?” Two case examples are provided in Fig. 1. Shifting our focus away from UIP and toward these more critical clinical questions will not be easy, as the pathobiology of fibrotic ILDs and their corresponding histologic features that enable their distinction and predict their behavior and response to therapy are incompletely understood, but this shift in focus must occur if we ever expect to unlock the mysteries of fibrotic ILDs and improve our diagnostic precision and accuracy. With this new approach, criteria for grading disease activity (e.g., fibroblastic proliferative activity or inflammatory activity) and staging of disease (e.g., extent or severity of fibrosis) must also be developed, akin to methods used routinely for non-neoplastic liver disease. This assessment may require incorporation of histologic findings, radiologic findings, and/or other clinical or laboratory parameters, but regardless of its basis, a robust grading and staging system will be needed to follow patients and determine the effectiveness of new therapies.

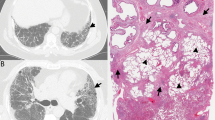
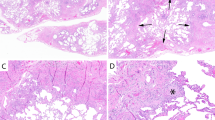
A a 74-year-old man and former smoker with gastroesophageal reflux disease (GERD) and (B) a 74-year-old man with rheumatoid arthritis. Do these biopsies show usual interstitial pneumonia (UIP)? Perhaps it is more important to ask “What is the most likely etiology and how should the patient be treated?”. In the former patient (A), the process is pauci-inflammatory with abundant fibroblast foci and focal peribronchiolar metaplasia, likely representing idiopathic pulmonary fibrosis and suggesting a role for anti-fibrotic therapy and control of GERD; in the latter patient (B), there is prominent chronic inflammation and follicular bronchiolitis but few fibroblast foci, consistent with rheumatoid arthritis-associated ILD and suggesting a role for immunomodulation. To the extent that current or future evidence supports using specific histologic features to predict therapeutic responsiveness, this information may provide more value to clinicians from a biopsy than simple assignment of a UIP or non-UIP diagnosis only.
Last, we must devise novel pathologic classifications and diagnostic criteria that that are evidence-based, reproducible, biologically and phenotypically relevant, easy to implement in clinical practice, and predictive of outcome and treatment response. This is the value our clinicians ultimately require from pathology, if we can but find a way to offer it. Despite ongoing challenges and imperfect terminology, we are practicing in an era of optimism and hope brought on by stunning advancements in molecular biology, imaging technology, and digital analytics38,39,40,41,42,43. There has never been a more exciting time to be a surgical pathologist, and the field of ILD is ripe for scientific breakthroughs. Advancements will hopefully come by leveraging the power of molecular diagnostics, aided by digital and computational pathology and augmented intelligence algorithms, tools that rest squarely in the hands of pathologists. We are uniquely positioned to revolutionize practice through development of more reliable and clinically meaningful terminology and diagnostic criteria for ILD aided by digital tools and discovery of new diagnostic, prognostic, and therapeutic biomarkers. Will we seize this opportunity and further cement pathology’s position at the heart of modern medicine, or leave this task to others? Only after the limitations of UIP are acknowledged and understood will pathologists be able to provide maximum diagnostic value from biopsies to clinical colleagues today and facilitate future advancements in the field. While a rose by any other name would smell as sweet, it’s time for pulmonary pathologists to move beyond UIP and devise a better way to classify, stage, and treat fibrosing ILDs.
References
Makela, K. et al. Analysis of the histologic features associated with interobserver variation in idiopathic pulmonary fibrosis. Am. J. Surg. Pathol. 42, 672–678 (2018).
Hashisako, M. et al. Interobserver agreement of usual interstitial pneumonia diagnosis correlated with patient outcome. Arch. Pathol. Lab. Med. 140, 1375–1382 (2016).
Hariri, L. P. et al. Pulmonary Pathology Society perspective on the 2018 American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society idiopathic pulmonary fibrosis clinical practice guidelines. Ann. Am. Thorac. Soc. 17, 550–554 (2020).
Smith, M. L. et al. Histopathologic assessment of suspected idiopathic pulmonary fibrosis: where we are and where we need to go. Arch. Pathol. Lab. Med. 144, 1477–1489 (2020).
Wuyts, W. A. et al. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur. Respir. Rev. 23, 308–319 (2014).
Walsh, S. L. F. et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir. Med. 4, 557–565 (2016).
Walsh, S. L. F. Multidisciplinary evaluation of interstitial lung diseases: current insights: number 1 in the series. “Radiol” edited Nicola Sverzellati Sujal Desai Eur Respir Rev 26, 170002 (2017).
Liebow, A. A. & Carrington, C. B. The interstitial pneumonias. In: M. Simon, E. J. Potchen, & M. LeMay (eds). Frontiers of pulmonary radiology: pathophysiologic, roentgenographic and radioisotopic considerations; proceedings of the symposium sponsored by Harvard Medical School, April 21–22, 1967. Vol. 1, 102-141 (Grune & Stratton: New York, 1969).
Travis, W. D. et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med 188, 733–748 (2013).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit Care Med. 198, e44–e68 (2018).
Lynch, D. A. et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir. Med. 6, 138–153 (2018).
Raghu, G. et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 202, e36–e69 (2020).
Wright, J. L., Churg, A., Hague, C. J., Wong, A. & Ryerson, C. J. Pathologic separation of idiopathic pulmonary fibrosis from fibrotic hypersensitivity pneumonitis. Mod Pathol 33, 616–625 (2020).
Churg, A., Ryerson, C. J. & Wright, J. L. Fibroblast foci and patchy fibrosis do not separate usual interstitial pneumonia from fibrotic hypersensitivity pneumonitis in transbronchial cryobiopsies. Arch. Pathol. Lab Med. 145, 1325–1326 (2021).
Churg, A. Hypersensitivity pneumonitis: new concepts and classifications. Mod Pathol 35, 15–27 (2022).
Fernandez Perez, E. R. et al. Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest 160, e97–e156 (2021).
Troy, L. K. et al. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir. Med. 8, 171–181 (2020).
Suehs, C., Bourdin, A., Vachier, I., Molinari, N. & Romagnoli, M. Transbronchial cryobiopsy in the diagnosis of interstitial lung diseases: methodologies and perspectives from the Cryo-PID and COLDICE studies. Ann. Transl. Med. 8, 1330 (2020).
Richeldi, L. et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2071–2082 (2014).
King, T. E. Jr. et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092 (2014).
Raghu, G., Anstrom, K. J., King, T. E. Jr, Lasky, J. A. & Martinez, F. J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 366, 1968–1977 (2012).
Raghu, G. et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am. J. Respir. Crit. Care Med. 192, e3–e19 (2015).
Kadura, S. & Raghu, G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur. Respir. Rev. 30, 210011 (2021).
Wells, A. U., Brown, K. K., Flaherty, K. R., Kolb, M. & Thannickal, V. J., IPF Consensus Working Group. What’s in a name? That which we call IPF, by any other name would act the same. Eur. Respir. J. 51, 1800692 (2018).
Collins, B. F. & Raghu, G. Antifibrotic therapy for fibrotic lung disease beyond idiopathic pulmonary fibrosis. Eur. Respir. Rev. 28, 190022 (2019).
Flaherty, K. R. et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 381, 1718–1727 (2019).
Flaherty, K. R. et al. Nintedanib in progressive interstitial lung diseases: data from the whole INBUILD trial. Eur. Respir. J. https://doi.org/10.1183/13993003.04538-2020 (2021).
Distler, O. et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N. Engl. J. Med. 380, 2518–2528 (2019).
Highland, K. B. et al. Efficacy and safety of nintedanib in patients with systemic sclerosis-associated interstitial lung disease treated with mycophenolate: a subgroup analysis of the SENSCIS trial. Lancet Respir. Med. 9, 96–106 (2021).
Kolb, M. R. & Flaherty, K. R. The justification for the progressive fibrotic phenotype. Curr Opin Pulm Med 27, 363–367 (2021).
Maslow, A. H. The psychology of science: a reconnaissance. 16 (Harper & Row: New York, NY, 1966).
Kolb, M., Raghu, G. & Wells, A. Prognostic impact of typical and probable usual interstitial pneumonia pattern in idiopathic pulmonary fibrosis: is the debate about biopsy a Star Wars saga? Eur. Respir. J. 55, 2000590 (2020).
Hariri, L. P. et al. The role of surgical lung biopsy in the diagnosis of fibrotic interstitial lung disease: perspective from the Pulmonary Fibrosis Foundation. Ann. Am. Thorac. Soc. 18, 1601–1609 (2021).
Raghu, G. et al. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir. Med. 7, 487–496 (2019).
Raghu, G. et al. A molecular classifier that identifies usual interstitial pneumonia in transbronchial biopsy specimens of patients with interstitial lung disease. Chest 157, 1391–1392 (2020).
Richeldi, L. et al. Utility of a molecular classifier as a complement to high-resolution computed tomography to identify usual interstitial pneumonia. Am. J. Respir. Crit Care Med. 203, 211–220 (2021).
Smith, M. L. The histologic diagnosis of usual interstitial pneumonia of idiopathic pulmonary fibrosis. where we are where we need go. Mod Pathol 35, 8–14 (2022).
Bowman, W. S., Echt, G. A. & Oldham, J. M. Biomarkers in progressive fibrosing interstitial lung disease: optimizing diagnosis, prognosis, and treatment response. Front. Med. (Lausanne) 8, 680997 (2021).
Kim, S. Y. et al. Classification of usual interstitial pneumonia in patients with interstitial lung disease: assessment of a machine learning approach using high-dimensional transcriptional data. Lancet Respir. Med. 3, 473–482 (2015).
Raciti, P. et al. Novel artificial intelligence system increases the detection of prostate cancer in whole slide images of core needle biopsies. Mod Pathol 33, 2058–2066 (2020).
Cui, M. & Zhang, D. Y. Artificial intelligence and computational pathology. Lab. Invest. 101, 412–422 (2021).
Makela, K. et al. Artificial intelligence identifies inflammation and confirms fibroblast foci as prognostic tissue biomarkers in idiopathic pulmonary fibrosis. Hum. Pathol 107, 58–68 (2021).
Kropski, J. A., Blackwell, T. S. & Loyd, J. E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 45, 1717–1727 (2015).
Acknowledgements
I thanks Dr. Maxwell L. Smith and Dr. Yasmeen M. Butt for reviewing the draft and providing helpful and insightful comments.
Author information
Authors and Affiliations
Contributions
Not applicable (B.T.L. is the sole author and is responsible for all content).
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Larsen, B.T. Usual interstitial pneumonia: a clinically significant pattern, but not the final word. Mod Pathol 35, 589–593 (2022). https://doi.org/10.1038/s41379-022-01054-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01054-2
This article is cited by
-
Diffuse idiopathic skeletal hyperostosis as a cause for dysphagia in a patient with ankylosing spondylitis
Wiener klinische Wochenschrift (2023)
-
Usual interstitial pneumonia (UIP): a clinically significant pathologic diagnosis
Modern Pathology (2022)
