
Abstract
Testicular adult granulosa cell tumor (AGCT) is a rare type of sex-cord stromal tumor that affects patients of a wide age range and has the potential for late metastasis. In the testis, the diagnosis of AGCTs often requires the exclusion of other more common types of sex-cord stromal tumors. Immunohistochemistry is of limited utility, being used mostly to confirm sex-cord lineage and to exclude other entities when morphology is not typical. Unlike ovarian AGCTs, which are molecularly homogeneous and harbor a specific activating FOXL2 mutation (c.7558C > T p.C134W) in >90% of cases, the molecular characteristics of testicular AGCTs remain largely unknown. In the current study, we analyzed 13 testicular AGCTs diagnosed at multiple institutions using massively parallel DNA sequencing to evaluate single nucleotide variants, copy number alterations, and structural variants. In all, 10/13 cases were sequenced successfully. Notably, the FOXL2 c.7558C > T (p.C134W) mutation was identified in only a single case (1/10, 10%). The remaining cases were molecularly heterogeneous, with largely nonrecurrent genetic variants. Putative driver events in individual cases included a well-characterized gain-of-function NRAS mutation, as well as inactivation of ATM and TP53, among others. The only highly recurrent finding was single copy loss of 22q (7/10 cases, 70%). Comparatively, the frequencies of FOXL2 c.7558C > T (p.C134W) and 22q loss in 12 metastatic ovarian AGCTs identified in our database were 92% (11/12) and 42% (5/12), respectively. The results of the present study suggest that testicular AGCTs are different from their ovarian counterparts in that they appear to be molecularly heterogeneous and only rarely harbor FOXL2 mutations.
Similar content being viewed by others
Introduction
Since the first description of an adult granulosa cell (AGCT) tumor in the testes almost 70 years ago1, relatively few case reports and small series of these tumors have been published2,3. Unlike AGCT of the ovary, which is the most common malignant sex cord-stromal tumor in adult females and constitutes 2–5% of all ovarian neoplasms, AGCT of the testis is very rare. In this location, AGCTs comprise less than 0.5% (<5/1000) of all sex-cord stromal tumors4 and their diagnosis is often one of exclusion, given the absence of pathognomonic histopathologic findings and specific adjunctive markers.
In men, AGCT commonly presents as a unilateral, slow-growing painless testicular mass2, and rarely exhibits symptoms secondary to overproduction of sex hormones4. Given the absence of a normal (benign) counterpart of AGCT in the testis (i.e., granulosa cells), and the clinical and epidemiological differences between AGCTs diagnosed in men and women, it is reasonable to hypothesize that ovarian and testicular AGCTs may be biologically dissimilar despite their shared histopathologic features. In the ovary, the overwhelming majority of AGCTs (~90–97%)5,6 harbor a specific activating mutation in the transcription factor FOXL2 c.7558C > T (p.C134W). In contrast, because of their overall rarity, the molecular events underpinning oncogenesis in AGCT of the testis remain largely unknown. In this study, we evaluated the histopathologic and molecular features of 13 testicular AGCTs (including cases reported previously in one of the largest series)2, using massively parallel DNA sequencing.
Materials and methods
This study was performed with the approval of the Institutional Review Board of Brigham and Women’s Hospital (MassGeneral Brigham/Partners Healthcare) and the collaborating institutions.
Identification and accrual of cases
The pathology databases of four institutions [Brigham and Women’s Hospital (BWH), Massachusetts General Hospital, El Camino Hospital (Mountain View, CA), and University of Massachusetts Baystate Healthcare] were queried for cases diagnosed as AGCT of the testis. Cases were retrieved from the archives and the slides were reviewed at the corresponding institutions by the submitting authors. Tumors in which the diagnosis was confirmed by re-review, and for which archival formalin-fixed paraffin-embedded material (blocks and/or slides) was available for molecular analysis, were included in the study.
Clinical data and pathology review of the cases
Clinical and demographic data were obtained from electronic medical records and pathology reports. A centralized re-review of selected slides was performed at BWH by two of the authors (SS and AMA). The following information was collected for each case4,7: age at diagnosis, tumor size, tumor site, overall cellularity, cytomorphology (epithelioid versus spindle cells, presence of nuclear grooves, and pleomorphism), architecture, number of mitoses per 10 high powered fields (HPF), presence of infiltrative growth, entrapment of normal structures (i.e., seminiferous tubules), and results of relevant immunohistochemical stains (when available). The architectural patterns used to describe the cases included: nests, solid sheets, cords, trabeculae, fascicles, or reticular growth. Nests were defined as solid groups of cells with discernible borders and included both small and large nests. Solid sheets were defined as large solid proliferations of neoplastic cells lacking distinct borders. Cords were defined as linear arrangements of tumor cells with a thickness of 1−2 cells, whereas trabeculae were defined as variably anastomosing elongated structures (length > width, but shorter than cords) that were usually >2 cells-thick. Fascicles were characterized by short bundles of neoplastic spindle cells. Reticular growth was characterized by interconnecting individual spindle cells within a loose stroma imparting a meshwork-like appearance. Overall cellularity was evaluated as low (ratio of cells to stroma <0.25), moderate (ratio of cells to stroma 0.25−0.5) and/or high (ratio of cells to stroma >0.5).
Cytomorphology was defined as epithelioid, spindled, or a combination thereof. The predominant cytomorphology was defined as the cytomorphology comprising >50% of the tumor volume on the slides reviewed. If a predominance of spindle or epithelioid cells could not be definitely established, cytomorphology was classified as mixed. Pleomorphism was subjectively evaluated as minimal when there was mild nuclear atypia and inconspicuous variation of nuclear size between neoplastic cells, moderate when there was mild to moderate nuclear atypia and only moderate focal variation in nuclear size, and severe when there was marked nuclear atypia and pronounced variation in nuclear size (including giant tumor cells). Infiltrative growth was defined as destructive (i.e., invasive) infiltration of adjacent structures, beyond just expansile growth. Definitionally, this also excluded non-destructive entrapment of benign seminiferous tubules, which was considered separately.
Massively parallel sequencing (OncoPanel) and identification of the comparator group
Massively parallel DNA sequencing was performed using a 447-gene targeted solid tumor assay (OncoPanel) as previously described by our group in validation studies8,9. In summary, areas of the tumor with sufficient cellularity (tumor nuclei/total nuclei ≥ 0.2, 20%) were marked by a pathologist (AMA) on a reference H&E-stained slide that was used to guide manual dissection of 2−10 formalin-fixed paraffin-embedded tissue sections (5 µm) per case. DNA was extracted with a commercial kit (Qiagen, Valencia, CA) following the manufacturer’s recommendations and DNA was fragmented by sonication. An input of 200 ng of DNA per sample (minimum threshold of 100 ng) was used to prepare the sequencing libraries (TruSeq LT library preparation kit; Illumina, San Diego, California). The selection of target sequences was achieved by hybridization to a set of custom-designed probes (Agilent SureSelect; Agilent Technologies, Santa Clara, CA). Sequencing was performed on an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA). Deconvolution of samples, read alignment, variant calling, and annotation was performed with a validated institutional informatics pipeline8,9,10. Additionally, in-house informatics algorithms were used for the detection of mutational signatures (POLE, APOBEC, smoking, UV) and mismatch repair status11. To reduce contamination with germline variants, events present at a frequency >0.1% in the gnomAD database (Broad Institute) were filtered out in this tumor-only assay. The reported variants were further reviewed and tiered for actionability and biological relevance by a molecular pathologist (LMS). Metastatic ovarian AGCTs sequenced by OncoPanel between 2013 and 2021 were identified in our database and used as a comparator.
Results
Clinical and histopathologic features
Thirteen cases of AGCT of the testis, diagnosed between 1976 and 2021, were included in this study. In one case (diagnosed in the early 1990s), the corresponding demographic information was not available. Consistent with previously reported cases, there was a wide age range at presentation (14–75 years) with a median age of 37 years (Table 1). All specimens were resections (i.e., either radical or partial orchiectomies), and the median tumor size was 2.2 cm (range 0.7–9.5 cm).
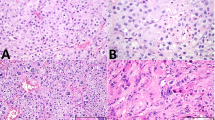
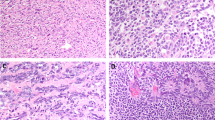
The histologic review was congruent with the morphologic spectrum seen in prior case studies and a series of testicular AGCTs1,2,3. All cases demonstrated high cellularity, either diffusely or focally (Table 2). The most common architectural patterns were nested (10 cases, ~77%) and solid/sheet-like growth (9 cases, ~69%), with other minor patterns including fascicular, trabecular, corded/tubular, and reticular (Table 2 and Fig. 1A). In several cases, the nested pattern was focal or multifocal. Two cases featured focal myxoid stroma, and Call-Exner bodies were identified in only one case (Fig. 1B). Cytomorphology was predominantly epithelioid in 4 cases (~31%), predominantly spindled in 1 case (~8%), and mixed in 8 cases (~62%). Nuclear pleomorphism was generally absent/mild (10 cases, ~77%) or moderate (2 cases, ~15%); only one case demonstrated significant pleomorphism, as well as necrosis. Nuclear grooves were present in all cases (Fig. 2A), although they were only focal in two cases. Similar to prior reports, mitoses varied greatly (from <1 to 107 per 10 HPF; median = 6 per 10 HPF), with atypical mitotic figures identified only in one case (Fig. 2B). Infiltrative growth was present in three tumors (~23%), mostly as invasion of the adjacent testicular parenchyma, rete testis, or hilar soft tissue (Fig. 3A). Entrapped benign seminiferous tubules were observed in 3 cases (~23%) (Fig. 3B). Lymphovascular invasion (LVI) was not identified in any of the cases. Overall, 3 cases (cases 5, 9, and 13) had a combination of clinicopathologic features suggestive of aggressive biology (Table 1)4,7.

A Representative image of predominantly solid sheet-like architecture, which was the dominant growth pattern (case 8). B Call-Exner bodies were identified in only one case (case 4); nuclear groves are also readily seen in this image.

A Nuclear grooves were present in all cases (case 8 is shown in this picture). B. Atypical mitoses were identified in only one case with prominent mitotic activity (case 13). The tumor featured scattered neoplastic cells with nuclear grooves and a predominant nested architecture. This tumor had biallelic inactivation of TP53 (see sequencing results) and overexpression of p53 by immunohistochemistry (Fig. 2B, inset; Clone D-O7, mouse monoclonal, dilution 1:500; DAKO, Carpinteria, CA).

A Invasion of rete testis was identified in one case (case 9). B Entrapment of benign seminiferous tubules was noted in three cases (case 6 is shown in this picture). Nuclear grooves are visible in both images at medium magnification.
Information on the immunohistochemistry performed as part of the original diagnostic evaluation was available for 8 cases (Table 2). All tested cases demonstrated positive staining for inhibin (7/7) and WT-1 (5/5). Moreover, expression of calretinin (3/4), SF1 (3/4), smooth muscle actin (SMA; 2/3), and keratin (only focal staining, 1/2) was seen in overlapping subsets of cases. In contrast, SALL4, OCT3/4, desmin, B-catenin, and EMA were invariably negative in all cases in which they were performed.
DNA sequencing
Of the 13 cases in the cohort, three failed sequencing according to standard quality assurance (QA) metrics (overall average number of reads <50; cases 4, 6, and 9). In the remaining 10 cases, the mean coverage was 188 reads across all targeted exons. Tumor mutational burden in the cohort ranged from 0.76 to 10.65 mutations per megabase (median 3.80). All cases were mismatch repair-proficient, and no specific mutational patterns (i.e., signatures) were identified due to the small mutational burden seen in the cohort.
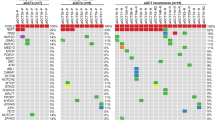
Pathogenic mutations with variant allele frequencies (VAFs) sufficient to suggest a contribution to tumorigenesis were identified in at least 5 of 10 cases (50%) that passed QA sequencing metrics, with no recurrent pathogenic mutations within the cohort. Additionally, one of the three cases that failed sequencing QA metrics harbored a pathogenic mutation BRCA2 mutation c.7558C > T (p.R2520*, case 6) known to be associated with hereditary breast and ovarian carcinoma. This BRCA2 variant had a VAF suggestive of germline origin, but its true biological significance (including presence or absence of loss of heterozygosity) could not be definitely determined. Notably, only one case (case 8) carried the FOXL2 c.7558C > T (p.C134W) mutation that is seen in ≥ 90% of ovarian AGCTs5,6 (Fig. 4 and Table 3). Interestingly, this mutation exhibited a VAF of 27%, suggestive of a sub-clonal event in the context of the high tumor cellularity (94%). Case 8 also harbored a pathogenic ATR co-mutation c.1869del (p.I624Ffs*17) with a VAF suggestive of possible germline origin. Importantly, the morphology of case 8 did not differ significantly from that of other cases in the series. A pathogenic TP53 c.743G > A (p.R248Q) mutation associated with a suspected deletion of the wild-type TP53 sequence was seen in case 13, consistent with loss of heterozygosity. Accordingly, immunohistochemistry demonstrated overexpression of the p53 protein in this case (Fig. 2B). Of note, this tumor had multiple morphologic findings indicative of aggressive biology, including a high mitotic count, atypical mitoses, moderate-to-severe nuclear pleomorphism, necrosis, and infiltrative growth. Additional cancer-relevant single-nucleotide variants detected in this series included mutations in ATM (c.8823_8824del p.Q2942Gfs*13; with copy neutral loss of heterozygosity; case 2), NRAS (c.183A > T p.Q61H; case 5) and CBFA2T3 (c.131dup p.P45Sfs*14; with loss of heterozygosity; case 7). Mutations in ATR (c.7817G > A p.R2606Q, classified as pathogenic in COSMIC) and ARID1B (c.2931C > A p.Y977*), identified in cases 1 and 3 respectively, were considered of indeterminate biological significance due to the absence of evidence of loss of heterozygosity and/or low VAFs.

For copy number alterations, only chromosome-level and arm-level events are illustrated. The X chromosome was excluded due to the absence of chromosome-level and arm-level copy number events detected in the cohort.
Recurrent copy number alterations were found in 9/10 cases (90%) that passed QA, most notably 22q arm-level loss which was identified in 7 of 10 cases (70%). Other alterations included arm-level loss of 16q (3/10; 30%), single copy loss of chromosome 10 (3/10; 30%), and whole chromosome gain of chromosomes 3 (4/10; 40%), 7 (4/10; 40%), 9 (3/10; 30%), and 12 (5/10; 50%). While three of these alterations resulted in loss of heterozygosity at loci of pathogenic mutations (ATM, TP53, and CBFA2T3), most copy-number losses identified herein were not associated with biallelic inactivation of tumor suppressors included in the sequencing panel. No structural alterations or cancer-relevant gene amplifications were detected.
Of note, the 2 cases with aggressive histopathologic features that underwent successful sequencing (cases 5 and 13) harbored an activating NRAS mutation and biallelic inactivation of TP53, respectively. However, from a molecular standpoint, these tumors were otherwise comparable to the rest of the testicular AGCTs included in this series. More specifically, their mutational burden and number of chromosome-level and arm-level copy number changes were not higher than those of the testicular AGCTs without aggressive histopathologic features.
The comparator group comprised 12 metastatic ovarian AGCTs from 12 individual female patients previously sequenced by OncoPanel. In contrast to their testicular counterparts, most of these tumors (11/12; 92%) harbored the FOXL2 c.7558C > T (p.C134W) mutation. The case that lacked FOXL2 c.7558C > T (p.C134W) harbored a CBL mutation (c.1259G > A p.R420Q, classified as pathogenic in COSMIC; VAF 0.44). Of note, none of the testicular AGCTs demonstrated pathogenic CBL mutations in this series. Copy number changes consistent with loss of 22q were present in 5 ovarian AGCTs (5/12, 42%).
Discussion
Testicular AGCT is an uncommon neoplasm that affects a wide age range of patients, but is diagnosed more frequently in adults2. They generally present as a unilateral slowly enlarging painless testicular mass (~90% patients), and less commonly include features of excess estrogen production such as erectile dysfunction, gynecomastia, or decreased libido (~10% patients)2. Ultrasonography typically reveals a solid hypoechoic mass12. Testicular AGCTs demonstrate a tendency for late metastases in ~16% of cases, most frequently to retroperitoneal lymph nodes, as well as to liver, lung, and bone2,3,13,14. Features suggestive of malignancy have been reported in 13–20% of tumors, including tumor size >4 cm, age at diagnosis >50 years, mitoses >30 per 10 HPF, rete testis or vascular invasion, necrosis, and cellular atypia4. However, the only factor that has demonstrated a statistically significant association with malignant behavior has been a tumor size >4 cm7.
Grossly, testicular AGCTs are solid, well-circumscribed lobulated masses, which are either unencapsulated or only partially surrounded by an incomplete pseudocapsule. They are thought to arise within the seminiferous tubules15, and usually exhibit expansile growth that displaces and compresses the adjacent testicular parenchyma16. Microscopic architectural patterns include solid, cystic, microfollicular, palisading, pseudopapillary, gyriform, insular, trabecular, and pseudosarcomatous growth2. Small cystic spaces containing hyalinized basement membrane or eosinophilic material, known as “Call-Exner” bodies, can be occasionally observed3. Mitotic activity is markedly variable in the cases reported in the literature17. In these neoplasms, immunohistochemistry is of only limited utility and mostly used to exclude alternative diagnoses. Frequently positive immunostains include inhibin, keratins (usually focal), smooth muscle antigen, progesterone receptor, and estrogen receptor2. Epithelial membrane antigen, c-Kit/CD117, and germ cell tumor markers such as SALL4, OCT3/4, and PLAP are consistently negative. Of note, the overall histopathologic and immunophenotypic characteristics of this cohort were comparable to those described in prior studies.
From a developmental perspective, the occurrence of AGCT in the testis is somewhat surprising, given the absence of normal nonneoplastic counterparts (i.e., granulosa cells) in this organ. Consequently, it is reasonable to speculate that testicular AGCTs may differ in their origin and biology when compared to ovarian AGCTs. However, in the bipotential gonad, common precursor cells derived from the coelomic epithelium have the biologic potential to differentiate to either granulosa or Sertoli cells. In females, differentiation to granulosa cells is induced by activation of WNT signaling in the absence of SRY expression, and maintained by expression of FOXL26. In contrast, differentiation to Sertoli cells in males is induced in part by repression of WNT signaling, and maintained by expression of DMRT3 and SOX918,19. Nonetheless, there appears to be a certain degree of lineage plasticity, as trans-differentiation to granulosa cells can be induced by β-catenin-driven ectopic expression of FOXL2 in Sertoli cells20.
From a molecular perspective, the overwhelming majority (>90%) of ovarian AGCTs are driven by a FOXL2 activating mutation (c.7558C > T p.C134W)5,6. This mutation is thought to induce a constitutive activation of FOXL2 that leads to uncontrolled granulosa cell differentiation and growth16. Mouse models of ovarian AGCT have further implicated the WNT signaling pathway as CTNNB1 activation in the setting of loss of PTEN or activation of KRAS21. In contrast, much less is known about the molecular events that underlie testicular AGCTs. Earlier studies comprising a small number of cases have yielded disparate results regarding the presence of FOXL2 c.7558C > T (p.C134W) in testicular AGCTs. Specifically, three groups have reported on FOXL2 status in testicular AGCTs based on single-gene assays, including Hes and colleagues (3 cases), Schubert and colleagues (1 case), and Lima and colleagues (5 cases)22,23,24. Of these, only Lima and colleagues identified the FOXL2 c.7558C > T (p.C134W) mutation in 2 of 5 cases, one of which was in fact an ovarian AGCT arising in an intraabdominal ovary of a phenotypically male patient. Our study confirmed that FOXL2 c.7558C > T (p.C134W) is infrequent in testicular AGCTs (~10%, 1/10 cases). Moreover, we demonstrated that testicular AGCTs are molecularly heterogeneous and do not harbor highly recurrent pathogenic single nucleotide variants or structural variants detected by our targeted sequencing panel. Of note, one of the cases that underwent successful sequencing had no genetic alterations detectable with our panel (case 12). Interestingly, the putative drivers identified in these cases were diverse (e.g., ATM, NRAS, TP53), suggesting that, unlike ovarian AGCT, testicular AGCT is a heterogeneous diagnostic category that encompasses lesions with disparate biologic features. Importantly, none of the testicular AGCTs assessed herein harbored CTNNB1 mutations, FH mutations, KRAS mutations, or MDM2 amplification, which have been described in the more common types of testicular sex-cord stromal tumors25,26,27,28.
The most consistent molecular alteration observed across the tumors analyzed in this series was 22q loss, identified in 70% of the cases that underwent successful sequencing. Although this alteration is nonspecific, it has been previously observed in AGCT of the ovary at a similar rate of 63%29 and in conjunction with other copy number alterations seen in our cohort including 16q loss and 12 gain30. The metastatic ovarian AGCTs included as a comparator in the present study also demonstrated recurrent loss of 22q, but at a slightly lower frequency (42%). The loss of 22q is an event that has been observed in other cancers, and we hypothesize that this alteration could reflect the loss of heterozygosity of a gene present in this chromosomal region that is not targeted by our panel.
Fashedemi et al. have reported that, in ovarian AGCTs that harbor activating FOXL2 mutations, progression to high-grade histology is associated with TP53 inactivation31. In our series, one tumor with prominent mitotic activity (107 mitoses/10 HPF), large size (9.5 cm), marked nuclear pleomorphism, and invasive growth also had molecular evidence of TP53 inactivation, but lacked a FOXL2 c.7558C > T (p.C134W) mutation. Interestingly, the findings in this case raised the possibility of loss of heterozygosity of TP53 in the context of the presence of a pathogenic germline variant. The morphologic features of the only case with a FOXL2 c.7558C > T (p.C134W) mutation were similar to those seen in other tumors of this series and were not worrisome for malignancy.
Unlike AGCTs, testicular juvenile granulosa cell tumors (JGCTs) almost exclusively affect patients of up to 10 years of age, with ~50% diagnosed in infants 1 month-old or younger and >90% diagnosed in infants up to 6 months-old32. More than 90% of JGCTs follow an indolent clinical course, without disease recurrence after initial treatment32. From a molecular perspective, JGCTs lack the FOXL2 c.7558C > T (p.C134W) mutation6,33,34 that is characteristic of most AGCTs. In fact, re-review of rare JGCTs with FOXL2 c.7558C > T (p.C134W) mutations resulted in their reclassification as AGCT34. Molecular analyses of JGCTs have demonstrated the presence of pathogenic GNAS35, AKT136, and DICER134 variants in subsets of these tumors. Pathogenic variants in these genes, all of which are covered by our panel, were not detected in any of the testicular AGCTs analyzed in this study.
The present study has several limitations that need to be mentioned. Firstly, the series is relatively small and includes samples collected over a large time span (>30 years). However, given the rarity of this entity, the size of the present series is comparable to that of other series of testicular AGCTs. Secondly, a targeted sequencing panel was used for molecular analysis, which precludes the detection of uncommon and novel genetic variants when compared with whole genome or whole exome approaches. Nonetheless, the use of an extensively validated and well-established sequencing platform with known performance characteristics warrants that the reported variants are clinically and biologically relevant. Thirdly, benign tissue was not available at the time of the study to determine the germline status, limiting our interpretation of germline variants to algorithmic detection. Finally, metastatic (rather than primary) ovarian AGCTs were used as a comparator. However, prior studies have demonstrated that metastatic and primary ovarian AGCTs are comparable from a molecular standpoint37. Notwithstanding these shortcomings, the results presented herein represent the first comprehensive molecular assessment of testicular AGCTs. More specifically, this is the first series of testicular AGCTs analyzed with massively parallel sequencing and the largest one with molecular analyses to date, comprising cases diagnosed at multiple institutions and a subset reported in a prior series2.
In conclusion, our study suggests that testicular AGCTs might be molecularly different from their ovarian counterparts in that they seem to harbor a heterogenous group of potential drivers with a low frequency of activating FOXL2 mutations. Additional genomic and epigenomic analyses may be helpful to further characterize these rare tumors and explore whether they represent a unified biologic entity.
Data availability
The data generated during the current study are available from the corresponding author on reasonable request.
References
Laskowski, J. Feminizing tumors of the testis: general review with case report of granulosa cell tumor of the testis. Endokrynol. Pol. 3, 337–343 (1952).
Cornejo, K. M. & Young, R. H. Adult granulosa cell tumors of the testis: A report of 32 cases. Am. J. Surg. Pathol. 38, 1242–1250 (2014).
Jimenez-Quintero, L. P. et al. Granulosa cell tumor of the adult testis: A clinicopathologic study of seven cases and a review of the literature. Hum. Pathol. 24, 1120–1126 (1993).
Grogg, J. B. et al. Risk factors and treatment outcomes of 239 patients with testicular granulosa cell tumors: a systematic review of published case series data. J. Cancer Res. Clin. Oncol. 146, 2829–2841 (2020).
Shah, S. et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 360, 2719–2729 (2009).
Jamieson, S. et al. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod. Pathol. 23, 1477–1485 (2010).
Hanson, J. A. & Ambaye, A. B. Adult testicular granulosa cell tumor: A review of the literature for clinicopathologic predictors of malignancy. Arch. Pathol. Lab. Med. 135, 143–146 (2011).
Sholl, L. M. et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, 1–19 (2016).
Garcia, E. P. et al. Validation of oncopanel a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med. 141, 751–758 (2017).
Abo, R. P. et al. BreaKmer: Detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 43, 1–13 (2015).
Papke, D. J. et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod. Pathol. 31, 1882–1890 (2018).
Gaylis, F. D., August, C., Yeldandi, A., Nemcek, A. & Garnett, J. Granulosa cell tumor of the adult testis: Ultrastructural and ultrasonographic characteristics. J. Urol. 141, 126–127 (1989).
Hammerich, K. H. et al. Malignant advanced granulosa cell tumor of the adult testis: case report and review of the literature. Hum. Pathol. 39, 701–709 (2008).
Dieckmann, K.-P., Bertolini, J. & Wuelfing, C. Adult granulosa cell tumor of the testis: A case report with a review of the literature. Case Rep. Urol. 2019, 7156154 (2019).
Fang, X. et al. A novel mouse model of testicular granulosa cell tumors. Mol. Hum. Reprod. 24, 343–356 (2018).
Fang, X. & Li, Q. New insights into testicular granulosa cell tumors (Review). Oncol. Lett. 20, 293 (2020).
Norman, R. W., Sheridan-Jonah, A., Merrimen, J. & Gupta, R. Adult granulosa cell tumor of the testicle. Can. J. Urol. 20, 6640–6642 (2013).
Matson, C. K. et al. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476, 101–105 (2011).
Wilhelm, D. et al. Sertoli cell differentiation is induced both cell-autonomously and through prostaglandin signaling during mammalian sex determination. Dev. Biol. 287, 111–124 (2005).
Li, Y. et al. ß-catenin directs the transformation of testis Sertoli cells to ovarian granulosa-like cells by inducing Foxl2 expression. J. Biol. Chem. 292, 17577–17586 (2017).
Richards, J. S. et al. Either Kras activation or Pten loss similarly enhance the dominant-stable CTNNB1-induced genetic program to promote granulosa cell tumor development in the ovary and testis. Oncogene 31, 1504–1520 (2012).
Schubert, T. E. O. et al. Adult type granulosa cell tumor of the testis with a heterologous sarcomatous component: Case report and review of the literature. Diagn. Pathol. 9, 1–7 (2014).
Lima, J. F. et al. FOXL2 mutations in granulosa cell tumors occurring in males. Arch. Pathol. Lab. Med. 136, 825–828 (2012).
Hes, O. et al. Mutational analysis (c.402C>G) of the FOXL2 gene and immunohistochemical expression of the FOXL2 protein in testicular adult type granulosa cell tumors and incompletely differentiated sex cord stromal tumors. Appl. Immunohistochem. Mol. Morphol. 19, 347–351 (2011).
Perrone, F. et al. Frequent mutation and nuclear localization of β-catenin in sertoli cell tumors of the testis. Am. J. Surg. Pathol. 38, 66–71 (2014).
Necchi, A. et al. Genomic features of metastatic testicular sex cord stromal tumors. Eur. Urol. Focus 5, 748–755 (2019).
Colecchia, M. et al. The Leydig cell tumour Scaled Score (LeSS): a method to distinguish benign from malignant cases, with additional correlation with MDM2 and CDK4 amplification. Histopathology 78, 290–299 (2021).
Rizzo, N. M. et al. Comparative molecular analysis of testicular Leydig cell tumors demonstrates distinct subsets of neoplasms with aggressive histopathologic features. Mod. Pathol. 34, 1935–1946 (2021).
Hillman, R. T. et al. KMT2D/MLL2 inactivation is associated with recurrence in adult-type granulosa cell tumors of the ovary. Nat. Commun. 9, 2496, https://doi.org/10.1038/s41467-018-04950-x (2018).
Geiersbach, K. B. et al. FOXL2 mutation and large-scale genomic imbalances in adult granulosa cell tumors of the ovary. Cancer Genet. 204, 596–602 (2011).
Fashedemi, Y. et al. Adult granulosa cell tumor with high-grade transformation: Report of a series with FOXL2 mutation analysis. Am. J. Surg. Pathol. 43, 1229–1238 (2019).
Kao, C. S., Cornejo, K. M., Ulbright, T. M. & Young, R. H. Juvenile granulosa cell tumors of the testis: a clinicopathologic study of 70 cases with emphasis on its wide morphologic spectrum. Am. J. Surg. Pathol. 39, 1159–1169 (2015).
D’Angelo, E. et al. Prognostic significance of FOXL2 mutation and mRNA expression in adult and juvenile granulosa cell tumors of the ovary. Mod. Pathol. 24, 1360–1367 (2011).
Baillard, P. et al. Rare DICER1 and absent FOXL2 mutations characterize ovarian juvenile granulosa cell tumors. Am. J. Surg. Pathol. 45, 223–229 (2021).
Kalfa, N. et al. Activating mutations of the stimulatory g protein in juvenile ovarian granulosa cell tumors: a new prognostic factor? J. Clin. Endocrinol. Metab. 91, 1842–1847 (2006).
Bessière, L. et al. A hot-spot of in-frame duplications activates the oncoprotein AKT1 in juvenile granulosa cell tumors. EBioMedicine 2, 421–431 (2015).
Zannoni, G. F. et al. FOXL2 molecular status in adult granulosa cell tumors of the ovary: A study of primary and metastatic cases. Oncol Lett. 12, 1159–1163 (2016).
Acknowledgements
We would like to thank Melissa Osias (Project Coordinator, Center for Advanced Molecular Diagnostics, BWH), Mr. Mark Buchannan (Histotechnician, BWH), and Ms. Mei Zheng (Immunohistochemistry Laboratory Manager, BWH) for their contributions to this project.
Funding
This research was supported by funds from the Department of Pathology, Brigham, and Women’s Hospital.
Author information
Authors and Affiliations
Contributions
Concept, design, and coordination: A.M.A.; contribution of cases: K.M.C., M.S.H., A.S., C.O.; histopathological evaluation: S.S. and A.M.A.; analysis and interpretation of molecular data: L.M.S.; integration and analysis of clinical, molecular and histopathologic data: S.S. and A.M.A.; manuscript draft: S.S. and A.M.A.; intellectual contributions: all authors; manuscript editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval/consent to participate
This research was performed with the approval of the Institutional Review Board of Brigham and Women’s Hospital (Partners Healthcare/ Mass General Brigham).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Siegmund, S., Sholl, L.M., Cornejo, K.M. et al. Molecular assessment of testicular adult granulosa cell tumor demonstrates significant differences when compared to ovarian counterparts. Mod Pathol 35, 697–704 (2022). https://doi.org/10.1038/s41379-021-00977-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00977-6
