
Abstract
Testicular Leydig cell tumor (LCT), the most common sex-cord stromal tumor in men, represents a small fraction of all testicular tumors (~1 to 3%). Although most testicular LCTs are indolent and cured by radical orchiectomy, 5–10% have aggressive biology and metastatic potential. In primary LCTs, large size, cytologic atypia, necrosis, increased mitotic activity, and vascular invasion have been associated with clinically aggressive tumors. From a molecular perspective, the characteristics of aggressive LCTs and the differences between aggressive and nonaggressive LCTs remain largely unexplored. This study compares the genomic landscape of aggressive and nonaggressive testicular LCTs. Twenty-six cases were analyzed using next-generation DNA sequencing (NGS) and immunohistochemistry. Cases were classified as aggressive LCT if they met published criteria for malignancy in primary (i.e., testicular) tumors or if they had pathology-proven metastatic disease; otherwise, cases were considered nonaggressive. This multi-institutional series included 18 aggressive LCTs (14 primary/testicular, 4 metastatic) and 8 nonaggressive LCTs. Two cases (2/26, 8%; both aggressive LCTs) failed sequencing and had negative (i.e., uninformative) FH immunohistochemistry results. One additional primary aggressive LCT failed sequencing but had informative FH immunohistochemistry results. Combined NGS and immunohistochemical analysis demonstrated FH inactivation in 5/26 cases (19%). In addition, NGS demonstrated CTNNB1 mutations or biallelic APC inactivation in 9/23 cases (39%), copy number changes without recurrent mutations in 6/23 (26%) cases, and no alterations in 4/23 cases (17%). CTNNB1 mutations were present in both aggressive and nonaggressive LCTs. In contrast, FH inactivation and multiple copy number changes were only identified in aggressive LCTs. In conclusion, three distinct subgroups of aggressive LCTs were characterized by FH inactivation, Wnt pathway activation, and copy number changes without recurrent mutations, respectively. Nuclear translocation of β-catenin and Wnt pathway activation appear to be early driver events that provide an environment conducive for progression to aggressive biology in a subset of LCTs.
Similar content being viewed by others

Introduction
Testicular Leydig cell tumors (LCTs), the most common type of sex-cord stromal tumor in men, are rare overall and account for less than 3% of all testicular neoplasms, according to historical data [1]. Updated figures from the Surveillance, Epidemiology, and End Results Program show that the ratio of germ cell tumors to LCTs is ~237:1 (17998:76), suggesting that LCTs currently account for less than 1% of all testicular tumors [2]. This can be explained in part by the substantial increase in the incidence of germ cell tumors seen in the last few decades, especially among ethnic minorities [3,4,5]. The age at diagnosis of testicular LCT has a bimodal distribution, with a small peak in early childhood (~20% of cases) and a larger peak in young to middle-aged adults (~80% of cases) [2, 6, 7]. Although most testicular LCTs diagnosed in adult men are indolent and behave as benign neoplasms, a minor but clinically relevant subset may pursue an aggressive clinical course characterized by systemic spread and suboptimal response to chemotherapy and radiotherapy [7]. Data from prior studies suggest that aggressive (i.e., malignant) LCTs may account for up to 10% of all testicular LCTs, resulting in a disease-specific mortality of 6.6% [2, 7, 8].
Clinical and pathological criteria have been proposed to help identify aggressive primary LCTs in orchiectomies before they metastasize. [7, 9,10,11] Given the refractoriness of aggressive LCTs to chemotherapy and radiotherapy, their early identification is of paramount importance to determine which patients need a close follow-up and early surgical intervention. Large tumor size (≥3–5 cm), increased mitotic activity (>3 mitoses per 10 hpf), necrosis, lymphovascular invasion, and/or cytologic atypia are characteristically present in cases with malignant potential. In prior series, the presence of two or more of these features has been used to define aggressive (i.e., “malignant”) primary LCTs [7, 9,10,11].
The molecular correlates of aggressive biology remain poorly defined in testicular LCTs. Two primary FH-deficient adult LCTs have been reported in the literature, including one in a patient with a germline FH mutation and family history of hereditary leiomyomatosis and renal cell carcinoma (HLRCC) [12]. To date, FH inactivation has not been identified in metastatic cases [11, 13] and it is not known if FH deficiency is associated with aggressive biology in testicular LCTs [12]. Prior studies of a limited number of metastatic LCTs have demonstrated a high frequency of aneuploidy and MDM2/CDK4 amplification, with CTNNB1 mutations being present in a smaller subset of cases [9, 11]. In contrast, the genomic landscape of primary aggressive LCTs (i.e., non-metastatic LCTs with aggressive histopathologic features) remains largely unexplored. Moreover, a comprehensive comparative molecular analysis of aggressive and nonaggressive testicular LCTs has not been undertaken. In the present study, testicular LCTs spanning the entire biologic spectrum (i.e., from histologically indolent primaries to metastatic tumors) were profiled and compared using next-generation DNA sequencing and immunohistochemistry (IHC).
Materials and methods
Identification of cases and procurement of tissue
This research was approved by the Brigham and Women’s Hospital (BWH)/Partners Healthcare Institutional Review Board.
Institutional pathology databases (BWH, Indiana University, Mayo Clinic, University of Rochester, and Massachusetts General Hospital) and the personal consultation files of the authors were queried to identify aggressive LCTs diagnosed in adult male patients. Additional testicular LCTs that did not meet criteria for aggressive disease were identified in the pathology archives of BWH, Massachusetts General Hospital, and Indiana University.
Archival stained and unstained pathology slides were retrieved. New unstained tissue sections were obtained for sequencing and IHC when formalin-fixed paraffin-embedded tissue blocks were available.
Pathologic evaluation of the cases
Cases were classified as either aggressive or nonaggressive LCT as described below. The original slides and pathology reports were initially reviewed by the submitting collaborators. Subsequently, all the slides available for the study were centrally reviewed at BWH by two of the authors (NMR and AMA). Given that most cases had been originally seen in consultation, only one or a few representative slides were available for review at the time of the study.
Primary (i.e., testicular) tumors were classified as aggressive LCT if two or more of the following criteria were present: size ≥3 cm, mitoses ≥3/10 hpf, nuclear atypia, necrosis, lymphovascular invasion, and invasive growth [7, 11, 14]. All aggressive LCTs had been originally diagnosed as malignant LCT or LCT with malignant potential by expert genitourinary pathologists. Cases that did not fulfill the criteria for aggressive disease were classified as nonaggressive LCT.
DNA sequencing and IHC
Unbaked formalin-fixed paraffin-embedded tumor sections were macrodissected from glass slides, using a corresponding H&E-stained section marked by a pathologist as a guide. Cellularity was estimated as the percentage of tumor nuclei in the area marked for macrodissection, and minimum cellularity of 20% was required for sample acceptance. Next-generation sequencing (NGS) using a clinically validated 447-gene solid tumor panel (OncoPanel) was performed as previously described by Garcia et al. and Sholl et al. [15, 16]. Briefly, DNA was isolated with a standard commercial kit (Qiagen, Valencia, CA) following the manufacturer’s recommendations. A target input of 200 ng of DNA (threshold of 100 ng) was used for the preparation of the sequencing libraries (TruSeq LT library preparation kit; Illumina, San Diego, CA). The target genes were selected by hybridization to a set of custom-designed probes (Agilent SureSelect; Agilent Technologies, Santa Clara, CA) and sequencing was performed on the Illumina HiSeq 2500 System (Illumina, San Diego, CA). A validated institutional informatic pipeline was used for deconvolution of samples, read alignment, variant calling (single nucleotide variants [SNVs], indels, copy number variants [CNVs], structural variants) and annotation [15,16,17]. Because this is a tumor-only assay, variants present at a population frequency of >0.1% in the gnomAD database (Broad Institute) were filtered out to avoid contamination with germline variants. In general, variants present at a frequency of 3% or lower were considered not significant and filtered out. However, variants present at low frequencies but reported as clinically relevant in ClinVar were rescued for manual review. All reported variants were further reviewed and tiered for actionability/biologic relevance by a molecular pathologist (LMS). In-house developed and validated algorithms were used for the detection of mutational signatures (POLE, APOBEC, smoking, UV) and mismatch repair status [18].
IHC was performed with primary antibodies against fumarate hydratase (FH, clone J-13, mouse monoclonal, Santa Cruz Biotechnology, Inc., Dallas, TX; dilution 1:1000), S(2-succinyl)-cysteine (2SC, rabbit polyclonal, Cambridge Research Biochemicals, Billingham, UK; dilution 1:500), β-catenin (clone 14, mouse monoclonal, BD Biosciences, San Jose, CA; dilution 1:2000) and p16 (ENZ-ABS377, mouse monoclonal, Enzo Life Sciences, Inc., Farmingdale, NY; dilution 1:75) according to the manufacturers’ recommendations. Positive and negative control cases were run in parallel for IHC. The following staining patterns were considered a positive result when present in lesional cells: loss (absence) of FH, cytoplasmic and nuclear expression of 2SC, nuclear expression of β-catenin, and loss (absence) of p16.
Results
Clinicopathologic characteristics of the cases
Twenty-six cases (patients) diagnosed between 1975 and 2020 were included in the study: 18 aggressive LCTs (14 primary/testicular, 4 metastatic) and 8 nonaggressive LCTs (Table 1). For 2 aggressive LCTs, multiple metastases were available and analyzed in parallel. The median age at diagnosis was 60 years (range 21–80 years) overall, 68.5 years (range 27–80 years) for aggressive LCTs, and 37.5 years (range 21–65 years) for nonaggressive LCTs. For the primary tumors, median size was 3.6 cm (range 0.7–10 cm) overall, 4 cm (range 2–10 cm) for primary aggressive LCTs and 1.3 cm (range 0.7–4.2 cm) for nonaggressive LCTs. The median number of mitoses per 10 high-power fields (hpf) was 4 (range < 1–25) overall, 6 for aggressive LCTs (range 1–25) and ~1 for nonaggressive LCTs (range < 1–4). Metastatic sites included lung, foot, pelvic lymph nodes, and retroperitoneal lymph nodes.
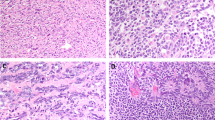
The architectural patterns of the tumors in the series included solid sheets, cords, nests, and microcystic growth, with most cases showing multiple patterns. Cytomorphology was somewhat variable (Fig. 1), especially in aggressive tumors, but all cases showed at least focal morphologic features characteristic of LCTs. The neoplastic cells had abundant eosinophilic cytoplasm with a granular texture. Focal areas of spindling and clear cytoplasmic change were only seen in aggressive LCTs.

A Some aggressive LCTs had minimal cytologic atypia with markedly increased mitotic activity (case 5). B Apoptotic cells with condensed eosinophilic cytoplasm, pyknosis, and karyorrhexis were common in LCTs with aggressive histologic features, as shown in the micrograph (case 5). C Some aggressive LCTs displayed striking cytologic atypia characterized by nuclear pleomorphism (case 12). Eosinophilic nuclear pseudoinclusions were also seen (inset C, case 12) D Rare aggressive LCTs showed focal spindling (case 10) and atypical mitotic figures (inset D, case 12). Please see “Materials and Methods” for a detailed description of the histopathologic features that define aggressive and nonaggressive LCTs.
In nonaggressive LCTs, nuclei were invariably round and regular with a single visible nucleolus and finely granular chromatin. In contrast, aggressive LCTs showed a broader spectrum of nuclear morphologies, with significant variations between cases and within each individual case. Nuclear morphology in aggressive LCTs ranged from minimally atypical round nuclei with slightly coarser chromatin than that seen in nonaggressive LCTs to giant hyperchromatic nuclei with eosinophilic macronucleoli, and/or intranuclear vacuoles (i.e., “pseudoinclusions”). Of note, prominent nucleoli with peri-nucleolar halos were seen, at least focally, in all FH-deficient aggressive LCTs (see sequencing and IHC results below). However, this finding was by no means specific, as conspicuous eosinophilic nucleoli and occasional peri-nucleolar halos were also present in several non-FH-deficient aggressive and nonaggressive LCTs. In aggressive tumors with marked nuclear atypia, areas with characteristic Leydig cell cytomorphology were also present.
Individual apoptotic cells with condensed eosinophilic cytoplasm, pyknosis and/or karyorrhexis were frequently present in aggressive LCTs but not in nonaggressive LCTs. Similarly, multinucleated tumor cells were easily found in aggressive LCTs but were not identified in nonaggressive neoplasms. Foci of tumor necrosis and atypical mitotic figures were only seen in aggressive LCTs.
Because the histomorphology of individual cases showed significant internal variation and only a limited number of representative slides were available at the time of the study, the features described above were not quantified.
Sequencing and IHC results
Twenty-three of the 26 cases included in the series underwent successful sequencing (23/26, 88%; Table 2). Of the three samples that failed sequencing or yielded insufficient DNA, all of which were primary aggressive LCTs, two also had negative (uninformative) IHC results. In all, NGS results were available for all (8/8) nonaggressive LCTs cases and for 15/18 aggressive LCT cases (11 primary, 4 metastatic), with multiple metastases analyzed in two aggressive LCTs (4 samples for case 1 and 2 samples for case 6) (Table 2). The median tumor mutational burden was 3.8 (range 3.0–11.4) overall, 4.2 (range 3.0–11.4) for aggressive LCTs, and 3.8 (range 3.0–7.6) for nonaggressive LCTs.
Analysis of aggressive LCTs demonstrated three distinct subgroups, characterized by FH mutations/indels, CTNNB1 or APC mutations/indels (Wnt pathway activation), and copy number instability without concurrent recurrent mutations (Fig. 2 and Table 2). In all, 4/15 (27%) aggressive LCTs (3 primary, 1 metastatic) harbored inactivating FH mutations, 5/15 (33%) aggressive LCTs (4 primary, 1 metastatic) harbored activating CTNNB1 (N = 4) or inactivating APC (N = 1) mutations, and 6/15 (40%) aggressive LCTs (5 primary, 1 metastatic) had copy number changes without recurrent mutations. One of the FH-deficient aggressive LCTs (case 4, primary) had a concurrent class II BRAF mutation (BRAF p.G469V) and TERT amplification. Although FH, CTNNB1, and APC-mutant LCTs had concurrent CNVs, these were overall less numerous than in aggressive tumors with only copy number changes. High-level co-amplification of MDM2/CDK4 was identified in 2/15 (13%) aggressive LCTs (1 primary, 1 metastatic), both of which had numerous additional copy number changes (cases 14 and 15). Of the 4 metastatic cases in the series, 1 was FH-deficient (case 1), 1 harbored an activating CTNNB1 mutation (case 6), and 2 demonstrated only numerous copy number changes (cases 13 and 14) including 1 case with MDM2/CDK4 co-amplification (case 14). Comparison of multiple metastases resected at different times from two individual patients showed that FH and CTNNB1 mutations were retained in all metastases (cases 1 and 4, respectively), which supports the driver role of these variants. The CNV profile of the different metastases from cases 1 and 6 demonstrated only a few differences between the samples.

Cases with metastases (m) are listed in chronologic order. Mutations/indels and amplifications/deep deletions are known clinically relevant variants. Immunostaining for 2SC and β-catenin was called present if moderate to strong (2+/3+) nuclear and cytoplasmic staining was observed. Abbreviations: A-LCT = aggressive Leydig cell tumor, NA-LCT nonaggressive Leydig cell tumor, Amp. amplification, CNV copy number variant, Deep Del. deep deletion (i.e., homozygous deletion), IHC immunohistochemistry, m metastases. aBorderline next-generation sequencing QC metrics (average reads 49). bFailed next-generation sequencing (or low DNA content). CNuclear expression. For the purpose of simplification, only chromosomes with arm-level and chromosome-level copy number changes are shown.
Tissue sections were available for FH IHC in all aggressive LCTs that failed sequencing and in 3/4 aggressive LCTs with FH mutations identified by DNA sequencing. IHC confirmed the concurrent loss of FH expression and 2SC upregulation in two cases with FH mutations, and loss of FH expression in one case with an FH mutation and only 1 slide available for IHC (case 3). Moreover, 1 of the 3 cases that failed sequencing had loss of FH and 2SC upregulation by IHC, resulting in a total of 5/18 (28%) aggressive LCTs with FH inactivation (Fig. 3). Three of 4 aggressive LCTs that harbored CTNNB1 or APC mutations had additional FFPE tissue available for β-catenin IHC, all of which demonstrated cytoplasmic and nuclear positivity (Fig. 4). The loss of p16 expression was also demonstrated by IHC in 3 aggressive LCTs (cases 6, 11, and 12) with deep deletions of CDKN2A (Fig. 5).

A FH-deficient aggressive LCT metastatic to the foot (case 1m3) B Microscopically, case 1m3 was not markedly atypical. Cells were eosinophilic with round nuclei and conspicuous nucleoli. Peri-nucleolar halos were seen in a subset of tumor cells. C FH expression was completely lost in tumor cells from case 1m3 (note the retained expression in endothelial cells). Lesional cells expressed the 2SC oncometabolite (inset C). D Case 16, which failed sequencing, had moderate-to-severe cytologic atypia; eosinophilic nucleoli with peri-nucleolar halos and nuclear pseudoinclusions were present in scattered tumor cells. E Immunohistochemistry demonstrated loss of FH expression and accumulation of the 2SC oncometabolite (inset E) in case 16.

A Macroscopic appearance of a nonaggressive testicular LCT with an activating CTNNB1 mutation (case 20). The tumor shows a well-circumscribed, smooth, gelatinous, tan cut surface without necrosis or hemorrhage. B Microscopically, case 20 demonstrated minimal cytologic atypia, but mitotic activity was identified (up to 4 mitoses per 10 HPFs). Small pinpoint nucleoli were present. C A significant subpopulation of the cells had cytoplasmic and nuclear expression of B-catenin. D APC-mutant aggressive LCT (case 9). Tumor cells contained variably sized nuclei with vesicular chromatin and prominent nucleoli. E. Nuclear and cytoplasmic β-catenin expression was seen in a significant subset of the tumor cells (case 9).

A Genome-wide copy number plot of case 14, showing multiple arm-level and chromosome-level copy number changes, as well as high-level TERT and MDM2 amplifications in chromosomes 5 and 12, respectively (CNVs are expressed as log2 ratios). B Microscopically, case 14 had only mild-to-moderate cytologic atypia despite being metastatic. C Case 12, a case with genomic instability and deep deletion of CDKN2A, demonstrated moderate nuclear pleomorphism and focal cytoplasmic clearing. D Immunohistochemistry, performed to confirm the sequencing results in case 12, demonstrated an absence of p16 expression in tumor cells with retained expression in stromal and endothelial cells. Abbreviations: CNVs copy number variants.
Analysis of nonaggressive LCTs demonstrated activating CTNNB1 mutations in 4/8 (50%) cases and no alterations in the remaining 4/8 (50%) cases (Fig. 2). Of note, the only CNV identified in nonaggressive LCTs was 22q loss in 2/8 cases (25%), including 2 of the 4 tumors with a CTTNB1 mutation. β-catenin IHC demonstrated nuclear and cytoplasmic positivity in all 4 (100%) CTNNB1-mutant cases (Fig. 2).
Considering the entire series (aggressive plus nonaggressive LCTs), 5/26 (19%) cases had FH inactivation identified by NGS and/or IHC, 9/23 (39%) cases harbored CTNNB1 or APC mutations, 6/23 (26%) had copy number changes without recurrent mutations, 4/23 (17%) cases had no molecular findings, and 2/3 cases failed NGS and retained FH expression by IHC. Comparison of the molecular findings in aggressive and nonaggressive LCTs revealed some interesting differences. All aggressive LCTs (both primary and metastatic) showed pathogenic genetic variants (either mutations/indels or CNVs), whereas half of the limited number of nonaggressive LCTs analyzed herein had no molecular alterations detected by a targeted sequencing panel (OncoPanel).
Copy number events were strikingly more numerous in aggressive than in nonaggressive LCTs, and 22q loss was the only CNV identified in the latter. Comparative evaluation of copy number changes demonstrated a few recurrent events involving chromosomes 2, 5, 7, 12, 18, 19, and 22 (Fig. 2). Additional recurrent copy number events were TERT amplification in 4 aggressive LCTs (3 primary, 1 metastatic), MDM2/CDK4 co-amplification in 2 aggressive LCTs (1 primary, 1 metastatic), and deep (two copy) deletion of CDKN2A in 3 aggressive LCTs (2 primary, 1 metastatic). TERT amplifications and deep deletions of CDKN2A were seen with concurrent CTNNB1, APC and FH mutations. In contrast, high-level amplification of MDM2/CDK4 was restricted to aggressive LCTs with copy number changes and absence of recurrent mutations, suggesting that this event is mutually exclusive with CTNNB1, APC and FH mutations. Also, the presence of MDM2/CDK4 co-amplification in both a metastasis (case 14) and a primary tumor (case 15) suggests that this might be an early event in a subset of aggressive LCTs.
IHC evaluation of sixteen additional nonaggressive Leydig cell tumors
Sixteen additional nonaggressive LCTs were evaluated with β-catenin, FH, and 2SC IHC (Table 3). The median tumor size was 1.4 cm (range 0.5–3.5 cm) and the median number of mitoses was <1/10 hpf (range < 1/10–3/10 hpf). All cases retained FH expression and were negative for abnormal 2SC expression (diffuse cytoplasmic and nuclear staining). Nuclear β-catenin expression was identified in 10/16 cases (63%) and was absent in 6/16 cases (37%). The staining pattern was multifocal or focal in most tumors, suggestive of sub-clonal activation of β-catenin (Fig. 6).

A, B Nonaggressive LCT (1.2 cm) with “foamy” cytoplasm. Nuclear positivity for β-catenin is seen in discrete cell clusters, suggestive of sub-clonal activation of β-catenin. C, D This nonaggressive LCT was 3.5 cm but did not have any additional worrisome features. Note how nuclear β-catenin expression is restricted to the cells on the left side of the micrograph, suggestive of sub-clonal β-catenin activation.
Discussion
The present study found that pathogenic genetic variants are more frequent and numerous in aggressive than in nonaggressive LCTs. Moreover, three distinct subsets of aggressive tumors were identified, characterized by inactivating FH mutations, Wnt pathway activation, and copy number changes without recurrent mutations.
Adult testicular LCTs comprise ~1% of all testicular neoplasms and commonly present during the 3rd to 4th decade of life [7, 19, 20]. Although the vast majority of testicular LCTs behave indolently, up to ~10% can metastasize and follow an aggressive clinical course [7, 14]. Low-stage aggressive (i.e., “malignant”) testicular LCTs have higher mortality than low-stage post-pubertal malignant germ cell tumors, most likely due to the relatively poor response of the former to systemic therapy [21]. Therefore, several systems have been proposed to try to identify aggressive primary LCTs with metastatic potential when they are still confined to the testis [7, 11, 14]. Histopathologic features classically associated with aggressive biology in LCTs include a relatively large size (>3–5 cm), lymphovascular invasion, infiltrative borders, tumor necrosis, increased mitotic activity (≥3 mitoses per 10 hpf) and cytologic atypia [7, 11, 14].
Although the molecular pathogenesis of testicular LCTs is beginning to be elucidated, there are still many uncertainties regarding the genomic changes that determine an aggressive clinical behavior in a subset of these tumors. In children, testicular LCTs appear to be driven by activating mutations of the luteinizing hormone receptor [22]. In contrast, adult testicular LCTs are likely more heterogenous and have different genetic drivers [12]. An aneuploid DNA content has been demonstrated in some adult testicular LCTs and this finding is particularly common in metastatic tumors [9]. Using a gene-specific approach, Carvajal-Carmona et al. identified the presence of pathogenic FH mutations in two adult testicular LCTs [12]. Recently, Necchi et al. have reported MDM2 amplifications and CTNNB1 mutations in 5/10 and 2/10 metastatic LCTs, respectively [13]. Moreover, Colecchia et al. performed a 161-gene NGS panel on a series of metastatic LCTs that includes cases from the prior study (Necchi et al.) and found MDM2 amplifications and CTNNB1 mutations in 3/10 and 2/10 tumors, respectively [11].
In the present series, FH mutations or loss of FH expression by IHC were found in 28% of aggressive LCTs (5/18 cases; 3 primary, 1 metastatic). Importantly, FH mutations were not seen in nonaggressive LCTs, suggesting that FH loss defines a subgroup of testicular LCTs with aggressive histopathologic features and metastatic potential. Microscopically, prominent nucleoli visible at 10× with occasional peri-nucleolar halos were present at least focally in all FH-deficient LCTs, but this was not a specific finding and other distinctive features were not identified in these cases. In the 2 FH-deficient testicular LCTs previously reported in the literature, FH inactivation was due to loss of heterozygosity in patients with germline FH mutations [12]. One of these patients had a confirmed family history of HLRCC. In this series, the only patient with an FH-deficient aggressive LCT for whom detailed clinical data were available did not have other tumors or a family history of HLRCC. This suggests that aggressive testicular LCTs can be the initial manifestation of HLRCC and their identification should prompt investigation of the germline status.
Another relevant finding of this study is that CTNNB1 mutations were identified in lesions that encompass the entire histopathologic spectrum, from nonaggressive primary LCTs to metastatic LCTs. Clinical data were available for two of the cases with CTNNB1-mutant nonaggressive LCTs. None of these patients had a family history of adenomatous polyposis coli or a personal history of other tumors, and both were disease-free at 4.1 and 4.6 years of follow-up, respectively. Given that activating CTNNB1 mutations were present across the entire biologic spectrum of testicular LCTs, Wnt pathway activation is likely an early driver event in these tumors. Evaluation of 16 additional nonaggressive LCTs demonstrated that slightly over half (~60%) of testicular LCTs without worrisome histologic features have nuclear expression of β-catenin, suggestive of underlying activating CTNNB1 mutations or inactivating APC mutations. Interestingly, the staining pattern observed in these LCTs was consistent with sub-clonal activation of β-catenin. Therefore, we hypothesize that nuclear translocation of β-catenin and Wnt pathway activation are early events that likely occur in sub-clones of “nonaggressive” LCTs, providing a favorable environment for biologic progression as additional genetic or epigenetic changes accumulate. Additional research is needed to explore this premise.
In this study, copy number alterations were strikingly more numerous in aggressive than in nonaggressive LCTs. A subset of aggressive LCTs was characterized by multiple CNVs, including large (i.e., arm-level and/or chromosome-level) copy number changes, and absence of recurrent pathogenic mutations. High-level amplification of MDM2 was only seen in two cases within this group of aggressive LCTs with copy number changes and in 13% of aggressive LCTs overall (2/15 cases successfully tested). The lower frequency of MDM2 amplification in this study compared to previous reports may be explained by differences in the study populations and by the inclusion of both primary and metastatic tumors herein [11, 13]. Of note, the detection of MDM2 amplification in both a primary aggressive LCT and a metastatic LCT suggests that MDM2 is probably an early event in a subset of aggressive LCTs with copy number changes and absence of recurrent mutations (i.e., FH or CTNNB1/APC mutations).
In this series, FH deficiency and CTNNB1 or APC mutations were mutually exclusive and appeared to define distinct molecular subtypes of LCTs. Similarly, MDM2 and CDK4 amplification did not overlap with FH deficiency, CTNNB1 mutations or APC mutations in the limited number of cases analyzed herein. In contrast, homozygous CDKN2A/CDKN2B deletion was seen in a case with a concurrent CTNNB1 mutation. TERT amplification was also identified in the case with an inactivating APC mutation (case 9) and in one of the FH-deficient cases (case 4). Arm-level CNVs, chromosome-level CNVs, high-level regional amplifications, and homozygous deletions were somewhat more frequent in cases with copy number changes and the absence of recurrent mutations. However, except for MDM2/CDK4 amplification, most CNVs were largely nonspecific and showed significant overlap with FH, CTNNB1, and APC mutations. Interestingly, one of the cases with multiple CNVs had molecular evidence of biallelic inactivation of CHEK2, suggesting that a deficiency of DNA double strand-break repair mechanisms could lead to copy number instability in some LCTs. The subset of aggressive LCTs that harbor copy number changes without recurrent mutations is most likely heterogeneous and might be amenable to further characterization by whole-exome sequencing, whole-genome sequencing, and epigenetic studies.
From a practical perspective, the results of this and prior studies suggest that IHC might be a useful surrogate marker of underlying genetic alterations in primary aggressive testicular LCTs [11, 13]. Because the different molecular subsets of aggressive LCTs appear to be mutually exclusive, it is reasonable to start with β-catenin IHC, as CTNNB1 or APC mutations are present in 40% of aggressive LCTs (6/15 successfully tested). If there is no nuclear β-catenin expression, further IHC for FH, 2SC, MDM2, and/or CDK4 might be informative [11]. Moreover, β-catenin IHC is also a useful surrogate marker of the underlying CTNNB1 mutational status in nonaggressive LCTs. Based on the small number of cases tested in this study, FH/2SC and β-catenin immunohistochemical stains appear to be sensitive markers of FH inactivation and CTNNB1 activation (or biallelic APC inactivation), respectively. However, because of the limited nature of the archival material, the specificity of these markers could not be assessed.
Prior studies have reported that Sertoli cell tumors frequently harbor CTTNB1 mutations and have diffuse and strong nuclear expression of β-catenin by IHC [23, 24]. In one of these series, several types of sex-cord stromal tumors were evaluated and nuclear β-catenin was not identified in any of the ten LCTs tested [23]. However, the results presented herein demonstrate that a subset of LCTs harbor activating CTTNB1 mutations (or inactivating APC mutations), consistent with data recently reported by Colecchia et al. and Necchi et al. [11, 13]. In this study, LCTs with CTNNB1 and APC mutations had nuclear expression of β-catenin that was most commonly multifocal (i.e., patchy). This expression pattern is highly suggestive of sub-clonal activation of β-catenin and is different from the diffuse nuclear expression seen in Sertoli cell tumors [23, 24].
This study has limitations that need to be mentioned. First, given the rarity of LCTs in general and of aggressive LCTs in particular, the number of cases in this series is somewhat limited, precluding the identification of additional rare molecular events. Second, non-neoplastic tissue was not available to determine whether any of the FH, CTNNB1, and APC mutations identified were of germline origin. Third, only one representative H&E slide was available for most cases at the time of the study, precluding a comprehensive histopathologic re-evaluation. Fourth, cases were evaluated using a targeted NGS panel which, albeit comprehensive, limits the identification of genetic variants to those present well-established cancer-relevant genes. Finally, outcome data were not available for the cases included in the series. However, the goal of this study was to explore the molecular features of cases that met the criteria for aggressive LCT, since the histopathologic and clinical characteristics of aggressive LCTs have been described in prior studies [7, 9, 14].
Despite the shortcomings mentioned above, the present work also has significant strengths. First, all cases were diagnosed by expert genitourinary pathologists, and some of the older tumors in this series were part of studies that originally described the histologic features of aggressive LCTs [9, 14]. Second, the number of aggressive testicular LCTs is large relative to prior multi-institutional studies. Third, the cases were profiled using a well-established and clinically validated NGS platform that targets a large number of cancer-relevant genes. Finally, lesions that span the entire histopathologic and biologic spectrum of testicular LCTs were analyzed, allowing a comparative evaluation of cases with nonaggressive and aggressive features.
In conclusion, adult testicular LCTs are biologically and molecularly heterogeneous. Cases with nonaggressive histopathologic features usually have either no genetic findings or only CTNNB1 mutations. In contrast, aggressive testicular LCTs have FH loss, CTNNB1/APC mutations (Wnt pathway activation), or multiple copy number changes without recurrent mutations, which characterize three distinct molecular subgroups of aggressive tumors. Larger multi-institutional studies with clinical follow-up are required to explore potential associations between molecular subtypes of aggressive LCTs and disease-specific outcomes.
Data availability
The data generated during the current study are available from the corresponding author on reasonable request.
References
Dilworth JP, Farrow GM, Oesterling JE. Non-germ cell tumors of testis. Urology. 1991;37:399–417.
Osbun N, Winters B, Holt SK, Schade GR, Lin DW, Wright JL. Characteristics of patients with Sertoli and Leydig cell testis neoplasms from a national population-based registry. Clin Genitourin Cancer. 2017;15:e263–66.
McGlynn KA, Devesa SS, Graubard BI, Castle PE. Increasing incidence of testicular germ cell tumors among Black men in the United States. JCO. 2005;23:5757–61.
Ghazarian AA, Trabert B, Devesa SS, McGlynn KA. Recent trends in the incidence of testicular germ cell tumors in the United States. Andrology. 2015;3:13–8.
Ghazarian AA, McGlynn KA. Increasing incidence of testicular germ cell tumors among racial/ethnic minorities in the United States. Cancer Epidemiol Biomark Prev. 2020;29:1237–45.
Emerson RE, Ulbright TM. Neoplasms of the testis. In: Chang L, MacLennan G, Bostwick D, editors. Urological surgical pathology. 4th ed. Philadelphia, PA: Elsevier; 2019. p. 800–7.
Fankhauser CD, Grogg JB, Hayoz S, Wettstein MS, Dieckmann K-P, Sulser T, et al. Risk factors and treatment outcomes of 1,375 patients with testicular Leydig cell tumors: analysis of published case series data. J Urol. 2020;203:949–56.
Heer R, Jackson MJ, El-Sherif A, Thomas DJ. Twenty-nine Leydig cell tumors: histological features, outcomes and implications for management. Int J Urol. 2010;17:886–9.
Cheville JC, Sebo TJ, Lager DJ, Bostwick DG, Farrow GM. Leydig cell tumor of the testis: a clinicopathologic, DNA content, and MIB-1 comparison of nonmetastasizing and metastasizing tumors. Am J Surg Pathol. 1998;22:1361–7.
McCluggage WG, Shanks JH, Arthur K, Banerjee SS. Cellular proliferation and nuclear ploidy assessments augment established prognostic factors in predicting malignancy in testicular Leydig cell tumours. Histopathology. 1998;33:361–8.
Colecchia M, Bertolotti A, Paolini B, Giunchi F, Necchi A, Paganoni AM, et al. The Leydig cell tumour Scaled Score (LeSS): a method to distinguish benign from malignant cases, with additional correlation with MDM2 and CDK4 amplification. Histopathology. 2021;78:290–9.
Carvajal-Carmona LG, Alam NA, Pollard PJ, Barclay E, Wortham N, Pignatelli M, et al. Adult Leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab. 2006;91:3071–5.
Necchi A, Bratslavsky G, Shapiro O, Elvin JA, Vergilio J-A, Killian JK, et al. Genomic features of metastatic testicular sex cord stromal tumors. Eur Urol Focus. 2019;5:748–55.
Kim I, Young RH, Scully RE. Leydig cell tumors of the testis. A clinicopathological analysis of 40 cases and review of the literature. Am J Surg Pathol. 1985;9:177–92.
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–8.
Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062.
Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas-Rudilla V, Lin L, et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 2015;43:e19.
Papke DJ, Nowak JA, Yurgelun MB, Frieden A, Srivastava A, Lindeman NI, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod Pathol. 2018;31:1882–90.
Carmignani L, Salvioni R, Gadda F, Colecchia M, Gazzano G, Torelli T, et al. Long-term followup and clinical characteristics of testicular Leydig cell tumor: experience with 24 cases. J Urol. 2006;176:2040–3.
Jou P, Maclennan GT. Leydig cell tumor of the testis. J Urol. 2009;181:2299–300.
Banerji JS, Odem-Davis K, Wolff EM, Nichols CR, Porter CR. Patterns of care and survival outcomes for malignant sex cord stromal testicular cancer: results from the National Cancer Data Base. J Urol. 2016;196:1117–22.
Liu G, Duranteau L, Carel JC, Monroe J, Doyle DA, Shenker A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. N Engl J Med. 1999;341:1731–6.
Zhang C, Ulbright TM. Nuclear localization of β-catenin in sertoli cell tumors and other sex cord-stromal tumors of the testis: an immunohistochemical study of 87 cases. Am J Surg Pathol. 2015;39:1390–4.
Perrone F, Bertolotti A, Montemurro G, Paolini B, Pierotti MA, Colecchia M. Frequent mutation and nuclear localization of β-catenin in sertoli cell tumors of the testis. Am J Surg Pathol. 2014;38:66–71.
Acknowledgements
We would like to acknowledge the following people for their significant help with this project: Ms. Mei Zheng (Immunohistochemistry Laboratory Manager, Department of Pathology, BWH, Boston, MA). Mr. Mark Buchannan (Histology technician, Department of Pathology, BWH, Boston MA). Mr. Derek C. Bernay (Immunohistochemistry Laboratory Technician, Department of Pathology, BWH, Boston, MA). Ms.Valerie Miakushko (Laboratory and Project Coordinator, Center for Advanced Molecular Diagnostics, BWH, Boston, MA) . Dr. Alexander O. Subtelny, MD, PhD (Department of Pathology, BWH, Boston, MA). Dr. Nicholas Baniak, MD (Department of Pathology, University of Saskatchewan, Saskatoon, Canada). Dr. Jennifer Gordetsky, MD (Department of Pathology, Vanderbilt University Medical Center, Nashville, TN). Dr. Neil I. Lindeman, MD (Director of the Center for Advanced Molecular Diagnostics, BWH, Boston, MA).
Author information
Authors and Affiliations
Contributions
Concept, design and coordination: AMA. Contribution of cases: MTI, JCC, KMC, MSH, and HM. Analysis of sequencing data: LMS. Interpretation of results and correlation of molecular and histopathologic features: AMA and NMR. Manuscript draft: AMA. Figures and tables: AMA and NMR. Intellectual contributions and manuscript review and editing: All authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rizzo, N.M., Sholl, L.M., Idrees, M.T. et al. Comparative molecular analysis of testicular Leydig cell tumors demonstrates distinct subsets of neoplasms with aggressive histopathologic features. Mod Pathol 34, 1935–1946 (2021). https://doi.org/10.1038/s41379-021-00845-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00845-3
This article is cited by
-
Familial syndromes associated with testicular and paratesticular neoplasms: a comprehensive review
Virchows Archiv (2024)
-
Molecular assessment of testicular adult granulosa cell tumor demonstrates significant differences when compared to ovarian counterparts
Modern Pathology (2022)
-
Clinicopathologic and molecular spectrum of testicular sex cord-stromal tumors not amenable to specific histopathologic subclassification
Modern Pathology (2022)
