
Abstract
The comprehensive genomic analysis of endometrial carcinoma (EC) by The Cancer Genome Atlas (TCGA) led to the discovery of four distinct and prognostically significant molecular subgroups. Molecular classification has the potential to improve risk-stratification when integrated with clinicopathologic features and has recently been included in national and international patient management EC guidelines. Thus, the adoption of molecular classification into routine pathologic and clinical practice is likely to grow significantly in the upcoming years. Establishing an efficient and standardized workflow for performing molecular classification on ECs, and reporting both the molecular and histologic findings in an integrative manner, is imperative. Here we describe our effort to implement rapid and routine molecular classification on all ECs diagnosed at our institution. To this effect, we performed immunohistochemistry as a surrogate marker for identifying genetic and/or epigenetic alterations in DNA mismatch repair (e.g., MLH1, PMS2, MSH6, MSH2), and TP53 genes. In addition, we have developed and employed a single-gene POLE SNaPshot assay, which is a rapid and analytically sensitive method for detecting select POLE exonuclease domain mutations (EDMs). We report our molecular testing workflow and integrative reporting system as well as the clinicopathologic and molecular features of 310 ECs that underwent routine molecular classification at our institution. The 310 ECs were molecularly classified as follows: 15 (5%) POLE mutant (POLEmut), 79 (25%) mismatch repair-deficient (MMRd), 135 (44%) no specific molecular profile (NSMP), and 81 (26%) p53 abnormal (p53abnl). This work provides an initial framework for implementing routine molecular classification of ECs.
Similar content being viewed by others
Introduction
EC is the most common gynecological cancer in the Western world and continues to increase in incidence1. Traditionally, the histologic subtype and clinicopathologic features such as patient age, tumor grade, stage, and lymphovascular invasion (LVI) have been used as prognostic indicators to direct surgery and adjuvant treatment. However, risk-stratification based on these factors is not entirely reliable and there is considerable interobserver variability in their assessment; thus developing additional strategies to guide surgical management, adjuvant therapy, and surveillance decisions is critical2,3,4,5,6.
Recently, there has been growing evidence that molecular classification of ECs can improve and further individualize risk-stratification. In the 2013 landmark EC study, TCGA identified four molecularly distinct endometrial cancer subgroups, each of which is associated with a different clinical outcome: (1) “ultramutated” tumors harboring exonuclease domain mutations (EDMs) in the DNA polymerase epsilon gene (POLEmut), 2) “hypermutated” tumors characterized by microsatellite instability (MSI-H/MMRd), 3) “copy number low” tumors, which lack TP53 and POLE mutations and are microsatellite stable (also referred to as “no specific molecular profile” or NSMP) and 4) “copy number high” tumors which have high numbers of copy number alterations indicative of chromosomal instability and recurrent TP53 mutations (also referred to as “serous-like” or p53abnl)7. This molecular classification has particular utility in the setting of high-grade EC, which exhibits poor diagnostic interobserver agreement by morphology alone8. Multiple independent retrospective and prospective studies have since demonstrated the molecular reproducibility and prognostic significance of these four subgroups as well as the potential for molecular classification to improve risk-stratification when integrated with clinicopathologic features9,10,11,12,13.
The integration of molecular classification with the clinicopathologic features of ECs is now included in the latest National Comprehensive Cancer Network (NCCN) and the joint European Society of Gynaecological Oncology (ESGO), European Society for Radiotherapy and Oncology (ESTRO), and European Society of Pathology (ESP) guidelines14,15. Prospective trials using an integrative molecular and clinicopathologic risk-stratification approach are underway to help refine management and prevent over- or under-treatment16. Initial results of the prospective PORTEC-3 trial showed the strong prognostic value of molecular classification in high-risk EC regardless of histotype, suggesting that de-escalation of adjuvant treatment should be considered for POLEmut tumors17. At this time, molecular classification has largely been performed in the clinical trial and/or research setting and has yet to be incorporated into routine clinical workflow and practice. Talhouk and colleagues proposed implementation of a step-wise algorithm for clinical testing referred to as the ProMisE (Proactive Molecular Risk Classifier for Endometrial Cancer) classifier9,11. While immunohistochemistry (IHC) is an effective surrogate marker for identifying the majority of aberrancies in DNA mismatch repair proteins (e.g., MLH1, PMS2, MSH2, and MSH6) and p53 in ECs, nucleic acid-based approaches (e.g., next-generation sequencing, Sanger sequencing, SNaPshot) are still required to assess POLE mutational status18,19.
As molecular classification becomes more widely adopted into clinical practice, effective integration of molecular testing into the routine diagnostic workflow for ECs is imperative. Herein we summarize our institutional experience in performing routine prospective molecular classification on all in-house ECs using a POLE hotspot SNaPshot assay and immunohistochemistry for p53 and MMR proteins. We also describe a roadmap for our implementation of this clinically applicable molecular classification and integrative reporting workflow.
Materials and methods
Case selection
Molecular classification was performed on all in-house ECs from June 2019 to March 2021 with available tissue for IHC and molecular testing. The molecular classification scheme utilized is based on the four molecular subgroups identified by TGCA studies: POLE mutant (POLEmut), mismatch repair-deficient (MMRd), p53 abnormal (p53abnl), and no specific molecular profile (NSMP)7. The cases that underwent molecular classification included all patients diagnosed with endometrial carcinoma seen by either a gynecologic oncologist or radiation oncologist at Stanford Medicine. The pre-operative endometrial biopsy, hysterectomy specimen, or secondary recurrent/metastatic disease underwent testing, depending on specimen availability and tumor cellularity.
Immunohistochemistry
IHC for p53, MLH1, PMS2, MSH2, and MSH6 was performed on all cases. A full list of immunohistochemistry reagents and conditions is provided in Suppl. Table 1. Methods of IHC interpretation are described in Suppl. Table 2. Appropriate internal positive controls (i.e., stroma and/or immune cells) were present and evaluated for all IHC stains.
MLH1 promoter hypermethylation
Tumors that exhibited absent nuclear MLH1 and PMS2 staining by IHC underwent MLH1 promoter hypermethylation testing at GoPath Laboratories (Buffalo Grove, IL). Tumors that showed an abrupt subclonal loss of MLH1 and PMS2 immunoreactivity with intact MSH2 and MSH6 expression did not undergo promoter hypermethylation analysis; however, this pattern has been repeatedly shown to be due to somatic/epigenetic inactivation of MLH1 (promoter hypermethylation) and interpreted as such20,21,22.
POLE SNaPshot assay
All cases underwent POLE mutational analysis using a hotspot SnaPshot assay performed on formalin-fixed paraffin-embedded (FFPE) tissue23. The POLE SNaPshot assay was initially conceived and designed in 2018 to cover the most common hotspot positions in published literature indexed in PubMed and publicly available cancer databases (COSMIC (https://cancer.sanger.ac.uk/cosmic; accessed December 2018)) and cBioportal (https://www.cbioportal.org; accessed December 2018). The assay covers the nine most common likely pathogenic and pathogenic POLE variants within exons 9, 11, 13, and 14 of the exonuclease domain, accounting for ~90–95% of all clinically relevant POLE EDMs variants reported in ECs to date7,24,25,26,27,28 (Table 1). See Devereaux et al. for a more detailed description of variant coverage and interpretation of pathogenicity23.
Briefly, the SNaPshot technique involves an initial PCR amplification of the relevant gene target regions, followed by multiplexed single-nucleotide primer extension as previously described29. The POLE gene reference sequence NM_006231.4 from NCBI was used for all primer design. Four primer sets were designed to amplify exons 9, 11, 13, and 14 and 15–20 nucleotide-long extension primers were designed to anneal immediately adjacent to the nucleotide positions of interest. Capillary electrophoresis using an ABI 3500xl: 24-capillary array (Applied Biosystems, Foster City, CA) was then utilized to detect the size of the extension product and the fluorescent signal of the SNaPshot products. The resulting data were analyzed using GeneMapper v5.0 with pre-specified detection parameters. Additional details of the POLE SNaPshot validation and methodology are described in Devereaux et al.23.
The analytic sensitivity (limit of detection) of the assay was conservatively approximated at a 10% variant allele fraction (VAF), with documented detection as low as 5% VAF for most variants23.
Statistics
Comparisons of the association between continuous clinicopathologic variables were performed using a chi-square test. A p value of <0.05 was considered significant.
Results
Implementation of routine and integrative molecular classification
Prior to initiating molecular classification of all ECs at our institution, a pathology workflow was developed to ensure both efficiency and standardization of case testing and integrative reporting. All EC cases first undergo traditional histologic evaluation. Subsequently, the most optimal, well-fixed FFPE tissue block exhibiting the highest tumor cellularity is selected for ancillary studies, including p53 and MMR protein (MLH1, PMS2, MSH2, and MSH6) IHC, which serves as a surrogate marker for alterations in those genes, as well as POLE hotspot mutational analysis by the SnaPshot assay. POLE mutational analysis is routinely performed in the molecular laboratory on a weekly basis and the results are reported directly in the electronic medical record. Based on prior validation studies, the POLE assay was conservatively determined to be able to reliably detect POLE variants down to a 20% tumor cellularity, which corresponds to a VAF of ~10% for a single allele mutation. Of note, molecular testing is still performed on tumors with estimated tumor cellularity of 5–20% given that there is some subjectivity in tumor cellularity estimates and some variants may still be detected below a 10% VAF. However, when the tumor cellularity is estimated to be below 20%, the report includes a disclaimer, warning of the risk of a false-negative result, and recommends a more sensitive molecular testing method (e.g., next-generation sequencing), if clinically indicated. A summary of the IHC and POLE results and the final molecular classification subgroup are integrated into the pathology report.
In the majority of EC cases (95%), the morphology is unambiguous and a diagnostic report is issued based on histologic findings to expedite clinical triage and management. Once POLE testing is complete, an addendum to the initial case report is issued to provide the results of the immunohistochemical and POLE molecular testing as well as assign a molecular classification subgroup. Of note, in select cases, all or a subset of the IHC are reported in the initial primary report when diagnostically indicated or on request; however, these results are then re-summarized in the addendum in the context of the final molecular status (Fig. 1A).

A In the majority of cases, the histotype is unambiguous and the diagnostic report is issued in week 1, and the subsequent results of the IHC and POLE molecular testing are reported in a molecular classification addendum in week 2. If diagnostically indicated or requested, IHC may be reported in the primary report with the findings re-summarized in the addendum in the context of the final molecular classification. B In a minor subset of cases, the morphology is ambiguous and/or is suspicious for an underlying POLE mutation; therefore, the diagnostic report is issued upon completion of testing in the context of the molecular classification during week 2.
Alternatively, a minority of cases (5%) exhibit a high-grade and/or ambiguous morphologic pattern that is diagnostically challenging and raises a broad differential (e.g., serous carcinoma or clear cell carcinoma) that can be further refined with ancillary IHC and/or POLE molecular testing. In these instances, the case is held until IHC and POLE testing is complete, and a diagnostic report is then issued taking into account both the morphologic findings and the molecular context (Fig. 1B). This workflow is designed to improve diagnostic reproducibility given that subset of high-grade ECs is known to have poor interobserver agreement by morphologic assessment alone8,30,31.
Molecular classification for each case is integrated into the pathology report using standardized text and formatting for both the purpose of efficient reporting and reader interpretation. The addendum report includes four parts: (1) a descriptive header referencing the molecularly-defined subgroups identified by TCGA studies, (2) descriptive interpretive comments for the POLE hotspot assay and each of the IHC results, (3) the individual results for each assay, and (4) a final molecular classification designation (e.g., POLEmut, MMRd, NSMP or p53abnl). The POLE molecular testing result comment in the surgical pathology report is derived from the finalized molecular report, which is then referenced for a more detailed description of the assay.
Of note, a small number of tumors may demonstrate more than one molecular feature and are referred to as “multiple-classifier” ECs. For instance, ECs may display MMRd and p53abnl (MMRd-p53abnl), POLEmut and p53abnl (POLEmut-p53abnl), or POLEmut and MMRd (POLEmut-MMRd) aberrations. In a “multiple-classifier” scenario we report a single final molecular classification subgroup for the EC, making note of the secondary classifier in the interpretive comment section. This reporting decision is based on the findings of Leon-Castillo et al., which show that genetic signatures, copy number alterations, and clinical outcome of the MMRd-p53abnl and POLEmut-p53abnl ECs hierarchically cluster with those of single classifier MMRd and POLEmut tumors, respectively, and are significantly different from the single classifier p53abnl tumors. This phenomenon is likely explained by the fact that the p53 classifier in these scenarios is a secondary passenger event and typically subclonal, which also tends to be reflected in the p53 IHC pattern32. Multiple classifier MMRd-POLEmut and POLEmut-MMRd ECs may also be observed, but show different genetic architectures/signatures and clinical outcomes based on whether the loss of POLE and MMR protein function is a primary or secondary event. For instance, ECs harboring pathogenic POLE variants that also demonstrate abnormal MMR IHC patterns typically show subclonal loss of MMR protein(s) and are genetically and biologically more similar to single-classifier POLEmut ECs. In contrast, MMRd tumors with secondary POLE variants typically show complete loss of MMR protein(s) by IHC, harbor nonpathogenic POLE variants, and are genetically and biologically akin to single-classifier MMRd ECs. Example addendum reports for “single classifier” POLEmut and “multiple classifier” MMRd-p53abnl ECs are shown in Supplementary Fig. 1.
Clinicopathologic characteristics of cohort
A total of 315 ECs were diagnosed in-house or as part of a secondary pathology review upon transferring care to our institute between June 2019 to March 2021. Of the 315 ECs, 310 had available tissue to perform both IHC and POLE molecular testing. Pre-operative endometrial biopsies (51%; 159/310) were the most commonly tested specimens, followed by hysterectomy (46%; 142/310) and metastasis or recurrence (3%; 9/310) specimens. The 310 ECs were classified into one of the four molecular subgroups: 15 (5%) POLEmut, 79 (25%) MMRd, 135 (44%) NSMP and 81 (26%) p53abnl. Multiple classifiers were detected in 1.3% (4/310) ECs, including 2 POLEmut-MMRd and 2 MMRd-p53abnl tumors. In all of the “multiple classifier” cases, the secondary alteration was subclonal by IHC. There were significant differences in age, histotype, grade, stage, and LVI among the molecular subgroups. A summary of the clinicopathological characteristics and the molecular classification details are shown in Table 2 and Fig. 2, respectively.

Diagram of the institutional cohort by molecular classification subgroups. All ECs are assessed for aberrancies in p53 and MMR proteins (MLH1, PMS2, MSH2, and MSH6) by IHC and undergo hotspot POLE mutational testing. Based on the molecular features, ECs are assigned to one of the four molecular subgroups as defined by TCGA studies: POLE mutant (POLEmut), mismatch repair-deficient (MMRd), p53 abnormal (p53abnl) and no specific molecular profile (NSMP). MLH1 promoter hypermethylation studies are performed on ECs showing non-subclonal MLH1/PMS2 loss. A small subset of tumors harbor more than on classifying features and are referred to as “multiple-classifier” ECs.
Of note, in 27 of the 310 (8.7%) of the cases that underwent POLE testing, tumor cellularity was estimated to be 5-15%, which is considered below the validated analytic sensitivity of the assay. The majority (81.5%; 22/27) of cases with low tumor cellularity were hysterectomy specimens, which usually contained predominately myometrial tissue whereas 18.5% (5/27) consisted of scant endometrial biopsy specimens. No POLE variants were detected by the SNaPshot assay in the 27 cases exhibiting low tumor cellularity; therefore, they were assigned to either the NSMP (22/27), MMRd (2/27), or p53abnl (3/27) subgroups based on the IHC results.
POLEmut group
Although all ECs routinely underwent MMR and p53 IHC at our institution prior to this molecular classification initiative, this was the first time the POLE status was routinely determined in our practice. A total of 15 ECs harboring pathogenic or likely pathogenic POLE mutations (POLEmut) were identified in this prospective institutional cohort. While POLEmut tumors represented 5% (15/310) of all ECs, they represented 6.4% (14/220) of all endometrioid ECs and 8% (4/49) high-grade endometrioid ECs or ambiguous high-grade carcinomas, not otherwise specified. Eleven tumors were FIGO grade 1–2, three tumors were FIGO grade 3, and one tumor showed features of a high-grade endometrial carcinoma that could not be definitively histotyped. The POLE SNaPshot assay detected the following POLE variants: p.Pro286Arg (c.857C>G; exon 9) (n = 6), p.Val411Leu (c.1231G>C/T; exon 13) (n = 5), p.Ala456Pro (c.1366G>C; exon 14) (n = 2), p.Pro436Arg (c.1307C>G; exon 13) (n = 1) and p.Phe367Ser (c.1100T>C; exon 11) (n = 1). Two of the POLEmut cases exhibited ‘multiple classifier’ features with subclonal loss of MSH6 or both MSH6 and MSH2; however, both were designated POLEmut in the final classification. This POLEmut designation was based on the fact that both ECs harbored known pathogenic POLE variants (p.Val411Leu and Phe367Ser) and showed subclonal loss of the MMR proteins by IHC, which is a feature consistent with a secondary passenger genetic alteration occuring in the setting of a POLE ultramutator phenotype (Fig. 3). Of the 12 POLEmut tumors that had undergone hysterectomy and staging, 75% (9) were early stage (FIGO I or II) and 25% (3) were advanced stage (FIGO III). In all of the FIGO stage III POLEmut tumors lymphovascular space invasion was either considered to be suspicious or definitive; one case was associated with ovarian metastasis (FIGO IIIA) and two cases with pelvic lymph node metastasis (FIGO IIIC1). All eleven cases that underwent hysterectomy with available follow-up were without evidence of disease after 1–20 month interval (median = 14). A summary of the clinicopathologic features of the POLEmut cases is presented in Table 3.

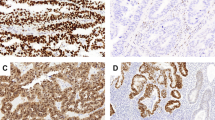
A POLEmut (p.Phe367Ser) EC exhibiting heterogeneous morphologic and immunohistochemical staining patterns, including A high-grade appearing areas with solid growth, numerous tumor infiltrating lymphocytes and B intact, albeit decreased, MSH6 expression as well as more low-grade areas with glandular growth (C) and loss of MSH6 expression by IHC (D). In the setting of a pathogenic POLE mutation resulting in an ultramutator phenotype, subclonal loss of MSH6 by IHC is likely due to a secondary or passenger genetic alteration.
Discussion
With molecular classification now incorporated into NCCN and ESGO/ESTRO/ESP endometrial carcinoma guidelines and the cumulative clinical trial data demonstrating its importance for risk stratification, it is likely that molecular status will increasingly become integrated into gynecologic oncology practice. In anticipation of this shift to a more integrative morphologic and molecular diagnostic approach, we have established and implemented a diagnostic workflow and reporting system and describe our institutional experience with performing routine molecular classification on ECs.
At present, molecular classification of ECs has largely been performed in the clinical trial and retrospective research setting and has yet to be incorporated into routine pathology practice. Determining an efficient, cost-effective, and technically feasible approach is required to stratify ECs into the four molecular subgroups. IHC has proven to be a useful surrogate marker for identifying the majority of loss of function alterations in mismatch repair genes (e.g., MLH1, PMS2, MSH2, and MSH6), and p53 in ECs, and IHC is available in most pathology settings. However, nucleic acid-based approaches are required to assess POLE mutational status and this has created a major barrier to routine identification of these tumors, particularly without employing a next-generation sequencing-based approach, which is both costly and time-consuming, or a Sanger sequencing approach, which has a low analytic sensitivity (20% VAF or >40% tumor cellularity for a single allele mutation). In order to fill this current gap in POLE molecular testing for ECs, commercial and lab-developed single-gene POLE assays that are sensitive, rapid, and cost-effective for detecting clinically relevant pathogenic POLE variants are likely to emerge in upcoming years and play a key role in more widespread adoption of EC molecular classification. At our institution, we developed and validated a novel POLE SNaPshot assay to interrogate pre-selected nucleotide positions within the EDM of POLE in order to perform molecular classification of ECs23.
We performed MMR and p53 immunohistochemistry and POLE mutational testing on all ECs diagnosed at our institute in order to better understand their molecular and clinicopathlogic features. At the time of project initiation molecular classification had not been incorporated into national or international EC guidelines and there was uncertainty regarding billing and insurance coverage of POLE molecular testing. Therefore, the cost of all POLE molecular testing was entirely funded by an institutional pathology departmental grant (value-based care grant) during this investigational and early implementation phase of molecular classification. Since the start of our testing initiative, molecular classification of EC has been included in the NCCN and ESGO/ESTRO/ESP guidelines, which we hope will lead to new and more consistent insurance policies related to the molecular testing of ECs in the future.
Determining an algorithm for molecular testing of ECs is likely to be institution-specific. Talhouk and colleagues developed and validated the ProMisE (Proactive Molecular Risk Classifier in Endometrial cancer) classifier which is based on a step-wise approach to molecular classification testing. The ProMisE classifier starts with an assessment of MMR proteins by IHC to enable prompt referral for hereditary cancer testing if positive. If MMR proteins are intact, POLE EDM mutational analysis is performed and, if negative, p53 IHC is performed9,11. This step-wise approach is cost-effective and enables stratification of ECs into the four group molecular subgroups in the majority of cases; however, POLEmut ECs may be missed if MMR IHC is performed first and shows loss, particularly if the subclonal pattern that may otherwise prompt POLE molecular testing is subtle and not appreciated. In addition, a step-wise testing approach may lead to delays in molecular classification.
Importantly, incorporating molecular testing into the routine clinical workflow has the potential to improve diagnostic reproducibility and prognostication and, therefore, oncologic management. Historically, there has been poor interobserver agreement in diagnosing high-grade ECs using morphologic assessment alone; however, molecular subtyping has the potential to aid in improving diagnostic reproducibility8,30,31. For instance, POLEmut tumors can exhibit ambiguous histomorphologic patterns that morphologically overlap with serous and clear cell carcinomas, but have much less aggressive tumor biology and better prognosis33,34,35. In the course of our study, we encountered high-grade ECs exhibiting an ambiguous morphologic pattern that were MMR proficient and p53 “wild-type” or “non-aberrant” by IHC. In these instances, a diagnostic report was not issued until the POLE mutational status was determined to enable interpretation of morphologic findings in context of the molecular status. Molecular status continues to become increasingly important for predicting prognosis across and within the different EC histotypes, including clear cell carcinomas, dedifferentiated/undifferentiated carcinomas, carcinosarcomas, and endometrioid carcinomas7,33,34,36,37,38.
Within our institutional cohort of 310 cases, the distribution of the different molecular subgroups is similar to that reported in the literature7,9,39,40. Specifically, 15 (5%) POLEmut, 79 (25%) MMRd, 135 (44%) NSMP, and 81 (26%) p53abnl ECs were detected. POLEmut tumors are enriched in and comprise 6.4% (14/220) of the endometrioid ECs and 8% (4/49) high-grade endometrioid ECs or ambiguous high-grade carcinomas, not otherwise specified. Notably, 25% (3/12) of the POLEmut ECs that underwent hysterectomy were high-stage (FIGO IIIA and IIIC). One of the limitations of this cohort is the short follow-up interval precluding any longitudinal analysis; however, we plan to continue to prospectively follow this cohort. Another potential limitation of our current approach is over 8% of our cases (n = 27) had low tumor cellularity (<20%); therefore, molecular testing of these low tumor cellularity cases via a method with a higher analytic sensitivity such NGS would have been beneficial to more definitively ensure a negative POLE status. Alternatively, enriching tumor cells by macrodissection off glass slides or coring blocks may also increase the tumor cellularity. In the future, we will consider implementing tumor macrodissection to enrich the tumor cellularity in these scenarios; however, this becomes a more time-intensive and technically skilled process. Lastly, while our lab-developed POLE assay is quite sensitive for detecting the more common pathogenic POLE variants in ECs, this assay will not detect rarer variants. Notably, one of the cases in our cohort exhibited a subclonal p53 aberrancy, however, no POLE hotspot mutations were detected by the SNaPshot assay and MMR proteins were intact by IHC. It is possible that this EC harbors a rare POLE mutation not detected by the assay. Alternatively, the possibility of a genetic alteration in one of the MMR proteins that still produce a wild-type immunoreactivity pattern by IHC cannot be excluded. Based on our experience, it is evident that there will be occasional ECs that could benefit from more extensive molecular characterization by next-generation sequencing to more firmly establish the ultimate molecular classification.
As molecular classification is performed more routinely, both efficient and effective integration of the molecular status with the clinicopathologic features becomes critical for oncologic management. At our institution, the molecular classification is reported within two weeks, which is a relatively short timeframe that has not delayed patient care. Since the implementation of molecular classification, the molecular subgroup has been incorporated into tumor board presentations and discussions. Currently, the multi-institutional PORTEC-4a trial is underway to evaluate the role of molecular status in determining the adjuvant treatment of ECs by comparing standard approaches versus individualized treatment based on the molecular risk profile of a patient’s tumor. Results of the PORTEC-4a trial are anticipated to show whether de-escalating treatment in cases with a favorable molecular profile is safe and cost-effective. Although gynecologic oncologists are still awaiting the PORTEC-4a outcome data and the publication of new recommendations and guidelines to change oncologic management, knowing the molecular status of ECs has still been valuable for the gynecologic and radiation oncologists at our institution. For instance, having the molecular status can help to resolve otherwise morphologically ambiguous high-grade tumors. In addition, assigning surveillance alone without adjuvant therapy is much more reassuring in the setting of a stage IA grade 2 endometrioid EC of the POLEmut subgroup, which is well-established to have an excellent prognosis, compared to a stage IA grade 2 endometrioid EC of the p53abnl subgroup. Lastly, the majority of gynecologic and radiation oncologists find that our standardized method of reporting molecular classification in an addendum is effective for communicating testing results. Consideration may be given to issuing a second, independent molecular classification report rather than an addendum report depending on the availability of electronic medical record settings and alerts, in order to avoid missing the information contained in a molecular addendum.
While a simplified, rapid molecular testing approach such as the one described herein may suffice in many instances, more extensive molecular testing by next-generation sequencing is still valuable and complimentary. For instance, a broader NGS gene panel that molecularly confirms the IHC findings determines additional genetic alterations, microsatellite instability, and/or tumor mutation burden (TMB) may be required for clinical trials, especially in the setting of metastatic or recurrent disease. While MMR and p53 IHC are excellent surrogate markers for molecular alterations in the MMR (MLH1, PMS2, MSH2, and MSH6) and TP53 genes, there is a small subset of cases that will be missed by IHC alone. For instance, some genetic alterations disrupt protein function, yet still result in a wild-type immunoreactive pattern by IHC41,42. In addition, NGS enables the detection of more POLE variants and their further characterization. For rare POLE variants, pathogenicity is best determined in the context of a high TMB (100 mutations/megabase) and the genomic architecture or signatures (e.g., COSMIC mutational signature 10, which exhibits strand bias for C>A mutations in TpCpT and T>G mutations in TpTpT contexts, is associated with pathogenic POLE variants)43. It remains to be seen whether single-gene POLE sequencing efforts will prevail or if we are moving toward targeted NGS panels for all or certain subsets of EC. In truth, this will largely be determined by turn-around requirements, cost, and insurance reimbursements.
In summary, we report our institutional experience performing routine molecular classification on all in-house ECs at our institute and implementing a clinically applicable molecular classification and integrative reporting workflow. To our knowledge, this is the first prospective implementation of molecular classification of ECs at diagnosis independent of a clinical trial.
References
Morice, P., Leary, A., Creutzberg, C., Abu-Rustum, N. & Darai, E. Endometrial cancer. Lancet 387, 1094–1108 (2016).
Creutzberg, C. L. et al. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicentre randomised trial. PORTEC Study Group. Post Operative Radiation Therapy in Endometrial Carcinoma. Lancet 355, 1404–1411 (2000).
Keys, H. M. et al. A phase III trial of surgery with or without adjunctive external pelvic radiation therapy in intermediate risk endometrial adenocarcinoma: a Gynecologic Oncology Group study. Gynecol. Oncol. 92, 744–751 (2004).
Creasman, W. T. et al. Surgical pathologic spread patterns of endometrial cancer. A Gynecologic Oncology Group Study. Cancer 60, 2035–2041 (1987).
Morrow, C. P. et al. Relationship between surgical-pathological risk factors and outcome in clinical stage I and II carcinoma of the endometrium: a Gynecologic Oncology Group study. Gynecol. Oncol. 40, 55–65 (1991).
Bendifallah, S. et al. Just how accurate are the major risk stratification systems for early-stage endometrial cancer? Br. J. Cancer 112, 793–801 (2015).
Cancer Genome Atlas Research, N., Kandoth, C. et al. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013).
Gilks, C. B., Oliva, E. & Soslow, R. A. Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. Am. J. Surg. Pathol. 37, 874–881 (2013).
Talhouk, A. et al. A clinically applicable molecular-based classification for endometrial cancers. Br. J. Cancer 113, 299–310 (2015).
Talhouk, A. & McAlpine, J. N. New classification of endometrial cancers: the development and potential applications of genomic-based classification in research and clinical care. Gynecol. Oncol. Res. Pract. 3, 14 (2016).
Talhouk, A. et al. Confirmation of ProMisE: a simple, genomics-based clinical classifier for endometrial cancer. Cancer 123, 802–813 (2017).
Stelloo, E. et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 28, 836–844 (2015).
Talhouk, A. et al. Molecular classification of endometrial carcinoma on diagnostic specimens is highly concordant with final hysterectomy: earlier prognostic information to guide treatment. Gynecol. Oncol. 143, 46–53 (2016).
Concin, N., et al. ESGO/ESTRO/ESP Guidelines for the management of patients with endometrial carcinoma. Virchows Arch https://doi.org/10.1007/s00428-020-03007-z (2021).
Network, N. C. C. Uterine Neoplasms (Version 1.2021), https://www.nccn.org/professionals/physician_gls/pdf/uterine.pdf Accessed March, 2021.
PORTEC-4a. Randomised trial of standard of molecular profile-based recommendation for radiotherapy after surgery for women with early-stage endometrial cancer. https:www.isrctn.com/ISRCTN1165025.
Leon-Castillo, A. et al. Molecular classification of the PORTEC-3 trial for high-risk endometrial cancer: impact on prognosis and benefit from adjuvant therapy. J. Clin. Oncol. 38, 3388–3397 (2020).
Stelloo, E. et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann. Oncol. 28, 96–102 (2017).
Singh, N. et al. p53 immunohistochemistry is an accurate surrogate for TP53 mutational analysis in endometrial carcinoma biopsies. J. Pathol. 250, 336–345 (2020).
Watkins, J. C., Nucci, M. R., Ritterhouse, L. L., Howitt, B. E. & Sholl, L. M. Unusual mismatch repair immunohistochemical patterns in endometrial carcinoma. Am. J. Surg. Pathol. 40, 909–916 (2016).
Pai, R. K. et al. Abrupt loss of MLH1 and PMS2 expression in endometrial carcinoma: molecular and morphologic analysis of 6 cases. Am. J. Surg. Pathol. 39, 993–999 (2015).
Grady, W. M., Rajput, A., Lutterbaugh, J. D. & Markowitz, S. D. Detection of aberrantly methylated hMLH1 promoter DNA in the serum of patients with microsatellite unstable colon cancer. Cancer Res. 61, 900–902 (2001).
Devereaux, K. A., et al. A Multiplex SNaPshot assay is a rapid and cost-effective method for detecting POLE mutations in endometrial carcinoma. Int. J. Gynecol. Pathol. In press (2021).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Campbell, B. B. et al. Comprehensive analysis of hypermutation in human. Cancer Cell 171, 1042–1056 e1010 (2017).
Church, D. N. et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J. Natl Cancer Inst. 107, 402 (2015).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Dias-Santagata, D. et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol. Med. 2, 146–158 (2010).
Lomo, L. et al. Histologic and immunohistochemical decision-making in endometrial adenocarcinoma. Mod. Pathol. 21, 937–942 (2008).
Hussein, Y. R. et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Mod. Pathol. 28, 505–514 (2015).
Leon-Castillo, A. et al. Clinicopathological and molecular characterisation of ‘multiple-classifier’ endometrial carcinomas. J. Pathol. 250, 312–322 (2020).
Bakhsh, S. et al. Histopathological features of endometrial carcinomas associated with POLE mutations: implications for decisions about adjuvant therapy. Histopathology 68, 916–924 (2016).
Conlon, N. et al. Endometrial carcinomas with a “serous” component in young women are enriched for DNA mismatch repair deficiency, lynch syndrome, and POLE exonuclease domain mutations. Am. J. Surg. Pathol. 44, 641–648 (2020).
Hoang, L. N. et al. Interobserver agreement in endometrial carcinoma histotype diagnosis varies depending on The Cancer Genome Atlas (TCGA)-based Molecular Subgroup. Am. J. Surg. Pathol. 41, 245–252 (2017).
DeLair, D. F. et al. The genetic landscape of endometrial clear cell carcinomas. J. Pathol. 243, 230–241 (2017).
Segura, S. E. et al. DNA mismatch repair-deficient endometrial carcinosarcomas portend distinct clinical, morphologic, and molecular features compared with traditional carcinosarcomas. Am. J. Surg. Pathol. 44, 1573–1579 (2020).
Espinosa, I., Lee, C. H., D’Angelo, E., Palacios, J. & Prat, J. Undifferentiated and dedifferentiated endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Am. J. Surg. Pathol. 41, 1121–1128 (2017).
McConechy, M. K. et al. Endometrial carcinomas with POLE exonuclease domain mutations have a favorable prognosis. Clin. Cancer Res. 22, 2865–2873 (2016).
Kommoss, S. et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 29, 1180–1188 (2018).
Hechtman, J. F. et al. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated with missense mutations in mismatch repair genes. Mod. Pathol. 33, 871–879 (2020).
Kobel, M. et al. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J. Pathol. Clin. Res. 2, 247–258 (2016).
Leon-Castillo, A. et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J. Pathol. 250, 323–335 (2020).
Funding
This study was funded in part by a Value-Based Care Award funded by the Pathology Department at Stanford University School of Medicine. Department of Pathology Value-Based Care Award and Gynecologic Pathology Division Funds, Stanford University School of Medicine.
Author information
Authors and Affiliations
Contributions
K.A.D., T.A.L. and B.E.H. conceived the work. All authors provided patient data. K.A.D., J.J.W., J.P. and B.E.H. analyzed the data. V.C. performed the statistical analysis. K.A.D. and B.E.H. drafted the paper. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was conducted under the approval of the Institutional Review Board at Stanford University School of Medicine in Stanford, CA, USA.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Devereaux, K.A., Weiel, J.J., Pors, J. et al. Prospective molecular classification of endometrial carcinomas: institutional implementation, practice, and clinical experience. Mod Pathol 35, 688–696 (2022). https://doi.org/10.1038/s41379-021-00963-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00963-y
This article is cited by
-
The P286R mutation of DNA polymerase ε activates cancer-cell-intrinsic immunity and suppresses endometrial tumorigenesis via the cGAS-STING pathway
Cell Death & Disease (2024)
-
Molecular profile in endometrial carcinoma: can we predict the lymph node status? A systematic review and meta-analysis
Clinical and Translational Oncology (2024)
-
Alteration in molecular properties during establishment and passaging of endometrial carcinoma patient-derived xenografts
Scientific Reports (2023)
