
Abstract
Xanthogranulomatous epithelial tumor (XGET) and keratin-positive giant cell-rich soft tissue tumor with HMGA2-NCOR2 fusion (KPGCT) are two recently described neoplasms with both distinct and overlapping clinical and histopathologic features. We hypothesized that XGET and KPGCT may be related and represent a histologic spectrum of a single entity. To test this, we sought to characterize the clinical, radiographic, immunohistochemical, ultrastructural and molecular features of additional tumors with features of XGET and/or KPGCT, which we refer to descriptively as keratin-positive xanthogranulomatous/giant cell-rich tumors (KPXG/GCT). The archives were searched for potential cases of KPXG/GCT. Clinical and imaging features were noted. Slides were assessed for histologic and immunohistochemical findings. Ultrastructural and next generation RNA sequencing-based analysis were also performed. Nine cases were identified arising in seven women and two men [median age of 33 years (range: 12–87)]. Median tumor size was 4 cm (range: 2.4–14.0 cm) and tumors presented in the thigh (2), buttock (1), forearm (2), groin (1), cranial fossa (1), ilium (1), and tibia (1). Morphologically, tumors were most frequently characterized by a fibrous capsule, with associated lymphoid reaction, enclosing a polymorphous proliferation of histiocytes, giant cells (Touton and osteoclast-types), mixed inflammatory infiltrate, hemorrhage and hemosiderin deposition, which imparted a variably xanthogranulomatous to giant cell tumor-like appearance. One case clearly showed mononuclear cells with eosinophilic cytoplasm characteristic of XGET. All cases expressed keratin and 7 of 9 were found to harbor HMGA2-NCOR2 fusions including cases with xanthogranulomatous appearance. One patient developed local recurrence and multifocal pulmonary lesions, which were radiographically suspicious for metastases. Shared clinical, histologic and immunohistochemical features, and the shared presence of HMGA2-NCOR2 fusions supports interpretation of KPXG/GCT as a single entity which includes XGET and KPGCT. Given limited clinical follow-up to date and rare cases with apparently aggressive findings, we provisionally regard these tumors as having uncertain biologic potential.
Similar content being viewed by others
Introduction
Xanthogranulomatous epithelial tumor (XGET)1 and keratin-positive giant cell-rich soft tissue tumors with HMGA2-NCOR2 fusion (KPGCT)2 are two recently described mesenchymal lesions. XGET presented most commonly in the superficial soft tissue or bone of young women1. These tumors were characterized by a fibrous capsule with associated lymphoid aggregates enclosing sheets of foamy histiocytes, Touton-type giant cells, osteoclast-type giant cells and admixed chronic inflammatory cells. Scattered keratin-positive mononuclear cells with distinctly eosinophilic cytoplasm were present, and gave the tumor its name. Tumors followed an indolent course though available follow-up was limited.
KPGCT were reported to present exclusively in superficial soft tissue with a predilection for young women2. Tumors were composed of sheets of mononuclear cells with admixed evenly distributed osteoclast-type giant cells and variably lobulated architecture. The periphery of the lobules were demarcated by fibrous bands and lymphoid aggregates. Stromal hemorrhage and hemosiderin deposition were also noted. Keratin expression and recurrent HMGA2-NCOR2 fusions were characteristic features. Indolent behavior was observed.
While some of the previously described features were unique to XGET and KPGCT, respectively, (i.e., xanthogranulomatous appearance and mononuclear cells with bright eosinophilic cytoplasm versus sheets of mononuclear cells with evenly distributed osteoclast-type giant cells and HMGA2-NCOR2 fusions), other microscopic, clinical and immunohistochemical features showed significant overlap. Both lesions were reported to contain fibrous bands/capsule with lymphoid aggregates, chronic inflammatory infiltrates and osteoclast-type giant cells. Further, the predominance of tumors arising in the superficial soft tissue of young women and expression of keratin in a subset of cells with variable highlighting of slender cytoplasmic processes were similar.
Given the overlapping features of reported cases, we hypothesized that XGET and KPGCT may represent different ends of the histologic spectrum of a single entity. To test this theory, we sought to characterize the clinical, radiographic, immunohistochemical, ultrastructural and molecular features of additional tumors with features of XGET and/or KPGCT, which we refer to descriptively as keratin-positive xanthogranulomatous/giant cell-rich tumors (KPXG/GCT).
Material and methods
Case selection, clinicopathologic evaluation, and radiographic interpretation
This study was approved by both institutional review boards. Two cases were identified during routine sign out, including the index case. Further, the pathology departmental archives of the Lauren V. Ackerman Laboratory at Washington University in St. Louis were search from 1988 to 2020 for cases of “juvenile xanthogranuloma,” “deep fibrous histiocytoma,” “reticulohistiocytoma” and “giant cell tumor of soft tissue” as well as keywords “xanthogranulomatous,” “Touton,” “giant cell rich,” “fibrohistiocytic” and “giant cell containing.” Two thousand nine hundred and two search results were screened and 310 cases were reviewed by two of the authors (CAD and JSAC) including ~220 cases previously diagnosed as juvenile xanthogranuloma and nine cases of “fibrous histiocytoma” or “xanthoma” of bone. Approximately 10% of reviewed potential cases originated in the bone, while the remainder arose in soft tissue. In addition, the molecular archive of the Department of Medicine and Pathobiology at Mount Sinai Hospital, University of Toronto, was searched for additional cases with HMGA2-NCOR2 fusions. Clinical data was retrieved from the electronic medical record and/or requested from referring physicians. Follow-up duration was calculated from pathologic diagnosis to last follow-up. Available slides, including previously performed immunohistochemical studies, were assessed for pathologic features and available imaging studies were reviewed by a musculoskeletal radiologist with expertise in oncologic imaging (JCB). Although initial attempts were made to classify tumors as XGET or KPGCT based on histologic features, the extent of overlapping features in our data set was such that the distinction was essentially arbitrary in several cases, and the decision was ultimately made to fully characterize all tumors under one umbrella to avoid artificial up front distinctions.
Immunohistochemical studies
Immunohistochemical studies were performed on 5 µm-thick formalin-fixed paraffin-embedded whole-tissue sections. The following immunohistochemical studies were performed using prediluted Ventana antibodies (per standard protocol): CD31 (JC70), SMA (1A4), keratin 5/6 (D5/16B4), keratin (AE1/AE3/PCK26), keratin 7 (SP52), anti-keratin (34betaE12), desmin (DE-R-11), INI-1 (MRQ27), GFAP (EP672Y), and S100 protein (polyclonal).
Electron microscopy
Formalin-fixed paraffin-embedded tissue was deparaffinized in xylenes, rehydrated through graded ethanol to 0.1M sodium cacodylate buffer then post-fixed in 2% osmium tetroxide/0.1M sodium cacodylate buffer for 1 h, dehydrated in graded ethanol to propylene oxide then infiltrated and embedded in PolyBed 812 (catalog# 08792-1; Polysciences, Hatfield, PA). Tissue blocks were sectioned at ninety nanometers, post stained first with 3% uranyl acetate and then Venable’s lead citrate and viewed with a JEOL model 1200EX electron microscope (JEOL, Tokyo, Japan). Digital images were acquired using the AMT Advantage HR (Advanced Microscopy Technology, Danvers MA) high-definition CCD, 1.3-megapixel TEM camera.
Next generation sequencing
Scrolls obtained from formalin-fixed paraffin-embedded tissue blocks were cut (4 at 10 microns) into Eppendorf tubes. RNA was extracted using ExpressArt FFPE Clear RNA Ready kits (Amsbio, Cambridge, MA). RNA-seq libraries were prepared with the TruSight RNA Fusion Panel (Illumina, San Diego, CA), as previously described3. Initial fusion gene analysis was performed using Illumina’s Local Run Manager (v. 1.3.0), which performs alignment using the STAR aligner and detects fusion using the Manta fusion caller. Additionally the run was analyzed using the fusion detection algorithm JAFFA (v.1.1), which performs alignment using BOWTIE2. The results from both algorithms were collated by Fusion Reporter (v.1.0). Subsequently, the DRAGEN RNA app, version 3.8.4, within the cloud-based BaseSpace genomics suite (Illumina, Inc. San Diego, CA, USA) was utilized to call fusions and generate raw counts for each of the target genes. Potential fusion locations were also manually reviewed using the Integrative Genomics Viewer (IGV), version 2.2.12 (Broad Institute)4,5,6.
Results
Clinical features
Seven tumors occurred in women and two in men with a median age of 33 years (range: 12–87) (Table 1). Median tumor size was 4 cm (range: 2.4–14.0 cm) and tumors presented in the thigh (2), buttock (1), forearm (2), groin (1), cranial fossa (1), ilium (1) and tibia (1). Five of six soft tissue tumors were centered in the subcutis. In two patients (Cases 1 and 2) the initial clinical impression was benign cyst with associated pain; Case 1 waxed and waned in size for years, while Case 2 clinically increased from 1 to 4 cm in 2 months. Three additional patients (Cases 3, 6, and 8) presented with rapidly enlarging masses over the course of weeks to months.
The two bone tumors presented as lytic lesions. Case 4 presented with tibial bone loss associated with a failed prosthesis and complicated by a periprosthetic fracture in a patient with history of total knee replacement for arthritis; this led to an initial clinical impression of particle disease. Case 5 presented with pain and a pathologic fracture through a lesion in the ilium. Case 7, while presenting in soft tissue, was suspected to be a local recurrence of a reported “benign tumor,” which was removed from the “left hip bone” 13 years prior. Unfortunately, the previous pathology was not available for review.
Radiological features
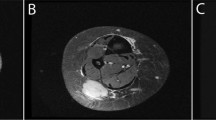
Four cases had preoperative imaging studies of the primary tumor available for review. Two of the cases had soft tissue masses (Cases 1 and 2, Fig. 1) and two had bone lesions (Cases 4 and 5, Fig. 2). Both soft tissue masses arose in the extremities (one in the medial thigh and one in the forearm) and were centered in the subcutaneous fat, extending from just below the skin surface nearly to the fascia. Both were assessed initially with ultrasound. Grayscale images of both lesions revealed a solid mass with hypoechoic, heterogeneous echotexture, slightly lobulated margins and vertical growth pattern. Color, power and pulsed Doppler interrogation showed both masses to have abundant internal vascularity with easily identifiable arterial waveforms. Case 2 underwent two ultrasounds 3 months apart and the mass increased substantially in size over this short duration from 1.3 × 1.0 × 1.1 cm to 2.6 × 1.5 × 2.3 cm—an approximately six-fold increase in estimated volume. Case 2 also underwent contrast-enhanced magnetic resonance imaging (MRI) shortly after the second ultrasound. On MRI, the mass was slightly hyperintense to skeletal muscle on T1-weighted sequences and hyperintense to muscle on T2-weighted, fat-suppressed sequences. There was reduced diffusion in the mass on diffusion-weighted images. After the intravenous injection of gadolinium contrast, the mass showed avid, uniform enhancement on the postcontrast sequences. Both masses were considered to represent solid, indeterminate lesions based on ultrasound and MRI, with the differential diagnosis including both benign and malignant entities, including sarcoma.

A–C Case 1. A Gross photograph showing a well-circumscribed subcutaneous tumor with yellow cut surface. B Grayscale sonogram longitudinal to the mass in the coronal plane shows a hypoechoic solid mass (arrow) centered in the subcutaneous fat, extending from the skin surface in a vertical growth pattern to the abut the fascia of the adductor muscle group (M). C Color Doppler image of the lesion in the transverse plane depicts extensive vascularity of the mass. D, E Case 2. D Axial T1-weighted MR image shows a mass that is slightly hyperintense to muscle and centered in the subcutaneous fat, separated from the deep peripheral fascia by a thin fat plane (arrow). Radius (R) and ulna (U) are labeled for orientation. E Axial T2-weighted, fat-suppressed MR image shows the lesion to be hyperintense to muscle with central areas of fluid signal intensity.

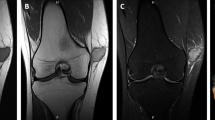
A, B Case 4. A AP and B lateral radiographs of the splinted right knee show an ill-defined lytic lesion (arrows) in the proximal tibia adjacent to the tibial component of a knee arthroplasty. Cortical stepoff (arrow in B) from the fracture is apparent on the lateral view. C–G Case 5. C Axial CT image of the pelvis with bone window obtained at initial presentation shows a lytic lesion in the right iliac wing with permeation of the outer table (arrow) and complete cortical destruction of the inner table. D Axial CT image with soft tissue window depicts heterogeneous attenuation within the lesion. E Coronal short tau inversion recovery (STIR) and (F) coronal T1-weighted MR images obtained following denosumab therapy. These show extensive interval expansion of the bone lesion, which now occupies the majority of the iliac bone and gives rise to a large soft tissue mass both medial and lateral to the bone. G AP pelvic radiograph obtained simultaneous to the MRI demonstrates near-complete erosion of the right iliac wing by a lytic, expansile bone lesion that thins the cortex and contains no discernible matrix.
Both bone lesions presented with pathologic fractures. Case 4 had a pathologic fracture of the proximal right tibia through an expansile, lytic lesion. Because the only images of this case consisted of AP and lateral radiographs obtained after casting, additional radiographic details are obscured by the cast. Of note, this lytic lesion abutted the tibial stem of a knee arthroplasty, and the lytic lesion was radiographically thought to represent osteolysis from particle disease. Case 5 presented with a lytic, expansile mass in the right iliac wing with pathologic fracture. This lesion had aggressive imaging features including expansion of the iliac bone, areas of cortical permeation and frank destruction of the inner table, and a heterogeneous soft tissue mass extending into the pelvis. The radiologic differential diagnosis included metastasis, hematologic malignancy and primary bone neoplasm. From an imaging standpoint, the mass did not originally involve an epiphyseal equivalent or extend to the articular surface of the hip, so giant cell tumor of bone was not considered in the radiologic differential. MRI and radiographs obtained 18 months after the initial CT to assess treatment response to denosumab showed marked enlargement of the bone lesion and an enormous soft tissue mass extending into the pelvis and gluteal muscles. The mass had lytic, expansile features with no internal matrix on CT or radiographs, and heterogeneously hyperintense material on T1-weighted and fluid-sensitive sequences.
Gross examination
Tumors were noted to be tan, yellow (Fig. 1), brown and/or maroon. Three cases were described as homogenous, 2 as lobular or nodular and 1 as encapsulated.
Initial pathologic diagnoses and histologic features
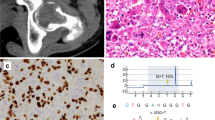
Initial pathologic diagnoses or impressions of referring pathologists included xanthogranulomatous epithelial tumor (1), juvenile xanthogranuloma (1), giant cell-rich neoplasm favor xanthogranuloma (1), benign fibrous histiocytoma (2), fibrohistiocytic process (1), tenosynovial giant cell tumor (1), giant cell tumor of soft tissue (1) and giant cell tumor of bone (1) (Table 2). The overall histologic impression varied from xanthogranulomatous (e.g., Cases 1 and 5) to mixed xanthogranulomatous and giant cell tumor-like (e.g., Case 3) to giant cell tumor-like (e.g., Cases 6 and 7) (Fig. 3). All six soft tissue lesions with evaluable periphery showed a fibrous capsule with associated variable peripheral lymphoid reaction (Fig. 4). Both skeletal lesions and the cranial floor tumor were fragmented to the point that assessment of a capsule was not possible, however one osseous case showed fibrous bands with lymphoid aggregates. Lobular or nodular architecture was seen in 6 tumors, while absent in 1 case and unknown in 2 cases. Necrosis, which appeared to be infarct-type, was present in 7/9 cases, and varied from focal to extensive. Because both osseous lesions presented with pathologic fractures, it was unclear if the necrosis present in these cases was entirely secondary to fracture, or if a component was due to tumor biology.

A Sheets of foamy histiocytes and scattered Touton-type giant cells impart a xanthogranulomatous appearance. A chronic inflammation infiltrate is prominent and scattered osteoclast-type giant cells are also present (Case 5). Photomicrographs of Case 3 show a tumor with both xanthogranulomatous (B) and giant cell-rich (C) areas. D Evenly distributed osteoclast-type giant cells set among mononuclear cells in Case 6 is histologically indistinguishable from conventional giant cell tumor of soft tissue in this field.

A Fibrous capsule with associated lymphoid reaction. B Nodular architecture with fibrous bands with associated lymphoid reaction. C Necrosis with zonal pattern consistent with infarct-type necrosis. D Hemosiderin deposition and mixed inflammatory infiltrate including eosinophils. E Area of storiform growth of spindle cells reminiscent of fibrous histiocytoma. F Atypical mitotic figure (white arrowhead). Also note mildly atypical mononuclear cell with increased nuclear to cytoplasmic ratio and bright eosinophilic cytoplasm (black arrowhead).
The median mitotic count was 6/10 hpf (range 1–7/10); an atypical mitotic figure was identified in single tumor (Case 5). Mononuclear cells with bright eosinophilic cytoplasm were clearly identified in Case 1 (Fig. 5). Similar cells were seen in other cases, particularly Case 5, however the cells were not as clearly identifiable. Osteoclast-type giant cells were present in all cases, and Touton-type giant cells were noted in 5 cases and varied from numerous to rare. Foamy histiocytes and a chronic inflammatory infiltrate were at least focally present in all cases. Scattered eosinophils were frequently observed and a subset of cases showed patchy neutrophilic infiltrates. Stromal hemorrhage (6/9) and hemosiderin deposition (6/9) were frequently seen, while metaplastic bone was uniformly absent. Areas with spindle cell morphology, and loose storiform and short fascicular architecture (benign fibrous histiocytoma-like) were noted in a subset of cases and Case 7 showed small mononuclear cells with epithelioid to plasmacytoid morphology and lymphovascular invasion (Fig. 6).

A Fibrous capsule with associated lymphoid aggregates enclosing a polymorphous cellular proliferation including foamy histiocytes. B Touton-type giant cells. Intermediate (C) and high (D) power photomicrographs of eosinophilic mononuclear cells.

A Case 2 shows histologic findings and a pattern of keratin AE1/AE3/PCK26 (inset) indistinguishable from cases in which HMGA2-NCOR2 fusions were detected. B Case 7 is characterized by sheets of small epithelioid to plasmacytoid mononuclear cells and numerous osteoclast-type giant cells. C In addition lymphovascular space invasion is noted and keratin AE1/AE3/PCK26 highlights many mononuclear cells with variable accentuation of the cytoplasmic periphery (D).
Immunohistochemical studies
In all cases there was a population of mononuclear cells which expressed keratin [AE1/AE3 (2) or AE1/AE3/PCK26 (7)] (Fig. 7); keratin highlighted long dendritic-like processes in a subset of cells, though the prominence of this finding was variable. In Case 7 accentuation of the cytoplasmic periphery was noted. Seven cases had available material for standardized testing. These 7 cases were positive for keratin AE1/AE3/PCK26 and 6 were positive for keratin 7. One case showed focal staining with keratin 5/6 and 4 cases showed scattered or rare cells highlighted by keratin 34βE12. S100 protein expression was seen in all 7 cases and was most frequently patchy. The morphology of the S100 protein-positive cells was frequently clearly dendritic however scattered rounded cells were also highlighted. SMA showed patchy positivity in the majority of cases (6/7), while desmin was negative (0/7). INI-1 was retained in all 7 tested cases and GFAP was negative (0/7). A variable combination of other immunohistochemical studies were performed as part of routine pathologic evaluation: CD68 (3), CD163 (2), CD1a (2), factor XIIIa (2) and myogenin (1). CD68 highlighted numerous histiocytes and also stained the giant cells. CD163 showed a similar pattern in 1 case but did not highlight giant cells in a second case. Scattered dendritic cells were highlighted with CD1a and numerous scattered cells were positive for factor XIIIa. Myogenin was negative.

Keratin AE1/AE3/PCK26 (A) and keratin 7 (B) are positive and variably highlight slender dendritic-like cytoplasmic processes. Focal expression of keratin 5/6 (C) and rare cells highlighted by keratin 34βE12 (D). E CD68 highlights numerous histiocytes and giant cells. CD163 highlights numerous histiocytes and is also positive in giant cells in one tumor (F) while negative in another (G).
Electron microscopy
Ultrastructural analysis was performed on cases 1–6 and showed the presence of thin organized intermediate filaments within the cytoplasm of at least rare cells in all 6 cases (Fig. 8). Basement membrane associated with scattered mononuclear cells with filaments was noted. A single cell with hemidesmosome-like structures was seen.

A Mononuclear cell in Case 1 filled with cytoplasmic filaments in parallel alignment and a basement membrane (arrowhead). B Mononuclear cell in Case 2 filled with similar cytoplasmic filaments.
Molecular findings
HMGA2-NCOR2 fusion was detected in 7/9 KPXG/GCT. The breakpoint for HMGA2 occurred at the 3′-end of exon 3 in 6 of 7 cases and the 3′-end of exon 2 in a single case. All had a breakpoint at the 5′-end of exon 16 of NCOR2. A single keratin-negative giant cell tumor of soft tissue with metaplastic bone formation was also sequenced and a fusion was not detected. Sequencing coverage of HMGA2 was poor, with maximal read depth of <100 (generally <50) for exons 2 and 3 in most cases, and identification of NCOR2 as the partner in short reads was complicated by the presence of tandem CTG repeats within exon 16, necessitating bioinformatics analysis through multiple pipelines and/or manual review in IGV to detect. One of the two cases in which a fusion was not detected had minimal coverage of HMGA2 with read depth <10 for exons 2 and 3, while the other had similar coverage as cases in which a fusion was detected.
Clinical follow-up
Six cases underwent simple excision. Two patients had excisional biopsy or curettage followed by resection/excision and 1 case was debrided. In most cases, margin status could not be determined. Seven cases had available follow-up, ranging from 1 week to 236 months (median: 13 months). Six cases showed no evidence of disease at last follow-up. Case 5, initially diagnosed as benign fibrous histiocytoma, was treated with denosumab. Serial pelvic MRI after 2 and 4 cycles of denosumab showed progressive disease, and the tumor was resected 6 months after initial curettage. The excision specimen showed increased areas of fibrosis associated with rare giant cells, compared to the curettage specimen. While this finding was reminiscent of denosumab treatment effect7,8, similar areas were focally present in the pre-denosumab specimen, ossification was not identified and the tumor was clearly enlarging during denosumab treatment. Radiographs at last follow-up showed no evidence of recurrence (7 months after excision).
Case 7, which potentially represented a soft tissue local recurrence of a bone tumor resected 13 years previously, was resected with positive margins. Fifteen months later, the patient presented with hip pain at the same site and was found to have a left femoral neck fracture subsequently treated with hemiarthroplasty; tumor was not identified at that time. Two years after this, the patient presented with local recurrence and multifocal lung lesions radiographically suspicious for metastatic disease, although no diagnostic biopsies had been performed at the time of last follow-up.
Discussion
Recently, Fritchie et al. described six cases of an unusual lesion of superficial soft tissue and bone with extensive xanthogranulomatous inflammation and keratin expression which they termed “xanthogranulomatous epithelial tumor”1. Shortly afterward, Agaimy et al. described 6 neoplasms of superficial soft tissue with significant morphologic overlap with giant cell tumor of soft tissue, keratin expression and the presence of HMGA2-NCOR2 fusions, for which the author proposed the term “keratin-positive giant cell-rich soft tissue tumors with HMGA2-NCOR2 fusion”2. We sought to describe the clinical, imaging, histopathologic, ultrastructural and molecular features of additional cases. Further, while both XGET and KPGCT were convincingly shown to be distinct from other well-characterized xanthogranulomatous, giant cell-rich, so-called fibrohistiocytic and keratin expressing lesions1,2, we hypothesized that XGET and KPGCT may represent morphologic variants of a single entity.
In our series, KPXG/GCT demonstrated a strong young female predominance (7 F:2 M, median age 33 years), and occurred most frequently in superficial soft tissues (5/9) and bone (at least 2/9), although involvement of deep soft tissue could be present. Among lesions presenting in soft tissue, the extremities were involved in 4/7 cases and rapid growth was reported in more than half. The demographic features and clinical presentation of our cohort were similar to those previously described1,2 with the exception of deep soft tissue sites.
Histologically, common findings included lobular or nodular architecture, osteoclast-type giant cells, stromal hemorrhage, hemosiderin deposition and a chronic inflammatory infiltrate. Of note, the cellular composition of our cases, showed a spectrum of patterns which ranged from extensively xanthogranulomatous to giant cell tumor-like. Morphologically, Case 1 was identical to cases described as XGET1. At the other end of the spectrum, Cases 6 and 7 showed a giant cell tumor-like appearance very similar to cases described as KPGCT2. Other cases showed mixed xanthogranulomatous and giant cell tumor-like features (e.g., Case 3). In addition, our cases frequently showed a fibrous capsule and peripheral lymphoid aggregates similar to XGET and KPGCT. This finding likely represents a diagnostically useful feature, though fragmentation may hinder assessment. Necrosis was a common finding in our cases, but infrequently seen in previously reported XGET and KPGCT. We suspect this difference in frequency may be due to sampling, the presence of pathologic fracture, or prior biopsy site changes in select cases.
All our cases showed keratin-positive mononuclear cells with variable highlighting of slender cytoplasmic processes similar to those seen in XGET and KPGCT. This is particularly significant as keratin-positive mononuclear cells are not at all typical of other well-characterized xanthogranulomatous, so-called fibrohistiocytic and bland giant cell-rich neoplasms of soft tissue and bone, with the exception of a subset of previously described giant cell tumors of soft tissue occurring in three adolescent to middle-aged females9 which may represent additional examples of KPGCT. In addition, variable staining with high molecular weight keratins (keratin 5/6 and 34βE12) was seen in a subset of our cases similar to described in XGET1. Of note, ultrastructural analysis showed findings, which while not entirely specific, were compatible with epithelial differentiation, as suggested by Fritchie et al.1.
The concept that the tumors in our study represent a spectrum that includes XGET and KPGCT, as suggested by clinical, morphologic and immunohistochemical similarities, is reinforced by molecular findings. HMGA2-NCOR2 fusions, as previously described in KPGCT, were detected in 7 of our cases, including tumors with typical XGET features, xanthogranulomatous, giant cell tumor-like and/or mixed features. While HMGA2-NCOR2 fusions were not detected in the original description of XGET, RNA-seq was only performed in 2 cases1 and visual inspection was required to detect the fusion in 1 case of KPGCT2. Manual inspection was also required to detect fusions in multiple cases in our study due to poor sequencing coverage of HMGA2, which makes it difficult for bioinformatics pipelines to identify consensus breakpoints. It is unclear if the poor coverage of HMGA2 may be due to underexpression of fusion transcripts secondary to rapid turnover or low transcription rates. Poor sequencing coverage can also result in false negatives when the neoplastic cell harboring the alteration of interest represents only a minority of the tissue nuclei, as is seen in tenosynovial giant tumor10. Moreover, the recruitment of apparently non-neoplastic cells (e.g., giant cells of osteoclast and Touton-types, histiocytes, lymphocytes) to the mass is reminiscent of the landscape effect seen in tenosynovial giant cell tumor11 and suggests a paracrine factor in the development of KPXG/GCT.
While the pathogenic mechanism of the HMGA2-NCOR2 fusion is unknown, chromosomal alternations involving high mobility group AT-hook 2 (HMGA2, HMGI-C) located at 12q14.3 have been described in a variety of mesenchymal neoplasms of bone, soft tissue and viscera. Many are benign, or less frequently locally aggressive, including uterine leiomyomas12,13,14, deep (aggressive) angiomyoma15,16, lipoma with or without bone and cartilage formation (osteochondrolipoma)14,17,18, myolipoma of soft tissue19, pulmonary chondroid hamartoma14, and soft tissue chondroma20. Rearrangements of HMGA2 are also implicated in a variety of mixed tumors including salivary gland pleomorphic adenoma21, myoid hamartoma of breast22, endometrial polyp14 and polypoid/mass-forming endometriosis23,24.
To our knowledge, outside of KPGCT, rearrangements involving nuclear receptor corepressor 2 (NCOR2) located at 12q24.3 have been reported in nine cases of mesenchymal neoplasia. A rearrangement of NCOR2 was noted as a likely incidental finding in a retroperitoneal MDM2-amplified dedifferentiated liposarcoma25 and a single case of GLI1-altered mesenchymal tumor was found to harbor a NCOR2-GLI1 fusion26. Interestingly, Panagopoulos et al. very recently reported 6 osteoclastic giant cell-rich tumor of bone with HMGA2-NCOR2 fusions, the illustrated cases of which show similar histologic features to the tumors presented in our series including the presence of foamy histiocytes though results of immunohistochemical studies for keratin were not reported27. Also of note, Brahmi et al. reported a case of a tenosynovial giant cell tumor in which a CSF1 rearrangement was not identified, while HMGA2-NCOR2 and NCOR2-SUPT3H fusion transcripts were detected28. The tumor illustrated in the representative photomicrograph is suggestive of tenosynovial giant cell tumor, however the report predates the recent original descriptions of XGET and KPGCT and the results of immunohistochemical studies for keratin(s) were not reported nor are such studies generally performed in the workup of suspected tenosynovial giant cell tumor. Whether the tumor descripted by Brahmi et al. represents an unusual tenosynovial giant cell tumor or a KPXG/GCT is unclear, however it is noteworthy that overexpression of CSF1 and IL-34 was detected and the tumor responded to CSF1R inhibition.
Concerning the two cases in which a HMGA2-NCOR2 fusion was not detected (Fig. 6), Case 2 is virtually identical to fusion-positive cases. Sequencing coverage of HMGA2 in this case was extremely poor and any possible fusion would have been below the limit of detection. Case 7, on the other hand, is somewhat of an outlier, both in its more aggressive clinical behavior and histological features of giant cell tumor-like morphology characterized by small epithelioid to plasmacytoid mononuclear cells. Sequencing in this case provided similar coverage as in cases in which an HMGA2 fusion was identified. Nevertheless, a false negative or variant fusion cannot be excluded and the morphology of this lesion is similar to a fusion-positive tumor described by Agaimy et al. and illustrated in their Fig. 3B, E2, supporting its inclusion as a KPXG/GCT.
Clinical follow-up was limited in many cases, however at least two relatively small subcutaneous tumors, including one with piecemeal excision, showed no evidence of recurrence during greater than 200 months of follow-up. Case 7, which may have already represented a recurrence at the time of presentation to our institution, went on to develop local recurrence and multifocal pulmonary lesions suspicious for metastases. It seems likely that relatively small superficial KPXG/GCT will behave in an indolent manner, however we provisionally regard KPXG/GCT as having uncertain biologic potential given the limited number of reported cases and findings of apparently aggressive behavior in a minority.
The histopathologic differential diagnosis for KPXG/GCT is well-represented by the original impressions put forth for cases in our cohort. KPXG/GCT with extensive xanthogranulomatous appearance can be distinguished from juvenile xanthogranuloma by the presence of a fibrous capsule with lymphoid aggregates in the former and infiltrative growth in the latter. KPXG/GCT can show areas of loose storiform growth of bland spindle cells reminiscent of fibrous histiocytoma, however, KPXG/GCT lacks limited infiltrative growth. Tenosynovial giant cell tumors are characterized by large synoviocyte-like epithelioid cells, variable desmin expression and rearrangements or deletions of CSF110,11,29, which are not present in KPXG/GCT. KPXG/GCT can be distinguished from inflammatory rhabdomyoblastic tumor by the presence of least focal infiltrative growth, nuclear atypia, and expression of desmin, myogenin and MyoD1 in the latter30,31,32.
In cases of KPXG/GCT with giant cell tumor-like morphology, the most important differential diagnostic considerations are giant cell tumor of soft tissue and giant cell tumor of bone. In addition to giant cell tumor-like morphology, histologic overlap also includes xanthoma cells and loose storiform (fibrous histiocytoma-like areas) as such findings can be seen in giant cell tumor of soft tissue9,33 and more commonly in giant cell tumor of bone. However, giant cell tumor of soft tissue and giant cell tumor of bone are not characterized by a peripheral lymphoid reaction, and bone formation is not a feature of KPXG/GCT2. Further, giant cell tumor of bone harbors characteristic mutations in histone 3, and immunohistochemical detection of H3F3A G34W mutation is an effective surrogate marker in >90% of giant cell tumor of bone34,35. Importantly, none of the above differential diagnostic considerations typically express keratin and are not expected to harbor HMGA2-NCOR2 fusions.
In conclusion, we present the clinical, immunohistochemical, ultrastructural and molecular findings of 9 cases of KPXG/GCT occurring in bone and soft tissue. The overlapping clinical and morphologic features, pattern of keratin expression and presence of HMGA2-NCOR2 fusions suggests that XGET and KPGCT represent morphologic variants of a single entity. Additional larger studies are needed to more fully characterize the clinical behavior of these tumors and we provisionally regard them as having uncertain biologic potential.
Data availability
Available upon request.
References
Fritchie, K. J., Torres-Mora, J., Inwards, C., Thway, K., Vaiyapuri, S., Jackson, R. et al. Xanthogranulomatous epithelial tumor: report of 6 cases of a novel, potentially deceptive lesion with a predilection for young women. Mod Pathol 33, 1889-1895 (2020).
Agaimy, A., Michal, M., Stoehr, R., Ferrazzi, F., Fabian, P., Michal, M. et al. Recurrent novel HMGA2-NCOR2 fusions characterize a subset of keratin-positive giant cell-rich soft tissue tumors. Mod Pathol 34, 1507-1520 (2021).
Dickson, B. C. & Swanson, D. Targeted RNA sequencing: A routine ancillary technique in the diagnosis of bone and soft tissue neoplasms. Genes Chromosomes Cancer 58, 75-87 (2019).
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. et al. Integrative genomics viewer. Nat Biotechnol 29, 24-26 (2011).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178-192 (2013).
Robinson, J. T., Thorvaldsdóttir, H., Wenger, A. M., Zehir, A. & Mesirov, J. P. Variant Review with the Integrative Genomics Viewer. Cancer Res 77, e31-e34 (2017).
Treffel, M., Lardenois, E., Larousserie, F., Karanian, M., Gomez-Brouchet, A., Bouvier, C. et al. Denosumab-treated Giant Cell Tumors of Bone: A Clinicopathologic Analysis of 35 Cases From the French Group of Bone Pathology. Am J Surg Pathol 44, 1-10 (2020).
Wojcik, J., Rosenberg, A. E., Bredella, M. A., Choy, E., Hornicek, F. J., Nielsen, G. P. et al. Denosumab-treated Giant Cell Tumor of Bone Exhibits Morphologic Overlap With Malignant Giant Cell Tumor of Bone. Am J Surg Pathol 40, 72-80 (2016).
Oliveira, A. M., Dei Tos, A. P., Fletcher, C. D. & Nascimento, A. G. Primary giant cell tumor of soft tissues: a study of 22 cases. Am J Surg Pathol 24, 248-256 (2000).
Ho, J., Peters, T., Dickson, B. C., Swanson, D., Fernandez, A., Frova-Seguin, A. et al. Detection of CSF1 rearrangements deleting the 3’ UTR in tenosynovial giant cell tumors. Genes Chromosomes Cancer 59, 96-105 (2020).
West, R. B., Rubin, B. P., Miller, M. A., Subramanian, S., Kaygusuz, G., Montgomery, K. et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci U S A 103, 690-695 (2006).
Mehine, M., Kaasinen, E., Mäkinen, N., Katainen, R., Kämpjärvi, K., Pitkänen, E. et al. Characterization of uterine leiomyomas by whole-genome sequencing. N Engl J Med 369, 43-53 (2013).
Quade, B. J., Weremowicz, S., Neskey, D. M., Vanni, R., Ladd, C., Dal Cin, P. et al. Fusion transcripts involving HMGA2 are not a common molecular mechanism in uterine leiomyomata with rearrangements in 12q15. Cancer Res 63, 1351-1358 (2003).
Tallini, G., Vanni, R., Manfioletti, G., Kazmierczak, B., Faa, G., Pauwels, P. et al. HMGI-C and HMGI(Y) immunoreactivity correlates with cytogenetic abnormalities in lipomas, pulmonary chondroid hamartomas, endometrial polyps, and uterine leiomyomas and is compatible with rearrangement of the HMGI-C and HMGI(Y) genes. Lab Invest 80, 359-369 (2000).
Rabban, J. T., Dal Cin, P. & Oliva, E. HMGA2 rearrangement in a case of vulvar aggressive angiomyxoma. Int J Gynecol Pathol 25, 403-407 (2006).
Rawlinson, N. J., West, W. W., Nelson, M. & Bridge, J. A. Aggressive angiomyxoma with t(12;21) and HMGA2 rearrangement: report of a case and review of the literature. Cancer Genet Cytogenet 181, 119-124 (2008).
Bartuma, H., Hallor, K. H., Panagopoulos, I., Collin, A., Rydholm, A., Gustafson, P. et al. Assessment of the clinical and molecular impact of different cytogenetic subgroups in a series of 272 lipomas with abnormal karyotype. Genes Chromosomes Cancer 46, 594-606 (2007).
Panagopoulos, I., Gorunova, L., Bjerkehagen, B., Lobmaier, I. & Heim, S. The recurrent chromosomal translocation t(12;18)(q14~15;q12~21) causes the fusion gene HMGA2-SETBP1 and HMGA2 expression in lipoma and osteochondrolipoma. Int J Oncol 47, 884-890 (2015).
Panagopoulos, I., Gorunova, L., Agostini, A., Lobmaier, I., Bjerkehagen, B. & Heim, S. Fusion of the HMGA2 and C9orf92 genes in myolipoma with t(9;12)(p22;q14). Diagn Pathol 11, 22 (2016).
Dahlén, A., Mertens, F., Rydholm, A., Brosjö, O., Wejde, J., Mandahl, N. et al. Fusion, disruption, and expression of HMGA2 in bone and soft tissue chondromas. Mod Pathol 16, 1132-1140 (2003).
Geurts, J. M., Schoenmakers, E. F., Röijer, E., Aström, A. K., Stenman, G. & van de Ven, W. J. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene 16, 865-872 (1998).
Panagopoulos, I., Gorunova, L., Andersen, H. K., Pedersen, T. D., Lømo, J., Lund-Iversen, M. et al. Genetic Characterization of Myoid Hamartoma of the Breast. Cancer Genomics Proteomics 16, 563-568 (2019).
Medeiros, F., Araujo, A. R., Erickson-Johnson, M. R., Kashyap, P. C., Dal Cin, P., Nucci, M. et al. HMGA1 and HMGA2 rearrangements in mass-forming endometriosis. Genes Chromosomes Cancer 49, 630-634 (2010).
Medeiros, F., Wang, X., Araujo, A. R., Erickson-Johnson, M. R., Lima, J. F., Meuter, A. et al. HMGA gene rearrangement is a recurrent somatic alteration in polypoid endometriosis. Hum Pathol 43, 1243-1248 (2012).
Muir, L. D., 2nd, Woelfle, J. D., Schowinsky, J. & Wilky, B. A. High grade sarcoma presenting as multifocal recurrent seromas after inguinal hernia repair: A case report. Rare Tumors 12, 2036361320975746 (2020).
Liu, J., Mao, R., Lao, I. W., Yu, L., Bai, Q., Zhou, X. et al. GLI1-altered mesenchymal tumor: a clinicopathological and molecular analysis of ten additional cases of an emerging entity. Virchows Arch (2021).
Panagopoulos, I., Andersen, K., Gorunova, L., Lund-Iversen, M., Lobmaier, I. & Heim, S. Recurrent Fusion of the Genes for High-mobility Group AT-hook 2 (HMGA2) and Nuclear Receptor Co-repressor 2 (NCOR2) in Osteoclastic Giant Cell-rich Tumors of Bone. Cancer Genomics Proteomics 19, 163-177 (2022).
Brahmi, M., Alberti, L., Tirode, F., Karanian, M., Eberst, L., Pissaloux, D. et al. Complete response to CSF1R inhibitor in a translocation variant of teno-synovial giant cell tumor without genomic alteration of the CSF1 gene. Ann Oncol 29, 1488-1489 (2018).
Cupp, J. S., Miller, M. A., Montgomery, K. D., Nielsen, T. O., O’Connell, J. X., Huntsman, D. et al. Translocation and expression of CSF1 in pigmented villonodular synovitis, tenosynovial giant cell tumor, rheumatoid arthritis and other reactive synovitides. Am J Surg Pathol 31, 970-976 (2007).
Martinez, A. P., Fritchie, K. J., Weiss, S. W., Agaimy, A., Haller, F., Huang, H. Y. et al. Histiocyte-rich rhabdomyoblastic tumor: rhabdomyosarcoma, rhabdomyoma, or rhabdomyoblastic tumor of uncertain malignant potential? A histologically distinctive rhabdomyoblastic tumor in search of a place in the classification of skeletal muscle neoplasms. Mod Pathol 32, 446-457 (2019).
Michal, M., Rubin, B. P., Kazakov, D. V., Michalová, K., Šteiner, P., Grossmann, P. et al. Inflammatory leiomyosarcoma shows frequent co-expression of smooth and skeletal muscle markers supporting a primitive myogenic phenotype: a report of 9 cases with a proposal for reclassification as low-grade inflammatory myogenic tumor. Virchows Arch 477, 219-230 (2020).
Cloutier, J. M., Charville, G. W., Mertens, F., Sukov, W., Fritchie, K., Perry, K. D. et al. “Inflammatory Leiomyosarcoma” and “Histiocyte-rich Rhabdomyoblastic Tumor”: a clinicopathological, immunohistochemical and genetic study of 13 cases, with a proposal for reclassification as “Inflammatory Rhabdomyoblastic Tumor”. Mod Pathol 34, 758-769 (2021).
O’Connell, J. X., Wehrli, B. M., Nielsen, G. P. & Rosenberg, A. E. Giant cell tumors of soft tissue: a clinicopathologic study of 18 benign and malignant tumors. Am J Surg Pathol 24, 386-395 (2000).
Behjati, S., Tarpey, P. S., Presneau, N., Scheipl, S., Pillay, N., Van Loo, P. et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45, 1479-1482 (2013).
Amary, F., Berisha, F., Ye, H., Gupta, M., Gutteridge, A., Baumhoer, D. et al. H3F3A (Histone 3.3) G34W Immunohistochemistry: A Reliable Marker Defining Benign and Malignant Giant Cell Tumor of Bone. Am J Surg Pathol 41, 1059-1068 (2017).
Funding
Washington University School of Medicine Department of Pathology and Immunology; Mount Sinai Hospital Department of Pathology and Laboratory Medicine.
Author information
Authors and Affiliations
Contributions
C.A.D. and J.S.A.C.—project conception and design, data gathering, data analysis, paper drafting, paper review/revision; J.C.B.—data gathering, data analysis, paper drafting, paper review/revision; R.B.—data analysis, paper drafting, paper review/revision; B.C.D.—data gathering, paper review/revision; R.E.S.—data gathering, data analysis, paper drafting, paper review/revision; E.G.D.—data gathering, data analysis, paper drafting, paper review/revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Study was approved by the institutional review boards of both participating centers.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dehner, C.A., Baker, J.C., Bell, R. et al. Xanthogranulomatous epithelial tumors and keratin-positive giant cell-rich soft tissue tumors: two aspects of a single entity with frequent HMGA2-NCOR2 fusions. Mod Pathol 35, 1656–1666 (2022). https://doi.org/10.1038/s41379-022-01115-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01115-6
