
Abstract
Major histocompatibility complex (MHC) class I is a membrane-bound protein complex expressed on nucleated human cells. MHC class I presents intracellular protein fragments to cytotoxic T cells and triggers an activation cascade upon neoantigen detection by these cells. MHC class I loss by tumor cells decreases tumor neoantigen presentation to the immune system and therefore represents a possible mechanism of immunotherapeutic resistance even among cancers that otherwise appear to be good candidates for checkpoint inhibition, such as mismatch repair (MMR)-deficient and PD-L1-positive malignancies. We herein assess MHC class I expression in a range of endometrial carcinomas, including MMR-deficient and PD-L1-positive cancers. Immunohistochemical staining for combined MHC class I A-, B-, and C-heavy chains was performed on 76 cases of endometrial carcinoma and was classified as present, subclonally lost, or diffusely lost. Tumoral PD-L1 expression, PD-L1 combined positive score, and CD3-positive T lymphocytes were also quantified. Forty-two percent of tumors showed loss of MHC class I expression, either in a subclonal (26%) or diffuse (16%) pattern. This included 46% of MMR-deficient and 25% of PD-L1-positive cancers. These findings suggest that tumoral MHC class I status may be an important factor to consider when selecting endometrial cancer patients for checkpoint inhibition.
Similar content being viewed by others


Introduction
Immunotherapy has recently become an option for individuals with advanced endometrial carcinoma and, while most often used for patients with mismatch repair-deficient tumors, can be employed irrespective of mismatch repair status in certain settings [1,2,3]. Although most endometrial carcinoma patients present at an low stage, late-stage endometrial cancers are associated with a poor prognosis and have few good traditional chemotherapeutic options [4]. Immunotherapies targeting the PD-1/PD-L1 checkpoint axis offer great promise for some patients, particularly those with highly mutated and thus immunogenic tumors that are highly infiltrated by lymphocytes [1, 5, 6]. However, a substantial proportion of eligible patients do not respond to checkpoint inhibition despite a seemingly susceptible immune milieu [1, 5, 6]. Elucidating mechanisms of treatment failure in these patients is key to selecting treatment-eligible patients and to identifying new combination or solo immunotherapy targets.
Checkpoint inhibitor-based immunotherapy works by facilitating existing adaptive anti-tumor responses [7]. Tumors evade immune attack through expression of the checkpoint ligand PD-L1 either directly on their cell surface or on tumor-associated macrophages [8]. The presence of PD-L1 in the tumor milieu enables an immunosuppressive interaction with surrounding PD-1-positive lymphocytes, leading to increased immune tolerance and allowing the tumor to propagate unimpeded by cytotoxic T cells. For anti-PD-1/PD-L1 therapy to be effective, cytotoxic T cells must therefore be both present and activated [9]. This occurs in a staged process, which begins in the tumor cell itself where MHC class I functions as a “flag pole” on which non-self antigens are displayed. A polymorphic MHC class I heavy chain and the constant beta2-microglobulin light chain join together within the endoplasmic reticulum of the tumor cell, after which an antigen joins with the heavy chain-light chain unit [7, 9, 10] .The trimer then relocates to the cell membrane. The interaction between the MHC class I-peptide complex and the T cell receptor on CD8 + T cells helps activate the cytotoxic response [10, 11].
Prior work has shown that loss of MHC class I expression occurs in a subset of endometrial carcinomas, including both mismatch repair-deficient and mismatch repair-intact tumors [12, 13]. Furthermore, recent investigations in other tumor types have shown that loss of MHC class I can happen in tandem with PD-L1 expression, potentially conferring resistance to anti-PD-1/PD-L1-based therapy in eligible candidates [14,15,16,17,18]. The relationship between PD-L1 and MHC class I expression has not, however, been previously investigated in endometrial cancer. We herein examine MHC class I expression in endometrial carcinomas with attention to mismatch repair and PD-L1 status.
Methods
Case selection
This study was approved by the University of Virginia Institutional Review Board (IRB #13310). Seventy-six patients who had undergone surgical resection for endometrial carcinoma were selected from the University of Virginia Surgical Pathology archive, including representatives from different tumor grades, stages, and mismatch repair subtypes. No serous carcinomas were included due to the absence of mismatch repair deficiency in this histotype in our case cohort. None of the tumors had undergone previous treatment by chemotherapy, radiotherapy, or immunotherapy. Stage was obtained through electronic chart review. Tumors were graded and assigned histologic classification at the time of diagnosis in accordance with the standardized International Federation of Gynecology and Obstetrics scheme, and both grade and histologic subtype were reviewed by a study pathologist (AMM) for diagnostic confirmation. For the purposes of statistical analysis, FIGO 3, dedifferentiated carcinoma, and carcinosarcoma were considered high-grade tumors and were analyzed together.
Mismatch repair status
Immunohistochemical staining for mismatch repair proteins was performed in the course of the institution’s universal Lynch syndrome screening algorithm as described by Mills & Longacre [19, 20]. Antibody details are as follows: MLH1: clone ES05, predilute, Leica Biosystems; PSM2: MRQ-28Mab, predilute, Cell Marque; MSH2: clone 25D12, predilute, Leica Biosystems; MSH6: clone 44 Mab, predilute, Cell Marque. Tumors demonstrating either dual MLH1/PMS2 loss or isolated PMS2 loss underwent MLH1-promoter hypermethylation testing by pyrosequencing (Epitech Bisulfite kit and MLH1 primer Kit, cat #97002, Pyromark Q24, Qiagen), which confirmed the presence of MLH1-promoter hypermethylation in all MLH1/PMS2-deficient cases. These cases were therefore classified as MLH1-promoter hypermethylated for analytical purposes. Tumors with loss of MSH2/MSH6, MSH6-only, or PMS2-only were designated as non-methylated mismatch repair-deficient. Tumors with retained immunohistochemical staining for all four mismatch repair proteins were designated as mismatch repair-intact.
MHC class I immunohistochemistry
Immunohistochemical staining for MHC class I expression was performed on whole sections. The probe targeted the classical MHC class I system, with detection of HLA-A, B and C heavy chains (anti-HLA class 1 ABC antibody [EMR8-5], cat# ab70328, Abcam, dilution 1:400). Tumoral staining was classified as “intact” (>90% of cells showing membranous and/or cytoplasmic expression of MHC class I), “subclonal loss” (10–90% MHC class I expression, with areas of retained MHC class I immediately juxtaposed with areas of negative tumor staining), or “diffuse loss” (<10% tumor cell expression of MHC class I). Occasional cases demonstrated retained MHC class I staining throughout the tumor but with varying intensity depending on the region; strongly staining regions were juxtaposed with mild to moderate staining regions. These tumors were classified as MHC class I intact due to the presence of appreciable membranous expression on all tumor cells. Interpretation was performed by AMM and LAF after an initial review at a double-headed microscope, and final classification was based on consensus opinion. Normal endometrial glands, stroma, and tumor-associated inflammatory cells served as internal positive controls.
PD-L1 immunohistochemistry
Immunohistochemical staining for PD-L1 (clone SP142, dilution 1:200; Spring Bioscience) was performed on whole sections and scored using the tumoral proportion and combined positive scores (CPS). Tumoral expression was classified based on the percentage of viable tumor showing partial or complete membranous staining of any intensity; this was scored on a continuous scale and was then sub-classified as 1–5%, 6–10%, 11–25%, 26–50%, or >50% for subsequent analyses. The CPS was calculated as follows: CPS = [(number of positive tumor cells, lymphocytes, & macrophages)/(total number of tumor cells)] × 100, with 100 representing the maxium possible score [21]. Each case given a score from 0 to 100, and a CPS of ≥1 was considered positive based on the FDA approval for anti-PD-1 therapy and treatment response in other tumors meeting this threshold [21,22,23]. Placental tissue served as an external positive control. PD-L1 tumoral proportion scores were previously reported for a subset of cases by Sloan et al. [24].
CD3 immunohistochemistry
Immunohistochemical staining for CD3 (Agilent technology, cat #A0452, polyclonal antibody, diluted 1:300) was performed on whole tissue sections. Intra- and peritumoral CD3 + lymphocytes were manually enumerated and averaged across three high-power fields selected to include the area of greatest staining density. Tonsillar tissue served as an external positive control. Average CD3 counts were previously reported for the majority of cases in a prior study [25].
Statistical analysis
Statistical analysis was performed using SPSS software (IBM SPSS Statistics 26, Aramonk, NY). Categorical variable comparisons were achieved using Pearson Chi-Square analysis and Fisher’s Exact Tests, continuous variable comparisons for exact CPS were achieved using Mann–Whitney U and Kruskal–Wallis with Bonferroni correction, and continuous variable comparisons with CD3 counts were achieved using Analysis of Variance testing.
Results
The tumors consisted of 68 endometrioid carcinomas (28 FIGO grade 1, 21 FIGO grade 2, 19 FIGO grade 3), 6 dedifferentiated carcinomas, and 2 carcinosarcomas. Most patients had stage I disease, though a range of stages were represented (61 stage I, 4 stage II, 10 stage III, 1 stage IV). Twenty-eight cases were mismatch repair-intact, 25 were mismatch repair-deficient due to MLH1-promoter hypermethylation, and 23 cases were mismatch repair deficient due to other causes (12 MSH2/MSH6 deficient, 7 MSH6 deficient, 1 MLH1/PSM2/MSH6 deficient, and 3 PMS2 deficient). There were no significant differences in grade or stage between mismatch repair groups (p = 0.11 and p = 0.76, respectively). The majority of tumors were of an endometrioid histotype. Of the other histotypes present, all six dedifferentiated tumors were mismatch repair-deficient, five of which were non-MLH1 promoter hypermethylated; one carcinosarcoma was mismatch repair deficient, MLH1-promoter hypermethylated; and one carcinosarcoma was mismatch repair-intact.
Relationship between mismatch repair status and MHC class I expression
Forty-two percent of tumors demonstrated loss of MHC class I expression, either in a subclonal (26%) or diffuse (16%) pattern (Table 1, Fig. 1). MHC class I expression patterns did not vary by tumor grade or stage (p = 0.625 and p = 0.643, respectively).

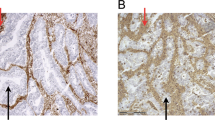
MHC class I was retained in the majority of endometrial carcinomas, as illustrated in case (a–b). A subset of cases, including the one shown in (c–d), demonstrated a subclonal loss pattern typified by areas of tumor with complete absence of expression juxtaposed with areas of retained staining. Another subset showed complete loss of MHC class I, as illustrated in the case in (e–f). All three cases illustrated are mismatch repair-deficient and all demonstrate intact MHC class I expression within background inflammatory cells and stroma. (A/C/E: H&E; B/D/F: MHC class I immunohistochemistry).
There was no difference in MHC class I expression pattern distribution between the three mismatch repair groups, nor was there a difference when comparing mismatch repair-intact tumors to the aggregate of mismatch repair-deficient tumors (p = 0.51 and p = 0.31, respectively) (Table 1). Thirty-six percent of mismatch repair-intact tumors and 46% of mismatch repair-deficient tumors showed either subclonal or diffuse MHC class I loss. Subclonal loss was demonstrated in 29% of mismatch repair-intact tumors and 25% of mismatch repair-deficient tumors, whereas diffuse loss was demonstrated in 7% of mismatch repair-intact tumors and 21% of mismatch repair-deficient tumors (Fig. 1).
When MLH1-promoter hypermethylated and non-methylated mismatch repair-deficient tumors were compared to each other, again, no differences were found in MHC class I expression patterns. Forty-four percent of MLH1-promoter hypermethylated tumors lost MHC class I expression (28% subclonal, 16% diffuse) in comparison to 48% of non-methylated mismatch repair-deficient tumors (22% subclonal, 26% diffuse) (p = 0.80).
Relationship between MHC class I expression and tumor size
Gross tumor size was available for 62 of 76 cases. The mean tumor size was 3.88 cm (SD 2.1 cm); there was no significant difference between the three mismatch repair groups or between the mismatch repair-intact and aggregated mismatch repair-deficient groups (p = 0.88, p = 0.98). There was no relationship between tumor size and MHC class I expression pattern analyzed by either intact expression, subclonal loss, and diffuse loss or as intact expression and any degree of loss (p = 0.38, p = 0.53).
PD-L1 immunohistochemical expression
PD-L1 tumor staining results were previously reported for the majority of the cases by Sloan et al. [24]. In the current series, tumoral PD-L1 expression at the ≥1% threshold was seen in 37% of endometrial carcinomas, and at the >5% threshold in 28% of cases (Table 2). Tumoral staining at the ≥1% and >5% thresholds was more common among mismatch repair-deficient cases than among the mismatch repair-intact cases (≥1% threshold: 50% vs. 14%, p < 0.01; >5% threshold: 31% vs. 4%, p < 0.01). Within the mismatch repair-deficient group, PD-L1 expression was more common among non-methylated mismatch repair-deficient cases than the MLH1-promoter hypermethylated cases at the ≥1% threshold though not the >5% threshold (≥1% threshold: 70% vs. 32%, p = 0.01; >5% threshold: 43% vs. 20%, p = 0.07).
Eighty-six percent of all endometrial carcinomas had a PD-L1 CPS of ≥1. As with tumoral PD-L1 expression, a CPS of ≥1 was more common among mismatch repair-deficient cases than among intact cases (100% vs. 61%, p < 0.001). Indeed, all mismatch repair-deficient cases had a CPS of ≥1. The median CPS was lower in the mismatch repair-intact group than in either the MLH1-promoter hypermethylated or non-methylated mismatch repair-deficient groups (1 vs. 15 and 1 vs. 20, p = 0.03 and p < 0.001). There was no difference in the median CPS between the MLH1-promoter hypermethylated and non-methylated mismatch repair-deficient cases (15 vs. 20, p = 0.49).
Relationship between MHC class I, PD-L1 expression, and CD3+lymphocytes
Tumors with fully intact MHC class I expression were more likely to express PD-L1 than tumors with some degree of MHC class I loss. This was true at both the ≥1% staining threshold (48% vs. 22%, p = 0.03) and at the >5% staining threshold (33% vs. 6%, p = 0.01) (Figs. 2 and 3). However, 25% of PD-L1-expressing tumors were found to demonstrate either subclonal (18%) or diffuse (7%) MHC class I loss. There was no difference between the subclonal and diffuse MHC class I loss groups in terms of PD-L1 expression at either the ≥1% or the >5% PD-L1 staining thresholds (25% vs. 17%, p = 0.68; 10% vs. 0%, p = 0.52). There was no relationship between tumor-associated CD3 + lymphocyte count and presence or absence of MHC class I expression (p = 0.642).

MHC class I loss of expression was not significantly associated with either mismatch repair status or PD-L1 CPS but was seen more frequently in cases with tumoral PD-L1 expression at the ≥1% and >5% thresholds. Some MMR-deficient cases showed complete loss of MHC class I, such as the PMS2-deficient tumor from a Lynch syndrome patient illustrated in (a–c) (a: H&E; b: MHC class I; c: PD-L1). The tumor cells were entirely PD-L1-negative in this tumor, however the background inflammatory cells were strongly PD-L1-positive. Other mismatch repair-deficient cases, such as the MSH2/MSH6-deficient tumor illustrated in (d–f), showed intact MHC class I expression with strong tumoral PD-L1 staining (d: H&E; e: MHC class I; f: PD-L1). This suggests that while the absence of MHC class I may interfere with the efficacy of anti-PD-1/PD-L1 checkpoint inhibition in some patients with mismatch repair-deficient tumors, in other immunotherapy candidates MHC class I is fully intact and anti-PD-1/PD-L1 drugs have more promise.

Loss of MHC class I was not unique to mismatch repair-deficient cancers. While many mismatch repair-intact tumors, like the one illustrated in (a–c) (a: H&E; b: MHC class I; c: PD-L1) showed intact MHC class I staining with absent PD-L1 expression, in others like the case in (d–e) (d: H&E; e: MHC class I; f: PD-L1), MHC class I was entirely lost. In this case, focal PD-L1 expression was appreciated on tumor-associated immune cells.
Discussion
Many malignancies thrive by altering their microenvironments and co-opting otherwise beneficial, adaptive biological processes. Evading the adaptive immune response to tumor-specific antigens is one strategy enlisted by malignancies to avoid destruction and represents the biological basis for clinically available immunotherapies. Endometrial carcinomas and their associated inflammatory cells can express a variety of targetable checkpoint molecules and immune-tolerizing enzymes including not only PD-L1, but also TIM-3, LAG-3, and IDO, provoking excitement about the potential for drugs blocking these targets [24, 26,27,28]. Endometrial carcinomas are particularly good candidates for immunotherapeutic interventions due to their high rates of mismatch repair deficiency, leading to elevated tumoral mutational burdens and neoantigen loads with a resultant increase in antitumoral inflammation [29]. A subset of advanced stage mismatch repair-intact endometrial carcinomas also respond to anti-PD-1 checkpoint inhibition, making these cancers one of the only carcinomas with an FDA-approved pathway to anti-PD-1 therapy irrespective of their mismatch repair signature [2, 5, 30,31,32].
However, the effectiveness of checkpoint-based immunotherapy is contingent on the presence of an activated adaptive immune response available to attack a tumor once its immune-inhibitory defenses are blocked. If tumors do not express MHC class I then they lack the ability to be targeted by cytotoxic T cells, and therefore therapeutic antibodies to downstream immunosuppressive molecules such as PD-1/PD-L1 may not be of value. Notably, loss of MHC class I expression should, in theory, render tumor cells more vulnerable to the host’s NK cells, as NK cells can become activated in the setting of missing MHC class I [33]. However, the clinical significance of anti-tumoral NK cell responses has yet to be well-elucidated in endometrial carcinoma. Early evidence suggests that these tumors are able to successfully evade NK cell-mediated cytotoxicity through other mechanisms [34]. Future studies investigating NK cell-directed immunotherapy will be of interest in MHC class I-deficient cancers [33, 35, 36].
Previous reports in a variety of tumor types, including endometrial carcinoma, have linked immune evasion to downregulation or mutation of the MHC class I structure [9, 13, 16, 17]. In a study of 486 patients with sporadic endometrioid endometrial carcinoma, de Jong et al. found that 41% of tumors downregulated their MHC class I expression [13]. This mainly occurred via changes in heavy chain expression, with only 1% of cases demonstrating expression changes due to both the heavy chain and the light chain. Furthermore, rates of downregulation did not differ significantly between mismatch repair-deficient and mismatch repair-intact tumors. Our study identified downregulation of MHC class I in a strikingly similar proportion—42%—of endometrial carcinomas, including many that initially appeared to be good candidates for immunotherapy based on mismatch repair deficiency and/or PD-L1 expression. However, while many PD-L1-positive cancers lose the ability to engage with cytotoxic T cells, plenty of others retain their MHC class I expression and remain strong candidates for immunotherapy that optimizes the cytotoxic T cell-mediated adaptive immune response. As was the case in the series by de Jong et al., we found no significant difference between mismatch repair-intact and deficient tumors.
Interestingly, while our prior studies have demonstrated differences in immunotherapeutic target expression between genetically driven and epigenetically driven (MLH1-promoter hypermethylated) mismatch repair-deficient endometrial carcinomas [24, 26,27,28], we did not find such a difference for MHC class I loss: non-methylated and MLH1-promoter hypermethylated mismatch repair-deficient tumors showed statistically comparable rates of MHC class I loss, and both were comparable to mismatch repair-intact tumors. These data suggest that tumors considered for immunotherapy ought to be evaluated for MHC class I expression irrespective of their mismatch repair status and mechanism of mismatch repair loss.
We also did not find a relationship between MHC class I expression and tumor-associated CD3 + lymphocytes. In their series, de Jong et al. observed lower rates of tumor-infiltrating CD8 + cytotoxic cells in cases with MHC class I loss [13]. Taken in concert with these findings, our results suggest that while cytotoxic T cells may be significantly reduced in the context of MHC class I loss, overall lymphocyte infiltration does not change—perhaps due to a larger contribution of effector or regulatory CD4 + T cells in cases with MHC class I loss. The histologic impression of robust tumor-associated lymphocytes thus does not guarantee an underlying intact MHC class I system, although identification of a CD8+ cytotoxic T cell-rich milieu may be valuable.
Our study adds to the findings from the de Jong series by incorporating PD-L1 data and contextualizing the MHC class I expression results in the era of immunotherapy. Tumors expressing PD-L1 in both ≥1% and >5% of tumor cells were more likely than PD-L1-negative cases to have fully intact MHC class I expression, though PD-L1 CPS was not associated with MHC class I expression. From a treatment perspective, it is important to note that a subset of PD-L1-positive cases (which might be considered better candidates for checkpoint inhibition, although expression isn’t currently codified as a criterion for drug access in this tumor type) had loss of MHC class I and therefore may be unlikely to respond to therapies aimed at enhancing the adaptive immune response.
The relationship between MHC class I and PD-L1 expression has been examined in numerous other tumor types aside from endometrial carcinoma, including head and neck squamous cell carcinoma, non-small cell lung carcinoma, hepatocellular carcinoma, Merkel cell carcinoma [16, 17, 37, 38]. As with our study, each of these series found that while MHC class I loss was not limited to PD-L1-positive cases, a subset of PD-L1-positive tumors demonstrated MHC class I loss. In vitro studies using human and mouse cell lines suggest that an inverse relationship between MHC class I and PD-L1 expression could be explained at a molecular level: the transcription factor IRF2 is involved in both increasing MHC class I presentation and decreasing PD-L1 presentation, and may play a role in cancers showing the MHC class I-negative, PD-L1-positive phenotype [15]. Future investigations of IRF2 in endometrial carcinoma may therefore be of interest.
None of the patients in our series had undergone prior treatment with chemotherapy, radiation, or immunotherapy. Downregulation of MHC class I expression via beta2-microglobulin mutation has been observed after anti-PD-1 therapy in melanoma, suggesting that malignancies may lose MHC class I expression as an adaptive response to checkpoint inhibition [39]. Although abnormalities in beta2-microglobulin appear to be uncommon in endometrial carcinomas based on data from the de Jong study, downregulation of HLA class I heavy chains has the same ultimate effect of decreasing MHC class I expression and appears to be relatively common in endometrial carcinoma based on our data and de Jong’s [13]. Studies comparing MHC class I expression in pre-and post-immunotherapy samples would therefore be of interest, as loss could emerge adaptively after initial exposure and/or confer resistance to a previously beneficial treatment.
Much of the current research on immunotherapy centers around finding the right combination of drugs for a particular patient [32]. The human immune system relies on complex interactions between a host of immunoactivating and immunoinhibitory molecules, and the PD-1/PD-L1 axis is just one of many that can be co-opted by malignancy. Many PD-L1-positive gynecologic cancers co-express other immunosuppressive molecules including LAG-3, TIM-3, VISTA, and IDO, and drugs targeting all of these—as well as immunostimulatory molecules such as OX40—are either clinically available or in clinical trials (NCT03538028; NCT02817633; NCT02812875; NCT02554812) [26,27,28, 40, 41]. Knowing that many tumors will simultaneously enlist more than one targetable method of immune evasion, investigators are focused on curating a multi-pronged immunotherapeutic approach involving combinations of these drugs. However, this kind of treatment layering is unlikely to have therapeutic benefit in cancers with MHC class I loss, because even after blocking immunosuppressive pathways, cytotoxic lymphocytes will remain unable to engage with the malignant cells in the absence of MHC class I. Indeed, this may explain why combination immunotherapy often fails to significantly improve response rates.
Developing a treatment strategy for tumors with MHC class I loss therefore becomes challenging. Histone deacetylase inhibitors have been studied as a possible intervention to promote protein upregulation in cancers where MHC class I loss is due to epigenetic factor, and show early promise. Ugurel et al. evaluated the impact of panobinostat, a histone deacetylase inhibitor, in two patients with metastatic Merkel cell carcinoma: one treated with pembrolizumab and the other with nivolumab [16]. Although neither patient in this study had a demonstrable clinical response, the single patient who had pre- and post-treatment biopsies had a measurable increase in MHC class I expression and tumor-infiltrating CD8 + T cells. This study highlights not only the need for similar studies in various tumor types, but also the need to assess clinical and pathologic changes in response to therapy.
Rational selection of patients who may be immunotherapy candidates and curation of tumor-specific treatments is important not only for optimizing responses, but also to minimize patient harm. Although less morbid than conventional chemotherapy, immunotherapy is associated with a host of autoimmune-like side effects that range from minor to fatal [42,43,44,45]. Immunotherapy overuse also carries significant financial implications for patients and for the healthcare system: the costs of single agent immunotherapy exceeds $100,000 annually [46]. Identifying cancers with MHC class I loss could be critical for preventing unnecessary administration of these drugs in patients who will not benefit.
There are several important limitations to this study. First, MHC class I expression was evaluated on a single tumor section. It is entirely possible that the proportion of cases showing subclonal loss could be underestimated in this context. Future studies evaluating MHC class I status across multiple tumor blocks may therefore be of interest. In addition, our series does not include clinical follow-up or treatment response data. Studies of MHC class I expression among immunotherapy responders and non-responders will be critical moving forward.
In summary, subclonal or diffuse loss of MHC class I expression occurs in a significant subset of endometrial carcinomas, including, but not limited to, mismatch repair-deficient and PD-L1-positive cases. Because MHC class I is vital for the presentation of tumor antigens to cytotoxic T-cells, immunotherapies that facilitate the adaptive immune response may be of little value in the context of MHC class I loss. MHC class I immunohistochemistry may therefore be an important biomarker for identifying immunotherapy candidates and preventing unnecessary administration of these drugs to patients who will not respond. Future studies directly evaluating the role of MHC class I in immunotherapeutic resistance will be of interest, as will work on the potential role of histone deacetylase inhibitors in upregulating MHC class I.
References
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Makker V, Rasco D, Vogelzang NJ, Brose MS, Cohn AL, Mier J, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20:711–8.
Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12:54.
Bestvina CM, Fleming GF. Chemotherapy for endometrial cancer in adjuvant and advanced disease settings. Oncologist. 2016;21:1250–9.
Ott PA, Bang YJ, Piha-Paul SA, Razak ARA, Bennouna J, Soria J, et al. T-cell–inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol. 2019;37:318–27.
Ott PA, Bang YJ, Berton-Rigaud D, Elez E, Pishvaian MJ, Rugo HS, et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1–positive endometrial cancer: results from the KEYNOTE-028 study. J Clin Oncol. 2017;35:2535–41.
Friedrich M, Jasinski-Bergner S, Lazaridou M-F, Subbarayan K, Massa C, Tretbar S, et al. Tumor-induced escape mechanisms and their association with resistance to checkpoint inhibitor therapy. Cancer Immunol Immunother. 2019;68:1689–1700.
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–74.
Kloor M, Becker C, Benner A, Woerner SM, Gebert J, Ferrone S, et al. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Res. 2005;65:6418–24.
Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11:823–36.
Mittica G, Genta S, Aglietta M, Valabrega G. Immune checkpoint inhibitors: a new opportunity in the treatment of ovarian cancer? Int J Mol Sci. 2016;17 https://doi.org/10.3390/ijms17071169.
Bijen CBM, Bantema-Joppe EJ, de Jong RA, Leffers N, Mourits MJE, Eggink HF, et al. The prognostic role of classical and nonclassical MHC class I expression in endometrial cancer. Int J Cancer. 2010;126:1417–27.
de Jong RA, Boerma A, Boezen HM, Mourits MJE, Hollema H, Nijman HW. Loss of HLA class I and mismatch repair protein expression in sporadic endometrioid endometrial carcinomas. Int J Cancer. 2012;131:1828–36.
Šmahel M. PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression. Int J Mol Sci. 2017;18:1331.
Kriegsman BA, Vangala P, Chen BJ, Meraner P, Bass AL, Garber M, et al. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J Immunol. 2019;203:1999–2010.
Ugurel S, Spassova I, Wohlfarth J, Drusio C, Cherouny A, Melior A, et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: a case series. Cancer Immunol Immunother. 2019;68:983–90.
Yoo SH, Keam B, Ock C-Y, Kim S, Han B, Kim J-W, et al. Prognostic value of the association between MHC class I downregulation and PD-L1 upregulation in head and neck squamous cell carcinoma patients. Sci Rep. 2019;9:7680.
Erdogdu IH. MHC Class 1 and PDL-1 Status of Primary Tumor and Lymph Node Metastatic Tumor Tissue in Gastric Cancers. Gastroenterol Res Pr. 2019;2019:4785098.
Mills AM, Liou S, Ford JM, Berek JS, Pai RK, Longacre TA. Lynch syndrome screening should be considered for all patients with newly diagnosed endometrial cancer. Am J Surg Pathol. 2014;38:1501–9.
Mills AM, Longacre TA. Lynch syndrome screening in the gynecologic tract: current state of the art. Am J Surg Pathol. 2016;40:e35–44.
Kulangara K, Zhang N, Corigliano E, Guerreo L, Waldroup S, Jaiswal D, et al. Clinical Utility of the Combined Positive Score for Programmed Death Ligand-1 Expression and the Approval of Pembrolizumab for Treatment of Gastric Cancer. Arch Pathol Lab Med. 2019;143:330–7.
Administration USF and D FDA approves pembrolizumab for advanced cervical cancer with disease progression during or after chemotherapy [Internet]. 2018. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm610572.htm.
Chung HC, Ros W, Delord J-P, Perets R, Italiano A, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 Study. J Clin Oncol. 2019;37:1470–8.
Sloan EA, Ring KL, Willis BC, Modesitt SC, Mills AM. PD-L1 Expression in Mismatch Repair-deficient Endometrial Carcinomas, Including Lynch Syndrome-associated and MLH1 Promoter Hypermethylated Tumors. Am J Surg Pathol. 2017;41:326–33.
Friedman LA, Ring KL, Mills. AMLAG-3 and GAL-3 in endometrial carcinoma: emerging candidates for immunotherapy. Int J Gynecol Pathol. 2020;39:203–12.
Moore M, Ring KL, Mills AM. TIM-3 in endometrial carcinomas: an immunotherapeutic target expressed by mismatch repair-deficient and intact cancers. Mod Pathol. 2019;32:1168–79.
Mills AM, Zadeh S, Sloan EA, Chinn Z, Modesitt SC, Ring KL. Indoleamine 2,3-dioxygenase in endometrial cancer: a targetable mechanism of immune resistance in mismatch repair-deficient and intact endometrial carcinomas. Mod Pathol. 2018;31:1282–90.
Friedman L, Ring K. Mills ALAG-3 and GAL-3 in endometrial carcinoma: emerging candidates for immunotherapy. Int J Gynecol Pathol. 2019;39:203–12.
Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015;1:1319–23.
Konstantinopoulos PA, Luo W, Liu JF, Gulhan DC, Krasner C, Ishizuka JJ, et al. Phase II study of avelumab in patients with mismatch repair deficient and mismatch repair proficient recurrent/persistent endometrial cancer. J Clin Oncol. 2019;37:2786–94.
Oaknin A, Duska LR, Sullivan RJ, Pothuri B, Ellard SL, Leath CA, et al. Preliminary safety, efficacy, and pharmacokinetic/pharmacodynamic characterization from GARNET, a phase I/II clinical trial of the anti–PD-1 monoclonal antibody, TSR-042, in patients with recurrent or advanced MSI-h and MSS endometrial cancer. Gynecol Oncol. 2019;154:17.
Rubinstein MM, Makker V. Optimizing immunotherapy for gynecologic cancers. Curr Opin Obstet Gynecol. 2020;32:1–8.
Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, et al. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 2014;122:91–128.
Degos C, Heinemann M, Barrou J, Boucherit N, Lambaudie E, Savina A, et al. Endometrial tumor microenvironment alters human NK cell recruitment, and resident NK cell phenotype and function. Front Immunol. 2019;10:877.
Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018;9:1869.
Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. 2019;10:1331.
Perea F, Sánchez-Palencia A, Gómez-Morales M, Bernal M, Concha A, Garcia MM, et al. HLA class I loss and PD-L1 expression in lung cancer: Impact on T-cell infiltration and immune escape. Oncotarget. 2018;9:4120–33.
Umemoto Y, Okano S, Matsumoto Y, Nakagawara H, Matono R, Yoshiya S, et al. Prognostic impact of programmed cell death 1 ligand 1 expression in human leukocyte antigen class I-positive hepatocellular carcinoma after curative hepatectomy. J Gastroenterol. 2014;50:65–75.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–29.
Mills AM, Peres LC, Meiss A, Ring KL, Modesitt SC, Abbott SE, et al. Targetable Immune Regulatory Molecule Expression in High-Grade Serous Ovarian Carcinomas in African American Women: a Study of PD-L1 and IDO in 112 Cases From the African American Cancer Epidemiology Study (AACES). Int J Gynecol Pathol. 2019;38:157–70.
Chinn Z, Stoler MH, Mills AM. PD-L1 and IDO expression in cervical and vulvar invasive and intraepithelial squamous neoplasias: implications for combination immunotherapy. Histopathology. 2019;74:256–68.
Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med. 2016;375:1749–55.
Läubli H, Balmelli C, Bossard M, Pfister O, Glatz K, Zippelius A. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J Immunother Cancer. 2015;3:11–015-0057-1. eCollection 2015
Cappelli LC, Gutierrez AK, Bingham CO 3rd, Shah AA. Rheumatic and musculoskeletal immune-related adverse events due to immune checkpoint inhibitors: a systematic review of the literature. Arthritis Care Res (Hoboken). 2017;69:1751–63.
Groisberg R, Hong DS, Behrang A, Hess K, Janku F, Piha-Paul S, et al. Characteristics and outcomes of patients with advanced sarcoma enrolled in early phase immunotherapy trials. J Immunother Cancer. 2017;5:100-017-0301-y.
The Value and Cost of Immunotherapy Cancer Treatments [Internet]. [cited 5 February 2020]. https://www.healthline.com/health-news/value-and-cost-of-immunotherapy#2.
Acknowledgements
The authors would like to thank the University of Virginia Biorepository and Tissue Research Facility for their skill and expertize in performing immunohistochemical staining for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Friedman, L.A., Bullock, T.N., Sloan, E.A. et al. MHC class I loss in endometrial carcinoma: a potential resistance mechanism to immune checkpoint inhibition. Mod Pathol 34, 627–636 (2021). https://doi.org/10.1038/s41379-020-00682-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00682-w
This article is cited by
-
High-plex spatial transcriptomic profiling reveals distinct immune components and the HLA class I/DNMT3A/CD8 modulatory axis in mismatch repair-deficient endometrial cancer
Cellular Oncology (2023)
-
MHC class I loss is associated with biliary/progenitor cell features and “cold” tumor-immune microenvironment in hepatocellular carcinoma
Virchows Archiv (2023)
-
Dual-sgRNA CRISPR/Cas9 knockout of PD-L1 in human U87 glioblastoma tumor cells inhibits proliferation, invasion, and tumor-associated macrophage polarization
Scientific Reports (2022)
-
Targeting immune checkpoints in gynecologic cancer: updates & perspectives for pathologists
Modern Pathology (2022)
-
Immune Checkpoint Inhibitors (ICI) in Advanced and Recurrent Endometrial Cancer
Indian Journal of Gynecologic Oncology (2022)
