
Abstract
Embryonal rhabdomyosarcomas (ERMS) account for 2–3% of cancers in pediatric and adolescent populations. They are rarer in adults. We and others have reported that ERMS arising in the uterine cervix may harbor mutations in the gene encoding the microRNA biogenesis enzyme, DICER1, but a large series of cases has not been published. In the uterus, distinguishing ERMS from adenosarcoma can be very challenging, even for expert pathologists, and DICER1 alterations have been identified in a variable subset of uterine adenosarcomas. We hypothesized that DICER1 genetic testing may be useful in distinguishing between ERMS and adenosarcoma. We conducted a central pathology review-based study of 64 tumors initially thought to be uterine ERMS or adenosarcoma; 19 neoplasms had a consensus diagnosis of ERMS, 27 of adenosarcoma and for 18, no consensus diagnosis was reached. The median age at diagnosis was 30 years (range 2.5–69) for ERMS, 57.5 years (range 27–82) for adenosarcoma, and 65.5 years (range 32–86) for no consensus cases. In our series, the DICER1 mutation prevalence differed between the three groups: DICER1 alterations were present in 18/19 (95%) ERMS, 7/27 (26%) adenosarcomas (p < 0.001), and 4/18 (22%) no consensus cases. A germline alteration was present in 6/12 ERMS patients tested versus 0/6 adenosarcoma patients. Thus, although DICER1 mutations are near ubiquitous in uterine ERMS and are significantly less common in uterine adenosarcoma, DICER1 testing is only of value in distinguishing between the two neoplasms when a DICER1 mutation is absent, as this is helpful in excluding ERMS. On review of the clinical and radiological features of the single DICER1 wild-type cervical ERMS, this was thought most likely to be of vaginal origin. Given the significant prevalence of DICER1 germline pathogenic variants in uterine ERMS, all patients with this diagnosis should be referred to a genetics service.
Similar content being viewed by others


Introduction
Rhabdomyosarcomas are soft-tissue sarcomas exhibiting skeletal muscle differentiation. Four histologic subtypes of rhabdomyosarcoma have been defined, namely embryonal (ERMS), alveolar, pleomorphic, and spindle cell/sclerosing, of which ERMS is the most common [1]. Rhabdomyosarcomas are most prevalent in the pediatric and adolescent populations and are estimated to comprise 2–3% of new cancer cases in these age groups in the United States [2]. Rhabdomyosarcomas in children are most commonly embryonal in type and preferentially involve the head and neck; in adults, the pleomorphic and embryonal types occur at roughly similar frequencies, and the extremities represent the most common site of disease [3]. Within the female genital tract, the vagina is the most frequent site of origin and vaginal rhabdomyosarcomas, which are almost all ERMS, typically occur in infant girls [4, 5]. ERMS originating in the uterine cervix differ from vaginal ERMS in that they often feature a later age of onset with approximately one-third of cases presenting in women aged 20 years and older [6]. The uterine corpus is an uncommon site for rhabdomyosarcoma, and most are of embryonal or pleomorphic subtypes.
Althugh most rhabdomyosarcomas are sporadic [4, 7], a genetic predisposition is suspected in a subset of cases. This is exemplified by the documented occurrence of rhabdomyosarcomas in the context of Li-Fraumeni syndrome (TP53 gene), retinoblastoma syndrome (RB1), Gorlin syndrome (PTCH1), Beckwith-Wiedemann syndrome (chr 11p15.5 region), and other tumor predisposition syndromes [8,9,10,11,12,13,14,15,16,17]. ERMS, especially cervical ERMS, has also emerged as an important component of DICER1 syndrome [18,19,20,21]. To date, 43 DICER1-related extracranial rhabdomyosarcomas from 40 patients have been reported, 31 (72%) of which involved the female genital tract (Supplementary Table S1). In a series of 80 next-generation sequenced rhabdomyosarcomas of various subtypes [22,23,24], the Cancer Genome Atlas Research Network identified only one DICER1-related neoplasm; this comprised an ERMS from a female patient harboring a somatic p.G1809R DICER1 “RNase IIIb hotspot” mutation with an allele frequency of 96%. The primary site was not reported [22].
Within the uterus (cervix or corpus), ERMS exhibits marked morphological overlap with adenosarcoma, especially when the latter exhibits rhabdomyoblastic differentiation or sarcomatous overgrowth [6, 25, 26]. This diagnostic challenge is highlighted in the literature with published cases of cervical ERMS having been misdiagnosed as adenosarcoma [4]. Traditionally considered a biphasic tumor, uterine adenosarcoma is now considered in most cases to be a mesenchymal neoplasm with “entrapment” of benign epithelial elements. A subset of adenosarcomas harbor the ESR1-NCOA2/3 fusion; the translocation is restricted to the mesenchymal component and is absent in the epithelial element [27]. In the uterus, a number of reports have now described DICER1 mutations in adenosarcoma [27,28,29,30]. These partially overlapping molecular abnormalities further augment the challenge of distinguishing between adenosarcoma and ERMS. The distinction is clinically important; whereas ERMS of the cervix is an established component of DICER1 syndrome, the association of adenosarcoma with DICER1 syndrome (and DICER1 mutations) is less well studied. Moreover, therapeutic management may differ for these two tumor types. Therefore, in this study, we investigated the prevalence of DICER1 alterations in a centrally reviewed series of 64 uterine tumors with an initial diagnosis of ERMS or adenosarcoma. We aimed to determine whether DICER1 genetic testing is useful in assisting in the distinction between ERMS and adenosarcoma.
Materials and methods
Case series and pathology review
This study was approved by the Research Ethics Board of the University Health Network in Toronto, ON, Canada (UHN CAPCR 16-5422) and the Institutional Review Board of the Faculty of Medicine of McGill University, Montreal, QC, Canada, no. A12-M117-11A and A08-M61-09B. Participants were recruited to the study in compliance with the second edition of the Canadian Tri-Council Policy Statement of Ethical Conduct of Research involving Humans.
To compare the involvement of DICER1 mutations in uterine ERMS and adenosarcoma, we reviewed the pathology of 64 uterine neoplasms where the diagnosis was thought to be adenosarcoma or ERMS. The tumors were reviewed independently by four pathologists of whom three (WGM, CJRS, BAC) are expert gynaecological pathologists and one, a pathology fellow (JYY). The pathology review was performed blinded to the molecular data, age at diagnosis, and site of origin. Features favoring adenosarcoma rather than ERMS included a prominent “phyllodes-like” architecture, stromal condensation surrounding glands, and glands throughout the neoplasm. For many, but not all cases, the skeletal muscle markers myogenin and myoD1 were available to aid in determining whether rhabdomyoblastic differentiation was present. A consensus diagnosis of “definitely ERMS” was rendered when: a) at least 3/4 pathologists indicated “definitely ERMS,” or b) 2/4 pathologists indicated “definitely ERMS,” plus one or more pathologists indicated “uncertain, favor ERMS.” A similar algorithm was used for a consensus diagnosis of “definitely adenosarcoma.” Other cases were classified as “uncertain” (no consensus) diagnoses.
Nineteen cases received a consensus diagnosis of ERMS, 27 cases of adenosarcomas and for the remaining 18 tumors, no consensus was achieved (Fig. 1). The adenosarcomas were further reviewed by one of the study pathologists (WGM) to assess for the presence of sarcomatous overgrowth and rhabdomyoblastic differentiation. Rhabdomyoblastic differentiation was diagnosed in the presence of obvious rhabdomyoblasts with abundant eosinophilic cytoplasm and/or the presence of nuclear immunoreactivity with myogenin and/or myoD1. Sarcomatous overgrowth was defined as areas of pure sarcoma without an epithelial component; in some cases, the areas of sarcomatous overgrowth exhibited rhabdomyoblastic differentiation (see below).

Abbreviation: ERMS, embryonal rhabdomyosarcoma.
Of the 64 tumors in this study, 6 have been published previously [31,32,33,34,35]. Fourteen of the ERMS arose in the cervix, two in the corpus, two involved the corpus and cervix and in one case, the origin within the uterus was not known. The median age at ERMS diagnosis was 30 years (range: 2.5–69 years). The site of the adenosarcomas was uterine corpus (n = 24) and cervix (n = 3); the median patient age at adenosarcoma diagnosis was 57.5 years (range 27–82 years). Sarcomatous overgrowth was present in five adenosarcomas, rhabdomyoblastic differentiation in one, and two cases exhibited both sarcomatous overgrowth and rhabdomyoblastic differentiation. The no consensus cases were diagnosed at a median age of 65.5 years (range 32–86 years); 1 originated in the cervix, 14 in the uterine corpus, and the origin within the uterus was not known for three cases.
DICER1 sequencing
Formalin-fixed, paraffin-embedded (FFPE) tumor tissue was obtained from all 64 neoplasms and matched non-tumoral tissue was available from 54 cases (Fig. 2). DNA was extracted from 3 to 7 10 µm sections of tissue using the QIAamp DNA FFPE Tissue Kit (QIAGEN, Toronto, ON, Canada). In brief, the DNA sequences containing the RNase IIIb hotspot mutation sites were screened in all 64 tumors by PCR amplification followed by Sanger sequencing. In addition, a custom-design Fluidigm Access Array [36] was successfully used to sequence the coding region and exon–intron boundaries of DICER1 (NM_177438) in DNA derived from 40 tumors (13 ERMS, 17 adenosarcomas, and 10 no consensus cases). A custom-design HaloPlexHS targeted capture (Agilent Technologies) was used to sequence the full DICER1 gene in a tumor-normal DNA pair from a single patient [37]. Variants were called in the Fluidigm- and HaloPlexHS-derived sequencing data using FreeBayes version 1.1.0.46-0 via the Galaxy toolshed (https://usegalaxy.org/) and annotation of functional consequences of variants was performed using wANNOVAR (http://wannovar.wglab.org/) [38]. Variants were manually visualized and assessed using the Integrative Genomics Viewer (http://software.broadinstitute.org/software/igv/). All mutations of interest were validated by Sanger sequencing. We performed PCR amplification and Sanger sequencing of the full DICER1-coding sequence in a single ERMS and two adenosarcomas that failed Fluidigm sequencing, that were RNase IIIb hotspot mutation-positive, and for which sufficient DNA was available (primer sequences are available on request). Sixteen cases only underwent screening for RNase IIIb hotspot mutations (Fig. 3). Germline versus somatic provenance of identified mutations was determined by site-specific PCR and Sanger sequencing of matched-normal DNA, where available (Fig. 2).

Ages at diagnosis and sites of origin are indicated, along with DICER1 mutation status (broken down by RNase IIIb hotspot and loss-of-function (LOF)), the tumor sequencing method employed, and the availability of matched-normal DNA. In the tumor type column, the presence of rhabdomyoblastic differentiation (RD) and sarcomatous overgrowth (SO) is indicated for adenosarcoma (AS) cases. Where a diagnosis of AS or embryonal rhabdomyosarcoma (ERMS) was favored for the no consensus cases, this has been indicated. Abbreviations: NI, none identified; NOS, not otherwise specified; LOH, loss of heterozygosity. Note: *On subsequent review, the imaging studies, age at diagnosis, and molecular findings all suggest that this is likely to be a primary vaginal tumor.

Boxes shaded in blue describe sequencing methods employed; green and gray colored boxes indicate positive and negative DICER1 testing results, respectively. The total number of samples relevant to each sequencing method or testing result is provided in square brackets and the number of samples from each pathology group (ERMS, adenosarcoma, or no consensus cases) is indicated below each box (see color key). Final DICER1 results are presented at the bottom in boxes with dashed lines.
Assessment for loss of heterozygosity (LOH) of the wild-type allele was performed in cases in which an RNase IIIb mutation was identified in the absence of a loss-of-function variant. This was achieved by PCR amplification of tumor DNA concurrently with the matched-normal DNA using primers specific to the regions flanking common single-nucleotide polymorphisms (SNPs) in the 3′-UTR of DICER1. The ~ 150–200 base-pair PCR products were analyzed by direct Sanger sequencing and the relative intensity of the peaks at the location of informative SNPs was compared between germline and tumor DNA to assess for potential LOH.
Results
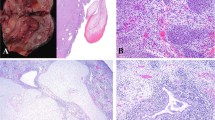
The results of the DICER1 genetic testing on the 19 tumors with a consensus diagnosis of ERMS are presented in Fig. 2 and Table 1. Also in Fig. 2 is a summary of the DICER1 results and the histological features of the 27 adenosarcomas and 18 no consensus cases (Tables 2 and 3, respectively). An algorithm illustrating the workflow leading to the final mutation tallies is shown in Fig. 3. Figure 4 provides an illustration of the typical features of ERMS, and Fig. 5 illustrates examples of adenosarcomas.

a Low power view showing hypocellular edematous areas and focal cellular areas. b Higher power view showing increased cellularity just deep to the cervical surface squamous epithelium (left of photomicrograph) resulting in a cambium layer. c Cellular areas composed of small cells with hyperchromatic nuclei. d Foci of cartilage are not uncommon in these neoplasms. e Positive cytoplasmic staining of cells with the muscle marker desmin and f the specific skeletal muscle marker myogenin.

a Low power view showing phyllodes-like architecture. b There is increased cellularity surrounding the glands, resulting in a cambium layer. c Mitoses are easily identified within the cambium layer. d Area of rhabdomyoblastic differentiation within adenosarcoma; these areas exhibited focal nuclear immunoreactivity with the skeletal muscle markers myogenin and myoD1 (not shown).
Embryonal rhabdomyosarcomas
All ERMS exhibited the classical histological appearance comprising polypoid neoplasms with hypocellular and hypercellular foci, the latter containing cells with ovoid to spindled hyperchromatic nuclei and scant cytoplasm [6]. Typically, there was increased cellularity just deep to the surface epithelium, resulting in a cambium layer. Foci of cartilage were present in 10/19 (53%) cases. In all cases, there was positive cytoplasmic staining of a proportion of cells with the pan-muscle marker desmin and at least some of the nuclei were positive with the specific skeletal muscle markers myogenin and/or myoD1 (Fig. 4).
The region encoding the RNase IIIb domain was sequenced in all 19 ERMS: 18/19 (95%) ERMS were found to harbor a DICER1 RNase IIIb hotspot mutation (hereafter “RNase IIIb positive”) and 1 tumor (5%) was RNase IIIb wild-type. Genomic DNA from the latter tumor was fully sequenced and no likely pathogenic variants in DICER1 were found. Of the 18 RNase IIIb hotspot mutations found, 12 were confirmed to be somatic, but previous studies have shown that RNase IIIb hotspot mutations are nearly always present in the tumor only [39, 40], and thus it is highly likely that all 18 of the mutations are somatic in origin. Of the 18 ERMS bearing one or more DICER1 alterations, 13 (68%) bore two DICER1 alterations consisting of one loss-of-function variant and one RNase IIIb hotspot mutation. No additional likely pathogenic DICER1 alterations were identified in the remaining five RNase IIIb-positive ERMS for which full gene sequencing was successful. However, LOH of the wild-type allele was not assessable in the latter five tumors owing to a lack of informative SNPs in the 3′-UTR of DICER1, so the “single hit” status of these five cases remains conjectural. A single cervical ERMS had no identifiable DICER1 alterations and subsequent review of the clinical, pathological and radiological features suggested it was most likely to represent a primary vaginal tumor (see discussion). These results suggest that all uterine (i.e., cervix and corpus) ERMS harbor at least one DICER1 alteration and that in most cases, a loss-of-function and an RNase IIIb somatic mutation is found.
Identifying patients with potential DICER1 syndrome is an increasingly important role of the pathologist and geneticist. In our study, six ERMS patients were found to bear a predisposing germline loss-of-function DICER1 pathogenic variant. A further seven ERMS harbored an additional loss-of-function DICER1 mutation or LOH, five of which were confirmed to be somatic in origin, thus ruling out DICER1 syndrome. The provenance of the other two loss-of-function alterations is not known (Table 1 and Fig. 2).
There were no pathological features that distinguished between the syndromic (i.e., germline DICER1 pathogenic variant plus RNase IIIb positive) ERMS cases compared with the cases with two somatic DICER1 mutations. Five of the six ERMS with germline variants arose in the cervix, and the other involved the cervix and corpus. Neither of the two tumors that involved the uterine corpus only (as opposed to the cervix only) was shown to have a germline DICER1 variant (Fig. 2 and Table 1).
Adenosarcomas
DICER1 testing revealed 7/27 (26%) adenosarcomas and 4/18 (22%) no consensus cases to be RNase IIIb positive (Fig. 2, Tables 2 and 3). Thus, DICER1 alterations were significantly less frequent in uterine adenosarcomas than in uterine ERMS (Fisher’s exact test p < 0.001). The full coding region was successfully screened in 19/27 adenosarcomas: three adenosarcomas, all of which were positive for RNase IIIb hotspot mutation, were found to harbor an additional loss-of-function or splice region DICER1 alteration. Two of the four RNase IIIb positive no consensus tumors had an additional inactivating DICER1 alteration. No likely pathogenic variants were identified in the other two no consensus tumors bearing a hotspot mutation; however, their loss of heterozygosity status could not be assessed. The 11 RNase IIIb hotspot mutations identified in adenosarcomas and no consensus cases were proven (n = 10) or considered (n = 1) to be somatic. In addition, we found three loss-of-function mutations, one splice region mutation and one LOH event, all of which were proven to be somatic in origin. Age at diagnosis of patients with an adenosarcoma bearing one or more DICER1 alterations ranged from 32 to 82 years with a median age of 58 years (versus a median age of 50 years for DICER1 wild-type neoplasms), and the age range for DICER1-mutated no consensus cases was 32–75 years (median 60.5 years).
No histological features distinguished DICER1 wild-type from DICER1-mutated adenosarcomas in our study (Fig. 5); two DICER1-mutated adenosarcomas exhibited sarcomatous overgrowth and one exhibited rhabdomyoblastic differentiation. In comparison, three DICER1 wild-type adenosarcomas exhibited sarcomatous overgrowth and two exhibited both sarcomatous overgrowth and rhabdomyoblastic differentiation (Fig. 2 and Table 2). Cartilaginous elements were not present in any of the adenosarcomas.
Literature review of DICER1 alterations in uterine adenosarcomas
Review of the published literature identified a total of 74 cases of uterine adenosarcoma that have undergone DICER1 genetic testing, 13 (18%) of which were reported to harbor one or more pathogenic somatic DICER1 alterations (Supplementary Table S2) [27,28,29,30]. One additional uterine adenosarcoma has been reported in the setting of a germline DICER1 pathogenic variant [41]. In a recent series of 19 adenosarcomas enriched for tumors exhibiting rhabdomyoblastic differentiation, 8/19 (42%) were found to harbor DICER1 mutations, 4 of which exhibited rhabdomyoblastic differentiation [30]. Combined with our data, DICER1 mutations have been identified in 20/101 (20%) uterine adenosarcomas. Where documented, rhabdomyoblastic differentiation was present in 15/81 (19%) cases of which 8 (53%) harbored DICER1 mutations; this represents a significantly higher prevalence of DICER1 mutations in adenosarcomas exhibiting rhabdomyoblastic differentiation, compared with those without this feature (Fisher’s exact test p value = 0.0034). Where information was available, 35/99 cases exhibited sarcomatous overgrowth, 8 (23%) of which were DICER1 mutated; this difference in prevalence is not statistically significant compared with cases lacking sarcomatous overgrowth (Fisher’s exact test p value = 0.61) (Supplementary Table S2). Of the 20 DICER1-mutated cases, four (20%) exhibited both sarcomatous overgrowth and rhabdomyoblastic differentiation; 7/18 (39%) DICER1-mutated cases with available information lacked these histological features. Thus, whereas rhabdomyoblastic differentiation is significantly more common in DICER1-mutated adenosarcomas, this feature is not a reliable indicator of the presence of DICER1 alterations.
Discussion
DICER1 functions canonically as an RNase III endoribonuclease in the RNA interference pathway where it is centrally involved in the biogenesis of microRNAs. These small non-coding RNAs negatively regulate gene expression by targeting their messenger RNA transcripts for degradation [39]. DICER1 syndrome is a rare, mainly pediatric tumor predisposition syndrome caused by biallelic mutations in the DICER1 gene (OMIM #601200). The predisposing DICER1 mutations are most often loss-of-function variants that inactivate one copy of DICER1. The second somatic mutations in most syndrome-related tumors are highly characteristic missense “RNase IIIb hotspot” mutations [39, 40]. The syndrome phenotype is pleiotropic and characterized by a unique assortment of hyperplastic and neoplastic disorders, including thyroid multinodular goiter, hamartomatous polyps, pleuropulmonary blastoma, ovarian Sertoli-Leydig cell tumor (SLCT) and pediatric cystic nephroma, among others [39]. Previous studies have shown that extracranial sarcomas associated with DICER1 syndrome are most frequently ERMS involving the urogenital tract [18, 42].
The pathological diagnosis of ERMS of the female genital tract is sometimes difficult with significant morphological overlap with other tumor types, especially adenosarcoma; this is further complicated by the fact that adenosarcoma can exhibit rhabdomyoblastic differentiation. Thus, we undertook a blinded pathological review of accessioned uterine cases submitted with a diagnosis of ERMS or adenosarcoma. The results of the review illustrate the diagnostic difficulties with no consensus in diagnosis achieved in a significant number of cases (28%); in many of these, different pathologists made a diagnosis of ERMS or adenosarcoma on the same case. We identified an overall DICER1 mutation prevalence of 95% in our cases of uterine ERMS and 26% in our uterine adenosarcomas. The genetic alterations identified were typical of those associated with DICER1 syndrome. One of the original cases with a consensus diagnosis of cervical ERMS lacked DICER1 alterations. This tumor was diagnosed at a younger age (2.5 years) than the other cases in the study and review of the radiology suggested the origin of the tumor to be the vagina. This tumor did not contain cartilage, which is a soft pointer against a cervical origin as a significant percentage of primary cervical ERMS (53% of cases in this study) contain cartilage while this is rarely a feature of primary vaginal ERMS. Similarly, when dealing with a uterine neoplasm where the differential includes ERMS and adenosarcoma, the presence of cartilage is a pointer favoring the former diagnosis since none of the adenosarcomas in our series contained cartilage. The association of vaginal ERMS and DICER1 is not well studied; three cases have been reported in association with a germline DICER1 alteration, one of which underwent somatic testing and was RNase IIIb positive (Supplementary Table S1). Our findings suggest that all uterine (i.e., cervix and corpus) ERMS are DICER1 related and cervical ERMS is especially likely to be owing to germline DICER1 pathogenic variants.
The potential use of genetic testing in the diagnostic work-up of another DICER1 syndrome-associated tumor, ovarian SLCT, has been proposed by our group [43]. In a recent study, we demonstrated that for ovarian sex cord-stromal tumors with an uncertain diagnosis (which is not uncommon given the significant morphological overlap between the various tumor types) and SLCT in the differential, DICER1 RNase III sequencing of the tumor may assist in differentiating non-SLCTs and well differentiated SLCTs (DICER1 wild-type) from moderately and poorly differentiated SLCTs, as the latter, based on our findings, are highly likely to be DICER1-mutated (30/30 tested cases) [43]. However, a recent study has shown that some moderately and poorly differentiated SLCTs in patients over the age of 50 years may instead harbor a somatic FOXL2 hotspot mutation, this mutation being characteristic of adult granulosa cell tumor [44]. Therefore, DICER1 mutations cannot unequivocally differentiate between adult granulosa cell tumor and SLCT, especially in older patients. In our SLCT study, we suggested that all patients with a diagnosis of ovarian SLCT should undergo germline DICER1 genetic testing [43].
A somewhat similar scenario exists in the uterus where, as already discussed, there is morphological overlap between ERMS and adenosarcoma. As we have shown, biallelic DICER1 alterations are frequent in uterine (cervix and corpus) ERMS [31,32,33,34,35, 45,46,47,48,49,50], and it would be of great diagnostic value if mutations did not occur in adenosarcoma. However, DICER1 mutations are present in an appreciable minority (20%) of uterine adenosarcomas [27, 28, 30] and it is clear that demonstration of a DICER1 mutation cannot distinguish between uterine ERMS and adenosarcoma. The presence of a DICER1 mutation in a uterine sarcoma with rhabdomyoblastic differentiation does not mean, on its own, that the tumor should be classified as rhabdomyosarcoma. Thorough tumor sampling and careful morphologic examination are required. The absence of a mutation is however helpful in excluding ERMS. Nevertheless, the frequency of DICER1 variants in these two tumors is very different. Importantly, whereas a diagnosis of ERMS, especially if arising in the cervix, potentially has important clinical implications for the patient and her family given its strong association with DICER1 syndrome, only rarely are uterine adenosarcomas associated with germline DICER1 pathogenic variants [41]. Occasional uterine carcinosarcomas also exhibit DICER1 mutations [51], but these too are non-germline; moreover, carcinosarcomas do not exhibit significant morphological overlap with ERMS, and they typically occur in elderly patients. Although we did not identify germline DICER1 alterations in two patients with uterine ERMS confined to the corpus, a 10 year-old girl with an RNase IIIb-positive uterine corpus ERMS has recently been reported to have DICER1 syndrome (Supplementary Table S1) [52].
In conclusion, we show that DICER1 alterations are significantly more prevalent in uterine ERMS compared with uterine adenosarcoma. However, DICER1 genetic testing is not useful in distinguishing between these tumor types, unless negative in which case ERMS is unlikely. Germline DICER1 pathogenic variants are likely to be sufficiently frequent in uterine ERMS arising in either the corpus or cervix that all such patients should be referred to medical genetic services for consideration of germline testing.
References
Fletcher C, Hogendoorn P, Merterns F, Bridge J (eds.). World Health Organisation classification of tumours of soft tissue and bone. International Agency for Research on Cancer Press: Lyon, France, 2013.
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64:83–103.
Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27:3391–7.
Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol. 2012;25:602–14.
Nasioudis D, Alevizakos M, Chapman-Davis E, Witkin SS, Holcomb K. Rhabdomyosarcoma of the lower female genital tract: an analysis of 144 cases. Arch Gynecol Obstet. 2017;296:327–34.
Li RF, Gupta M, McCluggage WG, Ronnett BM. Embryonal rhabdomyosarcoma (botryoid type) of the uterine corpus and cervix in adult women: report of a case series and review of the literature. Am J Surg Pathol. 2013;37:344–55.
Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. 1999;4:34–44.
Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest. 1995;95:1606–11.
Rodjan F, Graaf Pd, Brisse HJ, Verbeke JILM, Sanchez E, Galluzzi P, et al. Second cranio-facial malignancies in hereditary retinoblastoma survivors previously treated with radiation therapy: Clinic and radiologic characteristics and survival outcomes. Eur J Cancer 2013;49:1939–47.
Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Mugica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol. 2006;3:575.
Brioude F, Lacoste A, Netchine I, Vazquez MP, Auber F, Audry G, et al. Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr. 2013;80:457–65.
Hanks S, Coleman K, Reid S, Plaja A, Firth H, FitzPatrick D, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159.
Coffin CM, Cassity J, Viskochil D, Randall RL, Albritton K. Non-neurogenic sarcomas in four children and young adults with neurofibromatosis type 1. Am J Med Genet A. 2004;127a:40–3.
Oguzkan S, Terzi YK, Guler E, Derbent M, Agras PI, Saatci U, et al. Two neurofibromatosis type 1 cases associated with rhabdomyosarcoma of bladder, one with a large deletion in the NF1 gene. Cancer Genet Cytogenet. 2006;164:159–63.
Crucis A, Richer W, Brugieres L, Bergeron C, Marie-Cardine A, Stephan JL, et al. Rhabdomyosarcomas in children with neurofibromatosis type I: a national historical cohort. Pediatr Blood Cancer. 2015;62:1733–8.
Gripp KW, Scott CI Jr, Nicholson L, McDonald-McGinn DM, Ozeran JD, Jones MC, et al. Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. Am J Med Genet. 2002;108:80–7.
Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet Part C, Semin Med Genet. 2011;157:83–9.
de Kock L, Rivera B, Revil T, Thorner P, Goudie C, Bouron-Dal Soglio D, et al. Sequencing of DICER1 in sarcomas identifies biallelic somatic DICER1 mutations in an adult-onset embryonal rhabdomyosarcoma. Br J Cancer. 2017;116:1621–6.
Stewart CJ, Charles A, Foulkes WD. Gynecologic manifestations of the DICER1 syndrome. Surg Pathol Clin. 2016;9:227–41.
Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965.
Messinger YH, Stewart DR, Priest JR, Williams GM, Harris AK, Schultz KA, et al. Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer. 2015;121:276–85.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.
Oberg JA, Glade Bender JL, Sulis ML, Pendrick D, Sireci AN, Hsiao SJ, et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med. 2016;8:133.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31.
McCluggage WG. Mullerian adenosarcoma of the female genital tract. Adv Anat Pathol. 2010;17:122–9.
Clement PB. Müllerian adenosarcomas of the uterus with sarcomatous overgrowth. A clinicopathological analysis of 10 cases. Am J Surg Pathol. 1989;13:28–38.
Piscuoglio S, Burke KA, Ng CK, Papanastasiou AD, Geyer FC, Macedo GS, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol. 2016;238:381–8.
Howitt BE, Sholl LM, Dal Cin P, Jia Y, Yuan L, MacConaill L, et al. Targeted genomic analysis of Mullerian adenosarcoma. J Pathol. 2015;235:37–49.
Hodgson A, Amemiya Y, Seth A, Djordjevic B, Parra-Herran C. High-grade mullerian adenosarcoma: genomic and clinicopathologic characterization of a distinct neoplasm with prevalent TP53 pathway alterations and aggressive behavior. Am J Surg Pathol. 2017;41:1513–22.
Bean GR, Anderson J, Sangoi AR, Krings G, Garg K. DICER1 mutations are frequent in mullerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol. 2018;32:280–89.
Rio Frio T, Bahubeshi A, Kanellopoulou C, Hamel N, Niedziela M, Sabbaghian N, et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA. 2011;305:68–77.
Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N. Engl J Med. 2012;366:234–42.
Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat. 2011;32:1381–4.
Tomiak E, de Kock L, Grynspan D, Ramphal R, Foulkes WD. DICER1 mutations in an adolescent with cervical embryonal rhabdomyosarcoma (cERMS). Pediatr Blood Cancer. 2014;61:568–9.
de Kock L, Boshari T, Martinelli F, Wojcik E, Niedziela M, Foulkes WD. Adult-onset cervical embryonal rhabdomyosarcoma and DICER1 mutations. J Low Genit Trac Dis. 2016;20:e8–e10.
de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Soglio DB-D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014;128:111–22.
de Kock L, Hillmer M, Wagener R, Soglio DB, Sabbaghian N, Siebert R, et al. Further evidence that full gene deletions of DICER1 predispose to DICER1 syndrome. Genes Chromosomes Cancer. 2018; https://doi.org/10.1002/gcc.22728.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72.
de Kock L, Wu MK, Foulkes WD. Ten years of DICER1 mutations: provenance, distribution, and associated phenotypes. Hum Mutat. 2019;40:1939–53.
Mullen MM, Divine LM, Hagemann IS, Babb S, Powell MA. Endometrial adenosarcoma in the setting of a germline DICER1 mutation: a case report. Gynecol Oncol Rep. 2017;20:121–4.
de Kock L, Foulkes WD. Sarcoma and germ-line DICER1 mutations. Lancet Oncol. 2016;17:e470.
de Kock L, Terzic T, McCluggage WG, Stewart CJR, Shaw P, Foulkes WD, et al. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli-Leydig cell tumors. Am J Surg Pathol. 2017;41:1178–87.
Karnezis AN, Wang Y, Keul J, Tessier-Cloutier B, Magrill J, Kommoss S, et al. DICER1 and FOXL2 mutation status correlates with clinicopathologic features in ovarian sertoli-leydig cell tumors. Am J Surg Pathol. 2019;43:628–38.
Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000 Res. 2015;4:214.
Cowan M, Suntum T, Olivas AD, Perpich M, Applebaum MA, Lastra RR, et al. Second primary rhabdomyosarcoma of the uterine cervix presenting with synchronous ovarian Sertoli-Leydig cell tumor: an illustrative case of DICER1 syndrome. Gynecol Oncol Rep. 2018;25:94–7.
Diets IJ, Waanders E, Ligtenberg MJ, van Bladel DAG, Kamping EJ, Hoogerbrugge PM, et al. High yield of pathogenic germline mutations causative or likely causative of the cancer phenotype in selected children with cancer. Clin Cancer Res. 2018;24:1594–603.
Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. 2012;59:558–60.
Gullo I, Batista R, Rodrigues-Pereira P, Soares P, Barroca H, do Bom-Sucesso M, et al. Multinodular goiter progression toward malignancy in a case of dicer1 syndrome: histologic and molecular alterations. Am J Clin Pathol. 2018;149:379–86.
Moke DJ, Thomas SM, Hiemenz MC, Nael A, Wang K, Shillingford N, et al. Three synchronous malignancies in a patient with DICER1 syndrome. Eur J Cancer. 2018;93:140–3.
Chen J, Wang Y, McMonechy MK, Anglesio MS, Yang W, Senz J, et al. Recurrent DICER1 hotspot mutations in endometrial tumours and their impact on microRNA biogenesis. J Pathol. 2015;237:215–25.
Dural O, Kebudi R, Yavuz E, Yilmaz I, Bay SB, Schultz KAP, et al. DICER1 related embryonal rhabdomyosarcoma of the uterine corpus in a prepubertal girl. J Pediatr Adolesc Gynecol 2019; https://doi.org/10.1016/j.jpag.2019.12.002.
Acknowledgements
WDF was funded by the Canadian Institutes of Health Research, Foundation Grant FDN-148390. MAR is a Garber Family Post-doctoral Fellow in Hereditary Cancer and a Dr. David T.W. Lin Fellowship awardee. JYY was funded by a Physicians' Services Incorporated Foundation resident research grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
de Kock, L., Yoon, JY., Apellaniz-Ruiz, M. et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol 33, 1207–1219 (2020). https://doi.org/10.1038/s41379-019-0436-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0436-0
This article is cited by
-
Familial syndromes associated with testicular and paratesticular neoplasms: a comprehensive review
Virchows Archiv (2024)
-
Genomic characterization of DICER1-associated neoplasms uncovers molecular classes
Nature Communications (2023)
-
Mullerian adenosarcoma: clinicopathologic and molecular characterization highlighting recurrent BAP1 loss and distinctive features of high-grade tumors
Modern Pathology (2022)
-
Endometrial polyps are non-neoplastic but harbor epithelial mutations in endometrial cancer drivers at low allelic frequencies
Modern Pathology (2022)
-
DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma
Modern Pathology (2022)
