
Abstract
Merkel cell carcinoma (MCC) is a high grade primary cutaneous neuroendocrine carcinoma and is among the most aggressive cutaneous malignancies. The rising incidence of MCC, together with its often rapidly aggressive course, underscore a critical need to recognize the histopathologic and the immunohistochemical features that inform its accurate diagnosis. In the current review, we summarize the current state of knowledge regarding the accurate diagnosis of MCC and the exclusion of other entities in the differential diagnosis. We provide a comprehensive review of genomic studies that identified the molecular-genetic drivers of MCC as well as a summary of studies identifying prognostic biomarkers that can facilitate risk stratification. Importantly, Merkel cell polyomavirus (MCPyV) appears to be causative in most cases of MCC and represents both a diagnostic and prognostic marker. Finally, as staging of MCC has undergone critical refinements with the introduction of the 8th Edition of the American Joint Committee on Cancer staging system, we provide an update on MCC staging. In particular, the prognostic significance of the sentinel lymph node (SLN) in MCC necessitates a systematic approach to its evaluation and diagnosis to ensure accurate and consistent risk stratification for patients, and we therefore provide a comprehensive overview of SLN evaluation in MCC. Finally, the intimate relationship between MCC and the integrity of the host immune system has led to paradigm-shifting therapeutic advances with the successful application of immune checkpoint blockade to treat patients with advanced disease, and we therefore summarize those studies and the correlative studies in which predictive biomarkers have been identified.
Similar content being viewed by others

Diagnosis of Merkel cell carcinoma
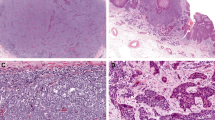
Accurate diagnosis of MCC begins with the recognition of its distinctive cytomorphology in the typical clinical context of a rapidly enlarging lesion on sun-exposed sites of elderly individuals [1, 2]. At scanning magnification (Fig. 1a), MCC typically grows as a dermal malignancy that often extends to involve the subcutis. An intraepidermal component may exist, but is infrequent and rarely extensive. The leading edge of MCC exhibits either infiltrative or pushing borders, and the tumor cells are usually accompanied by a variably dense lymphohistiocytic inflammatory infiltrate (Fig. 1b). Stromal mucin may also be present and represents an important diagnostic pitfall (see below). MCC tumor cells are arranged as sheets, cords, and trabeculae. The tumor cells of MCC show a characteristic neuroendocrine cytomorphology with scant cytoplasm and uniform round to oval nuclei with finely granular (‘salt and pepper’) chromatin and inconspicuous, small nucleoli. Mitotic figures and apoptotic bodies are often numerous (Fig. 1c), and thus, areas of geographic necrosis are similarly common. In most cases of MCC, the tumor cells are intermediate in size, but large cell and small cell morphologies also occur [2, 3].

Cytokeratin 20-positive Merkel cell carcinoma. a Dense proliferation of tumor cells diffusely effaces the dermis without involvement of the overlying epidermis (H&E, ×20). b Tumor cells arranged as sheets, cords, and trabeculae (H&E, ×100). c Higher magnification reveals cytologic features of Merkel cell carcinoma. The tumor cells contain minimal amounts of cytoplasm and medium sized oval nuclei with finely granular chromatin. The cells exhibit nuclear molding, numerous mitotic figures and apoptotic bodies (H&E, ×400). Merkel cell carcinoma is typically positive for d perinuclear dot-like cytokeratin 20 (×200), e synaptophysin (×200), f chromogranin (×200), but is negative for g TTF-1 (×200). Cytokeratin 20-negative Merkel cell carcinoma with small cell morphology. h Dense proliferation of tumor cells effaces the superficial dermis with ulceration of the overlying epidermis (H&E, ×20). i Higher magnification reveals small cell nuclear features of Merkel cell carcinoma. The tumor cells contain minimal amounts of cytoplasm, and in this case display small oval nuclei with finely granular chromatin. The cells exhibit nuclear molding, numerous mitotic figures and apoptotic bodies (H&E, ×400). Merkel cell carcinoma in this case is positive for j pancytokeratin cocktail (×200), but negative for k cytokeratin 20 (×200). The tumor cells are diffusely positive for l synaptophysin (×200) and m Neurofilament (×200) with perinuclear dot-like positivity, but is negative for (N) TTF-1 (×200)
Despite a relatively characteristic appearance, immunohistochemical (IHC) studies are required both to confirm the diagnosis and to exclude other entities with overlapping morphologies, including poorly differentiated primary cutaneous malignancies and metastases to the skin (Tables 1 and 2). MCCs show positivity for cytokeratins, frequently with some degree of perinuclear dot-like positivity. The most specific marker is cytokeratin 20 (CK20; Fig. 1d). A comprehensive review of the literature revealed 87.4% of MCCs (716/819) to be CK20+ (Table 1) [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. Because MCCs are neuroendocrine carcinomas, they show evidence of neuroendocrine differentiation, including synaptophysin (92.0%, 115/125; Fig. 1e); chromogranin (84.1%, 111/132; Fig. 1f) neuron specific enolase (97.5%, 39/40) and CD56 (88.2%, 30/34), respectively (Table 2). Neurofilament (NF) is an additional sensitive marker, with specificity for MCC over most other neuroendocrine carcinomas, and is expressed in 79.7% of MCC (Table 1). Given the strong reliance on CK20 in the diagnosis of MCC, CK20-negative MCCs represent a particularly difficult challenge (Fig. 1h–n, which also depicts a tumor with ‘pushing’ borders). For these cases, NF-positivity together with TTF-1 negativity (Fig. 1m, n) inform the diagnosis; MCPyV staining has limited sensitivity [34]. Notably, cytokeratin 7 (CK7) and Terminal deoxynucleotidyl transferase (TdT) have been reported to be positive in 17.6% (43/245) and 25.9% (29/112) MCCs, respectively, and represent important pitfalls when making the diagnosis of MCCs (Table 2). Recently, it was found that a substantial minority of MCPyV-negative MCC demonstrate divergent immunophenotypic findings including TTF-1 positivity, and absence of NF and/or CK20 [29, 32].
MCC occasionally presents with divergent differentiation. Although multiple reports describe glandular differentiation [35,36,37,38], the most commonly encountered divergent differentiation is squamous, often present in multiple foci throughout the tumor [36, 39,40,41,42,43,44] (Fig. 2a, b). Alternatively, distinct areas of invasive SCC or SCCIS may be present; this can represent collision phenomenon rather than transdifferentiation [45]. Rare forms of divergent differentiation include sarcomatoid or neuroblastic [2].

Merkel cell with divergent differentiation and mimics with other malignancies. Merkel cell may present with a, b evidence of squamous differentiation (H&E; ×200). Merkel cell carcinoma mimicking basal cell carcinoma (c) with peripheral palisading, mucinous stroma and retraction artifact (H&E, ×200). d Merkel cell carcinoma infrequently exhibits intraepidermal growth, but intraepidermal growth of MCC is rarely extensive when present (H&E, ×200)
The most commonly encountered differential diagnostic considerations include basal cell carcinoma (BCC), melanoma, hematologic malignancies, and metastatic neuroendocrine carcinomas secondarily involving the skin (most commonly small cell carcinoma of the lung) (Table 3 and Fig. 3a–c). At scanning magnification, BCC represents the closest mimic to MCC, with nodular to infiltrative aggregates of basaloid cells in the dermis. Morphologic clues that favor the diagnosis of BCC over MCC include a connection to the overlying epidermis, peripheral palisading of basaloid cells in the nests of BCC and retraction artifact between the basaloid nests and the characteristic stroma associated with BCC (Fig. 3a, c). MCC may disperse as single cells into stromal mucin, unlike BCC. Although the distinction is usually made after careful histopathologic examination, cases in which MCC shows features of BCC (including peripheral palisading and retraction artifact; Fig. 2c) have been described, and stromal mucin is not uncommon in MCC. In addition, some cases of BCCs may exhibit evidence of finely granular (neuroendocrine type) chromatin and/or an exceptionally high mitotic rate. For those rare instances in which the histomorphologic distinction between MCC and BCC is not definitive, an IHC panel including CK20 and CK5/6 efficiently facilitates this distinction: MCCs are CK20+ and CK5/6− (the exception being MCCs with squamous differentiation), whereas BCCs are CK20− and CK5/6+ [46]. Neuroendocrine markers may be expressed in BCC, and hence are not useful for this distinction [2]. As a high grade cutaneous malignancy with an often aggressive clinical course, cutaneous melanoma (CM) often enters into the differential diagnosis of MCC (Fig. 3d–f). Distinctive features of CM include extensive intraepidermal growth (Fig. 3e) which is far more common in CM than MCC, in which intraepidermal growth is infrequently observed, and rarely extensive when present (Fig. 2d), intracytoplasmic pigmentation, and nuclear cytomorphology as melanoma cells typically show coarse chromatin and a prominent nucleolus. Furthermore, rare cases of melanoma exhibit a small cell cytomorphology (Fig. 3f). These cases pose diagnostic difficulty and require IHC studies to confirm the diagnosis. A wide range of hematolymphoid malignancies (both primary and those secondarily involving the skin) might also be considered in the differential diagnosis with MCC—most notably those which show CD56-positivity, including NK/T cell lymophoma (Fig. 3g–i) and blastic plasmacytoid dendritic cell neoplasm. When considering these, a comprehensive IHC panel should be employed to confirm lymphoid or myeloid lineage and exclude MCC. In addition to CD56, MCC may express TdT or Pax5 in a minority of cases, but will be uniformly negative for most other lymphoid and myeloid markers [2, 3].

Differential diagnosis of Merkel cell carcinoma. a Basal cell carcinoma with nodular pushing borders (H&E, ×20) and b high density cellularity (H&E, ×100). c Basal cell carcinoma more commonly exhibits peripheral palisading, mucinous stroma and retraction artifact (H&E, ×400). d Cutaneous melanoma showing vertical growth phase (H&E, ×20). e Melanoma in situ showing extensive intraepidermal involvement with pagetoid migration (H&E, ×200). f Invasive melanoma may mimic MCC, particularly when it exhibits small cell features (H&E, ×400). g Cutaneous involvement by extranodal NK/T cell lymphoma showing effacement of the dermis (H&E, ×20). h Higher power shows perivascular distribution of the tumor cells (H&E, ×200). i Tumor cells grow as clusters and sheets with variable cytomorphologic size and atypia (H&E, ×400)
One of the more challenging histopathologic differential diagnostic consideration for MCC is excluding a cutaneous metastasis from a visceral high grade neuroendocrine carcinoma—most commonly small cell carcinoma of the lung (SCLC). Histopathologically, these carcinomas would be indistinguishable from MCC, necessitating the application of IHC to facilitate the distinction. In general, MCC is CK20+ (87%; 716/819), NF+ (80%; 400/502), and TTF-1-negative (7% positive; 35/476), whereas SCLC is CK20-negative (5% positive; 18/337) and NF-negative (0/143 positive), but TTF-1+ (88%; 190/217) (Tables 1 and 4). [5,6,7,8,9,10, 12, 15, 18,19,20,21,22,23,24,25, 28, 47]. When CK20 is expressed in SCLC, the pattern is typically focal [26]. MCPyV large T-antigen (LT-ag) also represents a specific but incompletely sensitive finding for MCC, as discussed below. Although MCPyV-negative MCC may deviate from the classic pattern as described above, in such cases TTF-1 is typically weak or absent [32] with rare exceptions [48, 49]. In contrast, TTF-1 classically demonstrates diffuse strong staining in most SCLC.
Small cell neuroendocrine carcinomas from nonpulmonary extracutaneous sites may also enter the differential diagnosis, especially when metastatic MCC of unknown primary is encountered. In this setting, CK20 (if greater than focal) and NF expression are specific for distinguishing MCC from neuroendocrine carcinomas arising at other sites [26], with the exception of small cell carcinomas arising from the salivary glands and uterine cervix [2, 26, 50]. The presence of HPV and absence of MCPyV is characteristic of uterine cervical SmCC [50, 51], whereas no specific marker distinguishes MCC from salivary SmCC [2].
Based purely on cytomorphologic overlap, a final (albeit rare) differential diagnostic consideration would be extraskeletal Ewing sarcoma (ES). Like MCC, ES consists of small tumor cells with oval nuclei and minimal cytoplasm. The tumor cells of ES are typically centered on blood vessels. Finally, IHC studies show that ES is only rarely positive for keratins and never for CK20. Instead, ES typically exhibits positivity for CD99, FLI-1, and S100 [52, 53].
Demographics and incidence of Merkel cell carcinoma
MCC is predominantly a disease of older Caucasian men on sun-exposed sites. A review of 14,414 patients with MCC in the National Cancer Database (1998–2012) revealed 62.1% of patients were men, 69.5% were ≥70 years of age and 96.4% were Caucasian. Moreover, 42.6% of cases arose on the head and neck, while an additional 23.6% arose on sun-exposed sites of upper limb and shoulder [1]. Risk factors for the development of MCC thus include old age, chronic sun exposure and immunosuppression. The incidence of MCC continues to rise. In the United States, alone, incidence of MCC has increased significantly over the past 3 decades: from 1.5 cases per million in 1986 to 4.4 cases per million in 2001 to 7.9 cases per million in 2011 [54,55,56].
Merkel cell carcinomagenesis: Virus or ultraviolet light exposure represent key drivers
The common co-existence of MCC in immunosuppressed patients suggested a relationship to an underlying pathogen. This hypothesis was confirmed as RNA sequencing on MCC tumors identified a novel human polyomavirus, MCPyV, in 8/10 (80%) of tumors [57]. Larger series have confirmed MCPyV is present in ~60–80% of MCC [58]. Only 11% (10/84) control tissues contained MCPyV DNA, and the majority of those showed comparatively lower copy numbers. Although highly variable between tumors, the genomic site of MCPyV DNA integration into MCC tumor cells is consistent for all neoplastic cells from any given tumor, arguing that integration of MCPyV represents an early event in MCC development [57]. MCPyV transformation of MCC cells relies heavily on two MCPyV-encoded proteins: LT-ag and small T-antigen (ST-ag) [2, 59,60,61]. LT-ag and ST-ag bind to a number of intracellular target proteins, culminating in cellular transformation [2, 60, 61].
Two critical DNA sequencing studies demonstrated the genetic pathways driving transformation of MCPyV-negative MCCs. DNA sequencing studies by Wong et al. [62] (32 MCCs, including 13 MCPyV+ and 21 MCPyV− tumors or cell lines) and Harms et al. [63] (16 MCCs, including 7 MCPyV+ and 9 MCPyV− tumors) confirmed fundamental differences between MCPyV+ and MCPyV− MCCs. Specifically, MCPyV− MCCs are characterized by a significantly higher mutational burden compared to MCPyV+ MCC, and most of those are UV-signature mutations. The most common mutations in MCPyV− MCCs impact TP53, RB1, and NOTCH family members [62,63,64]. Together with studies identifying MCPyV in ~80% of MCCs [57, 58], these findings created a molecular-genetic paradigm for MCC development that was binary: MCPyV− tumors driven by the progressive accumulation of UV-induced somatically acquired mutations, and MCPyV+ tumors driven by integration of the MCPyV and expression of oncogenic LT-ag and ST-ag proteins. It is important to note that in geographic regions with less UV exposure, MCCs are far more commonly MCPyV+, whereas in areas with high UV exposure, MCPyV-negative MCCs predominate [60].
Detection of Merkel cell carcinoma polyomavirus in tissue
The initial identification of MCPyV in MCC highlighted a relative frequency of 80% of MCPyV in primary MCC [57]. Subsequent studies (generally relying on DNA based modalities like PCR based amplification of MCPyV sequences) demonstrated a range of 40–100% MCCs to be MCPyV-positive [39, 65,66,67,68,69,70,71,72,73,74,75,76]. The variability in results across studies reflects a number of different factors, including geographical differences in MCPyV prevalence (lower in regions with higher UV exposure), preanalytical variables (tissue substrate [formalin fixed paraffin embedded versus fresh frozen], and analytical differences (primers used and their efficacy to amplify the intervening DNA template whose length also varied). Although MCPyV has also been reported by some investigators at low frequencies in normal skin and hematolymphoid cells as well as in other cutaneous tumors such as BCC and squamous cell carcinoma [3, 60, 77,78,79], the detection of MCPyV in a cutaneous tumor with neuroendocrine morphology is a relatively specific marker of MCC [2, 3].
A monoclonal antibody directed against a specific region of the T-antigen that is unique to MCPyV (the CM2B4 clone) supported the detection and visualization of MCPyV oncoprotein directly in tumor cells. Using CM2B4, most positive tumors display nuclear T-antigen expression, with variable cytoplasmic reactivity. IHC detection of MCPyV with CM2B4 ranges from 39 to 90% [39, 67, 75, 80,81,82]. The sensitivity of IHC depends on preanalytic variables (tissue fixation) as well as viral copy numbers in the tumor cells [44]. Studies comparing PCR to IHC detection of MCPyV in tissue typically show good agreement. As would be expected, most studies find PCR to be more sensitive than IHC. In cases where both approaches could be applied, PCR detected MCPyV in 76% (85/112) of MCCs, whereas IHC (using CM2B4 clone) detected MCPyV T-antigen in 56% (63/112) of tumors [39, 67, 75, 80,81,82]. In a landmark study, Moshiri et al compared the ability of two antibody clones (CM2B4 encompassing amino acids 116–129 in LT-ag, and Ab3 which recognizes amino acids 79–260 in LT-ag) and a PCR based amplification targeting the LT-Ag sequence in a series of 282 MCCs. They showed that the CM2B4 clone possessed the combined highest sensitivity (88%) and specificity (94%) compared with PCR (83% and 81%, respectively) and AB3 (98% and 45%, respectively). In addition, they confirmed that MCPyV-positive tumors have improved progression-free survival, disease-specific survival, and overall survival (OS) compared with MCPyV-negative MCCs, although this is not independent of stage at presentation [58]. Additional detection methods for MCPyV include RNA in situ hybridization and next generation sequencing [60].
Staging MCC
As MCC is an aggressive malignancy, 65% of patients present with disease localized to the cutaneous site, 26% present with regional lymph node metastases, and 8% present with distant metastases [1]. As has been shown for most other solid cancers, staging and prognosis in MCC reflect the extent of disease burden at presentation. Five year OS rates are ~51% for those with localized disease, 35% for patients with regional lymph node metastases, and 14% when distant metastases are present. These differences are the basis for the current TNM staging system [1] (Table 5).
T-categorization stratifies prognostic variables of the primary tumor. The two variables that define the T-category are [1] the clinical measurement of tumor size and [2] extension of tumor beyond the subcutis (fascia, cartilage, muscle and bone). The cutoffs for these are applied as follows: pT1 (≤2 cm), pT2 (>2 cm, but ≤5 cm), pT3 (>5 cm), and pT4 (primary tumor invades the underlying fascia, cartilage, muscle, or bone). Among clinically and/or pathologically node-negative patients, 5-year OS rates robustly reflect grouping primary MCC according to these T-categories: 55.8% (pT1); 41.1% (pT2/pT3), and 31.8% (pT4) [1]. Additional histopathologic attributes of the primary tumor also correlate with the probability of survival and are therefore also reported in pathologic descriptions of the primary tumor. In a study of 156 patients with MCC [83], univariate analyses showed the following primary tumor features correlated with patient survival: tumor thickness, tumor size, deepest anatomic level of involvement, tumor growth pattern, presence of lymphovascular invasion (LVI), presence of tumor infiltrating lymphocytes, and presence of solar elastosis. Multivariate models including these significant variables, showed that stage, tumor thickness, tumor growth pattern, and LVI independently associated with patient survival. When histopathologically confirmed negative lymph nodes were considered in isolation, histopathologic features that associated with survival included the deepest anatomic compartment of involvement, tumor growth pattern of the tumor, and tumor infiltrating lymphocytes [83].
For the purposes of staging, according to the 8th Edition of the AJCC, regional metastases of MCC are first categorized according to whether the nodal disease was identified by clinical or pathologic evaluation of the lymph node basin [1]. Some patients (typically because of underlying comorbidities) are staged only by clinical modalities (imaging and/or physical exam). Those with clinically evident lymph node metastases have worse prognosis than those with clinically occult lymph node involvement [1]. Patients without clinical evidence of lymph node involvement may have microscopic metastases requiring pathologic confirmation. As such, patients with clinically negative lymph nodes, altogether have an intermediate prognosis that is better than those with clinically evident nodal disease, but worse than those with confirmed pathologically negative lymph node disease. Among patients who are staged according to pathologic evaluation of their regional lymph nodes, survival for patients with pathologically confirmed lymph node positive (i.e., clinically occult) disease (pN1a) is worse than those with pathologically node-negative disease (pN0). Taken together, the sentinel lymph node (SLN) biopsy is recommended for staging of patients with MCC who are reasonable candidates for the procedure [1] to distinguish patients with clinically occult disease from those who are considered bona fide lymph node negative. In general, a combination of systematic histopathologic interrogation and ancillary IHC studies maximize sensitivity in the appraisal of SLNs in MCC [33] (Fig. 4). Given the inconsistent sensitivity of any given neuroendocrine marker, IHC examination of SLNs in MCC often relies on pancytokeratin and CK20 stains [2, 3]. The combination of histopathologic and IHC assessment further emphasizes the absence of a minimum size threshold for metastatic MCC deposits in the SLN to qualify as metastatic disease (pN1a). Of note, chronic lymphocytic leukemia, sometimes previously undiagnosed, may be incidentally encountered in SLN biopsy specimens given the increased risk for MCC in that population. A final variable captured in the updated 8th Edition AJCC staging system is the distinction of patients with regional lymph node disease without a known primary MCC (Stage IIIA) from patients with clinically evident regional lymph node disease with a known primary MCC (Stage IIIB) in recognition of the better prognosis for patients with clinically evident metastatic MCC of unknown primary compared with those with documented primary MCC and clinically evident metastases [84,85,86]. Recent studies have also emphasized the importance of both the pattern of SLN involvement as well as the extent of disease burden in the SLN as additional prognostic factors in MCC [87, 88]. In particular, patients with diffuse effacement of their SLN by MCC showed worse OS compared with those with ‘non-solid’ SLN involvement. Additional, larger scale studies are required to determine if the extent of SLN disease burden should be integrated into future MCC staging systems.

Sentinel lymph node processing in Merkel cell carcinoma. a Schematic of sentinel lymph node processing. Top shows recommended serial sectioning to maximize exposure of subcapsular space. All tissue is submitted for histopathologic evaluation. Schema for sentinel lymph node evaluation shown below. If after evaluation of the initial H&E (b), there is evidence of tumor (H&E, ×40 left and (H&E, ×200, right), the diagnosis of metastatic Merkel cell carcinoma is rendered. If the initial H&E is negative for tumor, then additional tissue levels and immunohistochemical studies for cytokeratin are performed to maximize sensitivity. c Antibodies for cytokeratin highlight intraparenchymal (top, left H&E ×100 and top, right pancytokeratin, ×100) deposits of Merkel cell carcinoma or alternatively subcapsular (bottom, left H&E ×100 and bottom, right pancytokeratin, ×100) deposits of Merkel cell carcinoma. There is no minimal size threshold to qualify as metastatic disease
Prognostic biomarkers in Merkel cell carcinomas
Numerous studies have interrogated an array of biomarkers as potential surrogates of patient survival [60]. Increased expression of c-Kit, a receptor tyrosine kinase, in primary MCC demonstrated a trend towards shorter survival compared to patients with reduced levels of c-Kit in the tumor cells (p = 0.07), although activating mutations in KIT have not been identified in MCC [89, 90]. Other biomarkers previously explored in MCC include: expression of nuclear survivin [91]; activating mutations in PIK3CA, the central kinase driving the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) oncogenic pathway [92]; expression of members of the Hedgehog signaling cascade [93,94,95]; vascular markers or growth factors [2]; and markers of cell proliferation and cell cycle entry [96, 97]. As noted above, MCPyV+ MCCs appear to have improved survival compared to MCPyV− MCCs [58, 76, 92, 98,99,100], although not all studies agree [101, 102], and this is not independent of stage at presentation [58].
p63 as a prognostic marker in MCC
One of the most thoroughly studied prognostic biomarkers in MCC is p63 [99, 103,104,105]. The first study included 47 primary MCCs in which p63 positivity (25/47; 53%) correlated with shorter survival compared to the p63-negative MCC (22/47; 47%; p < 0.0001) [104]. In an expanded cohort of 70 patients [103], the same group showed: (i) patients with p63-negative MCCs exhibited longer OS and disease-free survival compared to patients whose MCCs were p63-positive; (ii) in multivariate analyses, only p63 positivity and stage at presentation showed independent prognostic significance; and (iii) considering only patients presenting with MCC localized to the primary site (stage I–II), p63-positivity correlated with worse survival compared with p63-negative tumors (p < 0.0001) [103]. In contrast, Higaki-Mori et al. showed no differences in survival according to p63 expression in their cohort of MCC patients [99]. In a large cohort of MCC patients, Stetsenko et al. [106] determined p63 expression in 128 MCC patient tumors and found that p63-positivity was independently predictive of reduced MCC-specific survival (together with stage). In a key experiment, they grouped patients according to stage at presentation and found that p63 expression no longer segregated patient survival, and p63 expression itself did not differ among the clinical stage groups. Therefore, to the extent that patient stage is already determined, the utility of routine assessment of p63 expression in MCC as a prognostic marker remains controversial [106].
The immune system as a biomarker in MCC
The importance of an intact host immune system on the proclivity for MCC to develop has been well established [107,108,109,110]. The relationship between immune compromise and MCC patient survival was demonstrated in a study of 471 MCC patients [111]. Immune suppressed patients with MCC showed reduced MCC-specific survival (n = 41; 40% at 3 years) when compared with those patients without immune suppression (n = 430; 74% at 3 years), and the host immune status predicted MCC-specific survival independently of stage at presentation [111].
The same group [112] subjected 35 primary MCCs to gene expression studies and found that increased expression of CD8+ cytotoxic T-cell associated genes (for example, CD8A and granzyme genes) correlated with longer MCC survival. In a validation cohort of 146 MCCs (including primary MCC and regional and visceral distant metastases), higher CD8+ T-cell infiltration independently correlated associated with longer survival compared with tumors with reduced intratumoral CD8+ T-cell infiltration [112]. Sihto et al. [113] quantified the tumor-associated immune infiltrates in 116 MCCs and showed that higher densities of CD3+ T-cells significantly associated with longer patient survival. In a series of 62 primary MCCs, Feldmeyer et al. [114] leveraged automated image analysis to precisely quantify the density of CD3+ and CD8+ T-cells in discrete regions of the tumor (periphery, center, and hot spot) and showed that higher densities of CD3+ and CD8+ T cells at the tumor periphery correlated with longer OS, and patients whose tumors had higher densities of CD8+ T-cell at the tumor periphery had longer DSS compared with those with lower CD8+ T-cell densities [114].
Therapeutic advances in MCC
Together, these findings confirmed the close relationship between a competent and active tumor-associated immune T-cell infiltrate and MCC patient survival. Further, the plethora of neoantigens in MCC (either virally encoded proteins in MCPyV-positive cases or the myriad of UV-induced mutations in MCPyV-negative tumors) [59, 62, 63, 115] strongly implicated the efficacy of immune checkpoint blockade therapy in MCC. In the initial clinical trial, the PD-1 inhibitor pembrolizumab was administered to 26 previously untreated patients with advanced MCC [116], producing an objective response rate of 56%, including a complete response in 15% of patients (4/26) and a partial response in 38% (10/26). In a separate trial, 88 patients with stage IV MCC were treated with the PD-L1 inhibitor avelumab as second-line therapy. This study demonstrated an objective response rate of 31.8% (28 of 88 patients—including 8 complete responders and 20 partial responders), which was especially remarkable considering these patients had already failed at least one round of prior chemotherapy [117]. Of particular significance, many of these patient responses have been shown to be durable [118, 119]. However, thus far, biomarkers predictive of response to immune checkpoint blockade have been elusive. Responses have not correlated with MCPyV status or tumor cell expression of PD-L1. In a study leveraging the power of multiplex immunofluorescence, Giraldo et al. [120] showed that not only the density of PD-1+ and PD-L1+ cells, but also the relative geographic proximity between PD-1+ and PD-L1+ cells predicts clinical response to PD-1 inhibition in MCC, suggesting that the likely engagement of PD-1 with PD-L1 is necessary and therefore likely predictive that abrogation of that interaction to exert a clinical effect.
In summary, MCC is an aggressive cutaneous malignancy that, although rare, is increasing in incidence. MCC tumors are thought to arise via two distinct pathways: UV-associated and MCPyV-driven. Recognition of key morphologic and immunophenotypic features of MCC is critical to avoid confusion with other cutaneous tumors and metastases and allow for prompt staging and intervention. Continued investigations are needed into improved diagnostic markers for the subset of MCC with aberrant immunophenotypes, prognostic markers, and predictive markers for therapy response.
References
Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, et al. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the new 8th edition AJCC staging system. Ann Surg Oncol. 2016;23:3564–71.
Harms PW. Update on Merkel cell carcinoma. Clin Lab Med. 2017;37:485–501.
Tetzlaff MT, Nagarajan P. Update on Merkel cell carcinoma. Head Neck Pathol. 2018;12:31–43.
Acebo E, Vidaurrazaga N, Varas C, Burgos-Bretones JJ, Diaz-Perez JL. Merkel cell carcinoma: a clinicopathological study of 11 cases. J Eur Acad Dermatol Venereol. 2005;19:546–51.
Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, Papadimitriou CS. Immunohistochemical distinction between merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99–104.
Byrd-Gloster AL, Khoor A, Glass LF, Messina JL, Whitsett JA, Livingston SK, et al. Differential expression of thyroid transcription factor 1 in small cell lung carcinoma and Merkel cell tumor. Hum Pathol. 2000;31:58–62.
Chan JK, Suster S, Wenig BM, Tsang WY, Chan JB, Lau AL. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am J Surg Pathol. 1997;21:226–34.
Cheuk W, Kwan MY, Suster S, Chan JK. Immunostaining for thyroid transcription factor 1 and cytokeratin 20 aids the distinction of small cell carcinoma from Merkel cell carcinoma, but not pulmonary from extrapulmonary small cell carcinomas. Arch Pathol Lab Med. 2001;125:228–31.
Chu P, Wu E, Weiss LM. Cytokeratin 7 and cytokeratin 20 expression in epithelial neoplasms: a survey of 435 cases. Mod Pathol. 2000;13:962–72.
Hanly AJ, Elgart GW, Jorda M, Smith J, Nadji M. Analysis of thyroid transcription factor-1 and cytokeratin 20 separates merkel cell carcinoma from small cell carcinoma of lung. J Cutan Pathol. 2000;27:118–20.
Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310–5.
Johansson L, Tennvall J, Akerman M. Immunohistochemical examination of 25 cases of Merkel cell carcinoma: a comparison with small cell carcinoma of the lung and oesophagus, and a review of the literature. APMIS. 1990;98:741–52.
Kervarrec T, Tallet A, Miquelestorena-Standley E, Houben R, Schrama D, Gambichler T, et al. Diagnostic accuracy of a panel of immunohistochemical and molecular markers to distinguish Merkel cell carcinoma from other neuroendocrine carcinomas. Mod Pathol. 2019;32:499–510.
Kurokawa M, Nabeshima K, Akiyama Y, Maeda S, Nishida T, Nakayama F, et al. CD56: a useful marker for diagnosing Merkel cell carcinoma. J Dermatol Sci. 2003;31:219–24.
Leech SN, Kolar AJ, Barrett PD, Sinclair SA, Leonard N. Merkel cell carcinoma can be distinguished from metastatic small cell carcinoma using antibodies to cytokeratin 20 and thyroid transcription factor 1. J Clin Pathol. 2001;54:727–9.
Llombart B, Monteagudo C, Lopez-Guerrero JA, Carda C, Jorda E, Sanmartin O, et al. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology. 2005;46:622–34.
McCalmont TH. Paranuclear dots of neurofilament reliably identify Merkel cell carcinoma. J Cutan Pathol. 2010;37:821–3.
Metz KA, Jacob M, Schmidt U, Steuhl KP, Leder LD. Merkel cell carcinoma of the eyelid: histological and immunohistochemical features with special respect to differential diagnosis. Graefes Arch Clin Exp Ophthalmol. 1998;236:561–6.
Moll R, Lowe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. 1992;140:427–47.
Nicholson SA, McDermott MB, Swanson PE, Wick MR. CD99 and cytokeratin-20 in small-cell and basaloid tumors of the skin. Appl Immunohistochem Mol Morphol. 2000;8:37–41.
Ordonez NG. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am J Surg Pathol. 2000;24:1217–23.
Schmidt U, Muller U, Metz KA, Leder LD. Cytokeratin and neurofilament protein staining in Merkel cell carcinoma of the small cell type and small cell carcinoma of the lung. Am J Dermatopathol. 1998;20:346–51.
Scott MP, Helm KF. Cytokeratin 20: a marker for diagnosing Merkel cell carcinoma. Am J Dermatopathol. 1999;21:16–20.
Shah IA, Netto D, Schlageter MO, Muth C, Fox I, Manne RK. Neurofilament immunoreactivity in Merkel-cell tumors: a differentiating feature from small-cell carcinoma. Mod Pathol. 1993;6:3–9.
Sidiropoulos M, Hanna W, Raphael SJ, Ghorab Z. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831–8.
Stanoszek LM, Chan MP, Palanisamy N, Carskadon S, Siddiqui J, Patel RM, et al. Neurofilament is superior to cytokeratin 20 in supporting cutaneous origin for neuroendocrine carcinoma. Histopathology. 2019;74:504–13.
Sur M, AlArdati H, Ross C, Alowami S. TdT expression in Merkel cell carcinoma: potential diagnostic pitfall with blastic hematological malignancies and expanded immunohistochemical analysis. Mod Pathol. 2007;20:1113–20.
Yang DT, Holden JA, Florell SR. CD117, CK20, TTF-1, and DNA topoisomerase II-alpha antigen expression in small cell tumors. J Cutan Pathol. 2004;31:254–61.
Kervarrec T, Tallet A, Miquelestorena-Standley E, Houben R, Schrama D, Gambichler T, et al. Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and negative Merkel cell carcinoma. Mod Pathol. 2019.
Koljonen V, Haglund C, Tukiainen E, Bohling T. Neuroendocrine differentiation in primary Merkel cell carcinoma—possible prognostic significance. Anticancer Res. 2005;25:853–8.
Kontochristopoulos GJ, Stavropoulos PG, Krasagakis K, Goerdt S, Zouboulis CC. Differentiation between merkel cell carcinoma and malignant melanoma: an immunohistochemical study. Dermatology. 2000;201:123–6.
Pasternak S, Carter MD, Ly TY, Doucette S, Walsh NM. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum Pathol. 2018;82:232–8.
Su LD, Lowe L, Bradford CR, Yahanda AI, Johnson TM, Sondak VK. Immunostaining for cytokeratin 20 improves detection of micrometastatic Merkel cell carcinoma in sentinel lymph nodes. J Am Acad Dermatol. 2002;46:661–6.
Miner AG, Patel RM, Wilson DA, Procop GW, Minca EC, Fullen DR, et al. Cytokeratin 20-negative Merkel cell carcinoma is infrequently associated with the Merkel cell polyomavirus. Mod Pathol. 2015;28:498–504.
Koba S, Nagase K, Ikeda S, Aoki S, Misago N, Narisawa Y. Merkel cell carcinoma with glandular differentiation admixed with sweat gland carcinoma and spindle cell carcinoma: histogenesis of merkel cell carcinoma from hair follicle stem cells. Am J Dermatopathol. 2015;37:e31–6.
Martin B, Poblet E, Rios JJ, Kazakov D, Kutzner H, Brenn T, et al. Merkel cell carcinoma with divergent differentiation: histopathological and immunohistochemical study of 15 cases with PCR analysis for Merkel cell polyomavirus. Histopathology. 2013;62:711–22.
Narisawa Y, Koba S, Inoue T, Nagase K. Histogenesis of pure and combined Merkel cell carcinomas: An immunohistochemical study of 14 cases. J Dermatol. 2015;42:445–52.
Scurry J, Brand A, Planner R, Dowling J, Rode J. Vulvar Merkel cell tumor with glandular and squamous differentiation. Gynecol Oncol. 1996;62:292–7.
Busam KJ, Jungbluth AA, Rekthman N, Coit D, Pulitzer M, Bini J, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol. 2009;33:1378–85.
Iacocca MV, Abernethy JL, Stefanato CM, Allan AE, Bhawan J. Mixed Merkel cell carcinoma and squamous cell carcinoma of the skin. J Am Acad Dermatol. 1998;39:882–7.
Iwasaki T, Matsushita M, Kuwamoto S, Kato M, Murakami I, Higaki-Mori H, et al. Usefulness of significant morphologic characteristics in distinguishing between Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative Merkel cell carcinomas. Hum Pathol. 2013;44:1912–7.
Paik JY, Hall G, Clarkson A, Lee L, Toon C, Colebatch A, et al. Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum Pathol. 2011;42:1385–90.
Pulitzer MP, Brannon AR, Berger MF, Louis P, Scott SN, Jungbluth AA, et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Mod Pathol. 2015;28:1023–32.
Sirikanjanapong S, Melamed J, Patel RR. Intraepidermal and dermal Merkel cell carcinoma with squamous cell carcinoma in situ: a case report with review of literature. J Cutan Pathol. 2010;37:881–5.
Falto Aizpurua LA, Wang M, Ruiz HA, Sanchez JL, Chan MP, Andea AA, et al. A case of combined Merkel cell carcinoma and squamous cell carcinoma: Molecular insights and diagnostic pitfalls. JAAD Case Rep. 2018;4:996–9.
Panse G, McNiff JM, Ko CJ. Basal cell carcinoma: CD56 and cytokeratin 5/6 staining patterns in the differential diagnosis with Merkel cell carcinoma. J Cutan Pathol. 2017;44:553–6.
Ralston J, Chiriboga L, Nonaka D. MASH1: a useful marker in differentiating pulmonary small cell carcinoma from Merkel cell carcinoma. Mod Pathol. 2008;21:1357–62.
Hughes AJ, French M, Ah-Weng A, Singh M. Diffuse and strong TTF-1 positivity in a combined Merkel cell carcinoma. Am J Dermatopathol. 2019.
Ortiz Salvador JM, Subiabre-Ferrer D, Alegre, de Miquel V. Primary cutaneous neuroendocrine carcinoma with diffuse expression of thyroid transcription factor-1: report of two cases. Indian J Dermatol. 2019;64:251.
McCluggage WG, Kennedy K, Busam KJ. An immunohistochemical study of cervical neuroendocrine carcinomas: neoplasms that are commonly TTF1 positive and which may express CK20 and P63. Am J Surg Pathol. 2010;34:525–32.
Stoler MH, Mills SE, Gersell DJ, Walker AN. Small-cell neuroendocrine carcinoma of the cervix. A human papillomavirus type 18-associated cancer. Am J Surg Pathol. 1991;15:28–32.
Folpe AL, Hill CE, Parham DM, O’Shea PA, Weiss SW. Immunohistochemical detection of FLI-1 protein expression: a study of 132 round cell tumors with emphasis on CD99-positive mimics of Ewing’s sarcoma/primitive neuroectodermal tumor. Am J Surg Pathol. 2000;24:1657–62.
Jimenez RE, Folpe AL, Lapham RL, Ro JY, O’Shea PA, Weiss SW, et al. Primary Ewing’s sarcoma/primitive neuroectodermal tumor of the kidney: a clinicopathologic and immunohistochemical analysis of 11 cases. Am J Surg Pathol. 2002;26:320–7.
Fitzgerald TL, Dennis S, Kachare SD, Vohra NA, Wong JH, Zervos EE. Dramatic Increase in the Incidence and Mortality from Merkel Cell Carcinoma in the United States. Am Surg. 2015;81:802–6.
Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surgical Oncol. 2005;89:1–4.
Paulson KG, Park SY, Vandeven NA, Lachance K, Thomas H, Chapuis AG, et al. Merkel cell carcinoma: current United States incidence and projected increases based on changing demographics. J Am Acad Dermatol. 2017;78:457–463.e2.
Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–100.
Moshiri AS, Doumani R, Yelistratova L, Blom A, Lachance K, Shinohara MM, et al. Polyomavirus-negative Merkel cell carcinoma: a more aggressive subtype based on analysis of 282 cases using multimodal tumor virus detection. J Investig Dermatol. 2017;137:819–27.
Becker JC, Stang A, DeCaprio JA, Cerroni L, Lebbe C, Veness M, et al. Merkel cell carcinoma. Nat Rev Dis Prim. 2017;3:17077.
Harms PW, Harms KL, Moore PS, DeCaprio JA, Nghiem P, Wong MKK, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol. 2018;15:763–76.
Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell polyomavirus. Curr Opin Virol. 2015;11:38–43.
Wong SQ, Waldeck K, Vergara IA, Schroder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75:5228–34.
Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu YM, Dhanasekaran SM, et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma. Cancer Res. 2015;75:3720–7.
Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget. 2016;7:3403–15.
Andres C, Belloni B, Puchta U, Sander CA, Flaig MJ. Prevalence of MCPyV in Merkel cell carcinoma and non-MCC tumors. J Cutan Pathol. 2010;37:28–34.
Becker JC, Houben R, Ugurel S, Trefzer U, Pfohler C, Schrama D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Investig Dermatol. 2009;129:248–50.
Bhatia K, Goedert JJ, Modali R, Preiss L, Ayers LW. Immunological detection of viral large T antigen identifies a subset of Merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. Int J Cancer. 2010;127:1493–6.
Bhatia K, Goedert JJ, Modali R, Preiss L, Ayers LW. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. Int J Cancer. 2010;126:2240–6.
Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516–21.
Garneski KM, Warcola AH, Feng Q, Kiviat NB, Leonard JH, Nghiem P. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Investig Dermatol. 2009;129:246–8.
Houben R, Schrama D, Alb M, Pfohler C, Trefzer U, Ugurel S, et al. Comparable expression and phosphorylation of the retinoblastoma protein in Merkel cell polyoma virus-positive and negative Merkel cell carcinoma. Int J Cancer. 2010;126:796–8.
Kassem A, Schopflin A, Diaz C, Weyers W, Stickeler E, Werner M, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68:5009–13.
Loyo M, Guerrero-Preston R, Brait M, Hoque MO, Chuang A, Kim MS, et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int J Cancer. 2010;126:2991–6.
Sastre-Garau X, Peter M, Avril MF, Laude H, Couturier J, Rozenberg F, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48–56.
Shuda M, Arora R, Kwun HJ, Feng H, Sarid R, Fernandez-Figueras MT, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer. 2009;125:1243–9.
Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009;101:938–45.
Ly TY, Walsh NM, Pasternak S. The spectrum of Merkel cell polyomavirus expression in Merkel cell carcinoma, in a variety of cutaneous neoplasms, and in neuroendocrine carcinomas from different anatomical sites. Hum Pathol. 2012;43:557–66.
Mertz KD, Paasinen A, Arnold A, Baumann M, Offner F, Willi N, et al. Merkel cell polyomavirus large T antigen is detected in rare cases of nonmelanoma skin cancer. J Cutan Pathol. 2013;40:543–9.
Ota S, Ishikawa S, Takazawa Y, Goto A, Fujii T, Ohashi K, et al. Quantitative analysis of viral load per haploid genome revealed the different biological features of Merkel cell polyomavirus infection in skin tumor. PLoS ONE. 2012;7:e39954.
Jung HS, Choi YL, Choi JS, Roh JH, Pyon JK, Woo KJ, et al. Detection of Merkel cell polyomavirus in Merkel cell carcinomas and small cell carcinomas by PCR and immunohistochemistry. Histol Histopathol. 2011;26:1231–41.
Leitz M, Stieler K, Grundhoff A, Moll I, Brandner JM, Fischer N. Merkel cell polyomavirus detection in Merkel cell cancer tumors in Northern Germany using PCR and protein expression. J Med Virol. 2014;86:1813–9.
Leroux-Kozal V, Leveque N, Brodard V, Lesage C, Dudez O, Makeieff M, et al. Merkel cell carcinoma: histopathologic and prognostic features according to the immunohistochemical expression of Merkel cell polyomavirus large T antigen correlated with viral load. Hum Pathol. 2015;46:443–53.
Andea AA, Coit DG, Amin B, Busam KJ. Merkel cell carcinoma: histologic features and prognosis. Cancer. 2008;113:2549–58.
Chen KT, Papavasiliou P, Edwards K, Zhu F, Perlis C, Wu H, et al. A better prognosis for Merkel cell carcinoma of unknown primary origin. Am J Surg. 2013;206:752–7.
Foote M, Veness M, Zarate D, Poulsen M. Merkel cell carcinoma: the prognostic implications of an occult primary in stage IIIB (nodal) disease. J Am Acad Dermatol. 2012;67:395–9.
Tarantola TI, Vallow LA, Halyard MY, Weenig RH, Warschaw KE, Weaver AL, et al. Unknown primary Merkel cell carcinoma: 23 new cases and a review. J Am Acad Dermatol. 2013;68:433–40.
Erstine EM, Tetzlaff MT, Jia X, Aung PP, Prieto VG, Funchain P, et al. Prognostic significance of “nonsolid” microscopic metastasis in Merkel cell carcinoma sentinel lymph nodes. Am J Surg Pathol. 2019;43:907–19.
Ko JS, Prieto VG, Elson PJ, Vilain RE, Pulitzer MP, Scolyer RA, et al. Histological pattern of Merkel cell carcinoma sentinel lymph node metastasis improves stratification of Stage III patients. Mod Pathol. 2016;29:122–30.
Andea AA, Patel R, Ponnazhagan S, Kumar S, DeVilliers P, Jhala D, et al. Merkel cell carcinoma: correlation of KIT expression with survival and evaluation of KIT gene mutational status. Hum Pathol. 2010;41:1405–12.
Swick BL, Srikantha R, Messingham KN. Specific analysis of KIT and PDGFR-alpha expression and mutational status in Merkel cell carcinoma. J Cutan Pathol. 2013;40:623–30.
Kim J, McNiff JM. Nuclear expression of survivin portends a poor prognosis in Merkel cell carcinoma. Mod Pathol. 2008;21:764–9.
Nardi V, Song Y, Santamaria-Barria JA, Cosper AK, Lam Q, Faber AC, et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin Cancer Res. 2012;18:1227–36.
Brunner M, Thurnher D, Pammer J, Heiduschka G, Petzelbauer P, Schmid C, et al. Expression of hedgehog signaling molecules in Merkel cell carcinoma. Head Neck. 2010;32:333–40.
Carroll TM, Williams JS, Daily K, Rogers T, Gelb T, Coxon A, et al. Hedgehog Signaling Inhibitors Fail to Reduce Merkel Cell Carcinoma Viability. J Investig Dermatol. 2017;137:1187–90.
Kuromi T, Matsushita M, Iwasaki T, Nonaka D, Kuwamoto S, Nagata K, et al. Association of expression of the hedgehog signal with Merkel cell polyomavirus infection and prognosis of Merkel cell carcinoma. Hum Pathol. 2017;69:8–14.
Fernandez-Figueras MT, Puig L, Musulen E, Gilaberte M, Ferrandiz C, Lerma E, et al. Prognostic significance of p27Kip1, p45Skp2 and Ki67 expression profiles in Merkel cell carcinoma, extracutaneous small cell carcinoma, and cutaneous squamous cell carcinoma. Histopathology. 2005;46:614–21.
Henderson SA, Tetzlaff MT, Pattanaprichakul P, Fox P, Torres-Cabala CA, Bassett RL, et al. Detection of mitotic figures and G2+ tumor nuclei with histone markers correlates with worse overall survival in patients with Merkel cell carcinoma. J Cutan Pathol. 2014;41:846–52.
Haymerle G, Janik S, Fochtmann A, Pammer J, Schachner H, Nemec L, et al. Expression of Merkelcell polyomavirus (MCPyV) large T-antigen in Merkel cell carcinoma lymph node metastases predicts poor outcome. PLoS ONE. 2017;12:e0180426.
Higaki-Mori H, Kuwamoto S, Iwasaki T, Kato M, Murakami I, Nagata K, et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol. 2012;43:2282–91.
Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res. 2011;17:4806–13.
Hall BJ, Pincus LB, Yu SS, Oh DH, Wilson AR, McCalmont TH. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911–7.
Schrama D, Peitsch WK, Zapatka M, Kneitz H, Houben R, Eib S, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Investig Dermatol. 2011;131:1631–8.
Asioli S, Righi A, de Biase D, Morandi L, Caliendo V, Picciotto F, et al. Expression of p63 is the sole independent marker of aggressiveness in localised (stage I-II) Merkel cell carcinomas. Mod Pathol. 2011;24:1451–61.
Asioli S, Righi A, Volante M, Eusebi V, Bussolati G. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer. 2007;110:640–7.
Fleming KE, Ly TY, Pasternak S, Godlewski M, Doucette S, Walsh NM. Support for p63 expression as an adverse prognostic marker in Merkel cell carcinoma: report on a Canadian cohort. Hum Pathol. 2014;45:952–60.
Stetsenko GY, Malekirad J, Paulson KG, Iyer JG, Thibodeau RM, Nagase K, et al. p63 expression in Merkel cell carcinoma predicts poorer survival yet may have limited clinical utility. Am J Clin Pathol. 2013;140:838–44.
Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Merkel cell carcinoma and HIV infection. Lancet. 2002;359:497–8.
Heath M, Jaimes N, Lemos B, Mostaghimi A, Wang LC, Penas PF, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375–81.
Hemminki K, Liu X, Ji J, Sundquist J, Sundquist K. Kaposi sarcoma and Merkel cell carcinoma after autoimmune disease. Int J Cancer. 2012;131:E326–8.
Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717–21.
Paulson KG, Iyer JG, Blom A, Warton EM, Sokil M, Yelistratova L, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Investig Dermatol. 2013;133:642–6.
Paulson KG, Iyer JG, Tegeder AR, Thibodeau R, Schelter J, Koba S, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol. 2011;29:1539–46.
Sihto H, Bohling T, Kavola H, Koljonen V, Salmi M, Jalkanen S, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872–81.
Feldmeyer L, Hudgens CW, Ray-Lyons G, Nagarajan P, Aung PP, Curry JL, et al. Density, Distribution, and Composition of Immune Infiltrates Correlate with Survival in Merkel Cell Carcinoma. Clin Cancer Res. 2016;22:5553–63.
Schadendorf D, Lebbe C, Zur Hausen A, Avril MF, Hariharan S, Bharmal M, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur J Cancer. 2017;71:53–69.
Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N Engl J Med. 2016;374:2542–52.
Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17:1374–85.
Kaufman HL, Russell JS, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after >/=1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer. 2018;6:7.
Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, et al. Durable tumor regression and overall survival in patients with advanced Merkel cell carcinoma receiving pembrolizumab as first-line therapy. J Clin Oncol. 2019;37:693–702.
Giraldo NA, Nguyen P, Engle EL, Kaunitz GJ, Cottrell TR, Berry S, et al. Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J Immunother Cancer. 2018;6:99.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tetzlaff, M.T., Harms, P.W. Danger is only skin deep: aggressive epidermal carcinomas. An overview of the diagnosis, demographics, molecular-genetics, staging, prognostic biomarkers, and therapeutic advances in Merkel cell carcinoma. Mod Pathol 33 (Suppl 1), 42–55 (2020). https://doi.org/10.1038/s41379-019-0394-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0394-6
This article is cited by
-
SATB2, CKAE1/AE3, and synaptophysin as a sensitive immunohistochemical panel for the detection of lymph node metastases of Merkel cell carcinoma
Virchows Archiv (2023)
-
Genomic evidence suggests that cutaneous neuroendocrine carcinomas can arise from squamous dysplastic precursors
Modern Pathology (2022)
-
Evidence of an epithelial origin of Merkel cell carcinoma
Modern Pathology (2022)
-
Clinical-Pathological Evaluation and Prognostic Analysis of 228 Merkel Cell Carcinomas Focusing on Tumor-Infiltrating Lymphocytes, MCPYV Infection and ALK Expression
Endocrine Pathology (2022)
-
Investigation of the RB1-SOX2 axis constitutes a tool for viral status determination and diagnosis in Merkel cell carcinoma
Virchows Archiv (2022)
