
Abstract
Molecular diagnostics of sarcoma subtypes commonly involve the identification of characteristic oncogenic fusions. EWSR1-PATZ1 is a rare fusion partnering in sarcoma, with few cases reported in the literature. In the current study, a series of 11 cases of EWSR1-PATZ1 fusion positive malignancies are described. EWSR1-PATZ1-related sarcomas occur across a wide age range and have a strong predilection for chest wall primary site. Secondary driver mutations in cell-cycle genes, and in particular CDKN2A (71%), are common in EWSR1-PATZ1 sarcomas in this series. In a subset of cases, an extended clinical and histopathological review was performed, as was confirmation and characterization of the fusion breakpoint revealing a novel intronic pseudoexon sequence insertion. Unified by a shared gene fusion, EWSR1-PATZ1 sarcomas otherwise appear to exhibit divergent morphology, a polyphenotypic immunoprofile, and variable clinical behavior posing challenges for precise classification.
Similar content being viewed by others


Introduction
Molecular diagnostics of sarcoma subtypes commonly involves the identification of characteristic oncogenic fusions. For small round blue cell tumors, a classic ancillary criterion is the identification of EWSR1 rearrangements including EWSR1-ETS in Ewing sarcoma and EWSR1-WT1 in desmoplastic small round cell tumor. Recently, owing to advances in molecular technology, an increasing variety of EWSR1 fusion partners have been described [1]. The presence of a well-described oncogenic fusion is an important clinical tool, as underlying disease biology and clinical behavior are significantly impacted by the fusion driver. As an example, “fusion negative” Ewing sarcomas and Ewing sarcoma-like tumors that lack EWSR1-ETS fusion have a molecular profile that is distinct from EWSR1-ETS fusion positive cases [2]. Cataloging and describing the clinicopathological features of less common fusion subtypes is therefore critical for optimal patient management.
EWSR1-PATZ1 is a rare fusion partnering that has previously been described in isolated cases of spindled or small round cell sarcomas [3,4,5]. An intrachromosomal rearrangement between EWSR1 and PATZ1 leading to an in-frame fusion that contains the activating N-terminal domain of EWSR1 and the zinc finger domain of PATZ1 was demonstrated in the initial description [3]. Although presenting and pathologic features have been described in prior reports, outcomes or treatment response data are limited or lacking. In addition to the sparse cases reported in sarcoma, EWSR1-PATZ1 fusion has also been described in four cases of clinicohistopathologically diverse pediatric central nervous system tumors [6,7,8,9] and PATZ1 expression has been proposed as a diagnostic and prognostic biomarker for glioblastoma [10].
Herein, 11 cases of neoplasms with EWSR1-PATZ1 fusion discovered by a next-generation sequencing assay, predominantly in a sarcoma population are reported. The demographics and genomic features of these cases are summarized. An extended clinicopathological review was performed on three of these cases that were identified at two sarcoma referral centers.
Materials and methods
Next-generation sequencing detection of EWSR1-PATZ1
EWSR1-PATZ1 fusions were detected in tumor samples of three patients from two institutions of the authors by a commercial next-generation sequencing platform [11] followed by reverse transcription-polymerase chain reaction and Sanger sequencing confirmation. Retrospective chart review was performed for clinicopathological details of these three patients under Institutional Review Board approved protocols. Eight additional EWSR1-PATZ1 cases were identified across all tumors sequenced on the same commercial platform (Table 1). Comprehensive genomic profiling to include DNA and RNA sequencing was performed and analyzed as previously described [11].
Reverse transcription-polymerase chain reaction and Sanger sequencing
Total RNA was extracted from representative formalin fixed paraffin-embedded tumor tissue of Cases 1–3 using the RNeasy formalin fixed paraffin-embedded tissue kit (Qiagen, Germantown, MD). The genomic coordinates for designing reverse transcription-polymerase chain reaction oligonucleotide primers were determined using IGV (Integrative Genomics Viewer) software [12]. Specifically, for two-step reverse transcription-polymerase chain reaction analysis, 300 ng of total RNA was reverse transcribed in the presence of random hexamers using the PrimeScript reverse transcription-polymerase chain reaction kit (Takara-Clontech, Mountain View, CA) followed by amplification using 5U of Takara Ex Taq HS polymerase (Takara-Clontech) and the combination of the primers listed in Table 2. Polymerase chain reaction was conducted as follows: 94 °C for 30 s, followed by 35 cycles at 94 °C for 30 s, 62 °C for 30 s, and 72 °C for 60 s. At the end of the 35 cycles, a final extension at 72 °C for 3 min was performed. The polymerase chain reaction products were analyzed on 1.8% agarose gels and visualized by ethidium bromide staining. The integrity of the mRNA was assessed by independent reverse transcription-polymerase chain reactions using primers to the ubiquitously expressed hypoxanthine phosphoribosyltransferase gene. The negative controls were devoid of template or Taq polymerase.
The polymerase chain reaction products of Cases 2 and 3 were purified by agarose gel electrophoresis and the DNA extracted utilizing the NucleoSpin Gel and Polymerase Chain Reaction Clean-Up kit (Macherey-Nagel Bethlehem, PA). Subsequent direct sequencing using the 3500 Genetic Analyzer (Applied Biosystems) was performed. BLAST (http://blast.ncbi.nlm.nih.gov/) and MacVector (MacVector, Inc. Apex, NC) programs were employed for analysis of the sequence data. The RNA and cDNA yield from Case 1 was insufficient for further analysis.
Molecular cytogenetic analysis (fluorescence in situ hybridization)
Dual color fluorescence in situ hybridization studies for assessment of MDM2 amplification and deletion of the 9p21.3 (CDKN2A) locus were performed on 4-µm-thick unstained tissue sections of representative formalin-fixed, paraffin-embedded tumor tissue from Cases 1–3 using the following pairs of commercially available probes: MDM2 locus-specific and chromosome 12 centromere-specific probes, and CDKN2A locus-specific and chromosome 9 centromere-specific probes (LSI MDM2 [12q15] Spectrum Orange/CEP12 Spectrum Green Probes and LSI p16INK4a [9p21] Spectrum Orange/CEP 9 Spectrum Green probes, respectively; Abbott Molecular, Des Plaines, IL) using conventional methodology as previously described [13].
Hybridization signals were assessed in 100 nonoverlapping interphase nuclei with strong, well-delineated signals. Images were acquired using the Cytovision Image Analysis System (Applied Imaging, Santa Clara, CA). The cutoff level for MDM2 amplification was defined as a ratio of MDM2/CEP12 ≥ 3.0 and MDM2 average copy number as ≥5 and for deletion of the CDKN2A locus as ≥30% based on in-house validation studies.
Results
Case histories
Case 1
A 36-year-old female was diagnosed with a pulmonary embolism after presenting with shortness of breath. Initial imaging was notable for subcentimeter pulmonary nodules and in retrospect, a soft tissue mass in the musculature near the right scapula was discovered. Ultimately a positron emission tomography (PET) scan performed almost 1 year after the initial pulmonary embolism diagnosis demonstrated widespread metastatic disease including a large hypermetabolic >10 cm intramuscular lesion near the right scapula, multiple pulmonary nodules and myocardial masses, bilateral adnexal masses, and additional soft tissue thoracoabdominal wall metastases. Pathology review of a biopsy taken from the presumed chest wall primary site demonstrated a small round blue cell tumor. Molecular cytogenetic testing was negative for rearrangement of the EWSR1, FUS, CIC, FOXO1, and SS18 loci. However, polysomy (3–7 copies) for each probe set with the exception of SS18 was detected. Commercial genomic profiling was pursued to potentially aid in further classification and identification of targets for future clinical trial therapy.
The patient began systemic therapy with mesna, adriamycin, and ifosfamide. Restaging scans after two cycles demonstrated a mixed response, but overall progressing disease. At this time, results from genomic testing detected an EWSR1-PATZ1 fusion. The systemic regimen was altered to a more “Ewing-like” or “desmoplastic small round cell tumor-like” VAC/IE (vincristine, adriamycin, and cyclophosphamide alternating with ifosfamide and etoposide), given in a dose-dense fashion when able to from a count recovery perspective. She completed ~5 months of therapy with VAC/IE with an initial minor disease response followed by stabilization. Due to cumulative toxicity concerns as well as plateau of response, her systemic therapy was altered to gemcitabine/docetaxel. She completed five cycles of this therapy with again minor disease response followed by stabilization. Due to quality of life concerns on prolonged aggressive chemotherapy, her systemic treatment was altered to pazopanib in an attempt to maintain disease stabilization while pursuing clinical trial evaluations. Unfortunately she had progression on this therapy and clinical decline. She was placed on hospice management ~30 months after her initial presentation and expired soon thereafter.
Case 2
A 53-year-old man presented with a large left upper arm/axillary mass that had been slowly growing for several years. Imaging revealed an 8.5 cm mass centered in the upper arm as well as widespread metastatic disease including bulky adenopathy in the bilateral axillae and hilum and numerous pulmonary nodules. Biopsy of the large primary site revealed a malignant small round cell malignancy that was positive for EWSR1 rearrangement by fluorescence in situ hybridization. Pathological findings were felt to be most consistent with myoepithelial carcinoma. An EWSR1-PATZ1 fusion was detected by commercial genomic testing.
Given the initial working diagnosis of myoepithelial carcinoma, the patient began a systemic regimen of carboplatin plus paclitaxel taking into account reported activity in this disease [14]. He experienced an initial clinical response with a near partial response radiographically after four cycles of therapy (~20% decrease in target lesion size). However, while remaining on this same therapy, he developed substantial disease progression over the next 2 months. He began on palliative radiation therapy for a hilar mass causing obstructive symptoms, but expired shortly thereafter due to progressive disease.
Case 3
An 81-year-old female with a past medical history notable for hypertension, diabetes mellitus, chronic kidney disease, and hypothyroidism, presented to the neurosurgery clinic to discuss treatment options for a soft tissue mass that she noticed a few months before. The mass was located in the posterior neck and was causing severe pain. Magnetic resonance imaging revealed a heterogeneous well-encapsulated 3.5 × 3.4 × 2.5 cm enhancing soft tissue mass in the posterior neck between the C2 and C3 spinous processes. Radiologically there was remodeling of bone and absence of bone marrow edema suggestive of a slow-growing neoplasm. A marginal resection of the mass was subsequently performed and sent to pathology for evaluation. Commercial molecular testing identified the presence of an EWSR1-PATZ1 fusion. The overall findings raised the possibility of a myoepithelial tumor although ultimately due to its unusual features, it was felt that the neoplasm was an unclassifiable low-grade sarcoma. The surgical resection margins were negative for tumor. The patient did not receive any adjuvant therapy and repeat magnetic resonance imaging after 6 months showed no evidence of recurrent tumor. Currently there is no evidence of disease 19 months after the resection.
Cases 4–11
Eight additional cases were identified with EWSR1-PATZ1 fusion from sequencing studies performed using the same platform (Table 1). Three of the eleven cases are central nervous system tumors, including one previously reported [7], and seven are sarcomas. Case 8 was listed as a soft tissue sarcoma from a disease site in the brain and it was therefore unclear whether this was a central nervous system primary or a sarcoma primary with central nervous system metastasis. Of the seven sarcoma cases, there was an essentially even gender distribution and relatively wide age demographic (range 11–81 years). The working diagnosis at the time of molecular testing was undifferentiated sarcoma (one low grade) in five cases and one each of myoepithelial carcinoma and alveolar rhabdomyosarcoma. Similar to previous reports, the EWSR1-PATZ1-related central nervous system tumors were all identified in the adolescent/young adult population [6,7,8,9].
Histopathological characterization of EWSR1-PATZ1 sarcomas
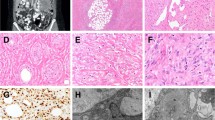
Biopsy or excision samples from three cases detailed underwent histopathological review. For Case 1, the biopsy revealed a high-grade malignant round cell neoplasm with fibrocollagenous bands (Fig. 1a–d). The neoplastic cells demonstrated mild pleomorphism with high nuclear-to-cytoplasmic ratios. Mitoses up to 4–5 per 10 high-power fields were noted. No significant necrosis was identified. The biopsy of Case 2 was composed of cells with an admixed round and spindle cell morphology and dense intratumoral fibrocollagenous stroma (Fig. 1e–h). Scattered cells had slightly atypical vesicular nuclei with small nucleoli, but lacked significant pleomorphism. Increased mitotic figures were not readily identified, though Ki-67 was positive in 20% of tumor nuclei. Significant tumor necrosis was present. For Case 3, the excision specimen showed a well-circumscribed mesenchymal proliferation composed of vast expanses of rounded cells focally interrupted by cystic spaces and a myxohyaline stroma (Fig. 1i–l). The cytological features varied in the degree of nuclear atypia; in the most cellular areas, the tumor cells were rather large with prominent nucleoli, whereas in less cellular areas they were smaller. Mitotic figures were scarce and no necrosis was evident. The proliferation index by Ki-67 was ~5%.

Case 1 (a–d H&E and immunohistochemistry). Needle biopsy demonstrating sheets of hyperchromatic round cells with mildly pleomorphic nuclei and high nuclear-to-cytoplasmic ratios and fibrocollagenous bands. No significant necrosis was identified. By immunohistochemistry the tumor was negative for S100 protein (c) and GFAP (d). Case 2 (e–h H&E and immunohistochemistry). Representative sections show the tumor to be composed of an admixture of round and spindled cells with dense intratumoral fibrocollagenous stroma. Individual nuclei exhibited fine chromatin with occasional small nucleoli. The neoplastic cells were immunoreactive for S100 protein (g) and GFAP (h). Case 3 (i–l H&E and immunohistochemistry). Representative sections show a mesenchymal proliferation composed of rounded cells interrupted by cystic spaces and a myxohyaline background. Cytologically the tumor cells varied slightly in size. Mitotic figures were scarce and no necrosis was identified. By immunohistochemistry the tumor was positive for S100 protein (k) and GFAP (l)
The immunohistochemistry findings are summarized in Table 3 (select stains illustrated in Fig. 1). Notably, neoplastic cells exhibited immunoreactivity for S100, muscle markers (e.g., desmin, myogenin, and MyoD1), GFAP and synaptophysin, and a lack of immunoreactivity for epithelial markers (e.g., keratin, AE1/AE3, and EMA) in at least two cases of the current study; findings analogous to prior reports of EWSR1-PATZ1 fusion positive sarcomas [3,4,5].
Identification of EWSR1-PATZ1 fusion cases
Paired-end sequencing data indicated a somatic intrachromosomal rearrangement of chromosome 22 in all cases (Fig. 2a). The genomic locations of the breakpoints at a base pair resolution were predicted by direct visualization of the mapped-sequencing data (Fig. 2b). An inversion of chromosome 22 material permits transcription of EWSR1 and PATZ1 in the same direction (Fig. 2c).

a Integrative genomics viewer visualization of alignments of paired-end sequencing reads from the whole exome sequence analysis of Case 3—split-screen showing representative mate pairs mapping to different regions of chr22q12.2. Dark red and blue sequences correspond to read pairs mapped in the reverse orientation compared to the reference genome. Green bars relate to sequences that align to the reference genome. The reading indicates that the 5′ part of EWSR1 on chromosome 22q12.2 was fused to the 3′ part of PATZ1 on chromosome 22q12.2 resulting in an EWSR1-PATZ1 fusion. b Genomic locations of paired-end sequencing read at base pair resolution obtained by next-generation sequencing for Cases 1–3. c Schematic diagram of chromosome 22q is illustrating the chromosomal location and transcription orientation of EWSR1 and PATZ1 before and after the intrachromosomal rearrangement. EWSR1 and PATZ1 have opposite directions of transcription and the distance between the two genes is ~2.0 Mbp
Material was available for RNA extraction from Cases 1–3. Reverse transcription-polymerase chain reaction and direct sequencing using customized primers targeting the predicted breakpoints was performed. Polymerase chain reaction products of expected sizes were detected in all three cases as shown by gel electrophoresis (Fig. 3a–c). One sample, Case 1, failed the direct sequencing of the polymerase chain reaction amplicon.

a Reverse transcription-polymerase chain reaction analysis of Case 1; lane M: 100 bp ladder (Thermo Fisher, Cat. no.15628019), lane 1: empty, lane 2: no template with primers E8-970/P1-1340, lane 3: no template with primers E8-1005/P1-1340, lanes 4 and 5: as lanes 2 and 3 with 2.0 µl of cDNA, b Case 2; lane M: 100 bp ladder (Thermo Fisher, Cat. no.15628019), lane 1: E8-1167 forward primer from EWSR1 exon 8 and reverse primer P-1665 from PATZ1 exon 1, lane 2: E8-1167 with P1-1689, c Case 3; lane M: 100 bp ladder (BioRad, Cat. no.1708202), lane 1: E8-1167 from EWSR1 exon 8 and P-1665 from PATZ1 exon 1, lane 2: E8-1167 with P1-1689, lane 3; E8-1167 with P1-1689, d Nucleotide sequence and deduced the amino acid sequence of the EWSR1-PATZ1 fusion transcript around the fusion point in Case 2, e The horizontal green bar indicates the EWSR1 intronic sequence included in the fusion between EWSR1 exon 8 (red bar) and PATZ1 exon 1 (blue bar). The exon numbers are based on the indicated RefSeq accession numbers. The dashed line shows the border between exon 9* composed of 28-nucleotide sequence and PATZ1 exon 1. The red and blue squares represent the exons of EWSR1 (NM_005243) and PATZ1 (NM_014323) genes, respectively. Solid bars indicate the exon borders, f Nucleotide sequence and inferred amino acid sequence of the EWSR1-PATZ1 fusion transcript in Case 3, g Schematic representation of the EWSR1 and PATZ1 genomic organization and fusion transcript. An 8* illustrates the novel exon created by the fusion of exon 8 of EWSR1 and sequences of PATZ1 exon 1 distal from nucleotide position 1606. The dashed line represents the border of exon 8*. The black triangle indicates the position of the in-frame deletion. The EWSR1-PATZ1 fusion created by an inversion of 22q12 removes the putative transcriptional repressor domain and the AT-hook domain at the N-terminus of PATZ1 and converts a transcriptional repressor into a transcriptional activator
Direct sequencing of the reverse transcription-polymerase chain reaction products of Case 2 (Fig. 3d) revealed a 28-nucleotide insertion between EWSR1 exon 8 and PATZ1 exon 1 at the nucleotide position 1631 (Fig. 3d, e). A basic BLAST analysis (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov) of the inserted sequence showed 100% identity with a genomic DNA sequence located within EWSR1 intron 8 near the 3′ end (GenBank accession number NG_023240). Analysis of this sequence at the genomic DNA level by the Human Splicing Finder software version 3.1 (http://www.umd.be/HSF/) and SROOGLE (http://sroogle.tau.ac.il/) was performed to identify splicing signals. A consensus acceptor splice site (aacttccatattag), a polypyrimidine tract and a potential lariat branch point (aatgaac) were identified. Finally, GeneMark (http://exon.gatech.edu/GeneMark/) software recognized the 28-nucleotide sequence of EWSR1 intron 8 as exonic encoding a ten-amino acid sequence (GKYSSLLKDS) inserted between amino acid 325 of EWSR1 and amino acid 324 of PATZ1.
Sanger sequencing of the reverse transcription-polymerase chain reaction products of Case 3 confirmed an in-frame fusion between exon 8 of EWSR1 and sequences of PATZ1 exon 1 at nucleotide position of 1606, A total of 944 nucleotides downstream of the translation initiation site (Fig. 3f, g). The sequence analyses also disclosed an in-frame deletion within the exon 1 of PATZ1 from 1611 to 1629 nucleotide position (Fig. 3f).
Genetic alterations in addition to the EWSR1-PATZ1 fusion
In addition to the EWSR1-PATZ1 fusion, comprehensive molecular profiling revealed a number of secondary genetic changes expected to contribute to oncogenesis (Table 1). Most notably, CDKN2A/CDKN2B loss was frequent, occurring in five of the seven sarcoma cases (71%) and one of the central nervous system tumors. Additional significant alterations were not recurrent in this series, but included single examples of oncogenic alterations in CTNNB1, DAXX, MDM2, NF2, RAF1, TLL2, and ZMYM3. All tumors were found to have a low mutational burden (<6 mutations/Mb) and to be microsatellite stable.
Molecular cytogenetic analysis (fluorescence in situ hybridization)
Fluorescence in situ hybridization analyses of Cases 1–3 confirmed the presence of MDM2 amplification and CDKN2A deletion seen by next-generation sequencing in Case 1 (Fig. 4a1, 4a2 respectively) with no evidence of either anomaly in Cases 2 and 3 (Fig. 4b, c). Specifically, Case 1 demonstrated an amplified MDM2/CEP12 ratio of 5.4 and homozygous deletion for the CDKN2A locus in 92% of the 100 interphase cells evaluated. Of note, the next generation sequencing profiling of Case 2 revealed a deletion involving CDKN2A exon 1; a finding below the resolution of fluorescence in situ hybridization with the commercial probe utilized [15].

a1 MDM2 amplification (orange signals; copy number control probe for CEP12 in green) is identified in Case 1, a2 Homozygous loss of the CDKN2A signals in the neoplastic cells (orange; copy number control probe for CEP9 in green) of Case 1. b, c For Cases 2 and 3, fluorescence in situ hybridization studies were negative for MDM2 amplification (Case 3 illustrated) and CDK2NA deletion (Case 2 illustrated)
Discussion
The identification of specific fusion transcripts has played a vital role in the classification of sarcomas. EWSR1-PATZ1 is a rare fusion partnering that currently resides in the World Health Organization (WHO) undifferentiated sarcoma category [16]. Descriptions of clinicopathologic features to include outcomes or treatment response data are limited or lacking, as is information regarding genetic alterations accompanying EWSR1-PATZ1. In the present study, the clinical, pathological, and genomic findings of a series of EWSR1-PATZ1 fusion related sarcomas are described. This report adds to a growing list of recurrent fusion pairings found in EWSR1-ETS negative sarcomas. Clinicopathological and molecular studies increasingly suggest that these non-EWSR1-ETS fusions define new disease entities distinct from Ewing sarcoma, at least for some of the more recurrent fusions like CIC-DUX4 and BCOR-CCNB3 [17,18,19]. Given the nonoverlapping clinicopathological and molecular findings in the present series as compared to classic Ewing sarcoma, as well as demonstration of a discrete expression profile by Watson et al. [4], suggests that EWSR1-PATZ1 sarcomas are distinct from Ewing sarcoma. In view of the wide range of clinical and pathological features observed in our series and prior reports, it is unclear whether EWSR1-PATZ1 is a “disease-defining” fusion or rather a promiscuous fusion partnering that might affect several distinct cancer types. At a minimum, this fusion driver can affect both primary sarcomas and primary central nervous system tumors.
Clinically, all patients in our series as well as eight previously reported [3,4,5], presented with soft tissue disease. Interestingly, slightly more than half of the cases (at least four of the seven current and four of the eight former) presented with a chest wall location (conceivably, two additional tumors labeled as coming from “head and neck” and “lung” could represent chest wall primary location as well). The age range of those diagnosed with EWSR1-PATZ1 sarcoma in the current and past reports is broad (1–81 years) with a mean of 42 years of age; gender distribution is near equivalent. Evidence of metastatic disease at the time of diagnosis characterized the clinical presentation of two patients (Cases 1 and 2 herein), a finding not previously described. However, development of metastatic disease 5–24 months subsequent to initial diagnosis was observed in two EWSR1-PATZ1 fusion sarcoma patients from earlier studies that included follow-up data [3, 5]. Moreover, all together at least three patients succumbed to their disease within 5–32 months from the time of diagnosis underlining the aggressive behavior impacting a subset of this entity. For Cases 1 and 2 with available systemic treatment outcomes, patients experienced modest responses to conventional systemic chemotherapies, not a typical “Ewing-like” chemosensitivity.
The histopathologic and immunophenotypic features described for EWSR1-PATZ1 sarcomas are fairly diverse [3,4,5]. Morphologically, the predominant cell shape has been described as small round, spindled, or a combination of round and spindled, often accompanied by fibrous stroma. Histologically, at least two of the three presently reviewed cases, Cases 2 and 3, exhibited relatively similar characteristics namely variably cellular areas composed of round to ovoid cells, with no significant pleomorphism or mitotic figures. Both cases shared S100 protein, GFAP, and desmin immunoexpression whereas keratins were negative. This immunohistochemical profile, albeit not definitive for, raised the possibility of myoepithelial carcinoma. Of note, expression of S100 protein, GFAP, muscle markers (e.g., desmin, myogenin, MyoD1), and CD34 and absence of immunoreactivity for epithelial markers were common to all cases for which these assays were performed (with the exception of Case 1 for S100 and GFAP) [3,4,5]. Although the diagnostic consideration of myoepithelial carcinoma is plausible, the histologic characteristics admittedly were not typical of this entity, despite its well-known broad architectural and cytological spectrum. Moreover, although a highly variable immunophenotype can be appreciated in individual myoepithelial tumor cases, S100 protein and an epithelial marker are consistently coexpressed and desmin and CD34 are consistently negative in myoepithelial tumors. Cases 1 and 2 behaved very aggressively whereas Case 3 in an indolent fashion. Cytologic features of moderate to severe atypia (e.g., prominent nucleoli, vesicular or coarse chromatin, and nuclear pleomorphism) relied upon for a diagnosis of malignancy in myoepithelial tumors were not striking in Cases 1–3. Foci of classical appearing-benign areas may be present within a malignant myoepithelial carcinoma; thus, it is conceivable that sampling could potentially contribute to an underestimate of malignant potential [20]. A comparison of transcriptional profiles of at least the S100+/GFAP+ EWSR1-PATZ1 fused cases to broadly accepted EWSR1-rearranged myoepithelial tumors would be of great interest. Although rearrangement of EWSR1 is observed in ~50% of myoepithelial tumors, a partnership with PATZ1 has not been reported [21,22,23,24,25].
PATZ1 is a transcription factor that is believed to be an important regulator of pluripotency and stemness [26]. In various cancers, PATZ1 expressional dysregulation has been implicated in oncogenesis, in some contexts as an oncogene and others as a tumor suppressor [10, 26,27,28,29,30]. In several malignancies, PATZ1 expression has been shown to oppose mesenchymal differentiation [10, 26]. In tumors harboring an EWSR1-PATZ1 fusion, the fusion event would be expected to lead to removal of the N-terminal repressor domain of PATZ1 and therefore result in aberrant overexpression of this transcriptional factor [3].
Notably, PATZ1 resides in close proximity, ~2 Mb away, from EWSR1 on chromosome 22. The EWSR1-PATZ1 fusion event is most likely the result of a submicroscopic intrachromosomal paracentric inversion, although genesis of this fusion via more complex structural alterations at least for some cases cannot be fully excluded [3, 5]. Various publications have addressed the limitations of engaging an ALK break-apart probe system for the detection of the non-small cell lung cancer associated inv(2)(p21p23) EML4-ALK fusion; this intrachromosomal paracentric inversion also considered to involve an extraordinarily short genomic distance at ~12 Mb [31,32,33,34,35,36]. Specifically, visualization of the subtle dissociation or “breaking apart” of the signals poses interpretative challenges and increased random colocalization of dissociated signals due to the short distance between the inversion involved genes makes differentiation between positive and negative patterns an additional interpretative complication that contribute to a comparatively low diagnostic sensitivity (as low as 40–70%) [31]. Therefore, it is probable that sensitivity of EWSR1 break-apart fluorescence in situ hybridization for the detection of the EWSR1-PATZ1 fusion is similarly compromised. The EWSR1 rearrangement was not detected by fluorescence in situ hybridization in Case 1, however, polysomy for this locus (3–6 copies of juxtaposed 5′EWSR1/3′EWSR1 signals) was observed. This is a novel finding for EWSR1-PATZ1 fusion sarcoma as previous cases with available data have reportedly shown diploid copy number (polysomy 22q12 also a highly unusual fluorescence in situ hybridization pattern for Ewing sarcoma [37]). The presence of polysomy may have further complicated recognition/detection of an EWSR1 rearrangement in Case 1. Analogous to other novel fusions, it would be expected that the true incidence of this recurrent event will be uncovered in time with the more widespread adoption of next-generation sequencing assays for diagnostic purposes.
Descriptions of insertions of intronic pseudoexons within fusion genes are uncommon. For example, insertions of intronic sequences in BCR-ABL and EWSR1-FLI1 fusions have been described in a few isolated cases [38,39,40]. Their identification however, has resulted in expansion of the spectrum of molecular transcript variant diversity. Interestingly in the present study, a novel intronic pseudoexon sequence inserted within the EWSR1-PATZ1 fusion of Case 2 was characterized by sequencing at the mRNA level. Specifically, a 28-nucleotide-long pseudoexon of EWSR1 inserted in a position of PATZ1 exon 1 that maintained the reading frame and did not produce a premature stop codon was identified. The novel EWSR1-PATZ1 chimeric transcript generated encodes a functional oncoprotein with an in-frame insertion of ten additional amino acids. The biological and clinical significance of this rearrangement, however, such as a potential difference in transactivation potential or correlation with a distinct clinical course are not known.
A unique and prominent finding of the genomically profiled EWSR1-PATZ1 neoplasms of the current series was loss or partial deletion of CDKN2A/CDKN2B in 71% (5/7) of the sarcomas and in one of the central nervous system tumors. Identification of recurrent secondary genetic events in fusion gene driven cancers has contributed to the understanding of the biology and behavior of many different tumor types. For example, inactivation of the 9p21 locus encoding tumor suppressors CDKN2A and CDKN2B is a frequent acquired event during BCR-ABL1 positive acute lymphoblastic leukemia progression and a poor prognostic marker of long-term outcomes [41, 42]. The incidence of homozygous loss of 9p21 (CDKN2A/CDKN2B) in Ewing sarcoma at around 12% represents a minor subgroup that has experienced conflicting conclusions regarding its utility as a reliable prognostic biomarker. In some studies, loss of CDKN2A in Ewing sarcoma has been shown to correlate with highly aggressive behavior and poor chemoresponse whereas in other studies, CDKN2A/CDKN2B loss was not significantly associated with event-free or overall survival [43,44,45,46,47]. In soft tissue myoepithelial tumor, the only recurrent aberration of known importance in a whole genome DNA copy number imbalance study of five cases lacking EWSR1 rearrangement or EWSR1-PBX1, FUS-PBX1, and TAF15-PBX1 fusions (as assessed by fluorescence in situ hybridization or reverse transcription-polymerase chain reaction), was homo- and heterozygous deletions of the 9p21 (CDKN2A/CDKN2B) locus [48]. Further profiling studies are needed to determine the prevalence and role of these tumor suppressor genes in soft tissue myoepithelial tumor.
Of potential prognostic significance, the tumors of Cases 1 and 2 exhibiting aggressive behavior with metastatic disease upon presentation and only weak to modest therapeutic responses both harbored CDKN2A/CDKN2B alterations in contrast to Case 3 that lacked this secondary genetic change and was clinically indolent. Specifically, Case 1 demonstrated homozygous deletion of the 9p21.3 locus and MDM2 amplification whereas a heterozygous CDKN2A exon 1 deletion accompanied the EWSR1-PATZ1 fusion in Case 2. The p53 pathway is negatively regulated by MDM2, which itself is sequestered and inhibited by p14ARF (encoded by the gene locus CDKN2A) [49, 50]. The tumor suppressor protein p16INK4A, also encoded by CDKN2A, exerts its biological effects through the pRb pathway [50, 51]. Consequently, deletion of CDKN2A usually results in loss of the growth inhibition provided by both the p53 and pRb pathways. Overexpression of MDM2 (often the result of MDM2 amplification) has been reported in a variety of tumors and may relate to an increased likelihood of distant metastases, decreased response to therapeutic intervention and poor clinical prognosis [52]. Retrospective genomic profiling for assessment of the prevalence of MDM2 amplification and coalterations of a series of 523 advanced cancer patients to include sarcomas and carcinomas revealed a low incidence (4%) of MDM2 amplification [53]. Of the 23 patients with MDM2 amplification, five (22%) were noted to have co-occurrence of CDKN2A/CDKN2B loss.
In summary, the current series of EWSR1-PATZ1 fusion-related sarcomas adds to the growing number of recurrent driver fusions found in EWSR1-ETS negative sarcomas. Clinicopathologically although EWSR1-PATZ1 fusion sarcomas appear to exhibit a roughly equal gender distribution and tendency to arise in the chest wall, the age range affected is quite broad and there is a spectrum of round to spindle cell morphology, polyphenotypic differentiation, and clinical behavior diversity posing challenges with precise classification. Herein, comprehensive molecular testing revealed a number of secondary aberrations expected to contribute to oncogenesis. Most notably, CDKN2A/CDKN2B loss was frequent, occurring in 5/7 (71%) sarcomas and 1/4 (25%) CNS tumors. CDKN2A/CDKN2B loss is considered a common mechanism for cell cycle dysregulation and may have prognostic significance in EWSR1-PATZ1 neoplasia.
References
Alava E de, Lessnick SL, Sorensen PH. Ewing sarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: International Agency for Research on Cancer; 2013. p. 306–9.
Brohl AS, Solomon DA, Chang W, Wang J, et al. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10:e1004475.
Mastrangelo T, Modena P, Tornielli S, Bullrich F, Testi MA, Mezzelani A, et al. A novel zinc finger gene is fused to EWS in small round cell tumor. Oncogene. 2000;19:3799–804.
Watson S, Perrin V, Guillemot D, Reynaud S, Coindre JM, Karanian M, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018;245:29–40.
Chougule A, Taylor MS, Nardi V, Chebib I, Cote GM, Choy E, et al. Spindle and round cell sarcoma with EWSR1-PATZ1 gene fusion. Am J Surg Pathol. 2019;43:220–8.
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131:833–45.
Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 2017;22:1478–90.
Alvarez-Breckenridge C, Miller JJ, Nayyar N, Gill CM, Kaneb A, D’Andrea M, et al. Clinical and radiographic response following targeting of BCAN-NTRK1 fusion in glioneuronal tumor. NPJ Precis Oncol. 2017;1:5
Siegfried A, Rousseau A, Maurage CA, Pericart S, Nicaise Y, Escudie F, et al. EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 2019;29:53–62.
Guadagno E, Vitiello M, Francesca P, Calì G, Caponnetto F, Cesselli D, et al. PATZ1 is a new prognostic marker of glioblastoma associated with the stem-like phenotype and enriched in the proneural subtype. Oncotarget. 2017;8:59282–300.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Lerman DM, Monument MJ, McIlvaine E, Liu XQ, Huang D, Monovich L, et al. Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children’s Oncology Group. Pedatr Blood Cancer. 2015;62:759–65.
Hoggard TM, Henderson-Jackson E, Bui MM, Caracciolo J, Teer JK, Yoder S, et al. Myoepithelial carcinoma with RB1 mutation: remarkable chemosensitivity to carcinoma of unknown origin therapy. BMC Cancer. 2017;17:250.
Savola S, Nardi F, Scotlandi K, Picci P, Knuutila S. Microdeletions in 9p21.3 induce false negative results in CDKN2A FISH analysis of Ewing sarcoma. Cytogenet Genome Res. 2007;119:21–6.
Fletcher CDM, Chibon F, Mertens F. Undifferentiated/Unclassified sarcomas. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: International Agency for Research on Cancer; 2013. p. 236–8.
Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53:622–33.
Antonescu CR, Owosho AA, Zhang L, Chen S, Deniz K, Huryn JM, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41:941–9.
Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–6. 44
Horick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183–96.
Brandal P, Panagopoulos I, Bjerkehagen B, Gorunova L, Skjeldal S, Micci F, et al. Detection of a t(1;22)(q23; q12) translocation leading to an EWSR1-PBX1 fusion gene in a myoepithelioma. Genes Chromosomes Cancer. 2008;47:558–64.
Brandal P, Panagopoulos I, Bjerkehagen B, Heim S. t(19;22)(q13; q12) translocation leading to the novel fusion gene EWSR1-ZNF444 in soft tissue myoepithelial carcinoma. Genes Chromosomes Cancer. 2009;48:1051–6.
Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–24.
Agaram NP, Chen HW, Zhang L, et al. EWSR1-PBX3: a novel gene fusion in myoepithelial tumors. Genes Chromosomes Cancer. 2015;54:63–71.
Flucke U, Mentzel T, Verdijk MA, Slootweg PJ, Creytens DH, Suurmeijer AJ, et al. EWSR1-ATF1 chimeric transcript in a myoepithelial tumor of soft tissue: a case report. Hum Pathol. 2012;43:764–8.
Ow JR, Ma H, Jean A, Goh Z, Lee YH, Chong YM, et al. Patz1 regulates embryonic stem cell identity. Stem Cells Dev. 2014;23:1062–73.
Chiappetta G, Valentino T, Vitiello M, Pasquinelli R, Monaco M, Palma G, et al. PATZ1 acts as a tumor suppressor in thyroid cancer via targeting p53-dependent genes involved in EMT and cell migration. Oncotarget. 2015;6:5310–23.
Fedele M, Franco R, Salvatore G, Paronetto MP, Barbagallo F, Pero R, et al. PATZ1 gene has a critical role in the spermatogenesis and testicular tumours. J Pathol. 2008;215:39–47.
Franco R, Scognamiglio G, Valentino E, Vitiello M, Luciano A, Palma G, et al. PATZ1 expression correlates positively with BAX and negatively with BCL6 and survival in human diffuse large B cell lymphomas. Oncotarget. 2016;7:59158–72.
Tian X, Sun D, Zhang Y, Zhao S, Xiong H, Fang J. Zinc finger protein 278, a potential oncogene in human colorectal cancer. Acta Biochim Biophys Sin. 2008;40:289–96.
Smuk G, Tornóczky T, Pajor L, Chudoba I, Kajtár B, Sárosi V, et al. Immense random colocalization, revealed by automated high content image cytometry, seriously questions FISH as gold standard for detecting EML4‐ALK fusion. Cytom A. 2018;93:653–61.
Demidova I, Barinov A, Savelov N, Gagarin I, Grinevitch V, Stroiakovaski D, et al. Immunohistochemistry, fluorescence in situ hybridization, and reverse transcription-polymerase chain reaction for the detection of anaplastic lymphoma kinase gene rearrangements in patients with non-small cell lung cancer: potential advantages and methodologic pitfalls. Arch Pathol Lab Med. 2014;138:794–802.
von Laffert M, Stenzinger A, Hummel M, Weichert W, Lenze D, Warth A, et al. ALK-FISH borderline cases in non-small cell lung cancer: implications for diagnostics and clinical decision making. Lung Cancer. 2015;90:465–41.
Selinger C, Cooper W, Lum T, McNeil C, Morey A, Waring P, et al. Equivocal ALK fluorescence in-situ hybridization (FISH) cases may benefit from ancillary ALK FISH probe testing. Histopathology. 2015;67:654–63.
Pekar-Zlotin M, Hirsch FR, Soussan-Gutman L, Ilouze M, Dvir A, Boyle T, et al. Fluorescence in situ hybridization, immunohistochemistry, and next-generation sequencing for detection of EML4-ALK rearrangement in lung cancer. Oncologist. 2015;20:316–22.
Li W, Zhang J, Guo L, Chuai S, Shan L, Ying J. Combinational analysis of FISH and immunohistochemistry reveals rare genomic events in ALK fusion patterns in NSCLC and responds to crizotinib treatment. J Thorac Oncol. 2016;12:94–101.
Sandberg AA, Bridge JA. Updates on cytogenetics and molecular genetics of bone and soft tissue tumors: Ewing sarcoma and peripheral primitive neuroectodermal tumors. Cancer Genet Cytogenet. 2000;123:1–26.
Roman J, Parziale A, Gottardi E, De Micheli D, Cilloni D, Tiribelli M, et al. Novel type of BCR-ABL transcript in a chronic myelogenous leukaemia patient relapsed after bone marrow transplantation. Br J Haematol. 2000;111:644–6.
Sorel N, Mayeur-Rousse C, Deverrière S, Roy L, Brottier-Mancini E, Guilhot F, et al. Comprehensive characterization of a novel intronic pseudo-exon inserted within an e14/a2 BCR-ABL rearrangement in a patient with chronic myeloid leukemia. J Mol Diagn. 2010;12:520–4.
Kovar H, Jugovic D, Melot T, Zoubek A, Lenoir G, Fink FM, et al. Cryptic exons as a source of increased diversity of Ewing tumor-associated EWS-FLI1 chimeric products. Genomics. 1999;60:371–4.
Williams RT1, Sherr CJ. TheINK4-ARF (CDKN2A/B) locus in hematopoiesis and BCR-ABL-induced leukemias. Cold Spring Harb Symp Quant Biol. 2008;73:461–7.
Iacobucci I, Ferrari A, Lonetti A, Papayannidis C, Paoloni F, Trino S, et al. CDKN2A/B alterations impair prognosis in adult BCR-ABL1-positive acute lymphoblastic leukemia patients. Clin Cancer Res. 2011;17:7413–23.
Wei G, Antonescu CR, de Alava E, Leung D, Huvos AG, Meyers PA, et al. Prognostic impact of INK4A deletion in Ewing sarcoma. Cancer. 2000;89:793–9.
Lopez-Guerrero JA, Pellin A, Noguera R, Carda C, Llombart-Bosch A. Molecular analysis of the 9p21 locus and p53 genes in Ewing family tumors. Lab Investig. 2001;81:803–14.
Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, et al. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol. 2005;23:548–58.
Lerman DM, Monument MJ, McIlvaine E, Liu XQ, Huang D, Bridge JA, et al. Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62:759–65.
Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4:1342–53.
Hallor KH, Teixeira MR, Fletcher CD, Bizarro S, Staaf J, Domanski HA, et al. Heterogeneous genetic profiles in soft tissue myoepitheliomas. Mod Pathol. 2008;21:1311–9.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45.
Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000.
Serrano M. The tumor suppressor protein p16INK4a. Exp Cell Res. 1997;237:7–13.
Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5:27–41.
Dembla V, Somaiah N, Barata P, Hess K, Fu S, Janku F, et al. Prevalence of MDM2 amplification and co-alterations in 523 advanced cancer patients in the MD Anderson phase 1 clinic. Oncotarget. 2018;9:33232–43.
Acknowledgements
The authors would like to thank Dr. Xiao-qiong Liu for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bridge, J.A., Sumegi, J., Druta, M. et al. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol 32, 1593–1604 (2019). https://doi.org/10.1038/s41379-019-0301-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0301-1
This article is cited by
-
Top 10 Histological Mimics of Neuroendocrine Carcinoma You Should Not Miss in the Head and Neck
Head and Neck Pathology (2023)
-
A novel LARGE1-AFF2 fusion expanding the molecular alterations associated with the methylation class of neuroepithelial tumors with PATZ1 fusions
Acta Neuropathologica Communications (2022)
-
Small round cell sarcomas
Nature Reviews Disease Primers (2022)
-
An international working group consensus report for the prioritization of molecular biomarkers for Ewing sarcoma
npj Precision Oncology (2022)
-
Multiscale-omic assessment of EWSR1-NFATc2 fusion positive sarcomas identifies the mTOR pathway as a potential therapeutic target
npj Precision Oncology (2021)
