
Abstract
In 2008, we presented three cases of ALK-positive histiocytosis as a novel systemic histiocytic proliferation of early infancy with hepatosplenomegaly and dramatic hematological disturbances. This series of 10 cases (including the original three cases) describes an expanded clinicopathological spectrum and the molecular findings of this histiocytic proliferation. Six patients had disseminated disease: five presented in early infancy with eventual disease resolution, and the sixth presented at 2 years of age and died of intestinal, bone marrow, and brain involvement. The other four patients had localized disease involving nasal skin, foot, breast, and intracranial cavernous sinus – the first three had no recurrence after surgical resection, while the cavernous sinus lesion showed complete resolution with crizotinib therapy. The lesional histiocytes were very large, with irregularly folded nuclei, fine chromatin, and abundant eosinophilic cytoplasm, sometimes with emperipolesis. There could be an increase in foamy histiocytes and Touton giant cells with time, resembling juvenile xanthogranuloma. Immunostaining showed that the histiocytes were positive for ALK, histiocytic markers (CD68, CD163) and variably S100, while being negative for CD1a, CD207, and BRAF-V600E. Next-generation sequencing-based anchored multiplex PCR (Archer® FusionPlex®) performed in six cases identified KIF5B-ALK gene fusion in five and COL1A2-ALK fusion in one. There was no correlation of gene fusion type with disease localization or dissemination. The clinicopathological spectrum of ALK-positive histiocytosis is broader than originally described, and this entity is characterized by frequent presence of KIF5B-ALK gene fusion. We recommend that every unusual histiocytic proliferative disorder, especially disseminated lesions, be tested for ALK expression because of the potential efficacy of ALK inhibitor therapy in unresectable or disseminated disease.
Similar content being viewed by others
Introduction
In 2008, we described a series of three patients with a novel type of systemic histiocytic proliferative disorder termed “ALK-positive histiocytosis” [1]. The patients were young infants presenting with pallor, hepatosplenomegaly, severe thrombocytopenia, and anemia. Two patients received a short course of chemotherapy with limited initial response, but all three patients eventually had disease resolution after many months. In one patient, skin lesions that subsequently developed had histological similarity to juvenile xanthogranuloma. However, unlike juvenile xanthogranuloma, the histiocytes in all three patients showed marked nuclear foldings and were uniformly immunoreactive for ALK. TPM3-ALK gene fusion was identified in the single case where materials were available for molecular analysis.
In this study, we describe the clinical, pathological, and molecular findings of 10 cases, including the original three patients with updated long-term follow-up. Significantly, a unique KIF5B-ALK gene fusion was identified in five cases, and one patient with an unresectable intracranial lesion showed a dramatic response to ALK inhibitor therapy.
Materials and methods
Case selection
Ten cases of ALK-positive histiocytosis from three hospitals (Royal Children’s Hospital, Melbourne, Australia; Queen Elizabeth Hospital, Hong Kong Special Administrative Region, China; and KK Women’s and Children’s Hospital, Singapore) were reviewed. These included the three cases previously published [1] and a previously published case of systemic juvenile xanthogranuloma for which materials could be obtained for study [2]. Clinical history was obtained from the patient charts, archival diagnostic materials were reviewed, and additional studies were performed.
Immunohistochemistry
Four μm-thick sections cut from formalin-fixed paraffin-embedded tissue blocks were used for immunostaining in a BenchMark ULTRA automated immunostainer (Ventana Medical System, Tucson, AZ). The antibodies used are listed in Table 1, with the OptiView DAB IHC Detection Kit (Roche) being used for detection. Negative controls and same-slide positive controls were employed.
Fluorescence in situ hybridization
For ALK gene fluorescence in situ hybridization, the sections were treated with the Vysis paraffin pre-treatment IV system (Vysis, Downers Grove, IL, USA). The tissue sections and ALK break-apart probes (Vysis) were co-denatured at 73 °C for 5 min and then incubated at 37 °C using the Thermobrite denaturation-hybridization system (Vysis). Fifty nuclei were scored, and ALK gene rearrangement was indicated by a split signal, defined as separation of signals by more than two signal widths. The result was considered positive if more than 20% of the assessed nuclei had split signals.
Nucleic acid extraction
Total RNA was extracted from scrolls cut from formalin-fixed and paraffin-embedded tissue blocks using the Promega ReliaPrep™ FFPE Total RNA Miniprep System (Promega, Madison, WI, USA) following the manufacturer’s recommended protocol. The quantity of RNA extracted was measured using the QuantiFluor® RNA System (Promega, Madison, WI, USA).
Gene fusion identification by anchored multiplex PCR (Archer® FusionPlex®)
To identify the partner gene fused with ALK, 200 ng of RNA was used for library preparation utilizing the Archer® FusionPlex® sarcoma panel kit (AK00328) following the manufacturer’s protocol (ArcherDX, Boulder, CO, USA). The prepared library was sequenced using an Ion Torrent PGM™ next-generation sequencer. The Hi-Q™ sequencing kit and Hi-Q™ OT2 kit were used following the manufacturer’s protocol (Life Technologies, Forster City, CA, USA). The data obtained was analysed by the Archer Data Analysis (version 4.1.1) portal (ArcherDX, Boulder, CO, USA).
Confirmatory reverse-transcriptase PCR and Sanger sequencing
The anchored multiplex PCR fusion gene results were confirmed by reverse transcriptase PCR (RT-PCR). 50 ng of sample RNA was used for one-step RT-PCR using the GoTaq 1-Step RT-PCR kit (Promega, Madison, WI, USA). Primers flanking the breakpoints for both KIF5B-ALK and COL1A2-ALK gene fusions are detailed in Table 2. The temperature profile for the one step RT-PCR was 45 oC for 15 min, 95 oC for 5 min, 40 cycles of 95 oC for 15 s, 60 oC for 15 s, and 72 oC for 20 s. The sizes of the PCR products were determined by inspection against a size ladder on 3% agarose gel. The bands of interest were excised and submitted for Sanger sequencing using the ABI3730xl DNA sequencer (Applied Biosystems, Foster City, CA) to confirm the breakpoint sequence.
Results
Clinical findings
The major clinical, pathological and molecular findings are summarized in Table 3, and detailed clinical histories are provided in the Supplementary Information. The patients included 4 males and 6 females, predominantly of young age, comprising 5 young infants, 2 children, 2 teenagers, and one adult. The patients included Caucasians and Asians (including Chinese, Thai, and Middle Eastern). Six cases had systemic (B1, B2, B3, P1, N1, and N2) and four localized disease (N3, N4, N5, and N6). Patients with systemic disease tended to be young children (five were younger than 3 months and one was 2 years 9 months old), who presented with abdominal distension (with hepatosplenomegaly in five), anemia and thrombocytopenia, and additional findings depending on site of involvement, e.g. skin nodules in patients B1 and N1, diarrhea and protein-losing enteropathy in patient N2. The localized cases presented with tumors in the following sites: nasal skin, intracranial cavernous sinus, foot and breast, and the patients tended to be older. All patients had disease resolution on follow up, except patient N2, who succumbed to systemic disease.
ALK-positive histiocytosis treated with crizotinib
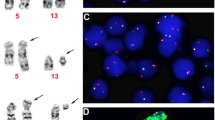
Patient N4 was a 15-year-old male presenting with a 3-month history of progressive diplopia and inability to move the right eye. He was found to have a 2.4 cm cavernous sinus lesion, and biopsy was complicated by asystole, likely secondary to a trigemino-cardiac reflex. Only a very small biopsy could be obtained. Histology showed a histiocytic infiltrate mixed with many lymphocytes, occasional plasma cells, and scattered Langhans-type giant cells (Fig. 1a). ALK immunohistochemistry was positive (Fig. 1b). The patient received crizotinib treatment and the cavernous sinus lesion (Fig. 1c) resolved completely 3 months later as assessed by imaging (Fig. 1d), and the child has remained well with full recovery of eye movements.

Histologic and radiologic features of patient N4 with cavernous sinus lesion. a Histiocytic infiltrate with scattered Langhans-type giant cells; there were many admixed lymphocytes (H&E, magnification ×200). b ALK immunoreactivity in histiocytic cells in a cytoplasmic staining pattern (magnification ×200). c, d Enhanced T1-weighted axial MR image of skull shows a right-sided cavernous sinus lesion (c, yellow arrow) that resolved completely following crizotinib treatment (d, green arrow)
Pathologic features
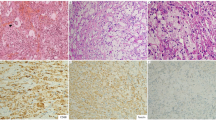
The proliferated histiocytes occurring in the various tissue sites were large cells with irregularly folded, deeply clefted or lobulated nuclei, fine chromatin and small nucleoli (Fig. 2a); occasional cells were multinucleated (Fig. 2b). They had abundant eosinophilic cytoplasm, which could show fine vacuoles. Occasionally they showed phagocytosis of lymphocytes (emperipolesis), polymorphonuclear leukocytes, normoblasts, red cells, or hemosiderin. Foamy histiocytes were absent, focal or extensive, and they showed irregularly folded nuclei similar to the non-foamy ones (Fig. 2c). They were often increased in repeat biopsies taken during the course of disease. There were sometimes admixed Touton giant cells. An exception was seen in case N5, wherein the histiocytes showed oval nuclei with distinct nucleoli (see later).

Histologic features of ALK-positive histiocytosis. a Patient P1 – characteristic histology of histiocytes of ALK-positive histiocytosis with irregularly folded, deeply clefted or lobulated nuclei, fine chromatin, and inconspicuous nucleoli; the histiocytes can be difficult to distinguish from the hepatocytes, which have round nuclei and more deeply eosinophilic cytoplasm. b Patient P1 – the histiocytic cells may be multinucleated. c Patient N1 – the histiocytes may have abundant vacuolated or foamy cytoplasm. d patient P1 – the histiocytic cells are dispersed in the sinusoids and aggregated in the portal tracts of the liver; the pattern of infiltration is best appreciated by ALK immunohistochemistry (inset). e Patient P1 – patchy bone marrow involvement by the histiocytic cells including multinucleate giant cell forms. f Patient N2 – diffuse bone marrow involvement by foamy histiocytes in the third bone marrow biopsy. g Patient N2 – intestinal involvement consists of diffuse lamina propria infiltrates of histiocytic cells which are ALK-immunoreactive (inset). h Patient N1 – the initial skin biopsy shows dermal infiltrates of histiocytes within intervening collagenous strands
In the involved liver (patients B1, B2, B3, P1), the histiocytes occurred singly and as tiny aggregates in the sinusoids, and as large aggregates in the portal tracts. Because of their large size and eosinophilic cytoplasm, the histiocytes could be difficult to distinguish from hepatocytes on casual examination (Fig. 2d) and often required ALK immunohistochemistry for identification (inset of Fig. 2d). Bone marrow involvement could be subtle (patients B2, B3), focal and patchy (patients P1, N2) (Fig. 2e) or extensive (third biopsy of patient N2) (Fig. 2f), while the pattern could not be determined in patient N1 because only an aspirate was available.
In patient N2, the duodenal biopsy showed diffuse dense infiltration of the lamina propria by histiocytes; the villous architecture was well preserved (Fig. 2g).
The skin lesions (patients B1, N1, N3) showed a non-circumscribed, non-epidermotropic infiltrate comprising sheets of histiocytes (Figs. 2h and 3a). Repeat skin biopsy at 8 months from patient N1 showed histiocytes with interspersed Touton giant cells and fibrosis with a storiform pattern (Fig. 3b). The breast lesion (patient N6) showed a non-circumscribed proliferation of histiocytes with focal spindling (Fig. 3c). The histiocytic nuclei showed deep foldings resulting in a cup-like morphology, and mitoses were numerous (Fig. 3d). The foot lesion (patient N5) showed fibrocollagenous tissue with interspersed islands of histiocytic cells (Fig. 3e) with different nuclear features of comparatively bland nuclei with single visible nucleoli (Fig. 3f) – this lesion from patient N5 had a different gene fusion from the other cases (see next section).

Histologic features of ALK-positive histiocytosis (continued). a Patient N1 – the histiocytic cells have ample palely eosinophilic to amphophilic cytoplasm, and ovoid nuclei with variably irregular nuclear contours and occasional nuclear grooves. b Patient N1 – a later biopsy at at 8 months showed interspersed Touton giant cells and fibrosis with a storiform pattern. c Patient N6 – the breast lesion showed a non-circumscribed proliferation of histiocytes. d Patient N6 – the histiocytes of the breast lesion had deep nuclear foldings with a cup-like morphology in some nuclei; mitoses are easily identified. e Patient N5 (COL1A2-ALK fusion) – the histiocytic infiltrate forms irregular islands and nests with surrounding dense collagen. f Patient N5 (COL1A2-ALK fusion) – the histiocytic cells have ample amphophilic cytoplasm that is finely vacuolated; the nuclei are round to oval and comparatively bland with a single small nucleolus and minimal nuclear contour irregularities. g Patient P1 – the histiocytic cells show strong cytoplasmic positive immunoreactivity for ALK. h Patient N2 – FISH using an ALK breakapart probe shows a positive result with separation of the green and red signals (arrows)
Immunostaining showed that the lesional histiocytes (including those with foamy cytoplasm) were ALK positive in a cytoplasmic pattern (Fig. 3g) and were also positive for histiocytic markers CD68 and CD163. S100 was positive in 4 of 8 cases (50%). CD1a, langerin and BRAF V600E were negative in all cases. Fluorescence in-situ hybridization utilizing breakapart probes to the ALK gene was performed in all but two cases (B2, B3), and all showed split signals (Fig. 3h).
Gene fusion study results
Suitable materials were available for next-generation sequencing-based anchored multiplex PCR in 6 cases. Five samples (P1, N1, N3, N4, and N6) showed KIF5B-ALK gene fusion, with identical breakpoints involving exon 24 of KIF5B and exon 20 of ALK in all five cases (Fig. 4a). One sample (N5) had a COL1A2-ALK fusion involving exon 51 of COL1A2 and a partial sequence of intron 18 followed by exon 19 of ALK (Fig. 4b). All the fusion sequences were confirmed by RT-PCR followed by Sanger sequencing. Cases B1, B2, B3, and N2 were analysed by RT-PCR for both the KIF5B-ALK and COL1A2-ALK gene fusions, but the RNA was too degraded to yield results. One case (B1) has previously been shown to harbor TPM3-ALK gene fusion [1].

Sequencing results. a Anchored multiplex PCR (Archer sarcoma panel) and confirmatory Sanger sequencing of case N1 showing strong evidence of fusion of KIF5B exon 24 with ALK exon 20. b Anchored multiplex PCR (Archer sarcoma panel) showing strong evidence of fusion of COL1A2 exon 51 with ALK exon 19, with intervening segment of ALK intron 18. c, d Confirmatory Sanger sequencing
Discussion
Clinicopathologic and genetic features of ALK-positive histiocytosis
Since the first publication on ALK-positive histiocytosis in 2008, there has only been one formal report on this entity, describing two neonates showing several features not observed in the original report, such as renal involvement, extensive marrow involvement, and presence of histiocytes with emperipolesis simulating Rosai-Dorfman disease [3]. These features are indeed observed in some of the newly encountered cases of this series. In a large series of tumors studied for the presence of ALK translocation, a 40-year-old man is reported to have systemic ALK-positive “non-Langerhans cell histiocytosis” involving cervical spinal cord, liver, peritoneum, bone, and skin [4]. Thus the clinicopathological spectrum of ALK-positive histiocytosis is wider than originally described [1]. The most common clinical picture is that of a very young infant presenting with hepatosplenomegaly and severe cytopenia, requiring transfusion support over many months. In spite of the worrisome clinical picture, there is gradual spontaneous recovery, possibly aided by chemotherapy. However, systemic disease can also occur in adults [4], and rare patients may succumb to systemic disease (such as patient N2 who had intestinal, bone marrow, and nervous system involvement). Other patients have localized disease, and remain well following resection, or as in the patient with inoperable cavernous sinus lesion, following treatment with ALK inhibitor crizotinib. The rapidity of the resolution in the latter patient suggests that this was the result of crizotinib rather than spontaneous resolution which in the other cases took a far longer period of time.
Since the first description of the typical translocation t(2;5)(p23;q35) in anaplastic large cell lymphoma and the subsequent cloning of the fusion genes ALK-NPM, the number of neoplasms recognized to show frequent ALK gene translocation has increased, including inflammatory myofibroblastic tumor [5], ALK-positive large B-cell lymphoma [6], and epithelioid fibrous histiocytoma [7]. A small percentage of pulmonary carcinomas [8], renal cell carcinomas [9,10,11], thyroid carcinomas [12, 13], colorectal carcinomas [14], pancreatic carcinomas [15], and Spitzoid tumors [16] have also been shown to exhibit ALK translocation. The remarkable and sustained response of ALK-positive neoplasms to ALK inhibitor, as observed in anaplastic large cell lymphoma [17], pulmonary carcinoma [18], and inflammatory myofibroblastic tumor [17, 19] underlines the importance of recognition of ALK gene translocation in neoplasms. In this series of ALK-positive histiocytosis, the encouraging response of the cavernous sinus lesion to ALK inhibitor treatment suggests that targeted therapy may also be considered in patients with systemic disease. Another case of systemic “non-Langerhans cell histiocytosis” with KIF5B-ALK fusion (consistent with ALK-positive histiocytosis) has similarly been reported to show prolonged robust response to ALK inhibitor [4].
Although ALK gene can be fused with many different partner genes in neoplasms [4], a distinctive partner gene is often implicated in a high proportion of cases for each tumor type, such as NPM in ALK-positive anaplastic large cell lymphoma [20], CLTC in ALK-positive large B-cell lymphoma [21], RANBP2 in epithelioid inflammatory myofibroblastic sarcoma [22], SQSTM1 and VCL in epithelioid fibrous histiocytoma [7, 23] and EML4 in pulmonary carcinoma [24]. This study on ALK-positive histiocytosis identifies a recurrent translocation between the KIB5B and ALK genes in the majority of cases by the technique of next-generation sequencing-based anchored multiplex PCR [25]. This technique identifies gene fusions (including novel gene fusions) in a PCR-based assay using primers targeting one fusion partner (from a panel of 26 genes), while the other gene partner is identified through next-generation sequencing in a manner agnostic of the identity and breakpoint of this binding partner.
The KIF5B-ALK gene fusion has thus far been found only in histiocytic neoplasms and a minority of ALK-positive lung carcinomas. It has a consistent breakpoint at exon 24 of KIF5B and exon 20 of ALK. KIF5B encodes the ubiquitous isoform of the heavy chain of kinesin-1, a motor protein involved in intracellular transport via microtubules [26], while ALK encodes a receptor tyrosine kinase. Exons 1–24 of KIF5B comprise the motor domain and most of the alpha helical coiled-coil that participates in kinesin dimerization, and is joined to the intracellular kinase domain of ALK starting from exon 20. As KIF5B is ubiquitously expressed, the resultant chimeric protein is likely a constitutively active kinase. KIF5B is an uncommon partner gene of ALK other than in ALK-positive histiocytosis [4]. In a study searching for targetable kinase alterations as oncogenic drivers in histiocytic neoplasms, two of 24 cases are found to harbor KIF5B-ALK gene fusion [27]. KIF5B-ALK is uncommonly found in lung carcinomas, with a different breakpoint in which exons 15 or 24 of KIF5B is fused to exon 20 of ALK [28, 29]. One of the solitary cases of ALK-positive histiocytosis in our series shows COL1A2-ALK gene fusion, a gene fusion that has not been described in any other neoplasm to date. COL1A2 encodes for the alpha 2 chain of type I collagen and point mutations of COL1A2 occur in osteogenesis imperfecta and related disorders of connective tissue [30].
Is ALK-positive histiocytosis a distinct entity?
ALK-positive histiocytosis appears to be a distinct clinicopathologic entity, and not merely a heterogeneous group of different histiocytic proliferations that share ALK gene translocation as oncogenic driver, at least for the systemic cases. Supporting features include: practically identical clinical presentation and outcome; proliferating histiocytes differing morphologically from those of other histiocytosis in showing marked nuclear foldings; ALK expression resulting from ALK gene translocation, often with KIF5B as partner gene; lack of typical clinical features of Erdheim-Chester disease (skeletal, cardiovascular and retroperitoneal involvement), and lack of evidence of BRAF V600E mutation (a common genetic alteration in Erdheim-Chester disease). The solitary cases probably also represent part of the spectrum of the entity, because the proliferated histiocytes show irregular nuclear foldings similar to the systemic cases (except the case with COL1A2-ALK fusion), and they show the same KIF5B-ALK fusion, a very uncommon type of ALK gene fusion in tumors [4].
Differential diagnosis
The histiocytes in ALK-positive histiocytosis may become foamy with time and Touton giant cells can be admixed, thus raising the differential diagnoses of juvenile xanthogranuloma (disseminated or localized) and Erdheim-Chester disease. Juvenile xanthogranuloma differs from ALK-positive histiocytes in that the lesional histiocytes (including foamy histiocytes) have round to oval nuclei, and S100 is uncommonly positive [31]. It is likely that some cases reported in the literature as disseminated juvenile xanthogranuloma may represent ALK-positive histiocytosis, as exemplified by case P1 [2, 32,33,34,35,36]. Erdheim-Chester disease, a disease predominantly of adults with mean age of 55–60 years, is a clonal systemic histiocytic proliferation most commonly involving bone, cardiovascular system and retroperitoneum, and histologically characterized by foamy histiocytes with small nuclei and Touton giant cells; the diagnosis is made with histology and phenotype of histiocytes in the appropriate clinical and radiological context [31, 37]. The majority of cases show BRAF-V600E mutation. ALK-positive histiocytosis differs in occurrence in much younger patients, lack of involvement of the typical anatomic sites, presence of localized disease in some patients, presence of non-foamy histiocytes with irregularly folded nuclei (sometimes exclusively), and lack of BRAF-V600E. It is unclear whether the two patients briefly mentioned in a report as having Erdheim-Chester disease which exhibit the KIF5B-ALK gene fusion (25-year-old with skin lesion rich in Touton giant cells and lacking BRAF-600E; 50-year-old with liver lesion) should be diagnosed as ALK-positive histiocytosis instead [27]. Since the lesional histiocytes of ALK-positive histiocytosis may show emperipolesis, Rosai-Dorfman disease enters into the differential diagnosis. The histiocytes of the latter differ in that the nuclei are round, with vesicular chromatin and distinct nucleoli. They are also consistently S100 positive. ALK-positive histiocytosis needs to be distinguished from histiocytic sarcoma, an aggressive neoplasm. The latter should show definite nuclear pleomorphism, readily identifiable mitotic activity, and is ALK negative. Epithelioid fibrous histiocytoma, a localized cutaneous tumor often showing ALK expression, has to be distinguished from ALK-positive histiocytosis involving skin [7, 23, 38]. This dermal tumor is more circumscribed and surrounded by epidermal collarettes. The cells have a more epithelioid morphology, although they may be small or spindled, and lack expression of S100 and CD68 [23]. The most common ALK gene fusion partners are SQSTM1 and VCL, and rarer partners include DCTN1, ETV6, PPFIBP1, and SPECC1L [7], indicating a lack of overlap with the specific gene partners observed in ALK-positive histiocytosis.
Conclusion
In summary, this series of ALK-positive histiocytosis documents an expanded clinicopathological spectrum of this condition to include both systemic and localized disease across a broad age range. We recommend testing for ALK expression or translocation in every unusual histiocytic proliferative disorder to aid in identification of this entity, which can potentially be effectively treated by ALK inhibitor therapy.
References
Chan JK, Lamant L, Algar E, et al. ALK+ histiocytosis: a novel type of systemic histiocytic proliferative disorder of early infancy. Blood. 2008;112:2965–8.
Chantranuwat C. Systemic form of juvenile xanthogranuloma: report of a case with liver and bone marrow involvement. Pediatr Dev Pathol. 2004;7:646–8.
Huang H, Gheorghe G, North PE, et al. Expanding the phenotype of ALK-positive histiocytosis: a report of 2 cases. Pediatr Dev Pathol. 2018;21:449–455.
Ross JS, Ali SM, Fasan O, et al. ALK fusions in a wide variety of tumor types respond to anti-ALK targeted therapy. Oncologist. 2017;22:1444–50.
Chun YS, Wang L, Nascimento AG, et al. Pediatric inflammatory myofibroblastic tumor: anaplastic lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer. 2005;45:796–801.
Laurent C, Do C, Gascoyne RD, et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma: a rare clinicopathologic entity with poor prognosis. J Clin Oncol. 2009;27:4211–6.
Dickson BC, Swanson D, Charames GS, et al. Epithelioid fibrous histiocytoma: molecular characterization of ALK fusion partners in 23 cases. Mod Pathol. 2018;31:753–62.
Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15:5216–23.
Kusano H, Togashi Y, Akiba J, et al. Two cases of renal cell carcinoma harboring a novel STRN-ALK fusion gene. Am J Surg Pathol. 2016;40:761–9.
Cajaiba MM, Jennings LJ, George D, et al. Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer. 2016;55:814–7.
Cajaiba MM, Jennings LJ, Rohan SM, et al. ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer. 2016;55:442–51.
Chou A, Fraser S, Toon CW, et al. A detailed clinicopathologic study of ALK-translocated papillary thyroid carcinoma. Am J Surg Pathol. 2015;39:652–9.
Kelly LM, Barila G, Liu P, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci USA. 2014;111:4233–8.
Yakirevich E, Resnick MB, Mangray S, et al. Oncogenic ALK fusion in rare and aggressive subtype of colorectal adenocarcinoma as a potential therapeutic target. Clin Cancer Res. 2016;22:3831–40.
Singhi AD, Ali SM, Lacy J, et al. Identification of targetable ALK rearrangements in pancreatic ductal adenocarcinoma. J Natl Compr Canc Netw. 2017;15:555–62.
Busam KJ, Kutzner H, Cerroni L, et al. Clinical and pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am J Surg Pathol. 2014;38:925–33.
Mosse YP, Voss SD, Lim MS, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: a Children’s Oncology Group Study. J Clin Oncol. 2017;35:3215–21.
Recondo G, Facchinetti F, Olaussen KA. et al. Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI?. Nat Rev Clin Oncol. 2018;15:694–708.
Theilen TM, Soerensen J, Bochennek K, et al. Crizotinib in ALK(+) inflammatory myofibroblastic tumors-Current experience and future perspectives. Pediatr Blood Cancer. 2018;65.
Downing JR, Shurtleff SA, Zielenska M, et al. Molecular detection of the (2;5) translocation of non-Hodgkin’s lymphoma by reverse transcriptase-polymerase chain reaction. Blood. 1995;85:3416–22.
De Paepe P, Baens M, van Krieken H, et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood. 2003;102:2638–41.
Marino-Enriquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–44.
Kazakov DV, Kyrpychova L, Martinek P. et al. ALK gene fusions in epithelioid fibrous histiocytoma: a study of 14 cases, with new histopathological findings. Am J Dermatopathol. 2018;40:805–14.
Rosenbaum JN, Bloom R, Forys JT, et al. Genomic heterogeneity of ALK fusion breakpoints in non-small-cell lung cancer. Mod Pathol. 2018;31:791–808.
Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–84.
Wilson MH, Holzbaur EL. Nesprins anchor kinesin-1 motors to the nucleus to drive nuclear distribution in muscle cells. Development. 2015;142:218–28.
Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6:154–65.
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15:3143–9.
Wong DW, Leung EL, Wong SK, et al. A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer. 2011;117:2709–18.
Reuter MS, Schwabe GC, Ehlers C, et al. Two novel distinct COL1A2 mutations highlight the complexity of genotype-phenotype correlations in osteogenesis imperfecta and related connective tissue disorders. Eur J Med Genet. 2013;56:669–73.
Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672–81.
Azorin D, Torrelo A, Lassaletta A, et al. Systemic juvenile xanthogranuloma with fatal outcome. Pediatr Dermatol. 2009;26:709–12.
Fan R, Sun J. Neonatal systemic juvenile xanthogranuloma with an ominous presentation and successful treatment. Clin Med Insights Oncol. 2011;5:157–61.
Hu WK, Gilliam AC, Wiersma SR, et al. Fatal congenital systemic juvenile xanthogranuloma with liver failure. Pediatr Dev Pathol. 2004;7:71–76.
Papadakis V, Volonaki E, Katsibardi K, et al. A rare case of neonatal systemic xanthogranulomatosis with severe hepatic disease and metachronous skin involvement. J Pediatr Hematol Oncol. 2012;34:226–8.
Takeuchi M, Nakayama M, Nakano A, et al. Congenital systemic juvenile xanthogranuloma with placental lesion. Pediatr Int. 2009;51:833–6.
Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124:483–92.
Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904–12.
Funding
This study was supported by the VIVA-KKH Pediatric Brain and Solid Tumor Program based at KK Women’s and Children’s Hospital, Singapore.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Chang, K.T.E., Tay, A.Z.E., Kuick, C.H. et al. ALK-positive histiocytosis: an expanded clinicopathologic spectrum and frequent presence of KIF5B-ALK fusion. Mod Pathol 32, 598–608 (2019). https://doi.org/10.1038/s41379-018-0168-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0168-6
This article is cited by
-
Seltene Differenzialdiagnose bei osteolytischer Läsion des Unterkiefers einer jungen Erwachsenen
Die Pathologie (2024)
-
Pathologic characteristics of histiocytic and dendritic cell neoplasms
Blood Research (2024)
-
Multisystem ALK-positive histiocytosis: a multi-case study and literature review
Orphanet Journal of Rare Diseases (2023)
-
Malakoplakia with aberrant ALK expression by immunohistochemistry: a case report
Diagnostic Pathology (2023)
-
Failure of crizotinib based systemic treatment in ALK positive histiocytosis involving the central nervous system: a case report and literature review
BMC Pediatrics (2022)
