
Abstract
Müllerian adenosarcomas are biphasic epithelial-mesenchymal tumors with benign epithelial and malignant mesenchymal components. The sarcoma component may be low or high grade; the latter is often seen in the presence of stromal overgrowth, which correlates with worse clinical outcome. Heterologous differentiation may also occur, usually in association with stromal overgrowth. DICER1 mutations have been reported primarily in a small subset of adenosarcomas with rhabdomyosarcomatous elements, but whether these are specific to the rhabdomyosarcomatous phenotype is unclear. In this study, we examined the clinical, pathologic, and genomic features of 19 müllerian adenosarcomas enriched for tumors with rhabdomyosarcomatous differentiation, as well as eight uterine carcinosarcomas with a rhabdomyosarcoma component. Somatic hotspot mutations in the RNase IIIb domain of DICER1 were identified in 8/19 (42%) adenosarcomas, of which four showed rhabdomyosarcomatous differentiation. DICER1 mutations were detected in 4/6 (67%) cases with a rhabdomyosarcoma component and in 4/11 (36%) cases without rhabdomyosarcoma. At least two DICER1 mutations were identified in 7/8 (88%) tumors, of which four had a truncating mutation. The hotspot DICER1 mutation in the remaining tumor was hemizygous and associated with loss of heterozygosity. Other less frequent recurrent somatic pathogenic alterations included Ras or PI3K/PTEN pathway aberrations (5/19 each, 26%), CDK4/MDM2 amplifications (3/19, 16%), and mutations in TP53 (3/19) and ARID1A (3/19). Two tumors demonstrated homozygous BAP1 deletion. One tumor harbored an ESR1-NCOA3 fusion gene. Carcinosarcomas with rhabdomyosarcomatous differentiation showed frequent mutations in TP53 (7/8, 88%) and the PI3K/PTEN pathway (6/8, 75%) but lacked DICER1 mutations. The findings highlight the importance of DICER1 mutations in müllerian adenosarcoma tumorigenesis and show that these alterations are not exclusive to heterologous rhabdomyosarcomatous differentiation.
Similar content being viewed by others

Introduction
Müllerian adenosarcomas are uncommon biphasic epithelial-mesenchymal tumors with benign or atypical epithelial elements and a malignant mesenchymal component. The uterine corpus is the most common primary site, but rare tumors have been reported in the cervix, ovary, fallopian tube, and vagina [1, 2]. Most patients have good clinical outcomes. A minority of tumors with poor clinical behavior often display deep myometrial invasion and/or the presence of stromal overgrowth, which is defined as > 25% of tumor volume comprising sarcoma alone and occurs in approximately 10% of tumors [1, 2]. Heterologous elements including fat, cartilage, bone, and rhabdomyoblasts are found in approximately 20% of tumors and are typically seen in the presence of stromal overgrowth [3]. Grading of the sarcoma component is controversial, but most cases are considered low grade. High-grade features of severe cytologic atypia, brisk mitotic activity, and necrosis are typically seen in association with stromal overgrowth, but can also occur in the absence of stromal overgrowth [4].
The pathogenesis of uterine adenosarcomas has not been fully characterized. Recent next-generation DNA sequencing studies have implicated recurrent alterations in the PI3K/AKT/PTEN pathway and MDM2/CDK4 amplification, with enrichment for TP53, ATRX, and PTEN mutations in high-grade tumors, and MYBL1, ATRX, and TP53 mutations, in addition to increased numbers of chromosomal copy number changes, in those with stromal overgrowth [4,5,6]. Recurrent FGFR2, KMT2C, and DICER1 mutations have been reported at a much lower frequency [4,5,6]. DICER1 encodes an endoribonuclease involved in miRNA processing, germline mutations of which predispose to Sertoli-Leydig tumors, childhood embryonal rhabdomyosarcomas, pleuropulmonary blastomas, and cystic nephromas [7]. Somatic DICER1 mutations are very rare in human cancers, but are enriched in other gynecologic tumors, including many with rhabdomyosarcomatous differentiation, including ovarian Sertoli-Leydig cell tumors, müllerian adenosarcomas, and a reported uterine carcinosarcoma [8, 9]. Of the four reported müllerian adenosarcomas with DICER1 mutations in which differentiation phenotype was described, three had a heterologous rhabdomyosarcoma component [4,5,6]. This apparent association between DICER1 mutations and malignant skeletal muscle-like differentiation in gynecologic tumors is peculiar, but whether the mutations are more specifically linked to the heterologous rhabdomyosarcomatous phenotype is not clear.
In this study, we examined the clinical, pathologic, and genomic features of 19 müllerian adenosarcomas specifically enriched for tumors with rhabdomyosarcomatous differentiation, in order to investigate the potential association of DICER1 mutations with skeletal muscle-like differentiation and to gain further insight into the pathogenesis of these rare tumors. The genomic results were additionally compared to eight uterine carcinosarcomas with rhabdomyosarcomatous differentiation.
Materials and methods
Study population
After obtaining institutional review board approval, the UCSF and El Camino Hospital Pathology archives were searched for all cases of müllerian adenosarcoma and uterine carcinosarcoma. A total of 19 adenosarcomas with available slides and tissue were identified (AS1-19). All H&E slides were reviewed by at least one gynecologic pathologist (A.R.S. and K.G.) to confirm the diagnosis. Each tumor was assessed for the presence of stromal overgrowth, the presence of heterologous differentiation, and the grade of the mesenchymal component. Of identified carcinosarcomas, eight with rhabdomyosarcomatous differentiation were selected. Clinical information was obtained from online electronic medical records.
Capture-based next-generation DNA sequencing
For capture-based next-generation DNA sequencing, matched normal and tumor tissue was selected from 18 adenosarcoma cases, and only tumor tissue was selected from one case (AS11). Pure sarcomatous areas were selected for analysis in all tumors. In tumors with stromal overgrowth, the areas of stromal overgrowth were analyzed, and in tumors with heterologous differentiation, the areas with rhabdomyosarcomatous differentiation were analyzed. In one case (AS3), areas with and without rhabdomyosarcomatous differentiation were selected for separate analysis. For carcinosarcomas with rhabdomyosarcomatous differentiation, only tumor tissue was sequenced.
Sequencing libraries were prepared from genomic DNA extracted from punch biopsies or macrodissected unstained sections from formalin fixed paraffin embedded tissue. Target enrichment was performed by hybrid capture using a custom oligonucleotide library. Capture-based next generation sequencing was performed at the UCSF Clinical Cancer Genomics Laboratory, using an assay (UCSF500 panel) that targets the coding regions of 480 cancer-related genes, select introns from approximately 40 genes, and the TERT promoter with a total sequencing footprint of 2.8 Mb (Supplemental Table S1). Sequencing was performed on a HiSeq 2500 (Illumina, San Diego, CA). Duplicate sequencing reads were removed computationally to allow for accurate allele frequency determination and copy number calling. The analysis was based on the human reference sequence UCSC build hg19 (NCBI build 37), using the following software packages: BWA: 0.7.10-r789, Samtools: 1.1 (using htslib 1.1), Picard tools: 1.97 (1504), GATK: 2014.4-3.3.0-0-ga3711, CNVkit: 0.3.3, Pindel: 0.2.5a7, SATK: 2013.1-10- gd6fa6c3, Annovar: v2015Mar22, Freebayes: 0.9.20, and Delly: 0.5.9 [10,11,12,13,14,15,16,17,18,19]. Only insertions/deletions (indels) up to 100 bp in length were included in the mutational analysis. Somatic single nucleotide variants and indels were visualized and verified using Integrated Genome Viewer. Genome-wide copy number analysis based on on-target and off-target reads was performed by CNVkit and Nexus Copy Number (Biodiscovery, Hawthorne, CA). Recurrent tumors were excluded from copy number analysis.
Immunohistochemistry
For immunohistochemistry, the following antibodies were used: BAP1 (C-4, 1:100, Santa Cruz Biotechnology, Santa Cruz, CA), desmin (D33, 1:5, Cell Marque, Rocklin, CA), and myogenin (F5D, 1:50, Cell Marque). Antigen retrieval was as follows: for BAP1, BOND ER2 (Leica Biosystems, Richmond, VA); for desmin, BOND ER1 (Leica Biosystems); and for myogenin, Ventana CC1 (Ventana, Tucson, AZ).
Statistical analysis
Statistical analysis was performed using Fisher’s exact tests and Mann-Whitney U tests where appropriate, using a significance level of p < 0.05.
Results
Clinicopathologic features of adenosarcomas
The clinicopathologic features of adenosarcomas in this study are shown in Table 1. Patient ages ranged from 25 to 92 years (mean 55 years). The most common presenting symptoms included vaginal bleeding and an abdominal mass. The majority of patients presented at stage 1 (n = 16). Of those with available clinical data (n = 13), eight patients received chemotherapy and/or radiation therapy and five patients received no adjuvant therapy. Two patients presented with pelvic recurrences, at 20 months (AS2) and 4 months (AS4) post-surgery, respectively. One patient presented with a vaginal cuff recurrence at 4 months post-surgery (AS5), which was treated with adjuvant radiation. Of eight patients with available follow-up data, four have no evidence of disease, two are alive with disease, and two died of disease (Table 1).
Two patients had synchronous uterine endometrioid adenocarcinoma (AS4 and AS16), and one had atypical endometrial hyperplasia (AS11). One patient had uterine serous carcinoma on an endometrial biopsy (AS12), and the following hysterectomy specimen showed no residual serous carcinoma (Table 1). While we considered a diagnosis of carcinosarcoma for these cases, the overall histopathologic features including the phyllodes-like growth pattern, the relatively low-grade appearance of the sarcoma component and the low-grade nature of the epithelial proliferation in most cases, are more characteristic of two separate synchronous malignancies. The existence of uterine adenosarcoma with synchronous endometrial hyperplasia or adenocarcinoma has been previously described [2, 20]. None of the patients in our series or those in the literature have clinically behaved like carcinosarcomas. The presence of DICER1 mutations in two such cases in our series further supports their classification as adenosarcoma rather than carcinosarcoma.
Histopathologic features of adenosarcomas
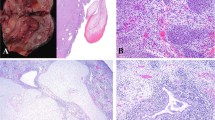
The hysterectomy specimen was reviewed in most cases (n = 16). In two cases (AS13 and AS14), only the recurrence specimens were available for our review. Tumors in hysterectomy specimens presented as exophytic, tan brown masses measuring from 1 cm to 20 cm. All tumors displayed the typical biphasic appearance of adenosarcoma, with benign or atypical glands and malignant stroma (Fig. 1a, b). All tumors displayed at least focal phyllodes-like architecture with intraglandular polypoid growth, stromal cuffing, stromal atypia, and mitotic activity. The stromal component was low grade in 11 cases (65%) and high grade in six cases (35%) (Fig. 1c, d). The two recurrent tumors were not graded or evaluated for stromal overgrowth. Of the six tumors with high-grade sarcoma, four (67%) showed stromal overgrowth. In total, stromal overgrowth was present in five tumors (29%), of which four showed high-grade sarcoma (80%). Heterologous rhabdomyosarcomatous differentiation was present in six tumors, and confirmed with immunohistochemical staining for desmin and myogenin (Fig. 2a, b). Most (5/6, 83%) of these showed histologic features of embryonal rhabdomyosarcoma, and one showed features of pleomorphic rhabdomyosarcoma (AS10). Four (67%) tumors with rhabdomyosarcoma had stromal overgrowth. The entire sarcoma component in both cases lacking stromal overgrowth was composed of rhabdomyosarcoma (Fig. 2c, d). An alternative diagnosis of embryonal rhabdomyosarcoma was considered, but both were classified as adenosarcoma based on the diffuse distribution of the glandular component throughout the tumor.

Morphologic features of müllerian adenosarcomas. (a) Histologic appearance of a typical müllerian adenosarcoma characterized by benign epithelium and a low-grade mesenchymal component showing phyllodes-like growth. (b) The mesenchymal component shows periglandular condensation with hypercellularity and cytologic atypia with occasional mitotic figures. The sarcoma component of müllerian adenosarcoma is low grade in most cases (c) but occasional cases have high-grade sarcoma with greater cytologic atypia and mitotic activity (d)

Müllerian adenosarcomas with rhabdomyosarcomatous differentiation. Heterologous differentiation in the form of rhabdomyosarcoma was seen in six cases (a) and confirmed with immunohistochemical staining for myogenin (b). In two cases that lacked stromal overgrowth, almost the entire sarcoma component was composed of rhabdomyosarcoma (c), as demonstrated by the positive staining with myogenin (d)
Genetic features of adenosarcomas
Somatic pathogenic genomic alterations are shown in Fig. 3, and all somatic mutations are listed in Supplemental Table S2. The mean target sequencing coverage was 595 (±186) unique reads per target interval. The most frequently identified alterations were in DICER1, which was mutated in eight (42%) of 19 tumors. All eight tumors harbored hotspot mutations at metal ion-binding sites (E1705, D1709, D1810, E1813) or adjacent residues (D1713, G1809) in the RNase IIIb domain (Table 2) [7, 21,22,23]. At least two DICER1 mutations were identified in 7/8 (88%) tumors, with four of these (AS1-3 and AS7) harboring a second inactivating (frameshift or stopgain) mutation. One tumor (AS5) had three additional missense mutations in addition to the hotspot. The relative allelic positions of these mutations (cis vs. trans) could not be definitively determined in any of the cases. In the only tumor without at least two DICER1 mutations, the hotspot mutation was hemizygous due to copy neutral loss of heterozygosity of chromosome 14q (Supplemental Figure S1). No germline DICER1 mutations were identified (Supplemental Table S4).

Pathogenic genomic alterations in müllerian adenosarcomas
Somatic aberrations in Ras or PI3K/PTEN pathway genes were identified in 5/19 (26%) adenosarcomas. In one case (AS14), a pathogenic germline frameshift PTEN variant (p.Y138fs) was associated with somatic loss of heterozygosity in the tumor. Other recurrent somatic alterations included TP53 mutations, CDK4/MDM2 amplifications, and ARID1A truncating mutations in 3/19 (16%) tumors each. Hotspot mutations in FBXW7 and truncating mutations in ATRX, ARHGAP35, and CTCF were detected in two tumors each (11% each). Deep deletions in BAP1 were also identified in two (11%) tumors, both of which showed loss of BAP1 immunohistochemical staining (Fig. 4a, b). In contrast, 11 additional tumors without BAP1 alterations retained BAP1 staining (Fig. 4c).

Adenosarcomas with BAP1 inactivation. Two cases with deep deletions in BAP1 (a) showed loss of BAP1 immunohistochemical staining (b), whereas 11 other cases without BAP1 alterations showed retained BAP1 staining (c)
One adenosarcoma (AS17) harbored an ESR1-NCOA3 gene rearrangement, resulting in fusion of the promoter and first three exons of ESR1 (amino acids 1–254) to exons 13–23 of NCOA3 (amino acids 801–1424). The fusion gene lacks the ligand-binding domain of ESR1 and preserves the nuclear receptor co-activator domain of NCOA3. This tumor otherwise had a simple genome without copy number alterations and lacked somatic mutations in genes on the panel (Fig. 3 and Supplemental Table S2).
Copy number analysis revealed recurrent gains of chromosomes 8 (6/19, 32%) and interstitial 1q (5/19, 26%) and recurrent loss of chromosome 22q (5/19, 26%).
The rhabdomyosarcomatous and non-rhabdomyosarcomatous areas of one tumor (AS3) were separately analyzed and harbored the same DICER1 and FBXW7 mutations, as well as chromosome 8 gain. No mutations were exclusive to either component. Loss of 17p was only identified in the rhabdomyosarcomatous component, whereas gain of distal 7p was only present in the non-rhabdomyosarcomatous component (Supplemental Figure S2).
Correlative analysis of genetic and histopathologic features of adenosarcomas
Four (50%) of the eight tumors with DICER1 mutations demonstrated rhabdomyosarcomatous differentiation (Fig. 3). Immunohistochemical staining for desmin and myogenin revealed no evidence of an occult rhabdomyosarcomatous component in the four tumors lacking histologic features of rhabdomyosarcoma (data not shown). DICER1 mutations were detected in 4/6 (67%) tumors with rhabdomyosarcomatous elements and in 4/11 (36%) tumors without rhabdomyosarcoma (p = 0.33) (Fig. 3). No association was identified between stromal overgrowth and DICER1 mutations, which were present in 3/5 (60%) tumors with stromal overgrowth and in 5/12 (42%) tumors without stromal overgrowth (p = 0.62). DICER1 mutations were also independent of sarcoma grade (p = 0.33), patient age (p = 0.56), and tumor stage (p = 0.42). Likewise, no associations were identified between any of the other mutations and tumor grade, presence of stromal overgrowth, and/or heterologous rhabdomyosarcomatous differentiation. Two of three TP53 mutations were in high-grade tumors with stromal overgrowth and rhabdomyosarcomatous differentiation (AS9, AS10), but one was identified in a low-grade tumor without overgrowth or heterologous elements (AS11).
Primary tumors with DICER1 mutations demonstrated less overall copy number alterations, including chromosomal gains, losses, amplifications, and deep deletions, than tumors without DICER1 mutations (p = 0.02) (Fig. 5). No significant associations were identified between overall copy number alterations and the presence of stromal overgrowth (p = 0.52), sarcoma grade (p = 0.47), or rhabdomyosarcomatous differentiation (p = 0.26). No significant associations were identified between specific recurrent copy number alterations (gain of 1q or 8 or loss of 22q) and DICER1 mutation, tumor grade, stromal overgrowth, or rhabdomyosarcomatous differentiation.

Pathogenic genomic alterations in carcinosarcomas with rhabdomyosarcomatous differentiation
Genetic features of carcinosarcomas with rhabdomyosarcomatous differentiation
Eight carcinosarcomas with heterologous rhabdomyosarcomatous differentiation were analyzed for comparison. In all cases, the presence of a rhabdomyosarcomatous component was confirmed with immunohistochemical staining for desmin and myogenin (data not shown). The mean target sequencing coverage was 628 (±115) unique reads per target interval. Most tumors harbored TP53 mutations and alterations in the PI3K/PTEN pathway (7/8, 88% each). Aberrations in cell cycle genes (MYC, MYCN, CCND1, and CCNE1) and chromatin remodeling genes (ARID1A and KMT2D) were identified in 4/8 (50%) tumors each. No DICER1 mutations were identified (Fig. 5 and Supplemental Table S3).
Discussion
DICER1 mutations are overall very rare in cancers [22], but both germline and somatic DICER1 mutations have been described in a spectrum of non-epithelial gynecologic tumors and rare uterine endometrioid carcinomas [8, 23]. Ovarian Sertoli-Leydig cell tumors and uterine cervical embryonal rhabdomyosarcomas are associated with inherited DICER1 syndrome, and there is a reported case of uterine adenosarcoma (without rhabdomyosarcoma) in a patient with germline DICER1 mutation [9, 24]. Somatic DICER1 mutations have been observed in ovarian sex cord stromal tumors, including Sertoli-Leydig cell tumors, gynandroblastomas, and Sertoli cell tumors, and in a few müllerian adenosarcomas and a rare uterine carcinosarcoma [5, 6, 8, 21, 22]. Some studies have suggested an intriguing possible relationship between DICER1 mutation and rhabdomyosarcomatous differentiation in the female genital tract. This includes increased risk for cervical embryonal rhabdomyosarcoma in DICER1 syndrome [9], as well as somatic DICER1 mutations in three of four ovarian Sertoli-Leydig cell tumors with rhabdomyosarcomatous differentiation, in one adult cervical rhabdomyosarcoma, in one carcinosarcoma with a rhabdomyosarcoma component, and in müllerian adenosarcomas with rhabdomyosarcoma elements [4, 6, 8]. Prior to our study, DICER1 mutations had been reported in a total of five müllerian adenosarcomas, with rhabdomyosarcomatous differentiation described in three of the four tumors in which the presence or absence of heterologous elements was evaluated [4, 6]. We identified hotspot DICER1 mutations in four of six müllerian adenosarcomas with rhabdomyosarcomatous differentiation, but also in an additional four adenosarcomas without heterologous rhabdomyosarcomatous differentiation. Our data therefore indicate that DICER1 mutations are frequent in müllerian adenosarcomas but are not exclusive to those with rhabdomyosarcomatous differentiation. The lack of somatic DICER1 mutations in carcinosarcomas with rhabdomyosarcomatous differentiation further suggests that these mutations are not associated with heterologous differentiation in this context.
All adenosarcomas in our study harbored at least two DICER1 mutations or a hemizygous DICER1 mutation, and all tumors had a hotspot mutation at a metal ion-binding or adjacent residue of the RNase IIIb domain. These hotspot mutations, which inhibit 5p miRNA processing and expression, have been previously described in sex cord stromal and other gonadal tumors and Wilms tumors and were also identified in four of the five DICER1-mutated adenosarcomas in prior studies [4, 6, 8, 21, 22, 25, 26]. In five tumors in our study, the hotspot mutation was associated with either a second inactivating mutation (four cases) or loss of heterozygosity (one case), suggestive of hemizygosity. Most sequenced DICER1-mutated tumors in previous studies were analyzed by Sanger sequencing for hotspot mutations, precluding identification of a second inactivating DICER1 mutation. However, more comprehensive sequencing in a small subset of these has also revealed additional somatic or germline inactivating mutations, which were shown in some studies to function in trans with respect to the hotspot [6, 22, 23, 25]. The presence of multiple DICER1 mutations per tumor in our study is also highly suggestive of biallelism, although this could not be confirmed due to spatial separation of the mutations. Our findings support the hypothesis that DICER1 acts neither like a typical tumor suppressor nor oncogene, requiring the combination of a specific hotspot mutation that affects enzymatic activity plus inactivation of the other allele, for tumor development [22, 26].
We identified an ESR1-NCOA3 fusion in one stage 1A low-grade adenosarcoma without stromal overgrowth or heterologous differentiation that otherwise had a simple genome without copy number changes. Piscuoglio et al. [6] recently also identified fusions involving ESR1 and NCOA2 or NCOA3 genes in two müllerian adenosarcomas without rhabdomyosarcomatous differentiation. NCOA2 fusion genes have been described in other soft tissue tumors, including mesenchymal chondrosarcoma, angiofibroma, and congenital/infantile spindle cell rhabdomyosarcoma, and NCOA2 and NCOA3 fusions have been reported in acute leukemias [27, 28]. The identification of recurrent NCOA2 gene fusions in congenital/infantile spindle cell rhabdomyosarcomas is particularly interesting in this context, raising the possibility that altered NCOA2 function may be associated with malignant skeletal muscle differentiation [29]. Hartmaier et al. [30] recently described rare fusions of ESR1 with various 3′ gene partners in ER-positive metastatic breast cancers, none of which involved NCOA2 or NCOA3. The breakpoints of these ESR1 fusion genes were reported to disrupt the ligand-binding domain, and the expressed proteins were constitutively hyperactive [30]. The ESR1 rearrangements in adenosarcomas in our study and the prior report by Piscuoglio et al. [6] also lack the ligand-binding domain. The findings suggest a possible role for ligand-independent estrogen receptor signaling in the pathogenesis of a subset of these tumors, which will require additional study.
Two (11%) adenosarcomas in our study demonstrated deletion and inactivation of BAP1 (BRCA-associated protein 1), a deubiquitinase and tumor suppressor involved in chromatin remodeling, gene expression, and DNA repair [31,32,33]. Germline or somatic BAP1 aberrations have been most frequently identified in melanocytic tumors, clear cell renal cell carcinomas, and malignant mesotheliomas, and a recent study identified homozygous BAP1 deletion in one of 19 müllerian adenosarcomas [6]. Based on data from The Cancer Genome Atlas, genomic BAP1 inactivation appears to be a rare event in sarcomas overall (<2%), but is typically due to homozygous deletion, as in this study [34, 35]. Our findings suggest that adenosarcomas are enriched for BAP1 inactivation compared to other sarcomas, but confirmation will require larger studies. Similarly, whether BAP1 loss in adenosarcomas is associated with poor prognosis, as has been suggested in other tumor types, remains to be seen [36, 37]. One BAP1 deleted case in our series showed high-grade sarcoma in the recurrence, presented with ovarian metastasis, and resulted in death from disease, whereas the second case presented at stage IA and showed no adverse prognostic features, but clinical follow-up was not available.
In contrast to a prior study, we did not identify a correlation between the presence of stromal overgrowth and copy number changes in adenosarcomas [5]. On the other hand, tumors with DICER1 mutations had significantly fewer copy number alterations than those lacking DICER1 mutations. Given lack of sufficient clinical follow-up data on our cohort, the significance of these findings is unclear. However, we note that three of five patients with tumor recurrence or metastasis demonstrated DICER1 mutations. Additional larger studies or meta-analyses will be required to determine the clinical impact and potential significance of DICER1 and other genetic alterations in müllerian adenosarcomas.
References
Eichhorn JH, Young RH, Clement PB, et al. Mesodermal (mullerian) adenosarcoma of the ovary: a clinicopathologic analysis of 40 cases and a review of the literature. Am J Surg Pathol. 2002;26:1243–58.
Clement PB, Scully RE. Mullerian adenosarcoma of the uterus: a clinicopathologic analysis of 100 cases with a review of the literature. Hum Pathol. 1990;21:363–81.
McCluggage WG. Mullerian adenosarcoma of the female genital tract. Adv Anat Pathol. 2010;17:122–9.
Hodgson A, Amemiya Y, Seth A, et al. High-grade mullerian adenosarcoma: genomic and clinicopathologic characterization of a distinct neoplasm with prevalent TP53 pathway alterations and aggressive behavior. Am J Surg Pathol. 2017;41:1513–22.
Howitt BE, Sholl LM, Dal Cin P, et al. Targeted genomic analysis of mullerian adenosarcoma. J Pathol. 2015;235:37–49.
Piscuoglio S, Burke KA, Ng CK, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol. 2016;238:381–8.
Foulkes WD, Priest JR, et al. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–72.
Conlon N, Schultheis AM, Piscuoglio S, et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol. 2015;28:1603–12.
Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol. 2012;25:602–14.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Prot. 2015;10:1556–66.
Rausch T, Zichner T, Schlattl A, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:1–33.
Ye K, Schulz MH, Long Q, et al. A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907 [q-bio.GN]. Available from: http://arxiv.org/abs/1207.3907.
Picard: A set of tools (in Java) for working with next generation sequencing data in the BAM: Broad Institute. http://broadinstitute.github.io/picard.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Street B, du Toit JP. Uterine adenosarcoma: report of a case with two further primary malignant tumors. Gynecol Oncol. 1981;11:252–8.
Witkowski L, Mattina J, Schonberger S, et al. DICER1 hotspot mutations in non-epithelial gonadal tumours. Br J Cancer. 2013;109:2744–50.
Heravi-Moussavi A, Anglesio MS, Cheng SW, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012;366:234–42.
Chen J, Wang Y, McMonechy MK, et al. Recurrent DICER1 hotspot mutations in endometrial tumours and their impact on microRNA biogenesis. J Pathol. 2015;237:215–25.
Mullen MM, Divine LM, Hagemann IS, et al. Endometrial adenosarcoma in the setting of a germline DICER1 mutation: A case report. Gyn Oncol Rep. 2017;20:121–4.
Wu MK, Sabbaghian N, Xu B, et al. Biallelic DICER1 mutations occur in Wilms tumours. J Pathol. 2013;230:154–64.
Wang Y, Chen J, Yang W, et al. The oncogenic roles of DICER1 RNase IIIb domain mutations in ovarian Sertoli-Leydig cell tumors. Neoplasia. 2015;17:650–60.
Jin Y, Moller E, Nord KH, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15; q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer. 2012;51:510–20.
Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. 2012;51:127–39.
Mosquera JM, Sboner A, Zhang L, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer. 2013;52:538–50.
Hartmaier RJ, Trabucco SE, Priedigkeit N, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy-resistant breast cancer. Annals Oncol. 2018;29:872–80.
Machida YJ, Machida Y, Vashisht AA, et al. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem. 2009;284:34179–88.
Yu H, Pak H, Hammond-Martel I, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA. 2014;111:285–90.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, et al. Histone H2A deubiquitinase activity of the polycomb repressive complex PR-DUB. Nature. 2010;465:243–7.
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disc. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Luchini C, Veronese N, Yachida S, et al. Different prognostic roles of tumor suppressor gene BAP1 in cancer: a systematic review with meta-analysis. Genes Chromosomes Cancer. 2016;55:741–9.
Wang XY, Wang Z, Huang JB, et al. Tissue-specific significance of BAP1 gene mutation in prognostic prediction and molecular taxonomy among different types of cancer. Tum Biol. 2017;39:1010428317699111.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Bean, G.R., Anderson, J., Sangoi, A.R. et al. DICER1 mutations are frequent in müllerian adenosarcomas and are independent of rhabdomyosarcomatous differentiation. Mod Pathol 32, 280–289 (2019). https://doi.org/10.1038/s41379-018-0132-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0132-5
This article is cited by
-
Familial syndromes associated with testicular and paratesticular neoplasms: a comprehensive review
Virchows Archiv (2024)
-
ATM mutation in aggressive uterine adenosarcoma in which systemic chemotherapies had remarkable effects
International Cancer Conference Journal (2023)
-
Mullerian adenosarcoma: clinicopathologic and molecular characterization highlighting recurrent BAP1 loss and distinctive features of high-grade tumors
Modern Pathology (2022)
-
Atypical uterine polyps show morphologic and molecular overlap with mullerian adenosarcoma but follow a benign clinical course
Modern Pathology (2022)
-
Endometrial polyps are non-neoplastic but harbor epithelial mutations in endometrial cancer drivers at low allelic frequencies
Modern Pathology (2022)
