
Abstract
Lipoblastoma-like tumor of the vulva was first described as a benign mesenchymal neoplasm of adipocytic differentiation having features of lipoblastoma, myxoid liposarcoma, and spindle cell lipoma. Prior studies of lipoblastoma-like tumor have evaluated PLAG1, HMGA2, and RB1 immunohistochemistry and DDIT3 rearrangement status, with results supporting its distinction from lipoblastoma and myxoid liposarcoma. However, absent RB1 expression was reported in a majority of tested cases, suggesting that lipoblastoma-like tumor may have underlying 13q alterations and be related to RB1-deleted soft tissue tumors. To further understand the molecular genetics of lipoblastoma-like tumor, we examined 7 cases by RB1 immunohistochemistry, DDIT3 and PLAG1 break apart FISH probes, RB1 enumeration FISH probe, and genomic copy number analysis by microarray. Patient age ranged from 21 to 56 years (median 35 years). Clinical follow up was available for 5 patients (71%) ranging 3–264 months (median 74 months). Microscopically, lipoblastoma-like tumor formed large lobules separated by thin and/or thick bands of fibrous tissue and had a prominent network of thin-walled vessels. Each tumor was predominantly composed of spindle cells and lipoblasts with variable quantities of mature adipocytes. RB1 immunohistochemistry exhibited a heterogeneous or “mosaic” pattern of weak and negative nuclear expression in all seven cases. DDIT3 and PLAG1 FISH were negative in each case. No evidence of RB1 regional gain or loss was identified by FISH. Genomic copy number analysis by chromosomal microarray showed a normal diploid profile in six tumors (86%). One tumor had copy number abnormalities consisting of an 11.9 megabase deletion from 1p13.3 to 1p11.2 and monosomy 14. Although lipoblastoma-like tumor has features of lipoblastoma, myxoid liposarcoma, and spindle cell lipoma, it is genetically different from these tumors. Furthermore, lipoblastoma-like tumor does not appear to have structural abnormalities of 13q resulting in deletion of RB1.
Similar content being viewed by others
Introduction
Lipoblastoma-like tumor of the vulva was first reported by Lae and colleagues in 2002 as a series of three cases [1]. The authors characterized lipoblastoma-like tumor as a mesenchymal neoplasm of adipocytic differentiation having morphologic features of lipoblastoma, myxoid liposarcoma, and spindle cell lipoma; they further suggested that lipoblastoma-like tumor is a benign tumor with a tendency to occur in young patients in which conservative complete excision is curative.
Since its description, nine additional cases of lipoblastoma-like tumor have been published [2, 3]. A series of eight cases by Mirkovic and Fletcher [3] evaluated PLAG1, HMGA2, and RB1 by immunohistochemistry and DDIT3 by fluorescence in situ hybridization (FISH). In addition to the morphologic features of lipoblastoma-like tumor, the authors concluded that essentially absent expression of PLAG1 and HMGA2 distinguished it from lipoblastoma and lack of DDIT3 rearrangement distinguished it from myxoid liposarcoma. However, RB1 expression by immunostain was negative in six of seven cases, leading them to suggest that lipoblastoma-like tumor may have underlying 13q alterations and be related to RB1-deleted soft tissue tumors, including spindle cell lipoma/pleomorphic lipoma, cellular angiofibroma, mammary-type myofibroblastoma [4], and, recently, superficial acral fibromyxoma [5].
To further understand the molecular genetics of lipoblastoma-like tumor, we examined a large series by immunohistochemistry, FISH, and genomic copy number microarray, specifically aiming to clarify the relationship of lipoblastoma-like tumor to tumors associated with RB1 deletion.
Materials and Methods
Cases of lipoblastoma-like tumor were identified by keyword search of the pathology databases of Mayo Clinic, Rochester, Minnesota, and Faculty of Medicine in Pilsen, Charles University, Czech Republic. No cases from the series by Lae et al. [1] were included. All available hematoxylin and eosin slides were retrieved and reviewed to confirm a diagnosis of lipoblastoma-like tumor. Internal institutional medical records, submitted consultation paperwork and referring pathologists and clinicians provided clinical and/or pathologic information for each patient. A representative formalin-fixed paraffin-embedded tissue block from each tumor was sectioned for immunohistochemistry, FISH and whole genome copy number analysis by single nucleotide polymorphism microarray (Oncoscan, Affymetrix, Santa Clara, CA).
Immunohistochemical analysis of RB1 was performed using heat-induced epitope retrieval of anti-human retinoblastoma protein (G3-245, 1:50 dilution, BD Biosciences, San Jose, CA) on the BenchMark XT automated detection system (Ventana Medical Systems, Tucson, AZ). Status of DDIT3 (12q13), PLAG1 (8q12.1), and RB1 (13q14) was evaluated by FISH using break apart probe sets for DDIT3 (Abbott Molecular, Abbott Park, IL) and PLAG1 (Agilent, Santa Clara, CA) and enumeration probes for RB1 (RB1 at13q14 and LAMP1 at 13q34) (Abbott Molecular). Samples were scored in a blinded manner by two technologists (50 intact nuclei each) according to previously described methods [6]. For genomic copy number analysis by microarray, tumor was macrodissected from unstained slides and processed for DNA extraction. DNA was calculated by Qubit fluorometric quantitation according to the manufacturer’s suggested protocol (Thermo Fisher, Waltham, MA). Data files were analyzed using Chromosomal Analysis suite software (ChAS, Affymetrix).
Results
Clinical findings
A total of seven cases of lipoblastoma-like tumor were identified (Table 1). Six cases were consultative and suggested diagnoses for these were myxoid liposarcoma (four cases), myxoid liposarcoma or spindle cell lipoma (one case), and myxoid spindle cell lipoma (one case). Patients ranged from 21 to 56 years of age (mean 35 years, median 35 years). Clinical presentation was a vulvar or groin mass in six patients and a painful vulvar cyst in the other. All seven patients underwent simple or conservative excision of their mass. Clinical follow up was available for five patients (71%) ranging from 3 to 264 months (mean 62 months, median 74 months). One patient experienced local recurrence approximately three months following excision and the remaining patients had no evidence of disease.
Gross and microscopic findings
Tumor size was known for all cases, ranging from 2 to 9 cm (mean 4.7 cm, median 5.5 cm). Microscopically, the tumors were relatively well delineated and formed large lobules partitioned by thick or thin bands of fibrous tissue (Fig. 1a, b). A variable degree of stromal collagenization was present, either as dense aggregates (Fig. 1b) or wispy fibers (Fig. 1c). All tumors had a prominent arborizing network of thin-walled vessels (Fig. 1d) and were composed predominantly of spindle cells and lipoblasts with variable number of intermixed mature adipocytes (Fig. 1e). The spindle cells had ample fibrillary eosinophilic cytoplasm, ovoid to pointed nuclei containing finely granular chromatin (Fig. 1f). Uni- and bi-vacuolated lipoblasts were present, but lacked nuclear enlargement and hyperchromasia (Fig. 1f). The cellularity of each tumor lobule was even in distribution without concentration at the periphery. Foci of red blood cell extravasation were occasionally seen. No necrosis or significant cytologic atypia or mitotic activity was identified.

Lipoblastoma-like tumor of the vulva formed large lobules separated by thick (a) or thin fibrous septa (b). Variable stromal collagenization was present, either as dense aggregates (b) or wispy fibers (c). A conspicuous network of thin-walled blood vessels was seen in all examples (d). Each tumor contained a mixture of spindle cells and lipoblasts and occasional interspersed mature adipocytes (e). The spindle cells had fibrillary eosinophilic cytoplasm, low grade cytologic features while the lipoblasts were cytologically bland and uni-or bivacuolated (f)
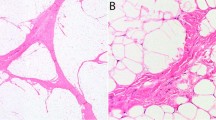
RB1 immunohistochemistry exhibited an alternating or “mosaic” pattern of weak and negative nuclear expression in all seven cases (Fig. 2a). The level of expression by tumor cells was slightly reduced when compared with that of non-neoplastic cells (Fig. 2b).

RB1 immunohistochemistry exhibited an alternating or “mosaic” pattern of weak and negative nuclear expression in all cases (a, b). The degree of expression by tumor cells was reduced when compared to non-neoplastic elements such as endothelium
Molecular cytogenetic findings
FISH was negative for DDIT3 and PLAG1 rearrangement in each of the 7 tumors. No evidence of regional gain or loss of RB1 was identified by FISH. Genomic copy number analysis by chromosomal microarray showed a normal diploid profile in six tumors (86%). One tumor had copy number abnormalities consisting of an 11.9 megabase deletion from 1p13.3 to 1p11.2 (Fig. 3) and monosomy of chromosome 14.

A total of six of seven cases (86%) had a normal diploid profile by chromosomal microarray. One case showed an ~12 megabase deletion spanning 1p13.3–1p11.2 (depicted). This same case also had monosomy of chromosome 14 (not shown)
Discussion
All 12 previously reported examples of lipoblastoma-like tumor are summarized in Table 2. The results of our study are in agreement with prior observations, and in aggregate, show lipoblastoma-like tumor to most commonly occur in young patients (range 13–56 years, median 27 years), as a variably sized vulvar mass (range 2–15 cm, median 5.6 cm). Recurrences have been few and are linked to resection margin involvement: of the 15 published cases with follow up, four recurred locally (27%), three of which had initially positive margins. Metastasis has not been reported.
The morphologic features of lipoblastoma-like tumor significantly overlap with lipoblastoma, myxoid liposarcoma, and spindle cell lipoma. In contrast to lipoblastoma-like tumor, lipoblastoma typically occurs in much younger children, arises on the trunk or extremities and exhibits a spectrum of immature and mature adipocytic differentiation, usually with “maturation” toward the periphery of the lobules [7]. At the genetic level, lipoblastoma is characterized by alterations of the PLAG1 proto-oncogene located at 8q12 with or without polysomy of chromosome 8 [8, 9], resulting in upregulation of PLAG1 protein which is detectable by immunohistochemistry. Mirkovic and Fletcher [3] identified PLAG1 expression in one of seven cases of lipoblastoma-like tumor, noting that the positive case occurred in an adult patient and was otherwise identical to other lipoblastoma-like tumors. While PLAG1 immunostaining was not performed in our study, we did not find PLAG1 alterations by FISH or microarray, supporting that lipoblastoma-like tumor is genetically distinct from lipoblastoma.
Myxoid liposarcoma tends to involve the proximal thigh of young to middle aged adults, lacks the circumscribed growth of lipoblastoma-like tumor and shows a reversed pattern of zonation than what is seen in lipoblastoma with loss of maturation at the periphery of its lobules [10, 11]. Genetically, myxoid liposarcoma carries t(12;16)(q13;p11)(FUS-DDIT3) in as many as 95% of tumors or the variant t(12;22)(q13;q12)(EWS-DDIT3) in the remaining cases [4]. Lipoblastoma-like tumor consistently lacks DDIT3 rearrangement, distinguishing it from myxoid liposarcoma. In our series, we did not find evidence of DDIT3 rearrangement in any case, either by FISH or microarray.
Spindle cell lipoma most commonly arises in posterior neck, shoulders, and back of older men, although it can occur in women and in unusual locations [12]. It consists of a protean admixture of bland spindle cells often arranged in small parallel or palisading groups, wiry collagen, myxoid matrix, and mostly mature adipocytes with only sparse lipoblasts [13, 14]. Spindle cell lipoma differs from lipoblastoma-like tumor by its lack of developed capillary vasculature and absent to very infrequent quantities of lipoblasts. Spindle cell lipomas have variably sized deletions involving the q-arm of chromosome 13 resulting in mono- or bilallelic loss of the tumor suppressor RB1 [15]. The same deletion is present in a spectrum of tumors with overlapping features including pleomorphic lipomas, cellular angiofibromas, most mammary-type myofibroblastomas [4] and recently, superficial acral fibromyxoma [5]. Loss of 13q13 results in absent RB1 expression by immunohistochemistry [16], a finding previously reported in six of seven cases of lipoblastoma-like tumor [3]. From this observation, the investigators suggested that lipoblastoma-like tumor may be linked to these RB1 deleted tumors. Our results are at variance with this proposal, as all cases of lipoblastoma-like tumor in our study had numerous tumor cells with some degree of retained nuclear staining intermixed with negative tumor cells. Furthermore, we did not identify 13q structural abnormalities by enumeration FISH or microarray in any case. Only one tumor had genomic copy number changes, consisting of deletion of 1p13.3 to 1p11.2 and monosomy 14; these alterations appear non-specific. Our findings indicate no evidence of structural abnormalities of 13q resulting in deletion of RB1 in lipoblastoma-like tumor as seen in 13q deleted tumors. Although RB1 gene deletions were not identified, other mechanisms may influence RB1 immunoexpression such as abnormal regulation of other components of the RB1 pathway, epigenetic aberrations, or changes in RB1 protein half-life.
An additional consideration in the differential diagnosis of lipoblastoma-like tumor is the myxoid variant of dermatofibrosarcoma protuberans. Dermatofibrosarcoma protuberans typically arises in the trunk or extremities of young to middle age adults, can have a richly myxoid extracellular matrix with prominent thin-walled vasculature [17] and potentially the appearance of adipocytes as a neoplastic constituent due to infiltration of subcutaneous tissue. However, dermatofibrosarcoma protuberans differs from lipoblastoma-like tumor by its lack of lipoblasts, frankly infiltrative growth into dermis and subcutis, and recurrent COL1A1-PDGFB gene fusion which is detectable in a majority of tumors, including its myxoid form [18].
Two individual cases of a lipoblastoma-like tumor involving the oral lip have been reported [19, 20]. These cases describe tumors that form small lobules or nests of mature adipocytes and lipoblasts associated with capillary vasculature and fibrous septa within the dermis. Although similarly named, the absence of a spindle cell component in the oral lip tumors is divergent from lipoblastoma-like tumor of the vulva, and the two tumors are considered separate entities.
In summary, the results of our study support that lipoblastoma-like tumor is genetically unrelated to lipoblastoma, myxoid liposarcoma and spindle cell lipoma. The significance of “mosaic” pattern RB1 expression is unclear, but does not appear related to structural alterations of RB1. Recognition of lipoblastoma-like tumor is important to potentially avoid misclassification and overtreatment of this benign tumor.
References
Lae ME, Pereira PF, Keeney GL, et al. Lipoblastoma-like tumour of the vulva: report of three cases of a distinctive mesenchymal neoplasm of adipocytic differentiation. Histopathology. 2002;40:505–9.
Atallah D, Rouzier R, Chamoun ML, et al. Benign lipoblastomalike tumor of the vulva: report of a case affecting a young patient. J Reprod Med. 2007;52:223–4.
Mirkovic J, Fletcher CD. Lipoblastoma-like tumor of the vulva: further characterization in 8 new cases. Am J Surg Pathol. 2015;39:1290–5.
Fletcher CD, Bridge JA, Hogendoorn P, et al. WHO classification of tumours of soft tissue and bone. Lyon: IARC Press; 2013.
Agaimy A, Michal M, Giedl J, et al. Superficial acral fibromyxoma: clinicopathological, immunohistochemical, and molecular study of 11 cases highlighting frequent Rb1 loss/deletions. Hum Pathol. 2017;60:192–8.
Sukov WR, Hodge JC, Lohse CM, et al. TFE3 rearrangements in adult renal cell carcinoma: clinical and pathologic features with outcome in a large series of consecutively treated patients. Am J Surg Pathol. 2012;36:663–70.
Coffin CM, Lowichik A, Putnam A. Lipoblastoma (LPB): a clinicopathologic and immunohistochemical analysis of 59 cases. Am J Surg Pathol. 2009;33:1705–12.
Bartuma H, Domanski HA, Von Steyern FV, et al. Cytogenetic and molecular cytogenetic findings in lipoblastoma. Cancer Genet Cytogenet. 2008;183:60–3.
Brandal P, Bjerkehagen B, Heim S. Rearrangement of chromosomal region 8q11-13 in lipomatous tumours: correlation with lipoblastoma morphology. J Pathol. 2006;208:388–94.
ten Heuvel SE, Hoekstra HJ, van Ginkel RJ, et al. Clinicopathologic prognostic factors in myxoid liposarcoma: a retrospective study of 49 patients with long-term follow-up. Ann Surg Oncol. 2007;14:222–9.
Creytens D. Lipoblastoma-like tumor of the vulva, an important benign mimic of myxoid liposarcoma. Int J Gynecol Pathol 2018 [Epub ahead of print].
Ud Din N, Zhang P, Sukov WR, et al. Spindle cell lipomas arising at atypical locations. Am J Clin Pathol. 2016;146:487–95.
Enzinger FM, Harvey DA. Spindle cell lipoma. Cancer. 1975;36:1852–9.
Michal M, Kazakov DV, Hadravsky L, et al. Lipoblasts in spindle cell and pleomorphic lipomas: a close scrutiny. Hum Pathol. 2017;65:140–6.
Dal Cin P, Sciot R, Polito P, et al. Lesions of 13q may occur independently of deletion of 16q in spindle cell/pleomorphic lipomas. Histopathology. 1997;31:222–5.
Chen BJ, Marino-Enriquez A, Fletcher CD, et al. Loss of retinoblastoma protein expression in spindle cell/pleomorphic lipomas and cytogenetically related tumors: an immunohistochemical study with diagnostic implications. Am J Surg Pathol. 2012;36:1119–28.
Mentzel T, Scharer L, Kazakov DV, et al. Myxoid dermatofibrosarcoma protuberans: clinicopathologic, immunohistochemical, and molecular analysis of eight cases. Am J Dermatopathol. 2007;29:443–8.
Jahanseir K, Xing D, Greipp PT, et al. PDGFB rearrangements in dermatofibrosarcoma protuberans of the vulva: a study of 11 cases including myxoid and fibrosarcomatous variants. Int J Gynecol Pathol 2017 [Epub ahead of print].
Jeong TJ, Oh YJ, Ahn JJ, et al. Lipoblastoma-like tumour of the lip in an adult woman. Acta Derm Venereol. 2010;90:537–8.
Val-Bernal JF, Hermana S, Sanchez R. Intradermal lipoblastoma-like tumor of the lip in an adult woman. Actas Dermosifiliogr. 2016;107:88–90.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Schoolmeester, J.K., Michal, M., Steiner, P. et al. Lipoblastoma-like tumor of the vulva: a clinicopathologic, immunohistochemical, fluorescence in situ hybridization and genomic copy number profiling study of seven cases. Mod Pathol 31, 1862–1868 (2018). https://doi.org/10.1038/s41379-018-0102-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0102-y
This article is cited by
-
Chromothripsis in lipoblastoma: second reported case with complex PLAG1 rearrangement
Molecular Cytogenetics (2023)
-
A large retroperitoneal lipoblastoma as an incidental finding: a case report
BMC Pediatrics (2021)
-
Lipoblastoma-like tumor of the spermatic cord: case report and review of the literature
Virchows Archiv (2021)
-
Primary Pleomorphic Lipoma of the Parotid Gland with Prominent Myxoid Change: Report of a Rare Case Mimicking Carcinoma Ex Pleomorphic Adenoma on Fine Needle Aspiration Cytology
Head and Neck Pathology (2020)
