
Abstract
In the current World Health Organization (WHO)-classification, therapy-related myelodysplastic syndromes (t-MDS) are categorized together with therapy-related acute myeloid leukemia (AML) and t-myelodysplastic/myeloproliferative neoplasms into one subgroup independent of morphologic or prognostic features. Analyzing data of 2087 t-MDS patients from different international MDS groups to evaluate classification and prognostication tools we found that applying the WHO classification for p-MDS successfully predicts time to transformation and survival (both p < 0.001). The results regarding carefully reviewed cytogenetic data, classifications, and prognostic scores confirmed that t-MDS are similarly heterogeneous as p-MDS and therefore deserve the same careful differentiation regarding risk. As reference, these results were compared with 4593 primary MDS (p-MDS) patients represented in the International Working Group for Prognosis in MDS database (IWG-PM). Although a less favorable clinical outcome occurred in each t-MDS subset compared with p-MDS subgroups, FAB and WHO-classification, IPSS-R, and WPSS-R separated t-MDS patients into differing risk groups effectively, indicating that all established risk factors for p-MDS maintained relevance in t-MDS, with cytogenetic features having enhanced predictive power. These data strongly argue to classify t-MDS as a separate entity distinct from other WHO-classified t-myeloid neoplasms, which would enhance treatment decisions and facilitate the inclusion of t-MDS patients into clinical studies.
Similar content being viewed by others


Introduction
Therapy-related myelodysplastic syndromes (t-MDS) are defined as MDS occurring as a complication of cytotoxic chemotherapy and/or radiation administered for an antecedent neoplastic or non-neoplastic disorder. According to the last World Health Organization (WHO)-classification, they belong to the group of therapy-related myeloid neoplasms (t-MNs) [1, 2]. From the first WHO-classification of myeloid disorders in 2001 [3], the WHO 2008 [4], to the current classification from 2016 [1, 2] patients with t-MDS have been considered as having a generally poor prognosis.
From the beginning, t-MDS patients were placed together into the large group of therapy-related myeloid neoplasms (t-MNs), independent of blast count and morphologic features such as cellularity or dysplasia. In the first WHO-classification, t-MNs were sub-classified based on causative agents: alkylating agent/ radiation-related versus topoisomerase-II inhibitor-related [3]. This subdivision was removed from the next WHO-classification [4], because in clinical practice it was difficult to apply, as many patients received combined chemotherapeutic regimens (alkylators, topoisomerase-II-inhibitors, antimetabolites, antitubulin agents) and/or radiation therapy.
The 2016 WHO-classification recognizes the fact that t-MNs can be sub-classified morphologically into t-MDS, t-MDS/MPN, and t-AML, but considers it best to distinguish them collectively from p-MNs as “a unique clinical syndrome” [1, 2], insinuating again that all t-MDS have a uniformly poor prognosis. If this were considered true, all treatable patients would need to receive a recommendation for an intensive/disease-modifying treatment approach, including allogeneic transplantation, chemotherapy, or hypomethylating agents.
Most prognostic tools for MDS have been developed excluding patients with t-MDS [5,6,7]. An exception is the MD Anderson Prognostic Scoring System (MDAPSS) [8]. Following the categorization of t-MDS as a non-separate subgroup of t-MN, most publications have analyzed a conglomerate of t-MDS and t-AML, usually including both treated and untreated patients [9,10,11,12,13,14,15,16,17]. Other publications [18,19,20] have focused on t-AML. In contrast, publications on t-MDS only are rare [21,22,23]. Not categorizing t-MDS as a subgroup of MDS limits proper clinical decision-making, interferes with epidemiological/ biological research, and supports the established practice of excluding t-MDS from clinical studies [24], thereby potentially preventing therapeutic improvements.
In cooperation with centers from the International Working Group for Prognosis in MDS (IWG-PM) as well as the U.S. MDS Clinical Research Consortium we have compiled a database comprising 2087 patients. After a detailed review of all ISCN formulas, strict criteria were applied regarding the cytogenetic data included in this analysis. Information existed on 1245 patients for overall survival (OS) and AML progression with complete reviewed data to apply IPSS-R and WHO-classification. For a comparison to primary MDS (p-MDS), we used data from 4593 patients from the IPSS-R database that was limited to the institutions contributing data to both projects.
These two very large databases on both t- and p-MDS enabled us to gather comparative prognostic data in t-MDS related to p-MDS, test the performance of currently existing tools for classification and prognostication, and finally improve the current stratification systems for use in t-MDS.
Material and methods
Eight different study groups in the US, Germany, Spain, Italy, Austria, and the Netherlands contributed 2087 patients in total. By the contributing centers all patients with a diagnosis of MDS according to WHO and/or FAB were included if they had a history of an antecedent disease leading to chemo- (including alkylating agents, topoisomerase-II inhibitors, antimetabolites, and antitubulin agents) and/or radiotherapy [1,2,3,4, 25, 26]. To test the applicability and performance of different scoring systems as well as classifications strict inclusion/exclusion criteria were applied. Selection criteria included information about primary disease, pretreatment (at least chemotherapy or radiation), valid data to calculate the IPSS-R, survival data, AML transformation, valid stratification variables age, sex, and year of diagnosis. Patients were excluded if age <16 years, AML-defining cytogenetic abnormalities [inv(16), t(15;17), t(8;21)], peripheral blasts >19%, normal karyotypes based on <10 metaphases analyzed, proliferative CMML, AML as a primary diagnosis, survival or time to AML < 2 months occurred, and if the primary disease was in progression, to focus on the prognostic impact of the MDS itself. Karyotype was documented within the ISCN formula [27] after cytogenetic review (performed by DH, FS, JG, and BH). Treatment in MDS-phase, including intensive AML-type chemotherapy and allogeneic stem cell transplantation, was not an exclusion criterion, but the analyses were repeated with untreated patients only. These results are given in the supplement.
FAB, WHO-2016, IPSS-R, and WPSS-R classifications were calculated. For the WHO-classification RCUD, RARS, and MDS del(5q) were grouped together as in the WPSS. To test and quantify differentiating abilities of these tools the stratified log-rank test and the stratified Dxy-coefficient [28] were applied. Dxy is a concordance coefficient varying between −1 and 1, with 0 representing no monotone discriminative ability and 1 perfect monotone discrimination of a tool with respect to the time of interest (transformation free survival, overall survival, time to AML). As the main risk criterion, we used transformation-free survival since as a combined endpoint it described the clinically relevant disease-related risk more appropriately than overall survival. For completeness, Dxy values for overall survival and time to AML transformation were also included. In detail, time variables were defined as follows: all start with diagnosis of MDS and are censored, if no event occurred until the end of follow up. Transformation free survival ends when transformation or death without transformation occur. Time to AML ends with transformation but is censored in case of death without transformation. The results are based on stratified analyses compensating for possible confounding influences of sex, age, center, and year of diagnosis. Except for the influence of the primary diagnosis on the outcomes analyzed, we also stratified for primary diagnosis. Event time was calculated from time of MDS-diagnosis. Time to AML transformation was analyzed by censoring at time of death and in addition by treating death as competing event. Cumulative incidence curves for death with and without transformation are shown in the supplement.
All analyses were conducted with the statistics software R 3.4.3, including the package “survival” [29, 30]. Two-sided P values less than 0.05 were considered significant. In line with the essentially exploratory nature of the study, no adjustment for multiple testing was applied.
Data from t-MDS patients were compared with a cohort of 4593 untreated p-MDS patients from six different study groups within the IPSS-R database, including only patients from centers that also contributed cases to the t-MDS project.
Results
Patient characteristics
Of the total number of 2087 t-MDS patients, 1245 fulfilled all relevant selection criteria. Patients with high-risk (22%) and very high-risk (31%) IPSS-R score were more frequent among t-MDS than in the p-MDS group. Although less frequent, a considerable number (8% and 21% patients) had an IPSS-R of very low and low-risk, respectively (p < 0.001). Concordantly, as expected from previous publications [8], 30% t-MDS patients had a very poor and 15% a poor-risk cytogenetic score, and 27% had ≥5 abnormalities. Conversely, 2% and 37% patients were diagnosed with a very good or good-risk karyotype, respectively: 30% presented with a normal karyotype and 21% with only a single aberration. Patient characteristics as well as a comparison to p-MDS can be found in Table 1 and Supplementary Table 1 (the same analysis, limited to untreated patients is shown in Supplementary Table 2). Regarding WHO, the size of the four different risk groups was almost equal: RCUD, RARS, and MDS del(5q) 25%, RCMD 32%, RAEB-1 23%, and RAEB-2 21% and the distribution of the sub-groups were relatively similar to p-MDS (34%, 29%, 17%, and 20%). However, patients with RAEB-I were more frequent and RARS (6 vs. 13%), MDS del(5q) (1 vs. 4%), as well as MDS/MPN (CMML, excluding proliferative CMML) (4 vs. 10%) were less frequent (p < 0.001). The median overall survival was 18 months with a median follow up of 60 months for the t-MDS patients. The group of untreated p-MDS patients from the IPSS-R database had a median survival of 41 months. Median follow up was 49 months for this cohort, respectively.
The t-MDS patients’ primary diagnoses were a solid tumor in 54% and a hematological disease in 43%. The remaining 13% of patients had received treatment for a benign immunological disease. Treatment included chemotherapy only in 46%, radiation only in 19%, and both in 35% of the patients.
Application of classification and prognostic scoring systems
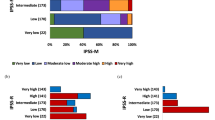
All classification and prognostic scoring systems applied were able to discriminate different risk groups within our t-MDS cohort, although the performance of the scoring systems was inferior when compared with p-MDS. The prognostic power measured by Dxy for the different classification and scoring systems in t- and p-MDS is given in Table 2 (the same analysis, limited to untreated patients only, is shown in Supplementary Table 3). The FAB-classification (Supplementary Fig. 1a) could only discriminate two different adjacent risk groups (RA vs. RAEB p < 0.001). There was no significant prognostic difference between RA and RARS (p = 0.8) as well as RAEB and RAEB-T (p = 0.291). However, these results are in line with results obtained in primary MDS and are thus not t-MDS specific. To examine the WHO-classification, we decided to use the categorization used within the WPSS, as there was no expectation regarding risk differences between RCUD, RARS, and MDS (del5q). With this combined low-risk group a better separation versus the remaining categories could be achieved, although there was no statistical significant difference between MDS with unilineage dysplasia (RCUD plus RARS plus MDS(del5q)) and MDS with multilineage dysplasia (p = 0.389), while RCMD vs. RAEB I (p < 0.001) and RAEB I vs RAEB II (p < 0.001) differed significantly (Fig. 1a). The same observation could be made in p-MDS (RCUD, RARS + MDS(del5q) vs. RCMD p = 0.403; RCMD vs. RAEB I p < 0.001; RAEB I vs RAEB II p < 0.001).

a outcome according to WHO 2016, b outcome according to IPSS-R, c Outcome according to cytogenetic IPSS-R risk categories.
The two prognostic scoring systems performed both very well. The IPSS-R could separate five different risk groups for all outcomes tested (Fig. 1b), while the WPSS separated five risk groups regarding OS, but only four regarding PFS and AML transformation (see Supplementary Fig. 1b). The influence of IPSSR(A) is shown in Supplementary Fig. 1c. Regarding these outcomes, the difference did not reach statistical significance between low and very low-risk, likely because the very low-risk group was relatively small (p = 0.146).
When we analyzed the performance of the cytogenetic component of the IPSS-R (cipssr), the prognostic power was already very high on its own. Only the difference between the very low and low-risk group did not reach statistical significance (p = 0.210), but this might very likely be a matter of the size of the very low-risk group (n = 24). The performance of the cipssr was at least equal in t-MDS compared with p-MDS (Fig. 1c).
Other possible, t-MDS specific influences like the primary diagnosis (Dxy 0.05) or type of prior treatment did not influence the different outcomes significantly. Only patients with other, non-malignant disease appeared to have a better outcome (Supplementary Fig. 1d, p = 0.051). Cumulative incidence of death with and without transformation as an addition to Fig. 1a–c are shown in Supplementary Fig. 2a–c.
Since the prognostic values of all classification and scoring systems as well as single variables, except the cipssr, were inferior compared with p-MDS (Dxy (IPSS-R) in t-MDS 0.38 for OS and 0.35 for time to AML), we analyzed if this was influenced by the fact that our cohort was a mixture of treated and untreated patients. This hypothesis could be verified since all scoring systems performed much better in the subsample of untreated t-MDS patients (see Fig. 2a–c and Supplementary Fig. 3a–c). In this subgroup the Dxy for the IPSS-R was not inferior (0.45 for OS and 0.48 for time to AML) when compared with the p-MDS cohort (0.40 for OS and 0.53 for time to AML) and for the cipssr 0.30 and 0.54 in t-MDS versus 0.23 and 0.28 in p-MDS (Supplementary Table 3).

a Outcome according to WHO-classification for treated patients. b Outcome according to WHO-classification for untreated patients. c Outcome according to WHO: p-MDS.
Although patients with t-MDS in general could be classified by the WHO-classification system developed for p-MDS, its performance differed within specific t-MDS subgroups. The prognostic power was almost comparable to p-MDS in patients with a solid tumor as primary disease as well as in patients after radiotherapy only. In patients with a history of a hematologic disease or after chemotherapy, the prognostic power was lower. However, even in these subgroups, patients with different outcomes could be separated (see Fig. 3a–c and Supplementary Fig. 4a–c).

a Outcome according to WHO: primary disease hematologic. b Outcome according to WHO: primary disease solid tumor c Outcome according to WHO: p-MDS.
Discussion
In this collaborative IWG-PM project, we were able to assemble the largest database on t-MDS to date, offering the analysis of well-characterized and clinically annotated data with a long follow up. Our analyses of the data presented here showed that patients with t-MDS benefited in a major way from receiving differentiated classification and prognostic evaluation, distinct from t-MN. Our scrupulous evaluation of the patients’ earlier treatment for malignant or non-malignant disease contributed to our case-finding.
T-MDS has been relatively neglected regarding classification and differentiated prognostication. All versions of the WHO-classification, including the latest from 2016, do not classify t-MDS within the group of MDS [1, 2]. Instead, t-MDS patients are currently still placed together with t-AML and t-MPD in a combined category of therapy-related myeloid neoplasms [1, 2]. Moreover, most established scoring systems were developed excluding t-MDS patients. The only t-MDS specific score was published by Quintas-Cardama et al. including variables such as performance status and age, which determine patient-related but not disease-specific risk features, and performance status might not be readily available in all databases [23].
Until now, only one paper analyzed the impact of the WHO-classification for p-MDS in t-MDS [21]. This publication by Singh et al. [21] included 155 patients with t-MDS or t-AML of whom only 81 patients were t-MDS. No differences were found in median survival times among patients classified into the different WHO subgroups and Singh and coworkers described a uniformly poor outcome in t-MDS regardless of morphologic classification. These results might be explained partially by the difference in size compared with our patient cohort. Furthermore, the paper includes patients not in remission from their primary disease. We excluded such patients to obtain a cleaner estimation of the specific MDS-related risk.
Earlier publications on t-MDS demonstrated mostly high-risk karyotypes in these patients, mainly including chromosome 5 and 7 abnormalities as well as complex karyotypes, in more than 90% of patients [9, 31, 32]. Although these features are still a hallmark of t-MDS in general, they do not represent all patients with a history of chemo- and/or radiotherapy. Our data demonstrated an unexpectedly high percentage of good-risk and normal cytogenetics, which are concordant with other more recently published data [13, 19, 23]. Regarding these early t-MDS publications, a reporting bias may have contributed since in some cases conspicuous cytogenetics might have been required for the question about previous treatments.
Even in the most recent WHO-classification [1, 2] the prognosis of t-MN is described as being generally poor and that prognosis of these patients is influenced strongly by karyotype as well as by the primary disease. Although this important information is given in the Revised Fourth Edition of the WHO series on histological and genetic typing of human tumors it remains purely descriptive, as it provides no consequences for the resulting t-MN classification. Chromosome 5 and/or 7 abnormalities, TP53 mutations, and complex karyotype are indicated by the WHO as having a particularly poor outcome, while patients with balanced translocations were stated to have a good prognosis, albeit not as good as in p-MN. However, exceptions to these generalizations are notable. In our database we observed some patients with isolated del(5q) and a history of exposure to mutagenic agents that have a good prognosis, as is the case in p-MDS [33]. Conversely, considered typical for t-MDS is the group with balanced translocations involving chromosome band 11q23, the localization of the MLL/KMT2A-gene [9, 33,34,35,36,37]. These translocations can, in contrast to the typical good risk “AML defining translocations” t(8;21), t(15;17), and inv(16), occur as t-MDS and are associated with an extremely poor prognosis as previously described [9, 33,34,35,36,37]. These categorization problems relate in part from combining t-MDS and t-AML leading to the use of AML cytogenetic classification systems for the entire group of t-MN. In our study we have demonstrated that the influence of cytogenetics is very high in t-MDS and the prognostic power of the cipssr is in t-MDS at least as good as in p-MDS, due to a high proportion of abnormal karyotypes. Without a proper morphological classification and separation from t-AML, t-MDS patients are also withheld the most powerful tool for prognostic evaluation.
In addition, prognostic factors for t-MDS, other than cytogenetics, are not recognized by the WHO-classification. In practice, this means that a patient with a blast count of 0% (t-MDS) is in the same risk category as a patient with a blast count of 99% (t-AML). Also, with the current WHO-classification irrelevant for therapeutic decision-making is whether a patient has dysplastic (t-MDS) or proliferative disease (t-MDS/MPD). These issues have major implications for patient therapy. Thus, proper morphologic and cytogenetic classification as well as separation from t-AML, provide t-MDS patients a most powerful method for treatment considerations.
Our data demonstrated that t-MDS patients can be subdivided by diagnostic procedures into groups with clearly varying prognoses. These findings are underlined by results from smaller group analyses focusing on the value of prognostic scoring systems [22, 23, 38]. It should be considered in particular that sporadic MDS cases might be assigned to the group of t-MN based on their therapeutic history although this could be only coincidental, and they actually belong to p-MDS. Observational data on MDS patients cannot prove a causal link between therapy and a later developing MDS. This fact is already implied by the term “therapy-related”. As we cannot differentiate between the two situations this should not be taken as an argument for a less scrupulous diagnostic classification of the concerned patients. Quite the contrary, such spontaneously developed MDS within the t-MDS group have the greatest disadvantage from not being classified as p-MDS.
In addition, we note that the survival of t-MDS patients in different WHO groups was inferior to p-MDS, similar to findings in t- vs p-AML [18, 19]. This might have several reasons, including patient-related factors associated with the primary disease (relapse/progression of the primary disease, cumulative toxicity of primary and secondary therapy on bone marrow reserve as well as other organ function), as previously discussed [10, 19, 22, 39, 40]. A second point for discussion might be the retrospective nature of our, as well as previous studies. And third, in the present database, we can only compare data from a t-MDS group that is heterogeneous regarding treatment to an untreated group of p-MDS patients. As we have demonstrated, this influences the power of prognostic models and likewise will influence patient outcomes in general as well, although further analyses are needed to understand the exact nature of such effects. We find a relevant shift towards better risk patients in our t-MDS cohort if we look at untreated patients only. It is likely, that there will be a similar shift in the p-MDS cohort as well, since treated p-MDS patients were not included in our reference database (compare Figs. 4 and 5). Results in t- and p-MDS would possibly be better comparable if the analysis were restricted to untreated patients only. However, regarding the main issue of differentiating and sub-classifying tMDS exclusion of treated cases would bias those conclusions, since we observed a relevant shift towards older age and lower-risk disease among untreated patients (Fig. 5, Supplementary Table 2).

a Distribution of IPSS-R subgroups in p- and t-MDS. b Distribution of WHO- subtypes (according to WPSS + RAEB-T) in p- and t-MDS. c Distribution of cytogenetic IPSS-R subgroups in p- and t-MDS. d Distribution of number of aberrations in p- and t-MDS.

a Comparison of IPSS-R risk-groups between treated and untreated patients. b Comparison of IPSS-R cytogenetic risk-groups between treated and untreated patients. c Comparison of WHO subgroups between treated and untreated patients.
Further, in addition to patient and method-related factors, biological differences between the leukemic stem cells might contribute as well, since inferior outcome of patients with t-MDS seems not only limited to survival. Especially in the good and intermediate-risk group, we observe this phenomenon with regard to AML transformation (see Fig. 6a–c and Supplementary Fig 5a-c). It is possible that, even within each subgroup there is a shift to higher risk cytogenetic or molecular abnormalities. This will be an important comparison for future analyzes. The impact of cytogenetics appears to be even greater in t-MDS compared with p-MDS. The major reason might be that the proportion of patients with aberrant karyotype was higher in t-MDS. In p-MDS, about half of the patients presented with a normal karyotype and 71% belonged to the large cipssr good risk group, whereas in t-MDS it was only 30 and 37%, respectively (see Table 1).

a Comparison p- and t-MDS according to WHO-classification. b Comparison t- and p-MDS according to IPSS-R risk-categories. c Comparison t- and p-MDS according to cytogenetic IPSS-R categories.
Regarding molecular differences, Singhal et al. found that t-MDS patients with ≥15% ringed sideroblasts had a low frequency of SF3B1 mutations, but a much higher frequency of TP53 mutations compared with patients with p-MDS [41]. In line with this observation the same analysis showed a generally much higher frequency of TP53 mutations in patients with t-MDS and about half of the patients had <5% marrow blasts at the time of t-MDS diagnosis [41]. Clonal hematopoiesis or germline predisposition can be found in hematopoietic cells of patients who develop t-MN, even before treatment of the prior disease, representing a sign of increased chromosomal instability and high-risk disease [16, 41,42,43,44]. However, preceding clonal hematopoiesis or genetic predisposition can also be found before the development of p-MDS [45,46,47,48,49].
Pedersen-Bjergaard et al. [50] suggested 25 years ago different genetic pathways for t-MDS and t-AML due to their differing distribution of genetic abnormalities. While this is the case for some t-MNs, in other cases, as has been evident with more recently developed mutational data, a biological continuum exists from clonal hematopoiesis to t-MDS and “secondary” t-AML with or without a MDS pre-phase and MDS-related features [16, 20, 41, 42]. These different mechanisms of leukemogenesis occur in therapy-related as in primary MNs.
Our data showed that, transformation-free survival (TFS) was poorer in t-MDS subgroups vs those in p-MDS: range 8–22 months, compared with 13–63 months, respectively. The impact of blast count and the performance of morphological classifications in t-MDS was somewhat less than in p-MDS, but remained substantial, in contrast to earlier, smaller publications [21]. Thus, all patients can and should be subdivided into different diagnostic subcategories and risk groups.
Knowledge of the diagnostic and prognostic evaluation of t-MN patients according to pathogenesis and disease characteristics is required for selecting patients who can be cured or will at least benefit from active treatment [10, 24] and would facilitate clinical as well as epidemiological research. An updated classification should separate t-MDS from t-AML and t-MDS/MPN as although these diseases might share some overlapping features, they exhibit differences in clinical presentation and molecular and cytogenetic characteristics [51, 52]. As for t-MDS, the feasibility of further sub-classification has been demonstrated by publications on t-AML as well [9, 18, 19, 37, 53,54,55,56]. Regarding t-MDS/MPN, only 4% of the patients in our database presented with t-CMML dysplastic, as its frequency is lower in t- compared with p-MDS [57]. Patients with t-CMML proliferative were excluded from this analysis. Our data indicate for all t-MDS an increased influence of karyotype on prognosis compared with other prognostic variables. This finding is similar to that demonstrated for p-CMML and t-CMML wherein the frequency of cytogenetic abnormalities is much higher in t-CMML, while the frequency of most molecular abnormalities seems to be comparable [57]. Based on this important publication, Patnaik et al. suggest that, due to the unique biological pattern and dismal prognostic impact, t-CMML should be considered as separate subtype in the classification scheme for both CMML and t-MN [57].
Based on our findings, we believe this newly established disease category t-MDS deserves further p-MDS-like subcategorization. We propose an approach using the WHO classification for p-MDS, but preceding each subgroup with a t-; for example, t-MDS-SLD, t-MDS-MLD, etc. We suggest restricting the use of prognostic systems to t-MDS patients in remission from their primary disease so as not to confound the results of risk factors for the MDS. For those not in remission it would be important to incorporate the confounding risk of the primary disease into clinical decision-making.
In summary, our data demonstrated that classification tools established in p-MDS were effective for stratifying subgroups in t-MDS and indicated the high prognostic relevance of cytogenetics in t-MDS. These findings from the largest t-MDS database to date should initiate a discussion of a potential revision of the WHO-classification and encourage clinicians to use the existing tools for risk assessment and treatment decisions for patients with therapy-related disease.
References
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. (eds). WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2017.
Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–302.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25:3503–10.
Kantarjian H, O’Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113:1351–61.
Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52.
Kim SA, Hong J, Park WC, Shin DY, Koh Y, Kim I, et al. Better treatment outcomes in patients with actively treated therapy-related myeloid neoplasms harboring a normal karyotype. PLoS ONE. 2018;13:e0209800.
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39:348–54.
Hartmann L, Nadarajah N, Meggendorfer M, Höllein A, Vetro C, Kern W, et al. Molecular characterization of a second myeloid neoplasm developing after treatment for acute myeloid leukemia. Leukemia. 2019. https://doi.org/10.1038/s41375-019-0633-3.
Kuzmanovic T, Patel BJ, Sanikommu SR, Nagata Y, Awada H, Kerr CM, et al. Genomics of therapy-related myeloid neoplasms. Haematologica. 2019. https://doi.org/10.3324/haematol.2019.219352.
Guru Murthy GS, Hamadani M, Dhakal B, Hari P, Atallah E. Incidence and survival of therapy related myeloid neoplasm in United States. Leuk Res. 2018;71:95–9.
Shih AH, Chung SS, Dolezal EK, Zhang SJ, Abdel-Wahab OI, Park CY, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98:908–12.
Gillis NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18:112–21.
Schroeder T, Kuendgen A, Kayser S, Kröger N, Braulke F, Platzbecker U, et al. Therapy-related myeloid neoplasms following treatment with radioiodine. Haematologica. 2012;97:206–12.
Kern W, Haferlach T, Schnittger S, Hiddemann W, Schoch C. Prognosis in therapy-related acute myeloid leukemia and impact of karyotype. J Clin Oncol. 2004;22:2510–1.
Kayser S, Döhner K, Krauter J, Köhne CH, Horst HA, Held G, et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117:2137–45.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76.
Singh ZN, Huo D, Anastasi J, Smith SM, Karrison T, Le Beau MM, et al. Therapy-related myelodysplastic syndrome: morphologic subclassification may not be clinically relevant. Am J Clin Pathol. 2007;127:197–205.
Zeidan AM, Al Ali N, Barnard J, Padron E, Lancet JE, Sekeres MA, et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS Clinical Research Consortium. Leukemia. 2017;31:1391–7.
Quintás-Cardama A, Daver N, Kim H, Dinardo C, Jabbour E, Kadia T, et al. A prognostic model of therapy-related myelodysplastic syndrome for predicting survival and transformation to acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2014;14:401–10.
Borate U, Norris BA, Statler A, Fu R, Bucy T, Sekeres MA. Representation of therapy-related myelodysplastic syndrome in clinical trials over the past 20 years. Blood Adv. 2019;3:2738–47.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–99.
Valent P, Orazi A, Steensma DP, Ebert BL, Haase D, Malcovati L, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017;43:73483–500.
McGowan-Jordan J, Schmid M. ISCN 2016: An international system for human cytogenomic nomenclature. S. Karger, Basel, 2016.
Harrell FE, Lee KL, Mark DB. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15:361–87.
Core Team, R: a Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2018, https://www.R-project.org/.
Therneau T, A Package for Survival Analysis in S. Version 2.38, 2015, http://CRAN.R-project.org/package=survival.
Le Beau MM, Albain KS, Larson RA, Vardiman JW, Davis EM, Blough RR, et al. Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no. 5 and 7. J Clin Oncol. 1986;4:325–45.
Pedersen-Bjergaard J, Philip P, Larsen SO, Andersson M, Daugaard G, Ersbøll J, et al. Therapy-related myelodysplasia and acute myeloid leukemia. Cytogenetic characteristics of 115 consecutive cases and risk in seven cohorts of patients treated intensively for malignant diseases in the Copenhagen series. Leukemia. 1993;7:1975–86.
Kuendgen A, Tuechler H, Nomdedeu M, Haase D, Garcia-Manero G, Komrokij RS, et al. Frequency and prognostic significance of cytogenetic abnormalities in 1269 patients with therapy-related myelodysplastic syndrome - a study of the international working group (IWG-PM) for myelodysplastic syndromes (MDS). Blood. 2016;128:112.
Pui CH, Relling MV, Rivera GK, Hancock ML, Raimondi SC, Heslop HE, et al. Epipodophyllotoxin-related acute myeloid leukemia: a study of 35 cases. Leukemia. 1995;9:1990–6.
Andersen MK, Johansson B, Larsen SO, Pedersen-Bjergaard J. Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica. 1998;83:483–8.
Zhang Y, Poetsch M, Weber-Matthiesen K, Rohde K, Winkemann M, Haferlach T, Gassmann W, et al. Secondary acute leukaemias with 11q23 rearrangement: clinical, cytogenetic, FISH and FICTION studies. Br J Haematol. 1996;92:673–80.
Bloomfield CD, Archer KJ, Mrózek K, et al. 11q23 balanced chromosome aberrations in treatment-related myelodysplastic syndromes and acute leukemia: report from an international workshop. Genes Chromosomes Cancer. 2002;33:362–78.
Ok CY, Hasserjian RP, Fox PS, Stingo F, Zuo Z, Young KH, et al. Application of the international prognostic scoring system-revised in therapy-related myelodysplastic syndromes and oligoblastic acute myeloid leukemia. Leukemia. 2014;28:185–9.
Nazha A, Seastone DP, Keng M, Hobson S, Kalaycio M, Maciejewski JP, et al. The revised international prognostic scoring system (IPSS-R) is not predictive of survival in patients with secondary myelodysplastic syndromes. Leuk Lymphoma. 2015;56:3437–9.
Ganser A, Heuser M. Therapy-related myeloid neoplasms. Curr Opin Hematol. 2017;24:152–8.
Singhal D, Wee LYA, Kutyna MM, Chhetri R, Geoghegan J, Schreiber AW, et al. The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia. 2019;33:2842–53.
Katagir S, Makishima H, Azuma K, Nannya Y, Saitoh Y, Yoshizawa S, et al. Predisposed genomic instability in pre-treatment bone marrow evolves to therapy-related myeloid neoplasms in malignant lymphoma. Haematologica 2019. https://doi.org/10.3324/haematol.2019.229856.
Gibson CJ, Lindsley RC, Tchekmedyian V, Mar BG, Shi J, Jaiswal S, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35:1598–605.
Schulz E, Valentin A, Ulz P, Beham-Schmid C, Lind K, Rupp V, et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J Med Genet. 2012;49:422–8.
Furutani E, Shimamura A. Genetic predisposition to MDS: diagnosis and management. Hematol Am Soc Hematol Educ Program. 2019;2019:110–9.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl J Med. 2014;371:2477–87.
Genovese G, Jaiswal S, Ebert BL, McCarroll SA. Clonal hematopoiesis and blood-cancer risk. N. Engl J Med. 2015;372:1071–2.
Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17:5–19.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Pedersen-Bjergaard J, Pedersen M, Roulston D, Philip P. Different genetic pathways in leukemogenesis for patients presenting with therapy-related myelodysplasia and therapy-related acute myeloid leukemia. Blood. 1995;86:3542–52.
Bacher U, Haferlach C, Alpermann T, Schnittger S, Kern W, Haferlach T. Patients with therapy-related myelodysplastic syndromes and acute myeloid leukemia share genetic features but can be separated by blast counts and cytogenetic risk profiles into prognostically relevant subgroups. Leuk Lymphoma. 2013;54:639–42.
Zhang L, Wang SA. A focused review of hematopoietic neoplasms occurring in the therapy-related setting. Int J Clin Exp Pathol. 2014;7:3512–23.
Kayser S, Krzykalla J, Elliott MA, Norsworthy K, Gonzales P, Hills RK, et al. Characteristics and outcome of patients with therapy-related acute promyelocytic leukemia front-line treated with or without arsenic trioxide. Leukemia. 2017;31:2347–54.
Pulsoni A, Pagano L, Lo Coco F, Avvisati G, Mele L, Di Bona E, et al. Clinicobiological features and outcome of acute promyelocytic leukemia occurring as a second tumor: the GIMEMA experience. Blood. 2002;100:1972–6.
Ornstein MC, Mukherjee S, Mohan S, Elson P, Tiu RV, Saunthararajah Y, et al. Predictive factors for latency period and a prognostic model for survival in patients with therapy-related acute myeloid leukemia. Am J Hematol. 2014;89:168–73.
Aldoss I, Pullarkat V. Therapy-related acute myeloid leukemia with favorable cytogenetics: still favorable? Leuk Res. 2012;36:1547–51.
Patnaik MM, Vallapureddy R, Yalniz FF, Hanson CA, Ketterling RP, Lasho TL, et al. Therapy related-chronic myelomonocytic leukemia (CMML): Molecular, cytogenetic, and clinical distinctions from de novo CMML. Am J Hematol. 2018;93:65–73.
Acknowledgements
We thank the International Working Group for Prognosis in MDS and the U.S. MDS Clinical Research Consortium as well as all participating centers for the contribution of patient data to compile the databases analyzed in this paper. MTV was supported by AIRC 5 × 1000 call “Metastatic disease: the key unmet need in oncology” to MYNERVA project (project #21267 http://www.progettoagimm.it). PV was supported by the Austrian Science Fund, grant F4704-B20. Open access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kuendgen, A., Nomdedeu, M., Tuechler, H. et al. Therapy-related myelodysplastic syndromes deserve specific diagnostic sub-classification and risk-stratification—an approach to classification of patients with t-MDS. Leukemia 35, 835–849 (2021). https://doi.org/10.1038/s41375-020-0917-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-0917-7
This article is cited by
-
The clinical outcomes of haploidentical stem cell transplantation (haplo-HSCT) for patients with therapy-related myelodysplastic syndrome: comparable to de novo myelodysplastic syndrome
Clinical and Experimental Medicine (2024)
-
Therapy-related MDS dissected based on primary disease and treatment—a nationwide perspective
Leukemia (2023)
-
Assessment and validation of the molecular international prognostic scoring system for myelodysplastic syndromes
Leukemia (2023)
-
The International Consensus Classification of myelodysplastic syndromes and related entities
Virchows Archiv (2023)
-
The complex karyotype in hematological malignancies: a comprehensive overview by the Francophone Group of Hematological Cytogenetics (GFCH)
Leukemia (2022)
