
Abstract
In 2022, two novel classification systems for myelodysplastic syndromes/neoplasms (MDS) have been proposed: the International Consensus Classification (ICC) and the 2022 World Health Organization (WHO-2022) classification. These two contemporary systems exhibit numerous shared features but also diverge significantly in terminology and the definition of new entities. Thus, we retrospectively validated the ICC and WHO-2022 classification and found that both systems promoted efficient segregation of this heterogeneous disease. After examining the distinction between the two systems, we showed that a peripheral blood blast percentage ≥ 5% indicates adverse survival. Identifying MDS/acute myeloid leukemia with MDS-related gene mutations or cytogenetic abnormalities helps differentiate survival outcomes. In MDS, not otherwise specified patients, those diagnosed with hypoplastic MDS and single lineage dysplasia displayed a trend of superior survival compared to other low-risk MDS patients. Furthermore, the impact of bone marrow fibrosis on survival was less pronounced within the ICC framework. Allogeneic transplantation appears to improve outcomes for patients diagnosed with MDS with excess blasts in the ICC. Therefore, we proposed an integrated system that may lead to the accurate diagnosis and advancement of future research for MDS. Prospective studies are warranted to validate this refined classification.
Similar content being viewed by others


Introduction
Myelodysplastic neoplasms (MDS) are a diverse set of clonal myeloid neoplasms with dysregulated hematopoiesis, causing cytopenia and dysplastic hematopoietic cells. MDS is marked by recurrent chromosomal abnormalities, clinical and genetic diversity, and varying prognoses [1, 2]. The classification criteria for hematopoietic neoplasms, including MDS, were collated, beginning with the third edition of the World Health Organization (WHO) classification of hematologic malignancies in 2001 [3,4,5]. In 2016, the WHO, in collaboration with the Society for Hematopathology and the European Association for Hematopathology, published the revised fourth edition of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (WHO-2016) [6].
Recently, major advances in molecular technology and the development of next-generation sequencing (NGS) have deepened our understanding of MDS pathobiology [7, 8]. Revisions to the current classification system are needed for precise diagnosis, classification, and prognostication. Hence, the International Consensus Classification (ICC), developed with input from a global Clinical Advisory Committee of pathologists, hematologists, oncologists, and geneticists, was published in 2022 [9]. The ICC’s innovative changes in MDS include reclassifying MDS with 10–19% blasts as MDS/acute myeloid leukemia (AML), MDS with mutated SF3B1 without excess blasts as MDS-SF3B1 regardless of ring sideroblast count, and introducing novel molecular categories like myeloid neoplasms with mutated TP53, and MDS/AML with MDS-related gene mutations. Concurrently, the 5th edition of the WHO (WHO-2022) classification was released [10]. Emphasizing molecular features, tissue architecture, and histological appearance, WHO-2022 classification introduced new categories, including MDS with biallelic TP53 inactivation (MDS-biTP53), hypoplastic MDS (MDS-h), and MDS with fibrosis (MDS-f) [10]. These two systems exhibit several shared features but also differ significantly. The term MDS with increased blasts-2 remains in WHO-2022 classification but changes to MDS/AML in the ICC. Moreover, the ICC requires a 10% blast threshold to define AML with recurrent genetic abnormalities (excluding BCR::ABL and TP53), unlike WHO-2022, where most AML with defining genetic abnormalities can be diagnosed with increased blasts in peripheral blood (PB) or bone marrow (BM) (may be <20%). Additionally, a novel risk-scoring system, the Molecular IPSS (IPSS-M), combining clinical parameters, cytogenetic abnormalities, and somatic mutations of 31 genes, was established, stratifying MDS patients into six risk categories [11].
Few studies have concurrently explored the ICC and WHO-2022 classification within the IPSS-M context, evaluating their differences. This study retrospectively reviewed a cohort of 635 patients diagnosed with primary MDS using WHO-2016, WHO-2022, and ICC criteria. We elucidated differences in clinical characteristics, IPSS-M category distribution, genetic features, and outcomes among MDS subtypes per ICC and WHO-2022 classification and further compared these systems’ applicability in the IPSS-M framework.
Materials and methods
A total of 635 patients with primary MDS whose BM samples were adequately cryopreserved for deep-targeted sequencing were included, and available data for the revised IPSS (IPSS-R) [12] and IPSS-M [11] risk assessments were obtained. Patients with prior chemotherapy/radiotherapy or hematologic malignancies were excluded, considering the distinct mutational profiles of primary and secondary MDS [13, 14]. BM cellularity and the grade of fibrosis were evaluated by reticulin staining and confirmed by pathologists. The Research Ethics Committee of the National Taiwan University Hospital approved this study (approval numbers: 201709072RINC, 202109078RINB, and 20220705RINB), and all participants provided written informed consent in accordance with the Declaration of Helsinki.
Cytogenetic analyses were performed and interpreted according to the International System for Human Cytogenetic Nomenclature [15, 16]. The TruSight myeloid sequencing panel (Illumina, San Diego, CS, USA) and HiSeq platform (Illumina, San Diego, CA, USA) were used to analyze alterations in 54 myeloid-neoplasm-related genes [11, 17, 18] (Supplementary Table 1). Five of the residual genes (ETNK1, GNB1, NF1, PPM1D, and PRPF8) defined by the IPSS-M model were not included and evaluation of TP53 copy-neutral loss of heterozygosity were not avalible in the current study. Library preparation and sequencing were performed according to the manufacturer’s instructions. The median reading depth was 10550x. Somatic mutations were cataloged using cancer database v86, SNP database v151, ClinVar, PolyPhen-2, and SIFT algorithms for variant consequence evaluation. The variant analysis algorithm for diagnostics is previously described [19]. NGS limitations necessitated FLT3-ITD analysis via polymerase chain reaction (PCR) and fluorescence capillary electrophoresis and KMT2A-PTD analysis via PCR and Sanger sequencing [16, 20].
Statistical analysis
The Mann–Whitney U test was used for continuous variables, and Fisher’s exact test or the χ2 test was used for discrete variables. The Kruskal-Wallis test was used to identify any significant disparities in medians among three or more groups. Leukemia-free survival (LFS) was defined as the interval between the date of diagnosis and the last follow-up, documented leukemic transformation, or death from any cause, whichever occurred first. Overall survival (OS) was defined as the interval between the date of diagnosis and the last follow-up or death from any cause, whichever occurred first. Survival curves were plotted using Kaplan-Meier analysis, and statistical significance was calculated using the log-rank test. The Cox proportional hazards model was used for univariable and multivariable analyses. Allogeneic hematopoietic stem cell transplantation (HSCT) was used as a time-dependent covariate [21]. All P-values were two-sided and considered statistically significant at P < 0.05. All analyses were performed using IBM SPSS Statistics v23 for Windows.
Results
Case allocation according to the ICC and 2022-WHO classification
The classification of the 635 patients diagnosed as having primary MDS based on the WHO-2016 classification and case allocation using the ICC and WHO-2022 criteria are presented in Table 1 and Fig. 1. In the ICC, 74.5% of patients remained in the MDS group, while 25.5% of patients were reclassified to the MDS/AML group. In the WHO-2016 classification, the MDS-5q subgroup was renamed to MDS with del(5q) in the ICC and MDS with low blasts and isolated 5q deletion in WHO-2022 classification. Nine (20.0%) patients in both the MDS with ring sideroblasts and single lineage dysplasia (MDS-RS-SLD) and MDS-RS and multilineage dysplasia (MDS-RS-MLD) subgroups, all with wild-type SF3B1, were distinguished from those with mutated SF3B1 (Table 1).

Case allocation from 2016 World Health Organization classification to 2022 International Consensus Classification and 2022 World Health Organization classification of 635 patients with myelodysplastic syndromes/neoplasms.
Analysis of ICC vs. WHO-2022 classification differences showed that 39 (36.8%) and 55 (37.4%) individuals with MDS, not otherwise specified (NOS) with SLD or MLD, respectively, qualified for hypoplastic MDS (MDS-h) diagnosis. Meanwhile, nine (8.5%) in the MDS, NOS with SLD group, and seven (4.8%) in the MDS, NOS with MLD group (wild type SF3B1) were diagnosed as MDS with low blasts (LB) and RS in WHO-2022 classification (Table 1). Notably, three (2.0%) in the MDS, NOS with MLD group were diagnosed with MDS-SF3B1 in WHO-2022 classification. Among these, two patients had RUNX1 mutation and one had mutated SF3B1 with variant allele frequency (VAF) <10%, excluding them from MDS-SF3B1 in ICC. Thirteen (9.9%) diagnosed with MDS with excess blasts (EB) in ICC, having significant BM fibrosis, were classified as MDS-f in WHO-2022 classification. In WHO-2022 classification, PB blast percentage criteria remain (5–19% as increased blasts-2 [IB2]), but <10% is considered MDS with EB, not MDS/AML in ICC. Thus, nineteen (14.5%) with PB blast percentages of 5–9% were diagnosed as MDS-IB2 in WHO-2022 classification (Table 1).
Over 90% of patients with multi-hit TP53 mutations exhibit complex karyotypes [22, 23]. However, the WHO-2022 criteria consider MDS-biTP53 only in cases with two or more TP53 mutations or one mutation plus TP53 copy number loss. Conversely, the ICC considers TP53 mutation with complex karyotype as multi-hit TP53 if TP53 locus heterozygosity information is unavailable. Additionally, when PB or BM blast ranges from 10–19%, patients with not only multi-hit, but also single TP53 mutation would be classified as MDS/AML with mutated TP53. Thus, 14 patients (23.3%) in the MDS or MDS/AML group with mutated TP53 in the ICC were classified as MDS-IB or MDS-f in WHO-2022 classfication (Table 1, Fig. 1). Due to differing blast percentage criteria, three patients identified as AML with CEBPA mutation in the basic leucine zipper domain per ICC were placed in MDS-IB2 (14.5%, 15.2%, 19% blasts in BM) as per WHO-2022 criteria (Table 1, Fig. 1). The key diagnostic discrepancies between ICC and WHO-2022 classification due to different diagnostic criteria were summarized in Supplementary Table 2.
Comparison of the demographic features of patients with different MDS subtypes
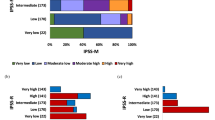
Clinical characteristics and genetic profiles of patients with various MDS subtypes per ICC/WHO-2022 classification are in Table 2, Supplementary Fig. 1, and Supplementary Table 3. In the ICC, all patients with mutated TP53 had high- or very high-risk IPSS-R/IPSS-M, whereas only one patient with MDS-SF3B1 had high-risk IPSS-M because of the presence of co-mutations including CBL, GATA2, and CEBPA. Moreover, MDS-EB subgroup patients were at higher risk according to IPSS-R or IPSS-M than MDS, NOS with SLD or MLD patients. All MDS/AML patients had intermediate to very high-risk IPSS-R. In the MDS/AML subgroup, 3.2% with MDS-related gene mutations and 5.0% with MDS/AML-NOS were moderate low-risk IPSS-M. MDS/AML patients with mutated TP53 showed the highest incidence of very high-risk IPSS-R or IPSS-M. In the WHO-2022 classification, patients classified as having MDS-LB and RS had similar IPSS-R and IPSS-M risk stratification distributions to those with MDS-LB, which were distinct from those with MDS-SF3B1. Patients with MDS-SF3B1 more frequently had very low, and low-risk IPSS-R (77.4%) and very low, low, and moderate low-risk IPSS-M (88.7%) than did patients with the other two subtypes (for IPSS-R: MDS-LB and RS vs. MDS-LB, 50.0% vs. 52.9%, P = 0.828, MDS-SF3B1 vs. MDS-LB and RS or MDS-LB, both P < 0.05; for IPSS-M, MDS-LB and RS vs. MDS-LB, 37.5% vs. 63.6%, P = 0.058, MDS-SF3B1 vs. MDS-LB and RS or MDS-LB, both P < 0.001; Table 2, Supplementary Figs. 1c and 1d). Patients with MDS-f exhibited similar BM blast percentage with MDS-IB1 (median blast 7% vs. 7%, P = 0.802), but significant less blast percentage than those with MDS-IB2 (median blast 7% vs. 13%, P < 0.001). The distributions of the IPSS-R and IPSS-M scores among patients with MDS-f were comparable to those among individuals with MDS-IB (Table 2, Supplementary Figs. 1c and 1d). Additionally, all patients with MDS-biTP53 had very high-risk IPSS-M (Table 2 and Supplemental Fig. 1d).
Fewer patients with MDS with mutated TP53 (8.7%, ICC), MDS/AML with mutated TP53 (8.1%, ICC), or MDS-biTP53 (10.9%, WHO-2022 criteria) underwent HSCT compared to those with other subtypes of MDS/AML, MDS-EB/IB, and MDS-f, despite being at a higher risk (Supplementary Table 3 and Table 2). This was attributed to a 22–43% early mortality rate at 3 months (Supplementary Table 3 and Table 2), no complete remission in patients before HSCT, and leukemic transformation within a median of 7.5 months post-MDS diagnosis, limiting transplantation feasibility in MDS-biTP53 patients.
Comparison of the genetic profiles among patients with different MDS subtypes
In the ICC, patients with MDS, NOS with SLD, and those with MDS, NOS with MLD exhibited similar mutation profiles (Supplementary Table 4a, Supplementary Fig. 2). Patients with MDS-SFB31 harbored distinct mutation patterns with higher frequencies of DNMT3A mutations (17% vs. 5%, P = 0.004) and TET2 mutations (34% vs. 12%, P < 0.001) compared to those with MDS, NOS with SLD and MLD (Supplementary Table 4a). Moreover, patients with mutated TP53 harbored a significantly higher frequency of TET2 mutations (26% vs. 9%, P = 0.019) but a lower frequency of ASXL1 (4% vs. 35%, P = 0.003) and RUNX1 (0% vs. 22%, P = 0.008) mutations compared to the EB subgroup (Supplementary Table 4a).
On the other hand, based on the WHO-2022 criteria, patients with MDS-LB and MDS-LB and RS exhibited comparable mutation profiles (Supplementary Fig. 3), whereas patients with MDS-SF3B1 had significantly different mutation patterns when compared to those of the former two subtypes. Patients with MDS-SF3B1 had significantly more DNMT3A (16% vs. 6%, P = 0.029) and TET2 mutations (32% vs. 14%, P = 0.004) but fewer STAG2 mutation (2% vs. 9%, P = 0.068) than did those with MDS-LB, while fewer of them harbored SRSF2 mutations when compared to those with MDS-LB and RS (3% vs. 25%, P = 0.014) (Supplementary Table 4b).
We evaluated the genetic profiles of the patients diagnosed with MDS-LB or MDS-h. With the exception of the U2AF1 mutation (MDS-LB vs. MDS-h, 9% vs. 1%, P = 0.017), patients in these two groups exhibited similar molecular landscapes. Among the patients with MDS-IB1, MDS-IB2, or MDS-f, patients with MDS-IB2 exhibited higher frequencies of NRAS (9% vs. 2%, P = 0.019), TET2 (19% vs. 10%, P = 0.059), and monoallelic TP53 (7% vs. 1%, P = 0.031) mutations than did those with MDS-IB1. At the same time, patients with MDS-f exhibited lower incidences of TET2 (0% vs. 19%, P = 0.020) and STAG2 mutations (8% vs. 27%, P = 0.050) compared to those diagnosed with MDS-IB. Mutational profile analysis indicated that patients with biallelic TP53 inactivation had specific genetic alterations, with different co-occurring patterns from those observed in patients with MDS-IB or MDS-f (Supplementary Fig. 3, and Supplementary Table 4b). Excluding SF3B1 and TP53, the molecular features including the presence or absence of mutations and their VAF were similar across WHO-2022 categories (data not shown).
Survival analysis and impact of HSCT
We performed pairwise comparisons of survivals among patients with each subtype of MDS, defined by the ICC and WHO-2022 classification, with the exclusion of the MDS-del(5q) subgroup, which comprised a very limited number of patients (n = 4). In the ICC, patients diagnosed with MDS, NOS with SLD or MLD had comparable prognoses (162.1 vs. 185.5 months, P = 0.274 for LFS; P = 0.269 for OS). Among the subtypes of MDS/AML, patients with MDS/AML-NOS had the best outcomes (MDS/AML-NOS vs. other MDS/AML, 14.5 vs. 8.4 months for LFS, P = 0.047; 28.1 vs. 14.2 months for OS, P = 0.008) (Supplementary Fig. 4). Furthermore, individuals carrying TP53 mutations experienced the worst outcomes, regardless of their blast percentage (MDS with mutated TP53 vs. MDS/AML with mutated TP53, 4.1 vs. 4.0 months for LFS, P = 0.396; 5.2 vs. 5.1 months for OS, P = 0.590) (Supplementary Fig. 4).
In the WHO-2022 classification, patients with MDS-h had a significantly longer median LFS and OS (185.5 months for both median LFS and OS) compared to those with MDS-LB and RS (38.3 months for median LFS [P = 0.013] and OS [P = 0.010]) but had a similar outcome to those with MDS-LB (170.2 months for median LFS [P = 0.152] and OS [P = 0.146]), and MDS-SF3B1 (114.6 months for median LFS [P = 0.549] and OS [P = 0.528]) (Fig. 2). Notably, patients with biTP53 mutations had worse outcomes (median LFS: 3.9 months and median OS: 4.5 months, all P < 0.001) than did those with MDS-f (median LFS:16.6 months, and median OS:17.7 months), MDS-IB2 (median LFS:10.0 months, median OS:17.7 months), and MDS-IB1 (median LFS:25.6 months and median OS:31.4 months). Patients with MDS-f had similar survival to those with MDS-IB2, and patients with MDS-IB2 had a shorter LFS (P = 0.003) and OS (P = 0.027) than those with MDS-IB1 (Fig. 2). Additionally, we compared patients with different blast ranges in those with MDS-f. It showed that patients with PB blast 5–19%, or BM blast 10–19% (15.5 months for LFS, 16.3 months for OS) had a trend of shorter LFS (P = 0.087) and OS (P = 0.081) compared to those with less blasts (21.9 months for LFS, 73.3 months for OS; Supplementary Fig. 5).

a Leukemia-free survival for patients with myelodysplastic neoplasms. b Overall survival for patients with myelodysplastic neoplasms.
Subgroup analysis of the impact of transplantation using time-dependent Cox regression analysis after adjusting for age revealed that in the ICC, HSCT could improve LFS (HR for LFS: 0.419, P = 0.019) in patients diagnosed with MDS with EB (Supplementary Table 5). Additionally, in the WHO-2022 classification, patients with MDS-IB1 may benefit from HSCT for LFS (HR for LFS: 0.450, P = 0.074); however, transplantation failed to improve the poor outcomes in patients with MDS-biTP53 (Supplementary Table 6).
Subtypes analysis based on the interaction between the ICC and WHO-2022 classification
Based on the ICC, we further classified patients by the diagnostic criteria of the WHO-2022 classification and explored the survival and genetic differences between each subtype. In the group of patients with MDS, NOS with SLD (n = 106) or MLD (n = 147), patients could be classified into the subtypes of MDS-SF3B1 (n = 3), MDS-LB and RS (n = 16), MDS-LB (n = 140), and MDS-h (n = 94), according to the WHO-2022 classification (Table 1). MDS-LB and RS were consolidated into the MDS-LB subgroup due to their comparable mutation profiles. Individuals diagnosed with MDS-h had a trend of better outcomes compared to those with MDS-LB (185.5 vs. 162.1 months, P = 0.075 for LFS; 185.5 vs. 101.1 months, P = 0.070 for OS) (Supplementary Fig. 6).
Patients diagnosed with MDS with EB (blast <10% in PB and BM) could be classified into the subtypes of MDS-IB1 (n = 99), MDS-IB2 (n = 19), and MDS-f (n = 13). Importantly, a blast percentage of 5–10% in PB implied shorter LFS (MDS-IB2 vs. MDS-IB1, 7.0 vs. 26.2 months, P = 0.004; MDS-IB2 vs. MDS-f, 7.0 vs. 21.9 months, P = 0.042). Patients with MDS-f did not show inferior outcome than those with MDS-IB1 (Supplementary Fig. 7). Moreover, patients with 5–10% blast in PB showed similarly poor outcomes as those classified as MDS/AML (blast percentage ≥10% in PB or BM; LFS 10.0 vs. 8.7 months, P = 0.586; OS 16.8 vs. 16.0 months, P = 0.824) (Fig. 3). Within the ICC framework, bone marrow fibrosis did not lead to further differentiation in survival outcomes across the genetic subgroups of MDS with mutated TP53, and MDS/AML with MDS-related gene mutations (Supplementary Figs. 8 and 9).

a Leukemia-free survival for patients with myelodysplastic neoplasms. b Overall survival for patients with myelodysplastic neoplasms.
Alternatively, under the WHO-2022 classification, patients with MDS-LB could be reclassified into subtypes based on the ICC, including MDS, NOS with SLD (n = 58) or MDS, NOS with MLD (n = 82) (Supplementary Table 7). Patients with MLD, despite similar mutation profiles, had significantly lower white blood cell (2.9 vs. 3.7 × 109 /L, P = 0.001), absolute neutrophil (1.5 vs. 2.2 × 109 /L), and platelet counts (54 vs. 101 × 109 /L, P = 0.006), with a trend towards shorter survivals than did SLD counterparts (77.3 vs. 170.2 months, P = 0.153 for LFS, P = 0.140 for OS). Furthermore, patients with MDS-h showed significantly longer survivals (median LFS and OS: 185.5 months; P = 0.037 for LFS, P = 0.034 for OS) compared to MLD, and similar outcomes (P = 0.828 for LFS; P = 0.824 for OS) to SLD (Supplemental Figure 10). Patients with MLD had a trend towards shorter survivals than did SLD counterparts.
For patients with biallelic TP53 inactivation in the WHO-2022 criteria, MDS (n = 17) or MDS/AML (n = 29) with mutated TP53 in the ICC (Supplementary Table 7) exhibited a similar prognosis (3.9 vs. 4.0 months for LFS, P = 0.211; 4.4 vs. 7.0 months for OS, P = 0.293) (Supplementary Fig. 11). Additionally, patients with MDS-IB2 could be classified as having MDS with EB (n = 19, peripheral blast 5–9%), MDS with mutated TP53 (n = 1, single TP53 mutation with VAF 41.2% and complex karyotype), MDS/AML with mutated TP53 (n = 8), MDS/AML with MDS-related gene mutations (n = 87), MDS/AML with MDS-related cytogenetic abnormalities (n = 12), MDS/AML-NOS (n = 19), and AML with CEBPA (n = 3) (Supplementary Table 7). Patients with MDS/AML and MDS-related gene mutations had a higher risk under IPSS-R or IPSS-M than did those with MDS with EB (92.0% vs. 63.2% for high or very-high risk IPSS-R, P < 0.001; 95.4% vs. 73.6% for high or very-high risk IPSS-M, P = 0.002) and MDS/AML-NOS (95.4% vs. 73.7% for high or very-high risk IPSS-M, P = 0002).
Survival analysis among these patients showed that those with MDS/AML with mutated TP53 (4.0 months for LFS, 4.1 months for OS) and with MDS-related cytogenetic abnormalities (10.6 months for LFS and OS) had the worst outcomes (MDS/AML with mutated TP53 vs. MDS/AML with MDS-related cytogenetic abnormalities, P = 0.399 for LFS, P = 0.131 for OS), followed by those with MDS-related gene mutations (vs. MDS/AML with mutated TP53; 10.7 months for LFS, P = 0.004; 20.7 months for OS, P < 0.001). Moreover, those with MDS/AML-NOS had the longest survivals (vs. MDS/AML with mutated TP53; 8.1 months for LFS, P = 0.052; 28.1 months for OS, P < 0.001) (Supplementary Fig. 12). In patients with MDS-f, survival differences were also noted among those with MDS with EB (n = 13, LFS 21.9 months, OS 22.2 months), MDS with mutated TP53 (n = 4, LFS 5.2 months, OS 5.9 months), and MDS/AML with MDS-related gene mutations (n = 6, LFS 12.7 months, OS 13.9 months) when classified by the ICC (pairwise comparison P value all <0.05 for OS, Supplementary Fig. 13).
Proposed integrated system based on the ICC and WHO-2022 classification
From our analysis, we propose: (1) MDS-h and MDS, NOS with SLD patients had better outcomes; (2) Patients with ≥5% blasts in PB show outcomes akin to MDS/AML, worse than do those with lower blasts; (3) When patients have a PB blast percentage ≥10%, not only multi-hit but also single-hit TP53 mutations confer detrimental effects. Therefore, it is crucial to differentiate these patients from those with MDS with mutated TP53; (4) In MDS-IB2 patients, further distinction is needed for those with MDS-related gene mutations and cytogenetic abnormalities due to differing prognoses; (5) Within the ICC framework, BM fibrosis’ survival impact was less pronounced, prompting the refinement of the two-class system. MDS-h and MDS, NOS with SLD patients were segregated from other low blasts MDS and individuals with 5–10% PB blasts were shifted from MDS with EB to MDS/AML. Univariable analysis linked bedside IPSS-M, IPSS-R, older age, and male sex with poorer outcomes (Supplementary Table 8). Patients were categorized into five subgroups using the refined system. Multivariable analysis showed IPSS-M (P < 0.001), older age (P < 0.001), and the refined system (P < 0.001) as independent predictors of LFS and OS, with HSCT improving LFS (HR, 0.492, P = 0.001) (Table 3).
Discussion
In a cohort of 635 MDS patients, we retrospectively assessed the clinicopathological significance and prognostic implications of the ICC and WHO-2022 classification in the context of IPSS-M. Differences in clinical characteristics, genetic features, and outcomes among MDS subtypes based on these two novel systems were observed. Apart from the substitution of MDS-SF3B1 for MDS-RS, additional innovative changes in the WHO-2022 criteria have included the introduction of MDS-biTP53, MDS-f, and MDS-h. Moreover, the ICC highlights molecular features in diagnosis and classification, introducing categories like MDS with mutated SF3B1 without excess blasts as MDS-SF3B1 regardless of RS percentage and MDS or MDS/AML with mutated TP53. It also introduces the concept of MDS/AML with MDS-related gene mutations. These systems share many features yet differ in terminology and defining new entities [9, 10, 24]. The threshold for myelodysplastic features is set at 10% across all hematopoietic cell lineages in both systems. While SLD and MLD distinction remains in MDS, NOS subclassification in the ICC is optional in WHO-2022 criteria. Considering the heterogeneous results from previous studies [25, 26], we believe that further analysis is warranted to assess the survival impact of the lineage of dysplasia. Our study also revealed distinct features in patients with MDS-SF3B1 compared to those with MDS-LB and RS, and MDS-LB, the latter two subtypes showing similar mutational landscapes.
Additionally, blast percentage criteria variances led to diagnostic discrepancies between MDS and AML, such as AML with CEBPA mutations (≥10% blasts in ICC; ≥20% in WHO-2022 criteria) or AML with mutated NPM1 (≥10% blasts in ICC; increased blasts percentage in WHO-2022 criteria). The term MDS with IB2 is retained in the WHO-2022 criteria, whereas it is modified to MDS/AML in the ICC to emphasize the continuum spectrum between MDS and AML. In the group of MDS/AML, patients could be further classified according to genetic profiles (TP53 mutation or MDS-related gene mutations) and cytogenetic abnormalities. Exploring these subtypes, we found PB blast percentage ≥ 5% linked to adverse outcomes, classifying it under the MDS/AML subtype. Identifying MDS/AML with MDS-related gene mutations or cytogenetic abnormalities aids in distinguishing patients’ survival rates.
MDS-h, a new entity in the WHO-2022 classification but not in ICC, comprises 10–27% of all MDS cases [27,28,29]. It is frequently observed in Asian MDS cohort or children cases [30, 31]. The different reported prevalence may result from (1) Variation in the criteria used to define hypocellular bone marrow across different studies; (2) Differences in patient enrollment criteria, including whether the study included de novo cases only or both de novo and secondary cases, as well as differences between French-American-British- and WHO-defined cohorts; (3) Variances in genetic and environmental backgrounds among the study populations; (4) The possibility of inadvertently including patients with aplastic anemia in the MDS-h cohort. In our study, 14.8% of patients were qualified for the diagnosis of MDS-h based on WHO-2022 definition (hypocellular marrow: <30% of normal cellularity in patients younger than 70 years and <20% in patients aged 70 and older). MDS-h patients show activated immune system features, especially effector T cells targeting hematopoietic stem and progenitor cells [27, 32]. They typically have severe cytopenia, fewer somatic mutations [28], higher immunosuppressive therapy response [33], and better outcomes with regards to the low-risk IPSS-R [34,35,36]. In our study, MDS, NOS patients with MDS-h showed a trend of improved survival compared to other low-risk MDS cases. Recognizing these patients aids in enhancing patient care through tailored treatment strategies.
BM fibrosis is correlated with higher white blood cell counts, BM blast percentages, and more pronounced dysmegakaryocytopoiesis in MDS. It is also associated with mutations in the TP53, SETBP1, and JAK2 genes [37, 38]. Additionally, several studies have recognized fibrosis as an independent factor for poor prognosis [37, 39,40,41,42]. We found that in the patients with increased blasts, BM fibrosis adversely affected outcomes in the WHO-2022 classification, and patients with MDS-f had fewer STAG2 and TET2 mutations. However, the negative impact of BM fibrosis appeared to be less prominent within the framework of the ICC.
Up to 10% of patients with primary MDS have mutations in TP53, which results in a heightened risk of AML transformation and dismal outcomes [43], particularly in the setting of multiple hits [23]. Only patients with multiple hits displayed specific associations with complex karyotypes, a few co-occurring mutations, high-risk presentations, and short survival [23]. Furthermore, monoallelic patients did not differ from TP53 wild-type patients in terms of outcome or response to therapy [23]. In this study, MDS-biTP53 patients had the shortest survival, with a distinct mutational landscape compared to MDS-IB or MDS-f patients (Supplementary Fig. 3 and Supplementary Table 4b). MDS or MDS/AML patients with TP53 mutations defined by the ICC had unfavorable prognoses. Patients with a single TP53 mutation and complex karyotype showed outcomes similar to those with multiple TP53 mutations, justifying classifying these cases into this entity. Cox regression analysis revealed HSCT did not improve outcomes in these patients, as per either ICC or 2022-WHO classification (Supplementary Table 5, 6), consistent with prior reports [44,45,46].
Through an expanded analysis of 7,017 patients on behalf of the International Consortium for MDS [47], Komrokji et al. documented that genetically defined entities (SF3B1, del5q, and biTP53) were unique, and the survival of patients with LB and RS (wild-type SF3B1) was similar to that of patients with MDS-LB. Our results are consistent with these findings. Although MDS-IB1 patients had longer OS, their LFS was comparable to MDS-IB2 in the Moffitt Cancer Center cohort. Survival analysis of our and the GenoMed4all cohort showed significant LFS and OS differences between subtypes [47]. Thus, the optimal cutoff value for the blast percentage requires further investigation. A unified classification system that included MDS-5q, MDS-SF3B1, MDS-h, MDS-SLD, MDS-MLD, MDS-LB, MDS-biTP53, MDS-f, and MDS-EB was proposed by the Moffitt Cancer Center [48]. According to our analysis, we tried to refine and tie two classification systems together. We posited that MDS-h subtype, distinguishable from other low blasts MDS, should be separately recognized. Patients with PB blast ≥5% should be identified due to poorer outcomes.
The study’s limitations include its retrospective nature, limited case number, exclusion of five of the residual genes defined by the IPSS-M model, unavailability of TP53 copy-neutral loss of heterozygosity, and heterogeneity in treatment regimens, though most high-risk MDS patients received hypomethylating agents or transplantation. The analysis for the impact of HSCT might be compromised due to patients’ comorbidities, pre-transplant treatments, and transplant modalities, et al. Prospective studies are needed to validate the refined MDS classification and assess HSCT’s impact on high-risk MDS patient outcomes.
In conclusion, the ICC and WHO-2022 classification effectively segregate this heterogeneous disease. However, coexisting diagnostic standards challenge clinicians in treatment and diagnosis and hinder clinical trials and research progress. Therefore, we propose an integrated classification system for accurate MDS diagnosis and effective risk-adapted treatment.
Data availability
The datasets generated during and/or analyzed during this study are available from the corresponding author upon reasonable request.
References
Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361:1872–85.
Sekeres MA, Taylor J. Diagnosis and treatment of myelodysplastic syndromes: a review. Jama. 2022;328:872–80.
World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopietic and Lymphoid Tissues (ed 3rd). Lyon: IARC Press; (2001).
WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. In: Swerdlow SHCE, Harris NL, et al., editor: Lyon: IARC; (2008).
Swerdlow SHCE, Harris NL, et al. eds. WHO Classification fo Tumours of Haematopoietic and Lymphoid Tissues.: Lyon: IARC; (2017).
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Spaulding TP, Stockton SS, Savona MR. The evolving role of next generation sequencing in myelodysplastic syndromes. Br J Haematol. 2020;188:224–39.
Tria FPT, Ang DC, Fan G. Myelodysplastic syndrome: diagnosis and screening. Diagnostics (Basel). 2022;12:1581.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet; beta version ahead of print]. Lyon (France): International Agency for Research on Cancer; 2022 [cited 2024 Jan 23th]. (WHO classification of tumours series, 5th ed.; vol. 11). Available from: https://tumourclassification.iarc.who.int/chapters/63.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Ossa JEA, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evidence. 2022;1:EVIDoa2200008.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Singhal D, Wee LYA, Kutyna MM, Chhetri R, Geoghegan J, Schreiber AW, et al. The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia. 2019;33:2842–53.
Kuzmanovic T, Patel BJ, Sanikommu SR, Nagata Y, Awada H, Kerr CM, et al. Genomics of therapy-related myeloid neoplasms. Haematologica. 2020;105:e98–e101.
International Standing Committee on Human Cytogenomic Nomenclature M-JJHRJMSSKG. ISCN 2020 an International System for Human Cytogenomic Nomenclature (2020): recommendations of the International Standing Committee on Human Cytogenomic Nomenclature including revised sequence-based cytogenomic nomenclature developed in collaboration with the Human Genome Variation Society (HGVS) Sequence Variant Description Working Group (2020).
Lee WH, Tsai MT, Tsai CH, Tien FM, Lo MY, Tseng MH, et al. Validation of the molecular international prognostic scoring system in patients with myelodysplastic syndromes defined by international consensus classification. Blood Cancer J. 2023;13:120.
Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41.
Lee WH, Lin CC, Tsai CH, Tseng MH, Kuo YY, Liu MC, et al. Effect of mutation allele frequency on the risk stratification of myelodysplastic syndrome patients. Am J Hematol. 2022;97:1589–98.
Tsai CH, Tang JL, Tien FM, Kuo YY, Wu DC, Lin CC, et al. Clinical implications of sequential MRD monitoring by NGS at 2 time points after chemotherapy in patients with AML. Blood Adv. 2021;5:2456–66.
Shiah HS, Kuo YY, Tang JL, Huang SY, Yao M, Tsay W, et al. Clinical and biological implications of partial tandem duplication of the MLL gene in acute myeloid leukemia without chromosomal abnormalities at 11q23. Leukemia. 2002;16:196–202.
Devillier R, Forcade E, Garnier A, Guenounou S, Thepot S, Guillerm G, et al. In-depth time-dependent analysis of the benefit of allo-HSCT for elderly patients with CR1 AML: a FILO study. Blood Adv. 2022;6:1804–12.
Haase D, Stevenson KE, Neuberg D, Maciejewski JP, Nazha A, Sekeres MA, et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia. 2019;33:1747–58.
Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26:1549–56.
Falini B, Martelli MP. Comparison of the international consensus and 5th WHO edition classifications of adult myelodysplastic syndromes and acute myeloid leukemia. Am J Hematol. 2023;98:481–92.
Zhang Y, Wu J, Xu Z, Qin T, Qu S, Pan L, et al. Impact of the international consensus classification of myelodysplastic syndromes. Br J Haematol. 2023;201:443–8.
Lee WH, Lin CC, Tsai CH, Tien FM, Lo MY, Ni SC, et al. Clinico-genetic and prognostic analyses of 716 patients with primary myelodysplastic syndrome and myelodysplastic syndrome/acute myeloid leukemia based on the 2022 international consensus classification. Am J Hematol. 2023;98:398–407.
Fattizzo B, Serpenti F, Barcellini W, Caprioli C. Hypoplastic myelodysplastic syndromes: just an overlap syndrome? Cancers (Basel). 2021;13:132.
Nazha A, Seastone D, Radivoyevitch T, Przychodzen B, Carraway HE, Patel BJ, et al. Genomic patterns associated with hypoplastic compared to hyperplastic myelodysplastic syndromes. Haematologica. 2015;100:e434–7.
Yue G, Hao S, Fadare O, Baker S, Pozdnyakova O, Galili N, et al. Hypocellularity in myelodysplastic syndrome is an independent factor which predicts a favorable outcome. Leuk Res. 2008;32:553–8.
Hasegawa D, Chen X, Hirabayashi S, Ishida Y, Watanabe S, Zaike Y, et al. Clinical characteristics and treatment outcome in 65 cases with refractory cytopenia of childhood defined according to the WHO 2008 classification. Br J Haematol. 2014;166:758–66.
Yao CY, Hou HA, Lin TY, Lin CC, Chou WC, Tseng MH, et al. Distinct mutation profile and prognostic relevance in patients with hypoplastic myelodysplastic syndromes (h-MDS). Oncotarget. 2016;7:63177–88.
Lopes MR, Traina F, Campos Pde M, Pereira JK, Machado-Neto JA, Machado Hda C, et al. IL10 inversely correlates with the percentage of CD8+ cells in MDS patients. Leuk Res. 2013;37:541–6.
Passweg JR, Giagounidis AA, Simcock M, Aul C, Dobbelstein C, Stadler M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care-SAKK 33/99. J Clin Oncol. 2011;29:303–9.
Kobayashi T, Nannya Y, Ichikawa M, Oritani K, Kanakura Y, Tomita A, et al. A nationwide survey of hypoplastic myelodysplastic syndrome (a multicenter retrospective study). Am J Hematol. 2017;92:1324–32.
Bono E, McLornan D, Travaglino E, Gandhi S, Gallì A, Khan AA, et al. Clinical, histopathological and molecular characterization of hypoplastic myelodysplastic syndrome. Leukemia. 2019;33:2495–505.
Calabretto G, Attardi E, Teramo A, Trimarco V, Carraro S, Mossuto S, et al. Hypocellular myelodysplastic syndromes (h-MDS): from clinical description to immunological characterization in the Italian multi-center experience. Leukemia. 2022;36:1947–50.
Melody M, Al Ali N, Zhang L, Ramadan H, Padron E, Sallman D, et al. Decoding bone marrow fibrosis in myelodysplastic syndromes. Clin Lymphoma Myeloma Leuk. 2020;20:324–8.
Fu B, Jaso JM, Sargent RL, Goswami M, Verstovsek S, Medeiros LJ, et al. Bone marrow fibrosis in patients with primary myelodysplastic syndromes has prognostic value using current therapies and new risk stratification systems. Mod Pathol. 2014;27:681–9.
Marisavljević D, Rolović Z, Cemerikić V, Bosković D, Colović M. Myelofibrosis in primary myelodysplastic syndromes: clinical and biological significance. Med Oncol. 2004;21:325–31.
Wang N, Xu H, Li Q, Fang X, Liu J, Sui X, et al. Patients of myelodysplastic syndrome with mild/moderate myelofibrosis and a monosomal karyotype are independently associated with an adverse prognosis: long-term follow-up data. Cancer Manag Res. 2020;12:5881–91.
Ramos F, Robledo C, Izquierdo-García FM, Suárez-Vilela D, Benito R, Fuertes M, et al. Bone marrow fibrosis in myelodysplastic syndromes: a prospective evaluation including mutational analysis. Oncotarget. 2016;7:30492–503.
Matteo Giovanni Della P, Luca M. Myelodysplastic syndromes with bone marrow fibrosis. Haematologica. 2011;96:180–3.
Montalban-Bravo G, Kanagal-Shamanna R, Benton CB, Class CA, Chien KS, Sasaki K, et al. Genomic context and TP53 allele frequency define clinical outcomes in TP53-mutated myelodysplastic syndromes. Blood Adv. 2020;4:482–95.
Britt A, Mohyuddin GR, McClune B, Singh A, Lin T, Ganguly S, et al. Acute myeloid leukemia or myelodysplastic syndrome with chromosome 17 abnormalities and long-term outcomes with or without hematopoietic stem cell transplantation. Leuk Res. 2020;95:106402.
Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376:536–47.
Bally C, Adès L, Renneville A, Sebert M, Eclache V, Preudhomme C, et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res. 2014;38:751–5.
Komrokji RS. Myelodysplastic neoplasms (MDS) classification from WHO 2017 to WHO 2022 and ICC 2022: an expanded analysis of 7017 patients on behalf of the International Consortium for MDS (ICMDS). 2023 European Hematogloy Association Hybrid Congress; 10 June; Frankfurt, Germany (2023).
Ball S, Singh AM, Ali NA, Aguirre LE, Jain AG, Schwabkey ZI, et al. A product of “Clash of Titans” or true reflection of disease biology? Validation of 2022 WHO and ICC classifications in a large dataset of patients with myelodysplastic syndrome. Blood. 2022;140:1118–20.
Acknowledgements
We would like to acknowledge the service provided by the Departments of Laboratory Medicine and Medical Research, National Taiwan University Hospital, and Tai-Chen Cell Therapy Center. Moreover, we would like to acknowledge the service provided by the DNA Sequencing Core of the First Core Laboratory, National Taiwan University College of Medicine. This work was partially supported with grants from the Hematology Society of Taiwan, Ministry of Science and Technology (Taiwan) (MOST 104–2314-B-002–128-MY4, 106-2314-B-002-226-MY3 and 108-2628-B-002-015, 109-2314-B-002-213 and 111-2314-B-002-279) and the Ministry of Health and Welfare (Taiwan) (MOHW 107-TDU-B-211-114009 and 111-TDU-B-221-114001).
Author information
Authors and Affiliations
Contributions
WHL was responsible for data collection and management, statistical analysis and interpretation, literature research, and manuscript writing. CHT, YYK, HIS, MHT, and YKC were responsible for next-generation sequencing data collection and interpretation. MCL was responsible for cytogenetic analyses. CCL, FMT, MYL, WCC, MKC, YTY, JLT, and HFT were responsible for data collection and management. SCY and CTY were responsible for grading of bone marrow fibrosis. BSK coordinated the study, analyzed and interpret data, wrote, and revised the manuscript. HAH designed, conceived, and coordinated the study, analyzed and interpret data, wrote, and revised the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, WH., Lin, CC., Tsai, CH. et al. Comparison of the 2022 world health organization classification and international consensus classification in myelodysplastic syndromes/neoplasms. Blood Cancer J. 14, 57 (2024). https://doi.org/10.1038/s41408-024-01031-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-024-01031-9
