
Abstract
Body temperature is an important determinant in regulating the activities of animals. In humans, a mild 0.5 °C hyperthermia can cause headaches, demonstrating that the maintenance of normal body temperature is a key for our health. In a more extreme example, accidental acute hypothermia can lead to severe shivering, loss of consciousness, or death, although the details of these mechanisms are poorly understood. We previously found that the TRPV4 ion channel is constitutively activated by normal body temperature. The activation threshold of TRPV4 is >34 °C in the brain, which enables TRPV4 to convert thermal information into cellular signaling. Here we review the data which describe how the deletion of TRPV4 evokes abnormal behavior in mice. These studies demonstrate that the maintenance of body temperature and the sensory system for detecting body temperature, such as via TRPV4, are critical components for normal cellular function. Moreover, abnormal TRPV4 activation exacerbates cell death, epilepsy, stroke, brain edema, or cardiac fibroblast activity. In this review, we also summarize the findings related to TRPV4 and disease.
Similar content being viewed by others

Introduction
The transient receptor potential ion channel was originally identified in mutant Drosophila photoreceptors [1]. In the Drosophila trp mutant, responses to light were acute and transient, and Ca2+ influx was significantly reduced. Hence, the ion channel responsible for these changes was termed “transient receptor potential”. Later, it was revealed that the TRP protein has six transmembrane domains and acts as an ion channel with high Ca2+ permeability. The TRP channels consist of six subfamilies including TRPC, TRPV, TRPM, TRPML, TRPP, and TRPA in mammals. Among these, eleven TRP channels are classified as thermosensitive TRP channels, which means they can be activated by various temperature ranges. TRPV4 is activated by nonnoxious heat stimuli (27–34 °C), and is related to various disease states. In this review, I summarize the important physiological roles of TRPV4 as a sensor of body temperature and/or mechanical stimuli, and discuss the relationships between TRPV4 activity and disease progression.
The TRPV4 ion channel
TRPV4 was identified as an osmotically-activated channel [2, 3]. Since the original discovery of TRPV4 as a stretch-activated channel [4], TRPV4 has also been shown to be activated by hypotonic stimuli [5, 6]. Cellular swelling activates phospholipase A2 (PLA2) [6, 7], and PLA2 produces arachidonic acid (AA) by hydrolysis of membrane lipids. AA is metabolized to epoxyeicosatrienoic acid (EET), which directly activates the TRPV4 channel by cytochrome P450 [7, 8]. TRPV4 is also a thermosensitive channel activated by warm stimuli [9, 10]. Both our lab and another group reported that TRPV4 is activated by physiological temperatures (>34 °C) in naïve cells [11, 12]. This heat activation of TRPV4 is required for endogenous signaling as suggested by the failure of TRPV4 heat activation in inside-out membrane patches [9].
Relationship between disease and TRPV4 as a thermosensitive ion channel
Clinical evidence suggests that brain temperature is a critical determinant of cognitive function. It has also been reported that therapeutic hypothermia leads to cognitive dysfunction in heart disease patients [13], and cooling of the prefrontal cortex in monkeys causes performance impairment during a delayed matching-to-sample task [14]. In rodents, brain cooling significantly impairs spatial learning [15]. These reports indicate that the maintenance of brain temperature is important for healthy brain function. However, the underlying molecular mechanisms for this regulation remain unknown.
We previously reported that physiological temperature activates TRPV4 and enhances neuronal excitability through TRPV4 activation in dissociated hippocampal neurons (Fig. 1) [12]. Furthermore, we revealed that brain temperature constitutively activates TRPV4 and enhances hippocampal activity in vivo [16] (Fig. 2). This was demonstrated in TRPV4-deficienct mice which displayed a significant decrease in social and depressive-like behaviors (Fig. 2) [16]. These results indicate that constitutive activation of TRPV4 by physiological brain temperature is necessary for normal brain function (Fig. 2). Indeed, it was reported that in a study involving whole-genome sequencing of 85 quartet families with autism, frame-shift mutations in TRPV4 were found in the patients with autism [17]. Furthermore, genome-wide association studies in control subjects and those with major depressive disorder revealed that TRPV4 mutations are a risk factor for depression [18]. These studies demonstrate that disruption of TRPV4 function might lead to psychiatric symptoms (Fig. 2).

The pictures show heat-evoked responses in a dissociated hippocampal pyramidal neuron. The hippocampal neuron (Top-left picture was the bright field image) was prepared from wild-type neonatal mice at P0, and cultured for 3 weeks. Fura2 fluorescence was measured upon application of the heat-ramp (from room temperature to 45 °C). The 340:380 nm ratio pseudo color expression was shown. An increase in cytosolic Ca2+ ([Ca2+]i) was observed at ~33 °C. Increased [Ca2+]i was not observed in the absence of extracellular Ca2+ or in TRPV4-deficient neurons, suggesting that functional TRPV4 was expressed in the hippocampal pyramidal neurons. TRPV4 is a thermosensitive ion channel activated at temperatures approaching the physiological core body temperature (>34 °C), therefore TRPV4 was thought to be an important molecule to regulate neural excitability in the hippocampus at body temperature [12]. Scale bar; 50 μm.

TRPV4 can be activated by normal brain temperature which leads to the enhancement of electrical activity. Thermal signal is converted to electrical activity. This results in synchronization of neuronal activity in the brain, and contributes to normal behavior. TRPV4KO mice lack this mechanism, lose synchronization, and display altered behaviors such as reduced depression-like behaviors and social behaviors (similar to psychiatric diseases).
Very recently, we revealed that brain ischemia evokes hyperthermia (an increase of ~2 °C) in ischemic regions [19]. This hyperthermia caused abnormal hyperactivation of TRPV4, and exacerbated brain edema [19]. Hence, a TRPV4 inhibitor may effectively suppress ischemia-induced brain edema. Hyperthermia also negatively affects neural development during fetal stages, and TRPV4 is involved in maternal “fever-associated” neural birth defects [20]. TRPV4 is also expressed in endothelial cells, and controls cerebral blood flow via activation of the Gq protein-coupled receptor [21]. Moreover, brain temperature and hyperthermia might affect blood flow through the activation of TRPV4. In addition, TRPV4 plays an essential role in thermal hyperalgesia in sensory nerves [22].
In sensory neurons, it has also been reported that TRPV4 is involved in chemotherapy-induced neuropathic pain [23]. Hence, the abnormal activation of TRPV4 under pathological conditions is strongly related to pain sensation. Specifically, many TRPV4 mutations cause peripheral neuropathy. Mutations in TRPV4 are associated with many severe diseases such as scapuloperoneal spinal muscular atrophy, Charcot–Marie–Tooth disease type 2C and skeletal dysplasia [24, 25]. TRPV4 gain-of-function mutations are responsible for Charcot–Marie–Tooth disease type 2C [24], in which mutated TRPV4 becomes sensitized at body temperature. Skin keratinocytes strongly express TRPV4 which is activated by body temperature causing Ca2+ influx. This leads to the enhancement of barrier function through increased E-cadherin expression and reorganization of the actin cytoskeleton [26]. During the winter season, skin dehydration is a common occurrence, and cold winter temperatures are thought to inhibit constitutive TRPV4 activation (by skin temperature) [27]. Recently, it has also been reported that TRPV4 expressed in skin keratinocytes causes Ca2+ influx in response to histaminergic pruritogens, leading to itch [28]. Hence, TRPV4 is a pruriceptor acting on skin keratinocytes. TRPV4 is also expressed in esophageal keratinocytes, and is associated with the exacerbation of Helicobacter pylori infection [29].
Physiological roles of TRPV4 in cerebrospinal fluid production
The highest level of TRPV4 expression is observed in the choroid plexus in the brain [3, 30]. It has been reported that TRPV4 in choroid plexus epithelial cells (CPECs) plays a pivotal role in the calcium homeostasis of cerebrospinal fluid (CSF) through α-klotho type 1. The calcium concentration of CSF is reduced in α-klotho type 1 KO mice, and α-klotho type 1 release from CPECs is enhanced by activation of TRPV4 [31], although the physiological significance of TRPV4 in CPECs remains unclear. The choroid plexus is involved in maintaining the brain environment in the lateral, third, and forth ventricles, and forms the blood–CSF barrier. The most important function of CPECs is CSF production. The generation of CSF is dependent on electrolyte transport from the basolateral to apical membranes of the CPECs and involves many ion transport proteins [30]. We found that TRPV4 in CPECs is activated by body temperature, and CPEC movement further activates TRPV4 through the production of EETs. TRPV4 activation leads to the activation of anoctamin 1 (ANO1), a Ca2+-activated chloride channel, and causes Cl− efflux [30]. This Cl− efflux can be a specific driving force for water efflux from CPECs. Hence, the choroid plexus can effectively generate a large amount of CSF without ineffectual energy loss. Another study revealed a different function of TRPV4 in the choroid plexus; they found that the activation of TRPV4 induced a marked decrease in filamentous actin, disintegrated the cell junctions, and affected basolateral-to-apical transport in the choroid plexus [32]. Taken together, these studies suggest that TRPV4 generates the specific driving force for CSF production.
Disease and TRPV4 as a mechanosensitive ion channel
TRPV4 can functionally act as an osmoreceptor [33]. Many cells such as vascular aortic endothelial cells, vascular smooth muscle cells, renal collecting duct cells, and lung epithelial cells, all of which endogenously express TRPV4, display hypotonicity-induced Ca2+ influx [33, 34]. The mechanism by which hypotonicity induces cellular swelling to activate TRPV4 seems to involve the production of lipid metabolites as described above [6, 7]. Furthermore, studies with TRPV4KO mice revealed that these animals have a reduced ability to regulate osmolality, and show impaired osmotically-induced release of antidiuretic hormone [35, 36].
Since TRPV4 is a mechanosensitive channel, TRPV4 can act as a sensor of shear stress or fluid flow. TRPV4 is highly expressed in aortic endothelial cells, microvascular endothelial cells, distal nephrons, and collecting duct epithelial cells, where it contributes to shear stress/fluid flow-dependent Ca2+ influx [33]. Furthermore, mechanical stimuli-induced TRPV4 activation significantly enhanced TGF-β1-evoked α-SMA fiber formation in cardiac fibroblasts [37]. These results suggest that mechanical stress and TGF-β1-mediated signaling synergistically promote myofibroblast differentiation through TRPV4 activation.
In the bladder, TRPV4 expression was observed in the urothelium and vascular endothelium, but not in other cell types [38]. Consistent with this expression pattern, TRPV4KO mice exhibited a lower frequency of voiding contractions. The amplitude of spontaneous contractions in explanted bladder strips derived from TRPV4KO mice was also significantly reduced. Moreover, intravesical stretch-evoked ATP release was significantly reduced in isolated whole bladders from TRPV4KO mice [38]. These studies revealed new roles for TRPV4 in voiding behavior, raising the possibility that TRPV4 plays a critical role in the urothelium-mediated transduction of intravesical mechanical pressure. This was later confirmed by our group. We established a cell stretch system to investigate stretch-evoked changes in intracellular Ca2+ concentration and ATP release. In TRPV4KO urothelial cells, the increase in intracellular Ca2+ in response to stretch stimulation was significantly attenuated compared to that in WT cells [39]. Potent ATP release occurred following stretch stimulation or administration of a TRPV4 agonist in WT urothelial cells, which was dramatically reduced in TRPV4KO cells [39]. Taken together, these results indicate that TRPV4 senses distension of the urinary bladder, which is converted into an ATP signal in the micturition reflex pathway during urine storage. Hence, urinary TRPV4 activation might be related to pollakiuria.
In the eye, it has recently been reported that trabecular meshwork cells express TRPV4 [40]. In glaucoma, the elevation of intraocular pressure is harmful to the eye, and antiglaucoma treatments aim to reduce intraocular pressure by decreasing the production of aqueous fluid in the anterior eye (inflow) or increasing its drainage through outflow pathways. The trabecular meshwork forms part of the outflow pathway toward Schelmm’s canal. In a glaucomatous eye model, intraocular pressure was significantly elevated, and TRPV4 inhibition restored intraocular pressure to normal levels [40]. These results suggest that TRPV4 regulates aqueous fluid outflow and intraocular pressure within the trabecular meshwork. However, the detailed mechanisms of how TRPV4 decreases intraocular pressure in glaucoma remain unknown, thus future studies are warranted.
Synergistic activation of TRPV4 by heat and mechanical stimuli exacerbates several diseases
As described above, TRPV4 can act as heat and/or stretch sensors. Notably, the weak artificial mechanical stimuli used in our protocol did not activate any mechanosensing activity in TRPV4-expressing HEK293 cells. In contrast, we observed significant mechanosensitive activity of TRPV4 in retinal Müller glia [41]. These results indicate that the Müller glia possess specific characteristics promoting the mechanosensitive properties of TRPV4. Based on our previous findings that TRPV4 is activated at temperatures above 34 °C [12], and that various ligands combined with increased temperature had synergistic–potentiation effects on TRVP4 activation [42], we hypothesized that body temperature significantly reduces the TRPV4 activation threshold against mechanical stimuli. Hence, we performed whole-cell patch clamp recordings by application of computer-programmed mechanical stimuli at different temperatures. We applied minimal artificial membrane stretch (10 mmHg) to Müller glial cells at 25 or 37 °C, and recorded the mechanical stimuli-evoked TRPV4 currents at each temperature. Compared with 25 °C conditions, physiological temperature (37 °C) significantly potentiated the mechanosensing activity of TRPV4 [41]. These results indicate that body temperature significantly elevates TRPV4 sensitivity to membrane stretch as a result of the synergistic effect of temperature and mechanical stimuli. In retinal detachment, significant Müller glial swelling (3 times the normal cell volume) has been observed among reactive glia [41, 43, 44]. Taken together, this suggests that retinal detachment-induced Müller glial swelling significantly activates TRPV4 channels. Furthermore, we revealed that this swelling-induced TRPV4 activation triggered Ca2+ influx and evoked MCP-1 release in Müller glia. Excessive amounts of MCP-1 recruit numerous macrophages near the retina, which target and kill photoreceptors [41] (Fig. 3). In addition to hypoxia, this is the critical mechanism by which photoreceptor death is exacerbated in retinal detachment (Fig. 3). Thus, TRPV4 inhibitors could suppress photoreceptor death in retinal detachment, and targeting TRPV4 in Müller glial might represent a novel therapeutic target for the prevention of photoreceptor cell death after retinal detachment.

Green cells represent the morphology of Müller glia in normal and retinal detachment (RD) retinas. In the normal retina, TRPV4 is activated by body temperature and contributes to the homeostatic function of Müller glial cells. In contrast, RD induces significant Müller glial swelling, causing further activation of TRPV4 by membrane stretch in addition to the activation induced by body temperature. TRPV4 activation induces Ca2+ influx, and evokes MCP-1 release from Müller glial cells. MCP-1 recruits numerous macrophages, which attack and kill photoreceptors (shown in gray). Thus, RD adversely impacts photoreceptor viability via TRPV4-dependent MCP-1 release from Müller glial cells. RPE retinal pigment epithelium, ONL outer nuclear layer, INL inner nuclear layer, and GCL ganglion cell layer. This figure is modified from my previous publication [41].
We have also reported that body temperature constitutively activates neural TRPV4 and regulates brain activity, as described above [12, 16, 45]. In another study, we revealed that the synergistic effects of temperature and mechanical stimuli on TRPV4 expressed in Müller glial cells are specifically required to regulate synaptic transmission in normal retina (paper in preparation). Based on these findings, our observations suggest that the contribution of Müller glial TRPV4 to synaptic transmission by retinal neurons through the effects of body temperature might apply to homeothermic animals in general [46]. Compared with cold-blooded animals, birds and mammals must perform advanced behaviors that require a finely tuned visual transmission system. Thus, homeostatic constitutive TRPV4 activation could contribute to this visual fine-tuning by taking advantage of a constant body temperature. However, temperature-dependent TRPV4 activation can negatively affect membrane stretch-dependent activation of TRPV4 in the presence of pathological cellular swelling.
Mechanical stimuli and Ca2+ entry are critically important for the maintenance of bone homeostasis. TRPV4 is expressed by both osteoblasts and osteoclasts [47] where it regulates bone formation and resorption [47, 48]. The mechanical stimulation of bones is necessary for regulating the balance between bone formation and resorption. TRPV4 has been clearly associated with bone formation in response to mechanical stimuli, and plays a pivotal role in osteoclast function. This is evident in TRPV4-deficient mice which possess increased trabecular bone mass/volume resulting in the impairment of bone resorption and a decreased number of osteoclasts [49].
Disease and body temperature
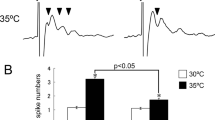
As described above, brain temperature is a critical determinant in the regulation of neuronal excitability [16]. Hence, acute hypothermia (such as in hypoglycemia, alcohol addiction, or cerebrovascular disorders, among others) causes severe shivering, lethargy, and reduction of respiration/heart function. The mechanisms by which hypothermia exerts its effects include: (i) reduction of cellular function and oxygen consumption; (ii) reduction of energy production and organ function; and (iii) peripheral circulatory disturbance. In acute hypothermia, both active external warming, as well as core rewarming are required to restore physiological functions. It is well known that therapeutic hypothermia is effective for several diseases such as ischemic stroke, traumatic brain injury, or cardiac arrest, due to the ability of therapeutic hypothermia to reduce oxygen consumption, cellular inflammation, and blood flow. In contrast to hypothermia, hyperthermia induces severe cell death over 43 °C. Based on this property, therapeutic hyperthermia is one strategy used in cancer treatment, although the mechanisms are poorly understood. Very recently, we reported that the temperature of epileptogenic foci was elevated by around 1 °C. This 1 °C hyperthermia caused the overactivation of TRPV4, triggering abnormal brain activity and exacerbating epileptogenic discharge (Shibasaki et al. in this special issue of Laboratory Investigation) [50].
Conclusion
The maintenance of body temperature is very important for our health. TRPV4 senses this body temperature and, more specifically, TRPV4 channels detect dynamic local temperature changes in the brain. In this special issue of Laboratory Investigation, we reveal for the first time that the range of local brain temperature can change by ~0.5 °C. (Shibasaki et al.) [50], and this small change exerts pivotal physiological effects depending on the tissue. These findings suggest that TRPV4 might represent a novel therapeutic target for preventing the progression of several diseases.
References
Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–23.
Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol. 2000;2:695–702.
Liedtke W, Choe Y, Marti-Renom MA, Bell AM, Denis CS, Sali A, et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell. 2000;103:525–35.
Lacampagne A, Gannier F, Argibay J, Garnier D, Le Guennec JY. The stretch-activated ion channel blocker gadolinium also blocks L-type calcium channels in isolated ventricular myocytes of the guinea-pig. Biochim Biophys Acta. 1994;1191:205–8.
Nilius B, Prenen J, Wissenbach U, Bodding M, Droogmans G. Differential activation of the volume-sensitive cation channel TRP12 (OTRPC4) and volume-regulated anion currents in HEK-293 cells. Pflugers Arch. 2001;443:227–33.
Goldenberg NM, Ravindran K, Kuebler WM. TRPV4: physiological role and therapeutic potential in respiratory diseases. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:421–36.
Ryskamp DA, Jo AO, Frye AM, Vazquez-Chona F, MacAulay N, Thoreson WB, et al. Swelling and eicosanoid metabolites differentially gate TRPV4 channels in retinal neurons and glia. J Neurosci. 2014;34:15689–700.
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–8.
Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem. 2002;277:47044–51.
Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J Neurosci. 2002;22:6408–14.
Chung MK, Lee H, Caterina MJ. Warm temperatures activate TRPV4 in mouse 308 keratinocytes. J Biol Chem. 2003;278:32037–46.
Shibasaki K, Suzuki M, Mizuno A, Tominaga M. Effects of body temperature on neural activity in the hippocampus: regulation of resting membrane potentials by transient receptor potential vanilloid 4. J Neurosci. 2007;27:1566–75.
Torgersen J, Strand K, Bjelland TW, Klepstad P, Kvale R, Soreide E, et al. Cognitive dysfunction and health-related quality of life after a cardiac arrest and therapeutic hypothermia. Acta Anaesthesiol Scand. 2010;54:721–8.
Fuster JM, Bauer RH. Visual short-term memory deficit from hypothermia of frontal cortex. Brain Res. 1974;81:393–400.
Moser EI, Andersen P. Conserved spatial learning in cooled rats in spite of slowing of dentate field potentials. J Neurosci. 1994;14:4458–66.
Shibasaki K, Sugio S, Takao K, Yamanaka A, Miyakawa T, Tominaga M, et al. TRPV4 activation at the physiological temperature is a critical determinant of neuronal excitability and behavior. Pflugers Arch. 2015;467:2495–507.
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21:185–91.
Wong ML, Arcos-Burgos M, Liu S, Velez JI, Yu C, Baune BT, et al. The PHF21B gene is associated with major depression and modulates the stress response. Mol Psychiatry. 2017;22:1015–25.
Hoshi Y, Okabe K, Shibasaki K, Funatsu T, Matsuki N, Ikegaya Y, et al. Ischemic brain injury leads to brain edema via hyperthermia-induced TRPV4 activation. J Neurosci. 2018;38:5700–9.
Hutson MR, Keyte AL, Hernandez-Morales M, Gibbs E, Kupchinsky ZA, Argyridis I, et al. Temperature-activated ion channels in neural crest cells confer maternal fever-associated birth defects. Sci Signal. 2017;10:eaal4055.
Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife. 2018;7:e38689.
Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M. Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem. 2004;279:35133–8.
Alessandri-Haber N, Dina OA, Yeh JJ, Parada CA, Reichling DB, Levine JD. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci. 2004;24:4444–52.
Nilius B, Owsianik G. Channelopathies converge on TRPV4. Nat Genet. 2010;42:98–100.
Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40:999–1003.
Sokabe T, Fukumi-Tominaga T, Yonemura S, Mizuno A, Tominaga M. The TRPV4 channel contributes to intercellular junction formation in keratinocytes. J Biol Chem. 2010;285:18749–58.
Kida N, Sokabe T, Kashio M, Haruna K, Mizuno Y, Suga Y, et al. Importance of transient receptor potential vanilloid 4 (TRPV4) in epidermal barrier function in human skin keratinocytes. Pflugers Arch. 2012;463:715–25.
Chen Y, Fang Q, Wang Z, Zhang JY, MacLeod AS, Hall RP, et al. Transient receptor potential vanilloid 4 ion channel functions as a pruriceptor in epidermal keratinocytes to evoke histaminergic itch. J Biol Chem. 2016;291:10252–62.
Mihara H, Suzuki N, Muhammad JS, Nanjo S, Ando T, Fujinami H, et al. Transient receptor potential vanilloid 4 (TRPV4) silencing in Helicobacter pylori-infected human gastric epithelium. Helicobacter. 2017;22:e12361.
Takayama Y, Shibasaki K, Suzuki Y, Yamanaka A, Tominaga M. Modulation of water efflux through functional interaction between TRPV4 and TMEM16A/anoctamin 1. FASEB J. 2014;28:2238–48.
Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–8.
Narita K, Sasamoto S, Koizumi S, Okazaki S, Nakamura H, Inoue T, et al. TRPV4 regulates the integrity of the blood-cerebrospinal fluid barrier and modulates transepithelial protein transport. FASEB J. 2015;29:2247–59.
O'Neil RG, Heller S. The mechanosensitive nature of TRPV channels. Pflugers Arch. 2005;451:193–203.
Sidhaye VK, Guler AD, Schweitzer KS, D'Alessio F, Caterina MJ, King LS. Transient receptor potential vanilloid 4 regulates aquaporin-5 abundance under hypotonic conditions. Proc Natl Acad Sci USA. 2006;103:4747–52.
Mizuno A, Matsumoto N, Imai M, Suzuki M. Impaired osmotic sensation in mice lacking TRPV4. Am J Physiol Cell Physiol. 2003;285:C96–101.
Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4-/- mice. Proc Natl Acad Sci USA. 2003;100:13698–703.
Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, et al. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52.
Gevaert T, Vriens J, Segal A, Everaerts W, Roskams T, Talavera K, et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J Clin Investig. 2007;117:3453–62.
Mochizuki T, Sokabe T, Araki I, Fujishita K, Shibasaki K, Uchida K, et al. The TRPV4 cation channel mediates stretch-evoked Ca2+influx and ATP release in primary urothelial cell cultures. J Biol Chem. 2009;284:21257–64.
Ryskamp DA, Frye AM, Phuong TT, Yarishkin O, Jo AO, Xu Y, et al. TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci Rep. 2016;6:30583.
Matsumoto H, Sugio S, Seghers F, Krizaj D, Akiyama H, Ishizaki Y, et al. Retinal detachment-induced muller glial cell swelling activates TRPV4 ion channels and triggers photoreceptor death at body temperature. J Neurosci. 2018;38:8745–58.
Gao X, Wu L, O'Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278:27129–37.
Machemer R. Experimental retinal detachment in the owl monkey. II. Histology of retina and pigment epithelium. Am J Ophthalmol. 1968;66:396–410.
Machemer R, Norton EW. Experimental retinal detachment and reattachment: I. Methods, clinical picture and histology. Bibl Ophthalmol. 1969;79:80–90.
Shibasaki K. TRPV4 ion channel as important cell sensors. J Anesth. 2016;30:1014–9.
Hanaoka N, Murakami Y, Nagata M, Horikawa K, Nagakura S, Yonemura Y, et al. Occupancy of whole blood cells by a single PIGA-mutant clone with HMGA2 amplification in a paroxysmal nocturnal haemoglobinuria patient having blood cells with NKG2D ligands. Br J Haematol. 2013;160:114–6.
Mizoguchi F, Mizuno A, Hayata T, Nakashima K, Heller S, Ushida T, et al. Transient receptor potential vanilloid 4 deficiency suppresses unloading-induced bone loss. J Cell Physiol. 2008;216:47–53.
McNulty AL, Leddy HA, Liedtke W, Guilak F. TRPV4 as a therapeutic target for joint diseases. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:437–50.
Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–65.
Shibasaki K, Yamada K, Miwa H, Yanagawa Y, Suzuki M, Tominaga M, et al. Temperature elevation in epileptogenic foci exacerbates epileptic discharge through TRPV4 activation. Lab Investig. 2019. https://doi.org/10.1038/s41374-019-0335-5.
Acknowledgements
The author would like to acknowledge the following funding sources: MEXT/JSPS KAKENHI JP15H05934 <Thermal Biology>, JP15H03000, JP18H03124, JP18K19418. Grants-in-Aid for Scientific Research from the Takeda Science Foundation, Sumitomo Foundation, the Brain Science Foundation, Narishige Neuroscience Research Foundation, Salt Science Research Foundation no. 14C2, the Ichiro Kanehara Foundation, and the Takano Life Science Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shibasaki, K. TRPV4 activation by thermal and mechanical stimuli in disease progression. Lab Invest 100, 218–223 (2020). https://doi.org/10.1038/s41374-019-0362-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0362-2
This article is cited by
-
Mechanosensitive TRPV4 channel guides maturation and organization of the bilayered mammary epithelium
Scientific Reports (2024)
-
Transient Receptor Potential Vanilloid 4: a Double-Edged Sword in the Central Nervous System
Molecular Neurobiology (2023)
-
Implications of Transient Receptor Potential Cation Channels in Migraine Pathophysiology
Neuroscience Bulletin (2021)
