
Abstract
Human relaxin-2 reduces hepatic fibrosis in mice. However, the effects of relaxin-2 on hepatic steatosis and fibrosis in animals with non-alcoholic fatty liver disease (NAFLD) remain to be elucidated. C57BL/6 mice fed a high-fat diet (HFD) or methionine–choline-deficient (MCD) diet were randomly assigned to receive recombinant human relaxin-2 (25 or 75 μg/kg/day) or vehicle for 4 weeks. In HFD-fed mice, relaxin-2 decreased systemic insulin resistance and reduced body weight, epididymal fat mass and serum leptin and insulin concentrations. In livers of HFD-fed mice, relaxin-2 attenuated steatosis and increased phosphorylation of insulin receptor substrate-1, Akt and endothelial nitric oxide synthase (eNOS), and activated genes that regulate fatty acid oxidation and suppressed acetyl-CoA carboxylase. Relaxin-2 had no direct anti-steatotic effect on primary mouse hepatocytes, but S-nitroso-N-acetylpenicillamine attenuated palmitic acid-induced steatosis and activated genes regulating fatty acid oxidation in hepatocytes. In mice fed an MCD diet, relaxin-2 attenuated steatosis, inflammation and fibrosis. Relaxin-2 increased eNOS and Akt phosphorylation and transcript levels of cytochrome P450-4a10 and decreased acetyl-CoA carboxylase in MCD-fed mouse livers. Moreover, expression levels of Kupffer cell activation, hepatic stellate cell activation and hepatocyte apoptosis were decreased in MCD diet-fed mice receiving relaxin-2. In conclusion, relaxin-2 reduces hepatic steatosis by activating intrahepatic eNOS in HFD-fed mice and further attenuates liver fibrosis in MCD diet-fed mice. Therefore, human relaxin-2 is a potential therapeutic treatment for NAFLD.
Similar content being viewed by others


Introduction
Non-alcoholic fatty liver disease (NAFLD) is now the most prevalent liver disorder worldwide [1]. NAFLD ranges from simple hepatic steatosis to non-alcoholic steatohepatitis (NASH), which can progress to liver fibrosis, cirrhosis and hepatocellular carcinoma [2, 3]. Once fat accumulates in hepatocytes, it sensitizes hepatocytes to injury, leading to cell death, recruitment of inflammatory cells and activation of hepatic stellate cells (HSCs) [4]. Unfortunately, no medications are currently approved for treatment of NAFLD, and the effectiveness of non-pharmacologic treatment is limited [5]. Thus, there is still an urgent need to identify or develop a therapeutic agent for this disease.
Relaxin-2, the major stored and circulating form of relaxin, is a peptide hormone named for its action in relaxing the female reproductive tract [6]. In 2002, the endogenous receptor for relaxin-2—relaxin/insulin-like family peptide receptor 1 (RXFP1)—was identified [7]. RXFP1 is present in a wide range of reproductive tissues and in heart, kidney, lung, liver, brain and blood cells [6]. In response to relaxin-2, RXFP1 couples to G-proteins to induce a complex pattern of cAMP production [8,9,10], thus contributing to phosphorylation of cAMP response element-binding protein (CREB) [11, 12]. Furthermore, relaxin-2 can activate endothelial nitric oxidase (eNOS), leading to production of nitric oxide (NO) [13, 14].
Relaxin-2 was found to have beneficial effects in several animal models of chronic liver injury. Bennett et al. [15] were the first to observe that relaxin-2 treatment decreased expression of smooth muscle actin and collagen I in HSCs. They further showed that cirrhotic liver and activated HSCs significantly expressed RXFP1 [16]. RXFP1 expression was low in normal rat liver but significantly greater in cirrhotic liver [17]. The same group also reported that relaxin-2 reduced fibrosis in models of progressive and established hepatic fibrosis [18, 19]. Furthermore, relaxin-2 decreased portal pressure by reducing contraction of HSCs and intrahepatic resistance [20]. Insulin resistance is important in hepatic steatosis, and Szepietowska et al. [21] showed that relaxin-2 was positively associated with insulin sensitivity in persons with diabetes. Preliminary data from patients with diabetes indicate that exogenous relaxin-2 administration may improve insulin sensitivity [22]. In mice fed a high-fat diet (HFD), relaxin-2 treatment improved systemic insulin resistance [23]; however, the effect on hepatic steatosis and intrahepatic insulin resistance were not investigated [23]. Taken together, these findings suggest that relaxin-2 has a potential therapeutic role in NAFLD. So far, there is no information on relaxin-2 on hepatic steatosis and fibrosis in animals with NAFLD. Therefore, in this study, we investigated the potential beneficial effects and mechanisms of relaxin-2 on livers of two mouse models: HFD-induced NAFLD with simple steatosis and methionine-choline-deficient (MCD) diet-induced NASH.
Materials and methods
Animals
Adult male C57BL/6 mice (National Laboratory Animal Center, Taipei, Taiwan) aged 8–10 weeks were used in all experiments. The mice were caged at 22 °C with a 12-h light-dark cycle and allowed free access to food. The study was approved by the Animal Experiment Committee of Taipei Veterans General Hospital and performed in accordance with the Guides for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences, USA.
Experimental design
Hepatic steatosis, obesity and insulin resistance were induced by feeding mice an HFD (D12492; 34.9% fat by weight, which yields about 60% calories from fat and has a caloric density of 5.24 kcal/g) for 8 weeks. Hepatic inflammation and fibrosis were induced by feeding mice an MCD diet (Dyet #518810, Dyets Inc., Bethlehem, PA) for 12 weeks. After 4 weeks of the HFD or 8 weeks of the MCD diet, mice were randomly assigned to receive recombinant human relaxin-2 (R&D Systems, Minneapolis, MN, USA), 25 or 75 μg/kg/day, or vehicle (double-distilled water, 100 μL) via subcutaneously implanted Alzet minipumps (1004 type; DURECT Corporation, Cupertino, CA, USA) for 4 weeks in conjunction with continuous HFD or MCD diet feeding. The food intake was estimated by measuring food pallet weights twice a week. The selected relaxin-2 doses were previously found to have anti-fibrotic effects on carbon tetrachloride-injured mice [19]. Mice fed a normal chow diet for 8 weeks or 12 weeks served as normal controls (n = 5). At 4 weeks after administration of relaxin-2 or vehicle, all groups of mice were sacrificed after overnight fasting (Supplementary Fig. 1).
Measurement of blood biochemistry, serum human relaxin-2, serum free fatty acids, insulin, leptin, and adiponectin
Alanine aminotransferase, triglycerides and cholesterol were measured in mouse sera with a standard auto-SMAC analyzer (Cobas 8000; Roche Diagnostics GmbH, Mannheim, Germany). Insulin levels were determined by using a mouse insulin ELISA kit (Crystal Chem Inc., Downers Grove, IL, USA). Serum leptin and adiponectin levels were measured with a mouse leptin ELISA kit (Cat. # EZML-82K) (EMD Millipore, St. Charles, MO, USA) and mouse adiponectin kit (EZMADP-60K) (EMD Millipore), respectively. Serum free fatty acid levels were determined with a Free Fatty Acid Assay Kit (Zen-Bio, Inc., Durham, NC, USA). Serum human relaxin-2 levels were measured with human relaxin-2 quantikine ELISA kit (R& D systems, Minneapolis, MN).
Measurement of hepatic steatosis
Triglyceride content in livers was measured by a triglyceride colorimetric assay kit (Cayman Chem Inc., Ann Arbor, MI, USA). Oil Red O staining was performed on cryosections (8 um) incubated in 60% isopropanol for 30 s and in Oil Red O solution (Muto Pure Chemicals, Co., Ltd., Japan) at 37 °C for 10 min. Specimen integrity was verified by staining with Mayer’s hematoxylin for 3 min.
Hepatic NOx
Liver tissues were homogenized and measured by using Nitrate/Nitrite Colorimetric Assay Kit (Cayman chemical, Ann Arbor, Michigan).
Glucose tolerance and insulin tolerance tests
Another set of HFD-fed mice treated with vehicle or relaxin-2 (n = 5 per group) and normal control mice (n = 5) underwent glucose tolerance and insulin tolerance testing. After a 16-h fast, glucose tolerance tests were performed after intraperitoneal injection of d-glucose (Sigma-Aldrich, Inc., St. Louis, MO, USA) at a dose of 2 mg/g body weight. For the insulin tolerance test, mice were injected with regular human insulin (Santa Cruz Biotechnology, Inc., CA, USA) at a dose of 0.75 U/kg body weight after a 6-h fast. Blood glucose levels were measured with a portable blood glucose meter (OneTouch Ultra 2, LifeScan, Johnson & Johnson, USA).
In situ zymography
Extracellular matrix degradation and protease activity were measured by in situ zymography, as previously described [19]. Briefly, cryopreserved liver sections were overlaid with a dye-quenched collagen (type 1 from Bovine skin) (DQTM Collagen, Thermo Fisher Scientific, Carlsbad, CA, USA)–agarose mixture and incubated for 16 h. Collagenase activity was observed with a fluorescence microscope (Olympus, AX-80).
Real-time quantitative reverse transcriptase-polymerase chain reaction (QPCR)
Total RNA was isolated by the acid guanidinium thiocyanate–phenol–chloroform extraction method [24]. RNA purity and concentration were determined spectrophotometrically. Two units of DNase (Cat.# M6101, Promega Corporation, Madison, WI) was added to 2 µg RNA. Complementary DNAs were synthesized by reverse transcription of 1 µg of the DNase-treated RNA according to the MMLV reverse transcriptase 1st-strand cDNA Synthesis Kit (EPICENTRE, Madison, WI). The cDNA solution was diluted to 100 µL and stored at −20 °C until assay. Real-time RT-PCR (QPCR) was performed for 10 min at 50 °C and 5 min at 95 °C. Subsequently, cycles was repeated which consisted of denaturation for 15 sec at 95 °C and annealing with extension for 1 min at 60 °C. The specificity of each PCR product was evaluated by melting curve analysis, and followed by agarose gel electrophoresis. Relative quantification of gene expression was performed by using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control.
The nucleotide sequences of the intron-spanning primers used in this study are shown in Supplementary Table S1. Quantitative gene expression was performed with an ABI PRISM 7900HT Sequence detection system (Applied Biosystems Inc. Foster City, CA, USA) using SYBR Green technology.
Western blot analysis
The specimens were homogenized on ice by using a Potter-type Teflon glass homogenizer in a solution containing RIPA buffer (50 mM Tris-HCl, pH 8.0, with 150 mM sodium chloride, 1.0% Nonidet P-40, 0.5% sodium deoxycholate), phosphatase inhibitor cocktail 1 (P-2850, Sigma Co.), protease inhibitor cocktail (P-8340, Sigma Co.) and phosphatase inhibitor cocktail 2 (R-5726, Sigma Co.). Homogenates were solubilized by vigorous vortex and sonication. The protein concentration of each sample was determined by the Bradford method. The protein (30–80 μg of protein/lane) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a PVDF-membrane (Millipore, MA, USA) by wet electro-blotting. The nonspecific sites on membranes were blocked with 5% non-fat dry milk in TBST. Thereafter, the blots were incubated with primary antibodies (Supplementary Table S2). After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA, USA) for 1 h. Subsequently, the blots were developed by enhanced chemiluminescence (ECL Western Blotting Analysis System, Amersham). The intensities of the interested bands were detected and analyzed by a BioSpectrum® Imaging System (UVP, LLC, CA, USA).
Immunohistochemical staining
Slides were incubated at 4 °C overnight with the primary antibodies shown in Supplementary Table S2. After overnight incubation, the slides were incubated with a secondary antibody for 30 min. The slides were colored using a super-sensitive polymer-HRP IHC detection system (BioGenex Laboratories Inc., Fremont, CA, USA) and then counterstained with Mayer’s hematoxylin. Images were captured with a microscope (Olympus, AX-80) and Olympus CellSens imaging software.
Immunofluorescent staining
The liver frozen sections were fixed by 4% paraformaldehyde, and permeabilized with 0.1% Triton. In the first day of double staining, vWF and RXFP1 antibodies (table S2) were incubated for 16 h at 4 °C. Then the sections were incubated with Alexa Fluor 488 Donkey anti-sheep IgG H&L (ab150177) at a 1/1000 dilution for vWF and Alexa Fluor 647 Donkey Anti-Rabbit IgG H&L (ab150075) at a 1/1000 dilution for RXFP1 for 120 min at 37 °C on day 2 and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Cell Signaling).
Sirius Red stain and TUNEL assay
Sirius red stain was performed by Picrosirius Red Stain Kit (Polysciences, Inc. Warrington, PA). The terminal deoxynucleotide transferase mediated dUTP nick-end labeling (TUNEL) assay was performed to evaluate apoptosis by an in situ cell death detection kit (Roche Diagnostics GmbH, Mannheim, Germany).
Hepatic hydroxyproline
The level was determined by using Hydroxyproline Colorimetric Assay Kit (BioVision, Inc., Milpitas, CA). Liver tissue 20 mg was homogenized and hydrolyzed with 200 μL 12 N HCl in a pressure-tight, teflon capped vial at 120 °C for 3 h. After transferring 10 μL of each hydrolyzed sample to a 96-well plate to dry at 65 °C, added 100 μL of the chloramine T reagent to each sample and standard and incubate at room temperature for 5 min. After adding 100 μL of the DMAB reagent to each well and incubated for 90 min at 60 °C, all samples were measured absorbance at 560 nm in a microplate reader.
In vitro study
Primary hepatocytes were isolated from male C57BL/6J mice (age 8–10 weeks) by modifying a previously described collagenase perfusion method [25]. At 24 h before plating the cells, cell culture plates were coated with 0.1% rat tail collagen (Sigma-Aldrich, Inc., St. Louis, MO, USA). Collagenase D and P were purchased from Roche (Mannheim, Germany). Primary hepatocytes were cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and antibiotics at 37 °C in a humidified atmosphere of 95% air and 5% carbon dioxide. Lipid accumulation in hepatocytes was induced by 0.66 mM palmitic acid. After exposure to control medium, palmitic acid with or without relaxin-2 (10–12, 10–10 or 10–8 M), palmitic acid with or without S-nitroso-N-acetylpenicillamine (SNAP; 10–5 M or 10–4 M; Santa Cruz Biotechnology, Inc., CA, USA) for 24 h, hepatocytes were measured for the degree of steatosis or collected for RNA extraction. For the steatosis study, 2 × 104 hepatocytes/well were seeded in 96-well plates. For RNA extraction, 2 × 105 hepatocytes/well were seeded in 6-well plates. The degree of steatosis in cells was determined by a steatosis colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA), and lipid accumulation was assessed by extracting Oil Red O and measuring absorbance at 490 nm (SpectraMax 250, Molecular device, CA, USA). For Oil Red O staining, hepatocytes were fixed in 4% paraformaldehyde (15 min) and stained with Oil Red O solution (30 min).
Primary hepatic stellate cells (HSCs) and Kupffer cells were isolated from male C57BL/6 mice as previously described [26] with minor modification. CD11b (+) Kupffer cells were selected by magnetic cell sorting using anti-CD11b Micro Beads (Miltenyi Biotec). The primary HSCs were seeded on the plastic dishes with Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and antibiotics for 7 days for in vitro activation. For RNA extraction and measurement of NOx production, 2 × 105 in vitro activated HSCs/well were seeded in the 12-well plates in the presence of relaxin-2 (100 ng/mL) or vehicle for 48 h while 2 × 105 Kupffer cells/well were seeded in the 12-well plates with RPMI-1640 in the presence of relaxin-2 (100 ng/mL), lipopolysaccharide (100 ng/mL) or vehicle for 24 h. For MTT measurement, 2 × 104 in vitro activated HSCs or Kupffer cells/well were seeded in 96-well plates in the presence of relaxin-2 (100 ng/mL) or Vehicle for 48 h (HSCs) or 24 h (Kupffer cells).
MTT assay
Cell viability was measured by methyl thiazolyl tetrazolium (MTT) assay (Sigma-Aldrich, Inc., St. Louis, MO, USA). MTT was dissolved with phosphate-buffered saline into a 5 mg/mL solution. After the addition of 20 μL of MTT solution and 2 h of incubation, the medium was aspirated, and the formed MTT violet crystal was dissolved with dimethyl sulfoxide. The amount of MTT metabolite was determined by using a microplate reader at an absorbance of 570 nm (SpectraMax 250, Molecular Devices, CA, USA).
Statistical analysis
For quantification of the staining in the histological and immunohistochemical images, five fields per slide were randomly selected. The percentages of positive stained areas were measured using Image J software (National Institutes of Health, Bethesda, MD) in an independent manner. The average of the five fields in each slide was calculated. Data were analyzed with GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA) and are expressed as mean ± standard deviation. The data were compared using Kruskal–Wallis one-way analysis of variance with the Dunn’s multiple comparisons test. A p-value of less than 0.05 was considered to indicate statistical significance.
Results
Relaxin-2 decreased body weight and epididymal fat weight, and improved systemic insulin resistance in HFD-fed mice
Body weight gain, epididymal fat weight, and serum triglyceride level were lower in HFD-R75 mice than in HFD-V mice. HFD-R25 mice exhibited a significant decrease in epididymal fat weight and serum triglycerides (Table 1). The intake of HFD did not differ between groups (HFD-V: 2.23 ± 0.16 g/day/mouse, HFD-R25: 2.22 ± 0.1 g/day/mouse, HFD-R75: 2.25 ± 0.33 g/day/mouse). In addition, the HFD-R75 group exhibited significantly higher quantitative insulin sensitivity check index, and lower serum insulin and leptin levels than the HFD-V group (Supplementary Fig. 2A). Glucose tolerance testing and insulin tolerance testing showed improved systemic insulin resistance in HFD mice given the higher dose of relaxin-2 (Supplementary Fig. 2B).
Relaxin-2 improved steatosis in livers of HFD mice
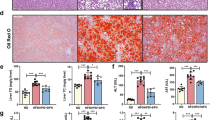
Hematoxylin and eosin staining and Oil Red O staining showed diminished lipid deposition in HFD mice receiving relaxin-2 (Fig. 1a, b). In addition, hepatic triglyceride level was significantly lower in the HFD-R75 group than in the HFD-V group (Fig. 1c). Only relaxin-2-treated mice can be detected human relaxin-2 in serum (Fig. 1d). Relaxin-2 treatment did not increase hepatic expression of relaxin-2 receptor (RXFP1) (Fig. 1e). RXFP1 expression has been observed on sinusoid endothelial cells but not on macrophages [20]. In addition, co-localization of RXFP1 and a sinusoidal endothelial cell marker, vWF, was demonstrated (Supplementary Fig. 3A). Interestingly, hepatic phosphorylation of eNOS was suppressed in the HFD-V group and restored in the HFD-R75 group (Fig. 1f, Supplementary Fig. 3B). Similarly, the hepatic nitrate/nitrite (NOx) levels in HFD-V mice were reduced as compared with those in the normal-V mice and increased in HFD-R75 group (Fig. 1f). In addition, relaxin-2 did not stimulate production of NOx from primary HSCs or Kupffer cells (Supplementary Fig. 4A, B). These findings suggest that relaxin-2 reduced hepatic steatosis and may induce sinusoidal endothelial cells in liver to produce NO through the relaxin-2 receptor.

Relaxin-2 improved hepatic steatosis and increased expression of phosphorylated endothelial nitric oxide synthase (p-eNOS) in mice fed a high-fat diet (HFD). a Hematoxylin and eosin (H&E) and Oil Red O staining of liver. Scale bar = 50 μm. b Quantification of the area stained by Oil Red O. c Hepatic triglyceride (TG) content. d Serum human relaxin-2 levels. e Western blots of relaxin/insulin-like family peptide receptor 1 (RXFP1) in livers. Loading protein: 40 μg/lane. f Hepatic nitrate/nitrite (NOx) levels and western blots of phosphorylated and total endothelial nitric oxide synthase (p/t-eNOS) in liver extracts. Loading protein/lane: 80 μg. N-V/N-R75: normal mice receiving vehicle or relaxin-2 (75 μg/kg/day); HFD-V/HFD-R25/HFD-R75: HFD-fed mice treated with vehicle or relaxin-2 (25 or 75 μg/kg/day). (#p < 0.05 vs. N-V; ##p < 0.01 vs. N-V; *p < 0.05 vs. HFD-V)
Relaxin-2 improved insulin sensitivity in liver and activated genes regulating fatty acid oxidation in HFD-fed mice
Although serum insulin concentration was lower in HFD-R75 mice (Supplementary Fig. 2A), expressions of insulin downstream signaling molecules such as phosphorylation of insulin receptor substrate-1 and phosphorylation of Akt were higher in HFD-R75 livers than in HFD-V livers; glucose transporter-2 expression was similar in these groups (Fig. 2a).

Relaxin-2 improved intrahepatic insulin sensitivity and increased lipid turnover in livers of HFD-fed mice. a Western blots of phosphorylated and total insulin receptor substrate (t/p-IRS1/2), total and phosphorylated forms of protein kinase B (t/p-Akt), glucose transporter-2 (GLUT2) and β-actin in livers. (40 μg/lane) b Western blots of total and phosphorylated forms of extracellular signaling–related kinase (t/p-ERK), cAMP response element–binding protein (t/p-CREB) and β-actin in livers. Loading protein: 40 μg/lane. c Hepatic cAMP levels. d Western blots of total and phosphorylated forms of acetyl-CoA carboxylase (t/p-ACC) and β-actin in livers. Loading protein: 40 μg/lane. e Hepatic gene expression related to lipid metabolism, such as fatty acid transport protein 1/4 (FATP1/4); liver X receptor alpha/beta (LXRα/β); carbohydrate response element-binding protein (ChREBP); sterol regulatory element–binding protein-1c (SREBP-1c); peroxisome proliferator-activated receptor α/γ1 (PPARα/γ1); carnitine palmitoyltransferase (CPT)-1a; cytochrome P450, family 4, subfamily a, polypeptide 10 (CYP4a10); acyl-CoA oxidase (AOX); microsomal triglyceride transfer protein (MTTP) and fatty acid synthase (FASN). The values are normalized to the values for GAPDH for each sample, and expression levels in the HFD-V group are arbitrarily assigned as 1. N-V: normal mice receiving vehicle; HFD-V/HFD-R25/HFD-R75: HFD-fed mice treated with vehicle or relaxin-2 (25 or 75 μg/kg/day). (#p < 0.05 vs. N-V; *p < 0.05 vs. HFD-V)
Relaxin-2 did not increase phosphorylation of ERK or CREB (Fig. 2b) or hepatic cAMP levels (Fig. 2c); however, it decreased the levels of total ACC (t-ACC) and slightly increased the ratios of p-ACC to t-ACC in livers of HFD-R25 and HFD-R75 mice (Fig. 2d). Interestingly, as compared with the HFD-V group, mice receiving relaxin-2 had significantly higher expression of peroxisome proliferator-activated receptor α (PPARα). In addition, HFD-R75 livers expressed higher transcript levels of cytochrome P450-4a10 (Cyp4a10), acyl-CoA oxidase (AOX), peroxisome proliferator-activated receptor γ 1 (PPARγ1) and fatty acid transport proteins 1 and 4 (FATP1 and FATP4) (Fig. 2e). These data suggest that relaxin-2 reduces hepatic steatosis by increasing fatty acid oxidation and decreasing t-ACC.
Relaxin-2 increased phosphorylated eNOS in gastrocnemius muscle in HFD-fed mice
In gastrocnemius muscle, relaxin-2 increased Akt phosphorylation without upregulation of p-insulin receptor substrate-1/2 or glucose transporter-4 (Supplementary Fig. 5A). In addition, relaxin-2 treatment increased phosphorylated eNOS in gastrocnemius muscle (Supplementary Fig. 5B). These findings suggest that relaxin-2 improved insulin sensitivity in liver and skeletal muscle, thereby improving systemic insulin resistance in HFD-fed mice.
Relaxin-2 increased expression of p-eNOS and PPARα in epididymal fat of HFD-fed mice
Similarly, expression of phosphorylated eNOS in epididymal fat was higher in the HFD-R25 and HFD-R75 groups than in the HFD-V group (Supplementary Fig. 6A). PPARα expression was significantly upregulated in the groups receiving relaxin-2 (Supplementary Fig. 6B). Thus, relaxin-2 may also promote fatty acid oxidation in epididymal fat, thus contributing to a decrease in fat mass.
Nitric oxide but not relaxin-2 had a direct anti-steatotic effect on hepatocytes
The effective dose of relaxin-2 is 1–100 ng/mL (1.6 × 10–10 to 1.6 × 10–8 M) for HSCs and sinusoidal endothelial cells [20, 27]. However, relaxin-2 10–12 to 10–8 M did not affect the cell viability of hepatocytes (Supplementary Fig. 7A) and had no anti-steatotic effect on hepatocytes (Supplementary Fig. 7B, C).
We found that relaxin-2 increased intrahepatic eNOS phosphorylation and therefore investigated the effect of SNAP (an NO donor) on palmitic acid–induced steatosis. SNAP 10–5 to 10–4 M did not affect hepatocyte viability (Fig. 3a). In addition, SNAP reduced steatosis induced by palmitic acid (Fig. 3b, c) and was associated with increased transcript levels of FATP4, PPARα, carnitine palmitoyltransferase (CPT)-1a, AOX and Cyp4a10 (Fig. 3d). Therefore, in HFD-fed mice, attenuation of hepatic steatosis might be attributable to relaxin-2–induced hepatic eNOS activation but not due to a direct effect of relaxin-2 on hepatocytes.

Nitric oxide donor decreased lipid accumulation in primary mouse hepatocytes by activating genes regulating fatty acid oxidation. a Cell viability after 24-h treatment with control medium (Ctrl) or different concentrations (10–5, 10–4, 10–3 M) of S-nitroso-N-acetylpenicillamine (SNAP) was determined by methyl thiazolyl tetrazolium (MTT) assay. b Optical density (OD) values for lipid accumulation and c Oil red O staining of primary mouse hepatocytes treated under different conditions. Ctrl: control medium, PA: 0.66 mM palmitic acid, PA + SNAP 10–5/10–4: 0.66 mM palmitic acid and SNAP 10–5/10–4 M. Scale bar = 50 μm. d Transcript expressions of fatty acid transport protein 4 (FATP4), peroxisome proliferator-activated receptor α/γ1 (PPARα/γ1), carnitine palmitoyltransferase (CPT)-1a, acyl-CoA oxidase (AOX) and cytochrome P450, family 4, subfamily a, polypeptide 10 (CYP4a10) in primary hepatocytes treated under different conditions. (#p < 0.05 vs. Ctrl; ##p < 0.01 vs. Ctrl; *p < 0.05 vs. PA; **p < 0.01 vs. PA) (n = 8 in each group)
Relaxin-2 attenuated steatosis in mice fed an MCD diet
Serum relaxin-2 only can be detected in relaxin-2-treated MCD mice (Supplementary Fig. 8A). After relaxin-2 treatment, MCD-fed mice had an improved NAFLD Activity Score [28] (Fig. 4a). The hepatic steatosis was significantly lower in MCD-R75 than in MCD-V mice (Fig. 4a). Relaxin-2 treatment increased hepatic NOx levels (Supplementary Fig. 8B). In addition, p-Akt and p-eNOS expressions were higher in livers of mice receiving relaxin-2 (Fig. 4b, c). RXFP1 expression was not different between MCD-V and MCD-R75 mice (Supplementary Fig. 8C). In addition, expressions of other relaxin-2 downstream signaling molecules, such as p-ERK and p-CREB, were not altered in livers of MCD mice receiving relaxin-2 (Supplementary Fig. 8D). Mediators of lipid metabolism that were altered in the HFD mice receiving relaxin-2 were examined. Relaxin-2 increased transcript expressions of FATP4 and Cyp4a10 (Fig. 4d). In addition, hepatic expression of t-ACC decreased after relaxin-2 treatment in MCD-fed mice without significant change in ratios of p-ACC to t-ACC (Fig. 4e). Taken together, these findings indicate that relaxin-2 treatment activated the eNOS/NO pathway in livers of mice fed an MCD with increased Cyp4a10 and decreased t-ACC, thereby attenuating hepatic steatosis.

Relaxin-2 ameliorated hepatic steatosis in mice fed a methionine-choline-deficient (MCD) diet. a Hematoxylin and eosin and Oil Red O staining of livers, NAFLD activity scores (NAS) and fibrosis staging and quantification of Oil Red O-positive areas in the four groups. Scale bar = 50 μm. NAS is based on the degree of steatosis, hepatocyte ballooning and lobular inflammation. b Western blots of total and phosphorylated endothelial nitric oxide synthase (t/p-eNOS) (80 μg/lane), c protein kinase B (t/p-Akt) (80 μg/lane) and β-actin in livers of the N-V, MCD-V and MCD-R75 groups. d Hepatic gene expression of fatty acid transport protein 4 (FATP4), peroxisome proliferator–activated receptor α/γ1 (PPARα/γ1), carnitine palmitoyltransferase (CPT)-1a, acyl-CoA oxidase (AOX) and cytochrome P450, family 4, subfamily a, polypeptide 10 (CYP4a10) in the MCD-V, MCD-R25 and MCD-R75 groups. e Western blots of total and phosphorylated acetyl-CoA carboxylase (t/p-ACC) in livers. (70 μg/lane) N-V: normal mice receiving vehicle; MCD-V/MCD-R25/MCD-R75: MCD mice treated with vehicle or relaxin-2 (25 or 75 μg/kg/day). (##p < 0.01 vs. N-V; *p < 0.05 vs. MCD-V; **p < 0.01 vs. MCD-V)
Relaxin-2 reduced chronic liver injury in MCD-fed mice
Alanine aminotransferase level was significantly higher in MCD-V mice than in the normal mice (Table 2). In addition, mice receiving relaxin-2 had less liver inflammation (MCD-R25: p = 0.1 vs. MCD-V; MCD-R75: p = 0.09 vs. MCD-V, Table 2). Furthermore, relaxin-2 decreased expression of F4/80 (Supplementary Fig. 9A, B) and hepatocyte apoptosis (Supplementary Fig. 9A). The hepatic transcript levels of tumor necrosis factor alpha and interleukin-6 were decreased in relaxin-2-treated MCD mice (Supplementary Fig. 9B). A noteworthy finding was that relaxin-2 treatment attenuated liver fibrosis in MCD-fed mice (Fig. 5a, b). The expression levels of alpha-smooth muscle actin (α-SMA) were decreased after relaxin-2 treatment (Fig. 5c). Tissue inhibitor of metalloproteinase (TIMP)-1 mRNA level in MCD-R75 mice was significantly lower than in MCD-V mice (Fig. 5d). In addition, matrix metalloproteinase (MMP)-13 expression was slightly higher in the MCD-R75 group (p = 0.13 vs. MCD-V). There was no significant change in the transcript expressions of collagen type 1α1 or MMP-2/9 in MCD mice after relaxin-2 treatment. In situ zymography revealed that relaxin-2 administration resulted in an increase in collagen-degrading activity in livers (Fig. 5e). Nevertheless, relaxin-2 did not directly inactivate Kupffer cells or HSCs (Supplementary Fig. 10). These findings suggest that relaxin-2 attenuated MCD diet-mediated liver fibrosis with reduction in hepatocyte apoptosis, Kupffer cells, and HSCs activation and with increased extracellular matrix degradation.

Relaxin-2 reduced liver fibrosis in mice fed a methionine–choline-deficient (MCD) diet. a Sirius red stained area of livers. Scale bar = 100 μm. b Hydroxyproline levels in liver. c Hepatic expression levels of mRNA and protein of α-smooth muscle actin (α-SMA) and immunohistochemistry staining of α-SMA with quantification of stained area (%) below. Loading protein: 30 μg/lane. Scale bar = 100 μm. d Hepatic gene expressions of collagen type 1α1 (Col1α1), tissue inhibitor of metalloproteinase (TIMP)-1 and matrix metalloproteinase (MMP)-2/-9/-13 in the four groups. e In situ zymography showing collagen-degrading activity in the four groups and quantification of dye-quenched areas. DAPI: 4′,6-diamidino-2-phenylindole. Scale bar = 50 μm. N-V normal mice receiving vehicle, MCD-V/MCD-R25/MCD-R75 MCD mice treated with vehicle or relaxin-2 (25 or 75 μg/kg/day). (#p < 0.05 vs. N-V; ##p < 0.01 vs. N-V; *p < 0.05 vs. MCD-V; **p < 0.01 vs. MCD-V)
Discussion
This is the first study to demonstrate that relaxin-2 exerted anti-steatotic and anti-fibrotic effects in mice with NAFLD. Our findings indicate that relaxin-2 activates the eNOS/NO pathway in liver to reduce hepatic steatosis. NO but not relaxin-2 had a direct anti-steatotic effect on hepatocytes. This effect is mainly attributable to activation of fatty acid oxidation genes and suppression of t-ACC. The reduction of lipid accumulation in relaxin-2–treated liver might help in improving intrahepatic and systemic insulin resistance. In addition, relaxin-2 treatment attenuated steatosis, inflammation, and fibrosis in livers of MCD-induced NASH. Fibrosis is the most important prognostic factor in NAFLD [29]. Our findings suggest that relaxin-2 is a promising agent for treatment of NAFLD, which still lacks an approved pharmacologic therapeutic agent currently. Moreover, relaxin-2 is a naturally occurring human peptide and a recombinant human relaxin-2, serelaxin, has been recently demonstrated to improve cardiac function in patients with heart failure [30] and renal dysfunction in human cirrhosis with good safety profile [31]. However, only short-acting intravenous and subcutaneous dosage forms of relaxin-2 are available currently [31], which may result in poor compliance in patients with NAFLD. Long-lasting recombinant relaxin-2 is anticipated.
The principal effects of relaxin-2 on cells are activation of eNOS/NO, ERK and the cAMP/CREB pathways [6, 11,12,13,14]. In this study, relaxin-2 treatment activated eNOS/NO but not ERK or CREB in liver. The hepatic cAMP levels remained unchanged too. Hepatocyte is the most predominant cell type in liver and expresses CREB [32] and ERK [33], but relaxin receptor does not express on hepatocytes [20]. This may explain why no change of CREB or ERK in the whole liver extracts after relaxin-2 treatment. However, immunohistochemistry indicated that p-eNOS expression was higher in the sinusoid region of relaxin-2–treated liver. Fallowfield et al. [20] elegantly demonstrated that RXFP1 was expressed in sinusoidal endothelial cells, but not in macrophages or hepatocytes, and that relaxin-2 induced NO production by sinusoidal endothelial cells. In our study, co-localization of RXFP1 and vWF was demonstrated and relaxin-2 cannot increase production of NOx from primary HSCs or Kupffer cells. Although we did not demonstrate if the relaxin-2-dependent reduced steatosis is abolished in mice after inhibiting RXFP1 in sinusoid endothelial cells, however; based on current evidences, relaxin-2 might bind RXFP1 on sinusoid endothelial cells, thereby activating eNOS there to produce NO.
Relaxin-2 administration had no effect on palmitic acid–induced steatosis. However, NO exerted a direct anti-steatotic effect on palmitic acid–induced steatosis. Expressions of FATP, PPARα, CPT1a, AOX, and Cyp4a10 were increased in hepatocytes after incubation of the NO donor SNAP and in livers of HFD-fed mice treated with relaxin-2. Similarly, in MCD-fed mice relaxin-2 treatment activated FATP and Cyp4a10 in liver. These data suggest that relaxin-2 promotes lipid turnover in hepatocytes by increasing intrahepatic NO in mice. Furthermore, in rat hepatocytes, the NO donors and the NO downstream signaling molecule cGMP inhibited ACC activity and fatty acid synthesis [34]. V-PYRRO/NO, a liver-selective NO donor, reduced liver steatosis in HFD-fed mice with increased the ratio of p-ACC to t-ACC [35]. In the present study, relaxin-2 treatment decreased t-ACC expression with slightly increased p-ACC/t-ACC ratio in mice fed an HFD and reduced t-ACC expression without significant change of p-ACC/t-ACC ratio in MCD mice, which may reduce ACC activity in liver. However, V-PYPRO/NO tended to decrease expressions of PPARα and CPT1 [35]. In addition, administration of another NO donor, arginine, improved hepatic steatosis induced by total parenteral nutrition in rats [36]. Therefore, the beneficial effects of the various NO donors on liver steatosis may be exerted along different pathways.
Insulin resistance is the core defect in NAFLD. Bonner et al. [23] first demonstrated that relaxin-2 administration improved insulin resistance in HFD-fed mice by increasing muscle glucose uptake; however, they did not evaluate fatty liver in their study. In this study, we found that relaxin-2 improved systemic insulin resistance as well as liver fat accumulation. In addition, p-Akt expression was increased in skeletal muscle and p-insulin receptor substrate-1 expression level was increased in livers of HFD-fed mice after relaxin-2 administration. These findings suggest that insulin sensitivity was improved in skeletal muscle and liver, which may contribute to the reduction in systemic insulin resistance.
The reduction in epididymal fat mass is likely attributable to improved systemic insulin resistance and the direct effect of NO on peripheral fat tissue. In epididymal fat of HFD-fed mice, relaxin-2 treatment increased p-eNOS expression and upregulated expression of PPARα, a major regulator of fatty acid oxidation. Fu et al. [37] reported that dietary arginine supplementation reduced fat mass in Zucker diabetic fatty rats with enhanced NO production. Furthermore, eNOS−/− mice had a lower metabolic rate and faster weight gain than did wild-type mice, because of decreased mitochondrial biogenesis in adipose tissue [38]. Therefore, relaxin-2 may reduce epididymal fat mass by activating eNOS, thereby contributing to decreases in body weight and serum leptin level.
Relaxin-2 was found to reduce liver fibrosis in carbon tetrachloride-injured rats and mice [19, 27]. Our results further showed that relaxin-2 had an anti-fibrotic effect on mice fed with an MCD diet. Williams et al. [27] showed that relaxin-2 decreased collagen deposition and TIMP-1 expression in culture-activated HSCs but did not alter expressions of type 1 collagen, MMP-13, or α-SMA. In an in vivo study, Bennett et al. [19] found that relaxin-2 decreased TIMP-2 and α-SMA expressions and increased MMP-2/13 expression levels in conjunction with increased collagen I–degrading activity. Similarly, in the present MCD diet-fed mice, relaxin-2 downregulated TIMP-1 expression, had a tendency to upregulate MMP-13 expression levels and increased overall extracellular matrix-degrading activity. Furthermore, relaxin-2 treatment also reduced the activation levels of Kupffer cells and HSCs in MCD diet-fed mice; however, relaxin-2 did not directly inactivate them. Higher NO production may contribute to the decreased Kupffer cell activation [39] with a decrease in hepatic TNFα expression, leading to lower hepatocyte apoptosis. Besides, decreased hepatic steatosis would attenuate hepatocyte apoptosis [40]. The reduction of Kupffer cell activation and hepatocyte apoptosis further contributed to the decreased HSC activation and live fibrosis in our study.
This study has limitations that warrant mention. In the analysis of weight reduction attributable to relaxin-2 in HFD mice, we lacked the equipment to measure energy expenditure and fecal fat content. In addition, the effect of relaxin-2 on pancreatic beta cells was not investigated.
In conclusion, relaxin-2 reduces liver steatosis in mice with NAFLD by activating the eNOS/NO pathway. In addition, relaxin-2 improves systemic insulin resistance and obesity in HFD-fed mice. In mice fed an MCD diet, relaxin-2 further attenuates liver fibrosis. Our results suggest that relaxin-2 has promise as a potential therapeutic agent for treatment of NAFLD.
References
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2Suppl 1):S99–S112.
McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:521–33.
Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol. 2004;40:578–84.
Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52.
Katan MB. Weight-loss diets for the prevention and treatment of obesity. N Engl J Med. 2009;360:923–5.
Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–80.
Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD, et al. Activation of orphan receptors by the hormone relaxin. Science. 2002;295:671–4.
Halls ML, Bathgate RA, Summers RJ. Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol Pharmacol. 2006;70:214–26.
Halls ML, Hewitson TD, Moore XL, Du XJ, Bathgate RA, Summers RJ. Relaxin activates multiple cAMP signaling pathway profiles in different target cells. Ann N Y Acad Sci. 2009;1160:108–11.
Halls ML, van der Westhuizen ET, Wade JD, Evans BA, Bathgate RA, Summers RJ. Relaxin family peptide receptor (RXFP1) coupling to G(alpha)i3 involves the C-terminal Arg752 and localization within membrane Raft Microdomains. Mol Pharmacol. 2009;75:415–28.
Singh S, Simpson RL, Bennett RG. Relaxin activates peroxisome proliferator-activated receptor γ (PPARγ) through a pathway involving PPARγ coactivator 1α (PGC1α). J Biol Chem. 2015;290:950–9.
Delghandi MP, Johannessen M, Moens U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal. 2005;17:1343–51.
Conrad KP, Novak J. Emerging role of relaxin in renal and cardiovascular function. Am J Physiol Regul Integr Comp Physiol. 2004;287:R250–61.
Zhang Q, Liu SH, Erikson M, Lewis M, Unemori E. Relaxin activates the MAP kinase pathway in human endometrial stromal cells. J Cell Biochem. 2002;85:536–44.
Bennett RG, Kharbanda KK, Tuma DJ. Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem Pharmacol. 2003;66:867–74.
Bennett RG, Mahan KJ, Gentry-Nielsen MJ, Tuma DJ. Relaxin receptor expression in hepatic stellate cells and in cirrhotic rat liver tissue. Ann N Y Acad Sci. 2005;1041:185–9.
Bennett RG, Dalton SR, Mahan KJ, Gentry-Nielsen MJ, Hamel FG, Tuma DJ. Relaxin receptors in hepatic stellate cells and cirrhotic liver. Biochem Pharmacol. 2007;73:1033–40.
Bennett RG, Heimann DG, Tuma DJ. Relaxin reduces fibrosis in models of progressive and established hepatic fibrosis. Ann N Y Acad Sci. 2009;1160:348–9.
Bennett RG, Heimann DG, Singh S, Simpson RL, Tuma DJ. Relaxin decreases the severity of established hepatic fibrosis in mice. Liver Int. 2014;34:416–26.
Fallowfield JA, Hayden AL, Snowdon VK, Aucott RL, Stutchfield BM, Mole DJ, et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology. 2014;59:1492–504.
Szepietowska B, Gorska M, Szelachowska M. Plasma relaxin concentration is related to beta-cell function and insulin sensitivity in women with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2008;79:e1–3.
Bani D, Pini A, Yue SK. Relaxin, insulin and diabetes: an intriguing connection. Curr Diabetes Rev. 2012;8:329–35.
Bonner JS, Lantier L, Hocking KM, Kang L, Owolabi M, James FD, et al. Relaxin treatment reverses insulin resistance in mice fed a high-fat diet. Diabetes. 2013;62:3251–60.
Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9.
Severgnini M, Sherman J, Sehgal A, Jayaprakash NK, Aubin J, Wang G, et al. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology. 2012;64:187–95.
Iwaisako K, Haimerl M, Paik YH, Taura K, Kodama Y, Sirlin C, et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor delta agonist. Proc Natl Acad Sci USA. 2012;109:E1369–76.
Williams EJ, Benyon RC, Trim N, Hadwin R, Grove BH, Arthur MJ, et al. Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut. 2001;49:577–83.
Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA. The NAS and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53:810–20.
Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61:1547–54.
Maggioni AP, Lopez-Sendon J, Nielsen OW, Hallen J, Aalamian-Mattheis M, Wang Y, et al. Efficacy and safety of serelaxin when added to standard of care in patients with acute heart failure: results from a PROBE study, RELAX-AHF-EU. Eur J Heart Fail. 2019;21:322–33.
Snowdon VK, Lachlan NJ, Hoy AM, Hadoke PW, Semple SI, Patel D, et al. Serelaxin as a potential treatment for renal dysfunction in cirrhosis: Preclinical evaluation and results of a randomized phase 2 trial. PLoS Med. 2017;14:e1002248.
Dell H, Hadzopoulou-Cladaras M. CREB-binding protein is a transcriptional coactivator for hepatocyte nuclear factor-4 and enhances apolipoprotein gene expression. J Biol Chem. 1999;274:9013–21.
Guegan JP, Fremin C, Baffet G. The MAPK MEK1/2-ERK1/2 pathway and Its Implication in hepatocyte cell cycle control. Int J Hepatol. 2012;2012:328372.
Garcia-Villafranca J, Guillen A, Castro J. Involvement of nitric oxide/cyclic GMP signaling pathway in the regulation of fatty acid metabolism in rat hepatocytes. Biochem Pharmacol. 2003;65:807–12.
Maslak E, Zabielski P, Kochan K, Kus K, Jasztal A, Sitek B, et al. The liver-selective NO donor, V-PYRRO/NO, protects against liver steatosis and improves postprandial glucose tolerance in mice fed high fat diet. Biochem Pharmacol. 2015;93:389–400.
Zheng JF, Wang HD, Liang LJ. Protective effects of nitric oxide on hepatic steatosis induced by total parenteral nutrition in rats. Acta Pharmacol Sin. 2002;23:824–8.
Fu WJ, Haynes TE, Kohli R, Hu J, Shi W, Spencer TE, et al. Dietary l-arginine supplementation reduces fat mass in Zucker diabetic fatty rats. J Nutr. 2005;135:714–21.
Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–9.
Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan-Stevenson V, Daum G, et al. Endothelial NO/cGMP/VASP signaling attenuates kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes. 2011;60:2792.
Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–101.
Acknowledgements
The authors thank Chai-Li Chen and Hsiu-Ying Chen for technical assistance.
Funding
The study was supported by grants from the Taipei Veterans General Hospital (V104B-029) and the National Science Council (MOST 104-2314-B-075 −023 -MY2, 106-2314-B-075-005-MY2) Taiwan.
Author information
Authors and Affiliations
Contributions
Study concept and design: KCL and HCL; acquisition of data: KCL, YCH, and HJS; analysis and interpretation of data: KCL, YCH, and CCC; drafting of the manuscript: KCL and YCH; critical revision of the manuscript for important intellectual content: YHH and HCL; statistical analysis: KCL, YCH and CCC; obtained funding: KCL and CCC; administrative, technical, or material support: YHH, MCH and HCL; study supervision: HCL.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lee, KC., Hsieh, YC., Chan, CC. et al. Human relaxin-2 attenuates hepatic steatosis and fibrosis in mice with non-alcoholic fatty liver disease. Lab Invest 99, 1203–1216 (2019). https://doi.org/10.1038/s41374-019-0240-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0240-y
This article is cited by
-
Emerging Therapeutic Targets for Portal Hypertension
Current Hepatology Reports (2023)
-
Interaction between estrogen receptor-α and PNPLA3 p.I148M variant drives fatty liver disease susceptibility in women
Nature Medicine (2023)
-
A relaxin-based nanotherapy for liver fibrosis
Nature Nanotechnology (2021)
