
Abstract
Inclusion body myositis (IBM) is the most common cause of primary myopathy in individuals aged 50 years and over, and is pathologically characterized by protein aggregates of p62 and mislocalized cytoplasmic TDP-43, as well as mitochondrial abnormalities in affected muscle fibers. Our recent studies have shown the accumulation of TDP-43 in mitochondria in neurons from patients with amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD), and revealed mitochondria as critical mediators of TDP-43 neurotoxicity. In this study, we investigated the association between mitochondria and TDP-43 in biopsied skeletal muscle samples from IBM patients. We found that IBM pathological markers TDP-43, phosphorylated TDP-43, and p62 all coexisted with intensively stained key subunits of mitochondrial oxidative phosphorylation complexes I–V in the same skeletal muscle fibers of patients with IBM. Further immunoblot analysis showed increased levels of TDP-43, truncated TDP-43, phosphorylated TDP-43, and p62, but decreased levels of key subunits of mitochondrial oxidative phosphorylation complexes I and III in IBM patients compared to aged matched control subjects. This is the first demonstration of the close association of TDP-43 accumulation with mitochondria in degenerating muscle fibers in IBM and this association may contribute to the development of mitochondrial dysfunction and pathological protein aggregates.
Similar content being viewed by others

Introduction
Inclusion body myositis (IBM) is the most common acquired myopathy in adults over 50 years of age, with a varied prevalence reported as 24.8–45.6 per million and a 2–3 times higher incidence rate in males compared with females [1,2,3]. IBM is characterized by slow progression, and there is often a delay of 5–10 years between disease onset and diagnosis [4,5,6]. The incidence of IBM appears to be increasing, which is likely a result of both improved diagnosis and the increasing ageing population [2]. Currently, there is no effective therapy for IBM [1]. Even with current immunotherapies, most patients become wheelchair dependent, affecting their quality of life and causing a high economic burden with a likely underestimated annual overall healthcare cost to IBM patients more than twice that of age-matched non-IBM counterparts [7,8,9,10].
IBM patients present with progressive, often asymmetrical, muscle weakness that predominantly affects the quadriceps muscles and finger flexors but also commonly involves biceps, triceps, facial, and swallowing muscles with dysphagia [11,12,13]. Disease pathogenesis is poorly understood, but both inflammatory and degenerative mechanisms may play a primary role [14, 15]. Histopathologically, IBM is characterized by inflammatory changes with endomysial inflammation, myofiber invasion by CD8+ T cells and cN1A autoantibodies, and myodegenerative pathologies including protein aggregates, rimmed vacuoles, and mitochondrial abnormalities [11, 14,15,16]. Accumulation of these protein aggregates, especially TAR DNA-binding protein 43 (TDP-43), within muscle fibers appears analogous to protein accumulations believed to be of pathophysiological importance in several central nervous system neurodegenerative disorders, such as amyotrophic lateral sclerosis (ALS) and frontotemporal degeneration (FTD) [17,18,19,20,21,22,23], suggesting the possibility that IBM may be pathogenetically related to these neurodegenerative diseases [19]. The pathogenic role of TDP-43 aggregates in IBM is unclear, though sarcoplasmic aggregation of TDP-43 has been shown to result in myofiber degeneration via endoplasmic reticulum stress and possibly calcium dysregulation [24].
Cells or mice expressing either wild type or mutant TDP-43 usually demonstrated abnormal mitochondrial morphology [25,26,27], transport [26, 27], and even function [26, 28, 29], suggesting mitochondria as likely targets of TDP-43. This notion is further supported by evidence showing that TDP-43 or truncated forms of TDP-43 can be present either inside or outside of mitochondria [30,31,32,33,34,35,36]. We and others have independently found that the portion of full-length TDP-43 inside of mitochondria can bind mitochondria-transcribed messenger RNAs (mRNAs) encoding subunits (ND3/6) of oxidative phosphorylation (OXPHOS) complex I to specifically impair its assembly and function [30, 36], whereas truncated TDP-43 lacking the M1 mitochondrial localization sequence [36] is restricted to the intermembrane space and has no effect on ND3/6 expression or mitochondrial function [30].
In this study, we sought to investigate the relationship between TDP-43 and mitochondria in IBM in an effort to elucidate the likely role of mitochondrial dysfunction in the IBM muscle degeneration.
Materials and methods
Fixed paraffin and frozen muscle samples
Frozen and formalin-fixed paraffin-embedded skeletal muscle tissues from diagnostic quadriceps muscle biopsies were obtained from University Hospitals Cleveland Medical Center under an approved Institutional Review Board protocol. Muscle tissue samples included 10 patients with IBM and 10 control subjects whose quadriceps muscle biopsy revealed no histopathological or histochemical abnormalities. All patients and controls were between ages 59 and 78 at the time of the biopsy. See Table 1 for information on the tissues used in this study. Archival paraffin-embedded samples from previously characterized cases of IBM with rimmed vacuoles and mitochondrial abnormalities were also used for validation of the immunohistochemistry analyses [37].
Immunohistochemistry
Serial adjacent sections of cross-sectioned muscle fibers were used to compare the same muscle fibers in both IBM and control samples. Slides that contained paraffin-embedded tissue sections were deparaffinized using xylene and rehydrated using graded ethanol. Slides were then incubated in tris buffered saline (TBS buffer, 50 mM Tris–HCl and 150 mM NaCl, pH = 7.6) for 10 min before antigen retrieval in 1x antigen decloaker (BioSB, cat#BSB 0021) using a TintoRetriever pressure cooker. Slides were then rinsed with running distilled water and incubated in TBS buffer for 10 min. Tissues were then incubated in 10% normal goat serum (NGS) in TBS for 30 min at room temperature in order to block nonspecific antibody-binding sites. Primary antibodies were applied on tissues for incubation overnight at 4 °C. The next day, the slides were rinsed and incubated with 1% and 10% NGS in TBS, respectively, followed by immunostaining via the peroxidase–antiperoxidase method and developed using DAB chromogen (BioCare Medical, cat#DB801L). Finally, slides were rinsed with distilled water, dehydrated using graded ethanol and xylene, and mounted with Permount. Antibodies used include TDP-43 (1:100, ProteinTech, cat#10782), phosphorylated TDP-43 at Serine 409/410 (1:200, ProteinTech, cat#22309-1-AP), p62 (1:50, Cell Signaling Technology, cat#5114S), and OXPHOS antibody cocktail (1:1000, Abcam, cat#ab110413). Total OXPHOS contains five mouse monoclonal antibodies, with each against complexes I–V: CI subunit NDUFB8, CII subunit SDHB, CIII subunit UQCRC2, CIV subunit CoxII, and CV subunit ATP5A.
Immunofluorescence
Slides were prepared similarly as immunohistochemistry through antigen retrieval. Slides were then rinsed with running distilled water and incubated in phosphate buffered saline (PBS) for 10 min. Tissues were incubated in 10% NGS in PBS for 45 min at room temperature in order to block nonspecific antibody-binding sites followed by primary antibody incubation overnight at 4 °C. The next day, the slides were also rinsed with 1% and 10% NGS, respectively in PBS, and incubated with secondary fluorescent antibodies for 2 h at room temperature in the dark. Finally, tissues were washed three times with PBS, stained with DAPI, and mounted with Fluoromount-G mounting medium (Southern Biotech, cat#0100-01). Additional antibodies used include Alexa Fluor 488 and 568 (1:250, Invitrogen, cat#A11034 and A11031). Sections were stained with DAPI (5 µg/mL) to visualize nuclei.
Immunoblot analysis
Immunoblot analysis of the frozen quadriceps muscle samples from IBM and control patients was performed. Frozen tissue samples were homogenized in 1x cell lysis buffer (Cell Signaling Technology, cat#9803S) with 1 mM phenylmethylsulfonyl fluoride (Millipore, cat#7110), protease inhibitor cocktail (Sigma Aldrich, cat#P8340), and phosphatase inhibitor (Sigma Aldrich, cat#P2805) on ice. After centrifugation at 14,000×g, supernatants were collected as Triton X-100 soluble fraction. The pellets were further solubilized in 1% SDS buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% SDS) with protease and phosphatase inhibitors on ice for 30 min, centrifuged at 14,000×g and the supernatants were collected as SDS-soluble fraction. The resulting pellets were solubilized in Urea buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mM Tris, 5 mM magnesium acetate, pH 8.5). After centrifugation, the supernatant was collected as urea-soluble fraction. The protein concentration was determined by BCA assay or Pierce™ 660 nm protein assay. Equal amounts (20 µg) of total protein extract were run on SDS–PAGE and transferred to Immobilon-P (EMD Millipore, IPVH 00010). Blots were blocked with 10% nonfat dry milk, and primary and secondary antibodies were applied as previously described [36]. The blots were developed using Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, WBKLS0500) and imaged by the ChemiDoc Imaging System (BioRad), GAPDH (Cell Signaling Technology, 2118), HSP60 (Abcam, ab46798), and Actin (EMD Millipore, MAB1501) were used as a loading control for blot development.
Quantification
Quantification of immunoblots was performed using Image Lab (BioRad, CA). Statistical analysis was done with Student's t-test using GraphPad Prism (GraphPad, CA). Data are means ± SEM. p < 0.05 was considered to be statistically significant.
Results
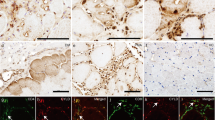
Immunohistochemical analysis of skeletal muscle tissue sections from IBM patients revealed the coexistence of TDP-43, pTDP-43, p62, and mitochondria using total OXPHOS antibody cocktail, which stains for mitochondrial OXPHOS complexes I–V in the same affected muscle fiber groups of IBM patients (Fig. 1). Of note, IBM muscle fibers positive for TDP-43, pTDP-43, or p62 exhibited strong immunoreactivity to OXPHOS mitochondrial markers (Fig. 1). Samples from control subjects all showed negative staining for p62 and pTDP-43, and weak staining for TDP-43 and OXPHOS mitochondrial markers (Fig.1).

Representative immunocytochemistry of TDP-43, pTDP-43, p62, and OXPHOS key subunits in affected muscle fibers in IBM. Adjacent sections show that the same affected IBM muscle fibers positively stained with TDP-43, pTDP-43, and p62 all exhibit intense mitochondrial staining using the OXPHOS antibody cocktail. In contrast, all age-matched control muscle fibers demonstrate relatively weak staining for all proteins. Cytoplasmic TDP-43 or pTDP-43 accumulation is only noted in IBM fibers
To investigate the likely colocalization between TDP-43 pathology and mitochondria, we performed double immunofluorescent analysis using skeletal muscle tissue sections from IBM patients. There was substantial overlap between pTDP-43 and OXPHOS mitochondrial markers (Fig. 2). Remarkably, high magnification and linescan analysis found some mitochondria to be completely colocalized with pTDP-43. However, due to low intensity of immunofluorescent staining for TDP-43, we were not able to assess the colocalization between TDP-43 and mitochondria (data not shown).

Representative double immunofluorescent staining of pTDP-43 and mitochondria in affected muscle fibers in IBM. The large-field and enlarged images show the colocalization between pTDP-43 and mitochondria in IBM muscle biopsy. The line-scan analysis along the solid white line depicted in the merged large-field image to the left is also shown. Green: pTDP-43; Red: mitochondria (stained by the OXPHOS antibody cocktail); Blue: DAPI
Immunoblot analysis of soluble quadriceps muscle tissue lysates prepared from IBM patients was further performed. Consistent with increased p62, pTDP-43 and the pathological forms of truncated TDP-43 (35 and 25 kDa forms) were all found to be increased in IBM cases, when compared with age-matched control subjects (Fig. 3). However, immunoblots of IBM skeletal muscles using the OXPHOS antibody cocktail demonstrated decreased levels of complexes I and III but unchanged complexes II, IV, and V (Fig. 3). To test whether there are biochemical differences in the soluble and insoluble pools of proteins from IBM skeletal muscles, we carried out immunoblot analysis of SDS and urea solubilized skeletal muscle extracts from Triton X-100 insoluble pellets. Consistent with immunoblots of the Triton X-100 soluble fraction, compared with age-matched control subjects, IBM cases showed greatly increased full-length and truncated TDP-43 or pTDP-43 in the SDS-soluble protein fraction (Fig. 4). Notably, truncated TDP-43 was only found in the urea solubilized SDS-insoluble protein fraction in IBM cases (Fig. 4).

Representative immunoblot analysis and quantification of TDP-43, pTDP-43, p62, and OXPHOS key subunits in the soluble protein pool of affected muscle fibers in IBM. Pathogenic pTDP-43 or truncated TDP-43 were significantly increased in the Triton X-100 soluble fraction derived from biopsied muscle fibers from IBM patients, while complexes I and III were significantly reduced (n = 10 per group). The levels were adjusted by GAPDH. Data are means ± s.e.m. Statistical analysis was done with Student's t-test. *P < 0.05, **P < 0.01, ns, not significant

Representative immunoblot analysis and quantification of TDP-43 and pTDP-43 in the insoluble protein pools of affected muscle fibers in IBM. Pathogenic pTDP-43 or truncated TDP-43 were greatly increased in both SDS and urea solubilized skeletal muscle extracts from Triton X-100 insoluble pellets from IBM patients (n = 10 per group). Data are means ± s.e.m. Statistical analysis was done with Student's t-test. *P < 0.05, **P < 0.01
Discussion
TDP-43 proteinopathy and mitochondrial abnormalities are two prominent pathological features of IBM. However, despite intensive effort devoted to understanding the underlying cause(s) of these two prominent IBM pathological features, limited study has been undertaken to identify their association. Here, we reported that pathological TDP-43 is highly colocalized with mitochondria in IBM affected myofibers. It is still unclear whether TDP-43 proteinopathy and mitochondrial abnormalities are interdependent IBM lesions. Of note, the intense mitochondrial staining was noted in muscle fibers negative for TDP-43, pTDP-43, or p62, suggesting that mitochondrial alterations may occur before TDP-43 inclusions or other pathological features of IBM. Noteworthily, although the mitochondrial staining by the OXPHOS antibody cocktail is increased in IBM, the levels of individual proteins recognized by the OXPHOS antibody cocktail were either decreased or unchanged, indicating that the enhanced mitochondrial staining in IBM muscle fibers should be attributed to increased mitochondria mass. As our patient samples are derived from pathologically confirmed cases of IBM 5–10 years after disease onset, future studies will be interesting to investigate the spatiotemporal relationship between TDP-43 proteinopathy and mitochondrial abnormalities during disease progression.
A growing body of evidence indicates mitochondria as important targets of TDP-43 [30,31,32,33,34,35,36]. However, there are considerable discrepancies as to its exact sub-mitochondrial localization. Full-length TDP-43 or truncated forms of TDP-43 have been reported either inside or outside of mitochondria [30,31,32,33,34,35,36]. The sub-mitochondrial localization of TDP-43 in IBM is still unknown. Consistent with our previous study reporting TDP-43 inside of mitochondria as highly phosphorylated TDP-43 in ALS and FTD [36], our results suggest that the species of TDP-43 highly associated with mitochondria are largely pTDP-43. Despite controversy on the effect of TDP-43 on mitochondrial function, we and other groups consistently demonstrated that TDP-43 overexpression caused mitochondrial abnormalities and neuronal dysfunction [26, 27, 29, 30, 32, 34, 36, 38]. The most recent independent study showed that truncated TDP-43 lacking the M1 motif was not present inside of the inner mitochondrial membrane and had no effect on mitochondrial function [30]. Consistently, we reported that the inhibition of mitochondrial TDP-43 by PM1 or the genetic deletion of PM1 alleviated TDP-43 toxicity on mitochondria and neurons, together suggesting that the mitochondrial localization likely plays an important role in mediating TDP-43 toxicity. Nevertheless, considering recent discrepant studies reporting the interaction of TDP-43 with different mitochondrial function and pathways, mitochondria-associated TDP-43 or truncated TDP-43 fragments noted in IBM may synergistically mediate toxicity on mitochondria and muscle fibers through multiple pathways involving but not limited to bioenergetics, mitochondrial dynamics, and ER/mitochondria tethering.
Similar to the previous study showing downregulated expression of complex I of mitochondrial respiratory chain as the initial feature [39], this study found that key subunits of complexes I and III were significantly decreased in IBM muscles. These findings are indeed consistent with our and other studies reporting complex I as the major target of TDP-43 inside of mitochondria [30, 36]. Therefore, it is possible that the changed assembly or dysfunction of OXPHOS complex I caused by mitochondrial-associated TDP-43 may play an unexpected but critical role in the onset and progression of IBM. The interplay between TDP-43 and OXPHOS complex I in IBM needs further detailed investigation. Of course, other OXPHOS complexes as well as mtDNA have also reported changes in IBM patients, suggesting the possible presence of other targets. Nevertheless, as mitochondria-encoded CoxII remains unchanged in IBM, the intact overall mitochondrial transcription and translation machineries or mitochondrial tRNAs and rRNAs should not be affected.
In this study, we provide timely evidence showing the close association between TDP-43 pathology and mitochondria in IBM affected skeletal muscles, and suggest mitochondria-associated TDP-43 as a likely contributor to mitochondrial and muscle dysfunction. Like mitochondrial dysfunction, TDP-43 proteinopathy is a prominent common pathological feature in various major neurodegenerative diseases including AD [40, 41], FTD [42, 43], and ALS [42, 43]. We believe that the further detailed investigation of the interplay between these two pathological features and their contribution to disease progress will provide new insights into these devastating diseases. Importantly, the suppression of TDP-43 mitochondrial association is sufficient to greatly prevent TDP-43-mediated neuronal toxicity [36], suggesting that targeting mitochondria-associated TDP-43 can be a novel therapeutic approach for IBM worthy of further translational exploration.
References
Naddaf E, Barohn RJ, Dimachkie MM. Inclusion body myositis: update on pathogenesis and treatment. Neurotherapeutics. 2018;15:995–1005.
Callan A, Capkun G, Vasanthaprasad V, Freitas R, Needham M. A systematic review and meta-analysis of prevalence studies of sporadic inclusion body myositis. J Neuromuscul Dis. 2017;4:127–37.
Dimachkie MM, Barohn RJ. Inclusion body myositis. Curr Neurol Neurosci Rep. 2013;13:321.
Catalan-Garcia M, Garrabou G, Moren C, Guitart-Mampel M, Hernando A, Diaz-Ramos A, et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci. 2016;130:1741–51.
Machado PM, Ahmed M, Brady S, Gang Q, Healy E, Morrow JM, et al. Ongoing developments in sporadic inclusion body myositis. Curr Rheumatol Rep. 2014;16:477.
Needham M, Corbett A, Day T, Christiansen F, Fabian V, Mastaglia FL. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J Clin Neurosci. 2008;15:1350–3.
Capkun G, Callan A, Tian H, Wei Z, Zhao C, Agashivala N, et al. Burden of illness and healthcare resource use in United States patients with sporadic inclusion body myositis. Muscle Nerve. 2017;56:861–7.
Peng A, Koffman BM, Malley JD, Dalakas MC. Disease progression in sporadic inclusion body myositis: observations in 78 patients. Neurology. 2000;55:296–8.
Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJ, Badrising UA. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain. 2011;134:3167–75.
Benveniste O, Guiguet M, Freebody J, Dubourg O, Squier W, Maisonobe T, et al. Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134:3176–84.
Oh TH, Brumfield KA, Hoskin TL, Stolp KA, Murray JA, Bassford JR. Dysphagia in inflammatory myopathy: clinical characteristics, treatment strategies, and outcome in 62 patients. Mayo Clin Proc. 2007;82:441–7.
Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain. 1989;112:727–47.
Dimachkie MM, Barohn RJ. Inclusion body myositis. Neurol Clin. 2014;32:629–46.
Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol. 2007;6:620–31.
Engel WK, Askanas V. Inclusion-body myositis: clinical, diagnostic, and pathologic aspects. Neurology. 2006;66:S20–9.
Weihl CC, Mammen AL. Sporadic inclusion body myositis—a myodegenerative disease or an inflammatory myopathy. Neuropathol Appl Neurobiol. 2017;43:82–91.
Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007.
Kusters B, van Hoeve BJ, Schelhaas HJ, Ter Laak H, van Engelen BG, Lammens M. TDP-43 accumulation is common in myopathies with rimmed vacuoles. Acta Neuropathol. 2009;117:209–11.
Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79:1186–9.
Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, et al. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009;40:19–31.
Olive M, Janue A, Moreno D, Gamez J, Torrejon-Escribano B, Ferrer I. TAR DNA-binding protein 43 accumulation in protein aggregate myopathies. J Neuropathol Exp Neurol. 2009;68:262–73.
Hernandez Lain A, Millecamps S, Dubourg O, Salachas F, Bruneteau G, Lacomblez L, et al. Abnormal TDP-43 and FUS proteins in muscles of sporadic IBM: similarities in a TARDBP-linked ALS patient. J Neurol Neurosurg Psychiatry. 2011;82:1414–6.
D’Agostino C, Nogalska A, Engel WK, Askanas V. In sporadic inclusion body myositis muscle fibres TDP-43-positive inclusions are less frequent and robust than p62 inclusions, and are not associated with paired helical filaments. Neuropathol Appl Neurobiol. 2011;37:315–20.
Rose MR, Group EIW. 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23:1044–55.
Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–9.
Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, et al. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Human Mol Genet. 2013;22:4706–19.
Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Human Mol Genet. 2014;23:1413–24.
Lu J, Duan W, Guo Y, Jiang H, Li Z, Huang J, et al. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res Bull. 2012;89:185–90.
Stribl C, Samara A, Trumbach D, Peis R, Neumann M, Fuchs H, et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J Biol Chem. 2014;289:10769–84.
Salvatori I, Ferri A, Scaricamazza S, Giovannelli I, Serrano A, Rossi S, et al. Differential toxicity of TDP-43 isoforms depends on their sub-mitochondrial localization in neuronal cells. J Neurochem. 2018;146:585–97.
Genin EC, Bannwarth S, Lespinasse F, Ortega-Vila B, Fragaki K, Itoh K, et al. Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol Dis. 2018;119:159–71.
Davis SA, Itaman S, Khalid-Janney CM, Sherard JA, Dowell JA, Cairns NJ, et al. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci Lett. 2018;678:8–15.
Woo JA, Liu T, Trotter C, Fang CC, De Narvaez E, LePochat P, et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat Commun. 2017;8:15558.
Izumikawa K, Nobe Y, Yoshikawa H, Ishikawa H, Miura Y, Nakayama H, et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci Rep. 2017;7:7709.
Hibiki Kawamata PP, Konrad Csaba, Palomo Gloria, Bredvik Kirsten, Gerges Meri, Valsecchi Federica, et al. Mutant TDP-43 does not impair mitochondrial bioenergetics in vitro and in vivo. Mol Neurodegener. 2017;12:37.
Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22:869–78.
Prayson RA, Cohen ML. Ubiquitin immunostaining and inclusion body myositis: study of 30 patients with inclusion body myositis. Hum Pathol. 1997;28:887–92.
Xu YF, Zhang YJ, Lin WL, Cao X, Stetler C, Dickson DW, et al. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6:73.
Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW, Turnbull DM. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol Appl Neurobiol. 2015;41:288–303.
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–45.
Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014;127:441–50.
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
Acknowledgements
This work was supported by the US National Institutes of Health (1R01NS089604 to X.W.) and the US Alzheimer’s Association (AARG-17-499682 to X.W.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huntley, M.L., Gao, J., Termsarasab, P. et al. Association between TDP-43 and mitochondria in inclusion body myositis. Lab Invest 99, 1041–1048 (2019). https://doi.org/10.1038/s41374-019-0233-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0233-x
This article is cited by
-
Cytotoxic immune cells do not affect TDP-43 and p62 sarcoplasmic aggregation but influence TDP-43 localisation
Scientific Reports (2023)
-
Feeder-supported in vitro exercise model using human satellite cells from patients with sporadic inclusion body myositis
Scientific Reports (2022)
-
Maintaining the balance of TDP-43, mitochondria, and autophagy: a promising therapeutic strategy for neurodegenerative diseases
Translational Neurodegeneration (2020)
