
Abstract
Activation of inflammation is an important mechanism in the development of nonalcoholic steatohepatitis (NASH). This study aims to delineate how mitophagy affects NLRP3 inflammasome activation in hepatic lipotoxicity. Mice were fed a high fat/calorie diet (HFCD) for 24 weeks. Primary rat hepatocytes were treated with palmitic acid (PA) for various periods of time. Mitophagy was measured by protein levels of LC3II and P62. NLRP3, caspase-1, interleukin (IL)-18, and IL-1β at mRNA and protein levels were used as indicators of inflammasome activation. Along with steatotic progression in HFCD-fed mice, ratio of LC3II/β-actin was decreased concurrently with increased levels of liver P62, NLRP3, caspase-1, IL-1β, IL-18, and serum IL-1β levels in late-stage NASH. PA treatment resulted in mitochondrial oxidative stress and initiated mitophagy in primary hepatocytes. The addition of cyclosporine A did not change LC3II/Τοmm20 ratios; but P62 levels were increased after an extended duration of PA exposure, indicating a defect in autophagic activity. Along with impaired mitophagy, mRNA and protein levels of NLRP3, caspase-1, IL-18 and IL-1β were upregulated by PA treatment. Pretreatment with MCC950, N-acetyl cysteine or acetyl-l-carnitine reversed inflammasome activation and a pyroptotic cascade. Additionally, mitophagic flux was partially recovered as indicated by increases in LC3II/Tomm20 ratio, parkin, and PINK1 expression, and decreased P62 expression. The findings suggest that impaired mitophagy triggers hepatic NLRP3 inflammasome activation in a murine NASH model and primary hepatocytes. The new insights into inflammasome activation through mitophagy advance our understanding of how fatty acids elicit lipotoxicity through oxidant stress and autophagy in mitochondria.
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) is a pathophysiologic syndrome with progression from nonalcoholic fatty liver (NAFL), to nonalcoholic steatohepatitis (NASH) to NASH-associated fibrosis and cirrhosis or cancer [1]. Its pathogenesis is complex and unclear. More importantly, there are no Food and Drug Administration-approved medications available for NASH treatment [2]. Therefore, exploration of the mechanism underlying the progression from NAFL to NASH may contribute to changes in current NASH treatments [2].
The pathologic characteristics of NASH are increased fatty acid content in hepatocytes and lipotoxicity through oxidant stress, endoplasmic reticulum stress, insulin resistance and hepatocellular death [3, 4]. Mitochondrial damage contributes to the progression from NAFL to NASH [5, 6]. As a critical site for fatty acid β-oxidation and tricarboxylic acid cycle for energy-coupling formation, mitochondria play a pivotal role in fatty acid consumption. Mitochondrial function and homeostasis, which are fundamental for the maintenance of normal energy, oxidant and metabolic status, are often impaired during overload of free fatty acids (FFA) [7]. Increased FFA content enhanced lipotoxicity through increased generation of reactive oxygen species (ROS) within steatotic hepatocytes [8]. One of ROS sources is from NADPH oxidase-2 (NOX-2) [9]. Mitochondria are one of the major sources of ROS (mtROS) that is generated by respiratory chain complex enzymes during energy formation [5, 6]. ROS, as the first hit, directly causes mitochondrial swelling and suppresses activity of the respiratory chain complex enzymes. As a result, ATP synthesis declines in hepatocellular mitochondria, and in turn, leads to apoptosis through a mitochondrial pathway [6]. Under physiological conditions, damaged mitochondria are removed through an autophagic mechanism, a process called mitochondrial autophagy or mitophagy [10]. Impaired autophagic machinery leads to accumulation of excessive ROS production and triggers cell death through apoptosis and necrosis [11]. However, the molecular interplay underlining this pathway in the progression from NAFL to NASH has not yet been elucidated.
Nod-like receptors (NLRs) are a group of innate immunity molecules that sense extracellular and intracellular environmental changes and elicit inflammatory responses. As one of major inflammasome molecules [12], NLRP3 is closely associated with many inflammatory diseases [13]. In a recent study we found that NLRP1/3 inflammasome levels are increased in both parenchymal and nonparenchymal cell types in a mouse model of NASH caused by HFCD plus high fructose and glucose in drinking water, and to regulate the mature and secretion of inflammatory IL-1β and IL-18 cytokines [14]. Reducing NLRP3 inflammasome activity significantly improved obesity-associated metabolic abnormalities [15,16,17]. Recent studies have demonstrated that normal mitophagic activity minimized NLRP3 inflammasome activation by decreasing mtROS generation in other systems [18, 19]. When mitophagy is impaired, damaged mitochondria will not be eliminated through action of PAMPs (pathogen-associated molecular patterns) or DAMPs (damage-associated molecular patterns) [20]. Deficiency of effective mitochondrial autophagy leads to excessive NLRP3 inflammasome activation, in turn, causing inflammation, tissue damage, organ dysfunction and eventually several diseases, such as type 2 diabetes and obesity [21]. However, the relationship between the mitophagic activity in hepatocytes and the activation of NLRP3 in NASH has not been characterized. Therefore, we hypothesize that mitochondrial autophagy plays an important role in the progression from NAFL to NASH, and assume that the compromised mitophagy may be a key step to increase oxidative stress in hepatocytes, which leads to the persistent activation of the inflammasome.
The overall aim of the present study is to investigate whether dysregulated lipid metabolism leads to impairment of mitophagy in steatotic hepatocytes, and in turn induces the activation of NLRP3 inflammasome and subsequent events contributing to a necroptotic cascade in the progression from NAFL to NASH. Our data provide direct evidence to support the theory that overwhelming FFA accumulation leads to mitochondrial damage with compromised mitophagy, which subsequently triggers inflammasome activation and results in necroptosis through a mitochondrial pathway. Elucidating the critical role of impaired mitophagy in a paradigm of lipotoxicity will deepen our understanding of molecular mechanisms in NASH initiation and progression, and aid in development of molecular targets for pharmacotherapeutic intervention.
Materials and methods
Animals and diets
Male 8-week-old C57BL/6 mice purchased from Shanghai Model Organisms Center (Shanghai, China) were maintained in light/dark (12 h light/12 h dark) cycle, temperature (22 °C) and humidity-controlled rooms with free access to drinking water. Mice were fed high fat/calorie diet (HFCD) (D12492i-rodent diet with 60 kcal% fat from Research Diets, New Brunswick, NJ, USA) or regular pellet chew diet (RD) for 24 weeks. All animal experimental procedures were approved by the Animal Ethic Committee of Fudan University School of Basic Medical Sciences and performed following the NIH Guidelines of Experimental Animal Handling and Use.
Serological and liver biochemical analysis
Mouse blood was collected from eye orbit bleeding, and serum was separated by centrifugation (2000 × g for 10 min at 4 °C). Serum levels of triglyceride (TG), total cholesterol (TC), alanine transaminase (ALT) and aspartate transaminase (AST) were determined by an automated biochemical analyzer (Beckman Coulter Company, Fullerton, CA, USA). Triglyceride levels in mouse liver were measured with commercially available detection kits (Applygen Technologies, Beijing, China) after being homogenized by a homogenizer (Shanghai Biheng Biotechnology Company, Shanghai, China). Serum IL-1β level was quantified with a mouse IL-1β ELISA kit from R&D Systems (Minneapolis, MN, USA), according to the manufacturer’s instruction.
Liver histopathologic examination
Fresh mouse liver tissue was fixed in 10% neutral formalin, embedded in paraffin, and sectioned in 4 μm thickness. Liver sections were stained in hematoxylin and eosin (H&E). The extent of liver fibrosis was visualized after Masson’s Trichrome staining (Sigma-Aldrich, St. Louis, MO, USA). Hepatocellular apoptosis in the liver was determined after TdT-mediated dUTP nick-end labeling (TUNEL) staining (Roche, Indianapolis, IN, USA). Mitophagy was indicated by double immunofluorescent staining of COX IV (cytochrome C oxidase IV as a mitochondrial marker) and LC3B in 5-μm-thick liver sections and observed under a confocal microscope (TCS SP8, Leica, Germany).
Immunofluorescent staining and confocal microscopic examination
Frozen liver sections were fixed in 4% neutral paraformaldehyde, permeabilized with 0.1% Triton X, blocked with 3% albumin, and were incubated overnight with primary antibodies, including anti-COX IV and anti-LC3B (information of all antibodies used is provided in Supplemental Table 1). Primary hepatocytes were seeded at 1.5 × 105 cells per well in six-well plates, treated with palmitic acid (PA, 200 μM) for 0, 1, 2, 6, 10 and 24 h. After the treatment, hepatocytes were washed three times with sterile phosphate buffered saline (PBS) and fixed with 4% neutral paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100 and blocked in 3% bovine serum albumin (BSA). They were then incubated overnight with primary antibodies, including anti-LC3B. Hepatocytes were further stained with MitoTracker (Invitrogen, Carlsbad, CA, USA) prior to PFA fixation. Secondary Alexa-488 or -594-conjugated antibodies (Life Technologies, Grand Island, NY, USA) were added for 1 h and DAPI was used for nuclear counterstaining. Stained cells were visualized under a confocal microscope within 24 h after mounting.
Hepatocyte culture and palmitic acid treatment
Isolation of rat hepatocytes was based on the two-step collagenase procedure as described by us previously [22]. Primary hepatocytes were cultured in William’s E medium supplemented with 10% FFA-free BSA at 37 °C with 5% CO2 [22]. PA (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in methyl alcohol at a stock concentration of 50 mM and added to William’s E medium containing 10% FFA-free BSA to make a final concentration of 200 μM in experiments. Primary hepatocytes were treated with PA for 0, 1, 2, 6, 10 and 24 h respectively. MCC950, a selective NLRP3 inhibitor from Selleck Chemicals (Houston, TX, USA) was dissolved in water as stock solution and added to culture medium to reach a final concentration of 50 μM. Cyclosporine A (CsA) (1 μM) were used as autophagy and mitophagy inhibitors. N-Acetyl cysteine (NAC, 20 mM) [23] and acetyl-l-carnitine (ALCAR, 1 mM) [24] were used as antioxidants.
MitoTracker and LysoTracker staining of mitochondria and lysosomes
To detect the mitophagic status and mitochondrial morphology in PA-treated primary hepatocytes, MitoTracker (Invitrogen, Carlsbad, CA, USA) was applied to stain the mitochondria in green and LysoTracker (Invitrogen, Carlsbad, CA, USA) was applied to visualize acidic lysosomes in red. Primary hepatocytes were plated at 1.0−1.5 × 105 cells/well in six-well plates. After being treated with PA at 200 μM for a different duration, hepatocytes were stained with MitoTracker in combination with LC3B antibody (Novus, Littleton, CO, USA) or with a LysoTracker probe, and thereafter were visualized under a confocal microscope.
Detection of cytosolic ROS
Intracellular ROS levels were measured using 2,7-dichlorodi-hydrofluorescein diacetate (DCFH-DA) as described previously [25, 26]. Primary hepatocytes were treated with PA at 200 μM for 0, 1, 2, 4, 6, 10, and 24 h and incubated with serum-free medium containing 10 μM of DCFH-DA for 30 min at 28 °C in dark. After incubation, hepatocytes were stained with cell-permeable nuclear dye Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) and images were acquired using the Operetta High Content Imaging System (PE Operetta, PerkinElmer, Inc., Waltham, MA, USA) with a ×20 high NA objective lens in a wide-field fluorescent mode. Primary hepatocytes were rinsed twice with William’s E medium and fluorescent DCF intensity was detected by a multimode plate reader at an excitation wavelength of 488 nm and emission wavelength of 525 nm.
Measurement of mitochondrial inner membrane potential
Primary hepatocytes were seeded at 1.5 × 105 cells per well in six-well plates. Mitochondrial inner membrane potential (MMP, ΔΨm) was measured using tetraethyl benzimidazolyl carbocyanine iodide (JC-1) as a fluorescent probe (Beyotime Biotechnology, Nantong, China) according to the manufacturer’s instructions [27]. Briefly, primary hepatocytes were pretreated with or without NAC or ALCAR followed by PA (200 μM) exposure. Treated hepatocytes were incubated with JC-1 staining solution (5×) for 20 min at 37 °C. Hepatocytes were then washed twice and resuspended in staining buffer (1×) containing JC-1. Fluorescent intensity of both mitochondrial JC-1 monomers (ex 490 nm/em 530 nm) and aggregates (ex 525 nm/em 590 nm) from hepatocytes was measured in a multimode plate reader. Data were normalized to carbonyl cyanide m-chlorophenyl hydrazone (CCCP)-treated positive control.
Detection of mitochondrial ROS
Mitochondrial ROS (mtROS) was measured using MitoSOX (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Primary hepatocytes were treated with PA in different concentration for various durations up to 24 h, or treated with PA at 200 μM plus/minus NAC and ALCAR. After incubation, hepatocytes were washed with William’s E medium, loaded with MitoSOX at 5 μM for 20 min at 37 °C, and washed three times with serum-free William’s E medium. Afterwards, hepatocytes were stained with nuclear dye Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) and images were acquired using the Operetta high content Imaging System. Fluorescent intensity was determined using a multimode plate reader at excitation of 510 nm and emission of 580 nm wavelength [27].
Mitochondrial and cytosolic fractionation
A cell mitochondrial isolation kit (Beyotime Biotechnology, Nantong, China) was used for the isolation of mitochondrial and cytosolic fractions following a modified protocol described previously [28]. Briefly, primary hepatocytes were cultured, trypsinized, and resuspended in extraction buffer from the kit. The lysates were centrifuged at 1000 × g for 10 min at 4 °C. The supernatant was additionally centrifuged at 3500 × g for 10 min to enrich the mitochondrial fraction. The supernatant from the last centrifugation (cytosolic protein) and the final pellet (mitochondrial protein) were subjected separately to Western blot analysis of specific protein.
Measurement of cytosolic mtDNA
Primary hepatocytes were treated with PA at 200 μM for a different duration. Cellular fractionation was then performed using a mitochondrial isolation kit (Beyotime Biotechnology, Nantong, China) according to the manufacturer’s instructions. Isolation of cytosolic mtDNA was performed by using a mitochondrial isolation kit as previously described [29]. Relative mtDNA copy number was measured using quantitative real-time PCR with the SYBR Green kit (Roche Molecular, Pleasanton, CA, USA). mtDNA quantity was normalized by simultaneous measurement of the nuclear gene β-globin and mitochondrial NADH dehydrogenase subunit 1 (ND1) (primer sequences were listed in Supplemental Table 2). ND1 was measured by quantitative real-time PCR with same volume of the DNA solution. Nuclear DNA encoding β-globin was used for normalization. mtDNA copy number was calculated using the following equation: relative copy number = 2ΔCt (ΔCt = Ctβ-globin − CtND1) [30].
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) release in culture supernatants was measured by an LDH Cytotoxicity Detection Kit (Beyotime Biotechnology, Nantong, China) according to the procedures reported previously [31]. Briefly, primary hepatocytes were seeded at 1.0 × 104 cells per well in 96-well plates. After treatment with PA (50, 100, 200 and 500 μM) for 1, 2, 4, 6, 10, and 24 h, cells in culture plates were centrifuged at 400 × g for 5 min. Then 120 μL of cell-free supernatant (William’s E with 1% fetal bovine serum (FBS) as cell culture medium) was incubated with 20 μL of LDH substrate solution in a clear 96-well plate for 30 min. The absorbance in optical density (OD) at 490 nm was read in a microplate reader (FlexStation 3, Molecular Devices, San Jose, CA, USA). The percentage of LDH release was calculated as follows: sample concentration (U/mL) = [(the OD value of sample − the OD value of control)/(the OD value of standard enzyme − the OD value of blank)] × (concentration of standard enzyme). Fold change in LDH release was expressed as a relative value compared to the negative control.
Protein extracts and immunoblotting analysis
Liver samples or primary hepatocytes were treated with RIPA Lysis Buffer (Beyotime Biotechnology, Nantong, China) containing protease inhibitor cocktail (Pierce, Rockford, IL, USA) to extract protein. After protein content was determined with BCA reagent (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 50 μg of total protein was boiled in loading buffer and loaded into 8–15% SDS-PAGE gel. Proteins were transferred to PVDF membranes after electrophoresis, blocked with 5% non-fat dry milk, and incubated overnight separately with primary antibodies against LC3, P62, Parkin, PINK1, NLRP3, caspase-1, IL-18 and IL-1β (Supplemental Table 1), followed by appropriate secondary horseradish peroxidase (HRP)-conjugated antibodies. Immunoreactive bands were visualized using the ECL Western blotting protocol (Millipore, Billerica, MA, USA). Densitometric analysis of the bands was performed using the ImageJ software (NIH, Bethesda, MD, USA). β-Actin or Tomm20 was used as a loading control of cytosolic or mitochondrial protein [32].
Total RNA isolation and quantification
Total RNA was isolated from mouse liver tissue or cells using TRIZOL reagent (Invitrogen Corporation, Carlsbad, CA, USA) following a standard protocol as described previously [33] and the reverse transcription reaction was performed using a Prime-Script RT reagent Kit (Takara Bio, Dalian, China). Real-time PCR quantification was performed using SYBR green as a fluorescent indicator in a CFX96 Thermal Cycler from Bio-Rad. Briefly, 10 μL of reaction mix containing cDNA, KAPA SYBR FAST qPCR master mix, and primers at a final concentration of 200 nmol was used in the reaction system. The sequences of the primers used for quantitative PCR are given in Supplemental Table 2. Relative target gene expression was calculated using 2−ΔΔCt method and normalized by house-keeping gene β-actin control [33].
Caspase-1 activity assay
Caspase-1 activity was determined by an assay kit (Beyotime Biotechnology, Nantong, China) according to the procedures described previously [26]. Briefly, the protein was extracted from primary hepatocytes treated with or without PA plus/minus MCC950, and quantitated by the Bradford Protein Assay Kit (Beyotime Biotechnology, Nantong, China). A total of 30 μg of protein from each sample was incubated with 10 μL of Ac-YVAD-pNA (2 mM) substrate in a total volume of 100 μL assay buffer for 1, 2, 4, 6, 10 and 24 h at 37 °C. Absorbance was read at 405 nm wavelength in a microplate reader. Caspase-1 activity was normalized by protein content of each supernatant, and expressed as a relative value compared to the negative control.
Statistical analysis
Data are presented as means and standard errors. Normal distribution test was performed prior to ANOVA test. Statistical significance was determined with the nonparametric Kruskal−Wallis test and the Mann−Whitney U test when appropriate for multicomparison between given two groups. Statistical analysis was undertaken with SPSS 19.0 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism 6.0 (GraphPad Software, La Jolla, USA). A p value ≤ 0.05 is considered statistically significant.
Results
Nonalcoholic steatohepatitis induced by high fat/calorie diet (HFCD) in mice
Compared to mice fed regular diet (RD), body weight and liver weight (Supplemental Fig. 1A&B) increased significantly in mice fed HFCD over 24 weeks. Levels of serum ALT, AST, fasting blood glucose and liver TG were significantly higher than those in the control group (RD) from 6 to 24 weeks (Supplemental Fig. 1C–F). Liver fat accumulation is presented as microsteatosis to macrosteatosis and mixed size of lipid droplets. A few cells seem to enter into piece or focal death with loss of nucleus, breakage into pieces or inflammatory infiltration at the site of cell loss. Inflammatory infiltration and mild fibrosis are visible especially in the portal triads of liver sections with H&E and Masson’s Trichrome staining from mice fed HFCD (Supplemental Fig. 2). Apoptotic cells were visualized on sections from mice fed HFCD for 12 weeks, but barely positive in RD-fed mice. The apoptotic cell counts were positively correlated with the degree of liver steatosis and inflammation (Supplemental Fig. 1G and Supplemental Fig. 2). In summary, feeding HFCD for 24 weeks resulted in remarkable fat accumulation, hepatic cell death with elevation of serum aminotransferases and apoptotic cell count. Inflammatory infiltration and mild fibrosis were visible majorly in the portal triads. All these findings confirm that NASH was induced by feeding mice HFCD for 24 weeks.
Impaired mitophagy at late-stage steatohepatitis in HFCD-fed mice
In order to determine the presence of mitophagy in HFCD-fed mice, LC3B was stained to indicate autophagic activity; and a mitochondrial enzyme, cytochrome oxidase IV (COX IV), was costained with LC3B to indicate mitochondrial involvement. It appeared that LC3B-positive cells were seen, and colocalized with COX IV; and that the colocalization was enhanced in mice fed HFCD for 6 weeks (Fig. 1). However, during a longer timeframe, less LC3B-positive cells were confirmed to be colocalized with mitochondrial protein marker COX IV by confocal microscopic observation, which suggests that the mitochondrial autophagy level was initially increased at 6 weeks, but started to decline at 12 weeks and almost disappeared at 24 weeks of HFCD feeding (Fig. 1). Consistent with colocalization of LC3B with COX IV in immunohistochemical staining, levels of LC3II and mitochondrial proteins such as parkin and PINK1 were initially elevated at 6 weeks, and decreased at 12–24 weeks in HFCD-fed mice (Fig. 2A). P62 is a ubiquitin-binding protein involved in autophagy. Lysosomal degradation of autophagosome leads to a decrease in P62 levels during autophagy. Therefore, altered P62 levels in an opposite direction found in Western blot analysis indicated lysosomal incorporation into autophagosome during a later stage of autophagy (Fig. 2A). To evaluate the mitophagic activity, we determined mRNA or mitochondrial protein levels of PARK2/parkin and PINK1 by Western blot analysis using β-actin as a loading control. As shown in Fig. 2A, protein levels of parkin and PINK1 were elevated in mice during the first 6 weeks of HFCD feeding, confirming that mitochondrial autophagy activity was enhanced at an early stage of steatosis. By 12 weeks of HFCD feeding, both parkin and PINK1 at protein levels, as well as PARK2 and PINK1 at mRNA levels started to decline significantly (Supplemental Fig. 1J&K) in comparison with those fed RD. By 24 weeks, P62 protein levels were significantly increased; whereas PARK2 and PINK1 mRNA levels were significantly suppressed, indicating that autophagic activity was declined along with increased steatotic extent and inflammation in HFCD-fed mice (Supplemental Fig. 1J&K). Consistent with changes of PARK2 and PINK1, mRNA levels of Atg5 and Atg7, markers of autophagy, were decreased after feeding HFCD for 12 weeks, confirming impaired autophagy at this stage (Supplemental Fig. 1H&I). In summary, it appeared that increased mitophagy did happen at an early stage of fat accumulation, and mitophagic activity was declined with increased severity of steatosis, necroptotic activity and inflammatory response in the liver over time, and may represent as deterioration in clearance of damaged mitochondria.

Colocalization of LC3B and COX IV in mouse liver. C57BL/6J mice were fed a control diet (regular diet, RD) or high fat-calorie diet (HFCD) for 6, 12, and 24 weeks. Mice fed for 2 weeks were used as baseline controls. Representative micrographs of fluorescent immunohistochemical costaining of LC3B (green) and COX IV (red) in frozen sections after feeding for 6, 12, and 24 weeks are shown. DAPI was used for nucleus staining (blue). Orange color (as indicated by yellow bars) in the merged panel represents the overlapping of LC3B staining (green) with COX IV (red). Images were recorded at original magnification (×400). Scale bars = 10 μm

Expression levels of autophagy-associated proteins and genes in mouse liver. A Parkin, PINK1, LC3, and P62 protein levels in mouse (RD and HFCD group) liver at 6, 12, and 24 weeks were analyzed by the Western blot analysis. B Western blot analysis of NLRP3, caspase-1, IL-18, and IL-1β protein levels in livers of mice fed a control diet (regular diet, RD) or high fat-calorie diet (HFCD). C Densitometric analysis of Western blot images. The densitometric ratio of Parkin, PINK1, LC3II, and P62 at 6, 12, and 24 weeks in total protein level (n = 3) using β-actin as a loading control. The densitometric ratio of NLRP3, caspase-1, IL-18, and IL-1β at 6, 12, and 24 weeks in total protein level (n = 3) using β-actin as a loading control. **p ≤ 0.01 compared to the controls (RD). NLRP3: NOD-like receptor-3
NLRP3 activation during late-stage NASH development
Activation of nod-like receptors, such as NLRP3, is an innate response to changes in extracellular and intracellular microenvironment. NLRP3 activation elicits a pyroptotic cascade that leads to cleavage of procaspase-1 into a matured caspase-1; in turn, the latter further catalyzes the cleavage of prointerleukin-1β (IL-1β) and prointerleukin-18 (IL-18). It is well-known that mature IL-1β and IL-18 are potent intermediates triggering inflammatory and fibrotic responses in the liver. As shown in Fig. 2B and Supplemental Fig. 3A–E, NLRP3 and ASC protein levels in mice fed HFCD deceased at 6 weeks. Accordingly, liver protein levels of cleaved caspase-1, IL-1β and IL-18 deceased in mice fed HFCD compared to those fed RD during the first 6 weeks (Fig. 2B, C). Both NLRP3 and its downstream proteins, i.e. cleaved caspase-1, IL-1β and IL-18, as well serum IL-1β levels (Supplemental Fig. 3F), were upregulated after 12 weeks of HFCD feeding, and were sustained at higher levels than those in mice fed RD (Fig. 2B, C). Mice on the RD diet do not show mitophagic activity (Figs. 1, 2A) or lipotoxicity, as demonstrated by steatohepatitis and liver necroptosis in both biochemical tests and histopathology (Supplemental Figs. 1&2).
PA treatment resulted in mitochondrial oxidative stress and impaired mitophagy
In order to further determine mitophagic status during lipotoxicity, primary hepatocytes were used as an in vitro model to assess lipotoxicity with overload of saturated fatty acids (palmitic acid, PA). After treatment with PA at 200 μM, JC-1 fluorescent intensity was significantly reduced during the first 10 h of PA exposure (Supplemental Fig. 6B), indicating there was a decrease in mitochondrial inner membrane potential (MMP). A decrease in ATP (Supplemental Fig. 6E) and MMP, together with a reduced mtDNA levels (Supplemental Fig. 6F) was a characteristic of mitochondrial dysfunction. Both cytosolic and mitochondrial levels of superoxide anions were increased as indicated by DCF and Mitosox fluorescent intensity 2 h after exposure to PA (Supplemental Figs. 4, 5, 6C&D). The increase in cytosolic and mitochondrial oxidant stress and MMP loss resulted in deterioration of hepatocellular viability as seen in elevated LDH release (Supplemental Fig. 6A).
In order to further confirm mitochondrial autophagic activity during lipotoxicity, LC3B and MitoTracker were used to determine autophagy over time during exposure to PA. As shown in Fig. 3, during PA exposure for the first 2 h, hepatocellular mitochondria maintained their abundance as reflected by MitoTracker, and there was increased green fluorescent staining of LC3B, which was colocalized with MitoTracker staining (Supplemental Fig. 5). However, mitochondrial MitoTracker staining became very faint although LC3B staining was detected, indicating that there was a notable loss of mitochondria at this time point. At the same time, protein levels of both LC3II and LC3I were decreased after an initial increase, and the ratio of LC3II/Tomm20 was decreased consistently after 2 h, indicating that mitophagic activity was gradually decreased [11], which was further confirmed by increased mitochondrial P62 protein levels [11] (Fig. 4A–C). Atg5 and Atg7 mRNA levels were decreased shortly followed with an initial increase, indicating a conversion from an increased autophagy level to a compromised status (Supplemental Fig. 7B). To further verify whether the mitophagy was impaired, mRNA levels of PARK2 and PINK1 were confirmed to follow the same trend as Atg5 and Atg7 during late-stages of PA exposure (Supplemental Fig. 7B).

PA treatment resulted in impaired mitophagy in primary hepatocytes. Representative confocal micrographs of MitoTracker (red) and LC3B (green) coimmunofluorescent staining in primary hepatocytes treated with PA for different durations (indicated on the left). DAPI was used for nuclear staining (blue). Orange color represents the overlapping of MitoTracker with LC3B (merged panel). Images were taken at original magnification (×400). Scale bars = 7.5 μm

NLRP3 inflammasome activation in PA-treated primary hepatocytes. A Western blot analysis of mitochondrial protein LC3B and P62 in primary hepatocytes treated with PA. Protein levels of NLRP3, caspase-1, IL-18, and IL-1β in primary hepatocytes treated with PA for different duration were assayed by Western blot analysis. The densitometric ratio of LC3II over Tomm20 (B) and P62 over Tomm20 in the mitochondrial fraction (C) (n = 3). **p ≤ 0.01 compared to the BSA controls or as indicated. The densitometric ratio of NLRP3 (D), caspase-1 (E), IL-1β (F), and IL-18 (G) over β-actin loading control at different time points (n = 3). *,**p ≤ 0.05 and 0.01 compared to the BSA controls or as indicated. H Activity of caspase-1 in cytosolic protein of PA-treated primary hepatocytes. **p ≤ 0.01 compared to the BSA controls or as indicated. I−K MCC950 is a selective NLRP3 inhibitor, which abrogated PA-induced inflammasome activation in primary hepatocytes. I Western blot analysis of protein levels of NLRP3, caspase-1, IL-18, and IL-1β in primary hepatocytes treated with PA plus or minus MCC950 for 6 h. J The densitometric ratio of NLRP3, caspase-1, IL-18, and IL-1β over β-actin (n = 3). *p ≤ 0.05 compared to the BSA controls. #,##p ≤ 0.05 and 0.01 compared to PA. K Activity of caspase-1 in cytosolic protein of PA and PA + MCC950-treated primary hepatocytes. *p ≤ 0.05 compared to the BSA controls, #p ≤ 0.05 compared to PA. PA: palmitic acid, Tomm20: translocase of outer mitochondrial membrane 20, NLRP3: NOD-like receptor-3, BSA: bovine serum albumin
High-resolution confocal microscopy was used to observe the colocalization of MitoTracker and LysoTracker in live primary hepatocytes after exposure to PA for 1 h. Green fluorescence of MitoTracker was compared with that from the control group (BSA), and overlapped red fluorescence from LysoTracker suggested an increase in the number of mitochondria for lysosomal degradation. With the extension of PA exposure time up to 10 h, green fluorescence gradually decreased, and the orange-yellow fluorescence, which reflected overlapping of green with the red fluorescence dimmed significantly, indicating that lysosomal degradation capability was accordingly compromised over time (Supplemental Fig. 7A). Therefore, these findings provided additional evidence for incapability of defective mitochondrial clearance. These results show that there was enhanced ROS production, mitochondrial damage, impaired mitophagy and reduced lysosomal degradation during lipotoxicity, and the final consequence was that damaged mitochondria were not cleared effectively from primary hepatocytes after PA exposure.
NLRP3 inflammasome activation in parallel with impaired mitophagy during lipotoxicity
Inflammasomes are sensing molecules that respond to microenvironmental changes in both intracellular and extracellular spaces. Overload of saturated PA is one of intracellular disturbances that lead to activation of inflammasomes, such as NLRP3 in microphages [34]. As shown in Fig. 4A & D, cytosolic NLRP3 protein levels were increased over time in exposure to PA. In turn, cytosolic protein levels of matured caspase-1, IL-1β and IL-18 were increased accordingly in primary hepatocytes exposed to PA (Fig. 4E–G), indicating that there was profound activation of inflammasome and a pyroptotic cascade concurrent with enhanced oxidant stress and impaired mitophagic activity. In addition, cytosolic caspase-1 activity was increased 2 h after PA exposure and further increased later on (Fig. 4H), consistent with an increase in cleaved caspase-1 protein levels 2−6 h after PA exposure, which confirmed enhanced NLRP3 inflammasome activation and pyroptotic activity along with PA exposure. Moreover, addition of MCC950, a selective NLRP3 inhibitor, abrogated PA-triggered NLRP3 activation, and subsequent cleavage of procaspase-1, pro-IL-1β and pro-IL-18, as well as increased caspase-1 activity in primary hepatocytes as demonstrated in Fig. 4I−K, and the findings further confirmed NLRP3 activation and the importance of subsequent pyroptosis in steatotic hepatocytes.
Antioxidant and mitochondria-protecting agent improved mitophagy
N-Acetyl cysteine (NAC) is widely used as an antioxidant in vivo and in vitro. Acetyl-l-carnitine (ALCAR) is reported to be a mitochondria-protective agent improving mitochondrial function, biosynthesis, and energy metabolism. In Fig. 5D−F, parkin and PINK1 protein levels were increased after pretreatment with NAC and ALCAR following PA exposure. The mitochondrial LC3II/Tomm20 ratio also increased along with mitochondrial P62 protein levels, indicating that mitophagic activity was partially restored (Fig. 5A−C). Cytosolic protein and mRNA levels of NLRP3 were decreased by NAC and ALCAR pretreatment, and the elevation of protein and mRNA levels of caspase-1, IL-1β, IL-18 and caspase-1 activity caused by PA exposure were reversed after pretreatment with NAC and ALCAR (Fig. 5G−L).

Antioxidant and mitochondrion-protecting agent improved mitophagy in primary hepatocytes. NLRP3 inflammasome activation was decreased in PA-exposed primary hepatocytes pretreated with antioxidant and mitochondria-protecting agent. A Mitochondrial protein LC3 and P62, D cytosolic protein Parkin and PINK1 in primary hepatocytes treated with PA, PA + NAC or PA + ALCAR were assayed by Western blot analysis. B, D, E, F The densitometric ratio of Parkin, PINK1, LC3II and P62 (e) (n = 3). G Western blot analysis of NLRP3, caspase-1, IL-18, and IL-1β protein levels in primary hepatocytes. H−K The densitometric ratio of NLRP3 (H), caspase-1 (I), IL-1β (J), and IL-18 (K) (n = 3). L Caspase-1 activity in PA-treated primary hepatocytes. **p ≤ 0.01 compared to the controls. ◊◊p < 0.01 compared to PA at 200 μM. #,##p < 0.05, 0.01 compared to PA + NAC. PA: palmitic acid, NAC: N-acetyl cysteine, ALCAR: Acetyl-l-carnitine, BSA: bovine serum albumin
Although the LC3II/Tomm20 ratio of hepatocytes 6 h after PA exposure was increased compared to the control, the mitochondrial protein P62 levels were increased in parallel (Fig. 5A−C). Similarly, in Fig. 4A−C, after PA treated for 6 h the LC3II/Tomm20 ratio and protein P62 levels of hepatocytes intended to be increased as well. To explore whether the increase of LC3II was caused by an increase in autophagy or decline in autophagosomal degradation (an increase in P62 level suggests decreased autophagosome degradation), primary hepatocytes were incubated with a mitophagy inhibitor, cyclosporine A (CsA), together with PA for 2−6 h (Fig. 6). Our results demonstrated that LC3II/Tomm20 ratios 2 h post PA treatment were reduced in primary hepatocytes after addition of CsA. P62 level was increased in parallel to LC3II/Tomm20 ratios 2 h after PA treatment (Fig. 6A, B). With the prolonging of PA exposure, the addition of CsA did not significantly change LC3II/Tomm20 ratios in PA-treated primary hepatocytes whereas P62 in the mitochondrial fraction was sustained (Fig. 6C−F). These findings indicate that increased mitophagic flux was not caused by an increase in its initiation but rather by a decrease in autophagosomal clearance, as reflected by accumulation of the autophagic substrate P62. Two hours after PA treatment we observed impaired mitophagy and elevated protein and mRNA levels of NLRP3. Concurrently, activity of caspase-1 (Fig. 4H, K), protein levels of caspase-1 and IL-1β were upregulated by PA treatment as well (Fig. 6E, F). Taken together, these data indicate that PA treatment resulted in impaired mitophagy, and in turn, compromised mitophagy contributed to NLRP3 inflammasome activation through mtROS in primary hepatocytes, although the details in molecular interplay remain to be investigated.

Change of mitophagy-associated protein levels and NLRP3 inflammasome activation in PA-exposed primary hepatocytes treated with a mitophagy inhibitor at different time points. A, C, E Cytosolic protein NLRP3, caspase-1, IL-18, IL-1β and mitochondrial protein LC3II, P62 in primary hepatocytes treated with PA, CsA or PA + CsA for 2 (A), 6 (C), and 10 (E) hours were assayed by Western blot analysis. B, D, F The densitometric ratio of mitochondrial LC3II and protein P62 levels over Tomm20, cytosolic protein NLRP3, caspase-1, IL-1β and IL-18 over β-actin (n = 3). *,**p ≤ 0.05, 0.01 compared to the BSA controls or as indicated. PA palmitic acid, NLRP3 NOD-like receptor-3, CsA cyclosporine A
Discussion
NASH is a dynamic stage of NAFLD and it may further progress to fibrosis and cirrhosis, and even hepatocellular carcinoma [1]. Therefore, factors affecting the progression from NAFL to NASH are the keys to reveal its pathogenesis in laboratory and clinical investigations. Autophagy is a self-saving process in clearance of cellular components. As a conserved self-protective mechanism, autophagy is maintained at a low level under a physiological condition [35]. Lipids are energy sources for hepatocytes through autophagy. Studies have shown that a defect of autophagy leads to lipid accumulation and insulin resistance in hepatocytes, which is one of the key factors in the progression from NAFL to NASH [36,37,38]. Mitochondrial autophagy is a part of autophagy, and impaired mitophagy has been demonstrated in the course of NAFLD in few studies [10, 39]. However, no studies have provided convincing evidence to demonstrate that impaired autophagy may be linked to profound inflammasome activation and subsequent hepatocellular death.
In the present study, the detection of mitophagy during NASH development confirmed that mitophagic activity was increased at an early stage (6 weeks) of HFCD feeding; whereas it was subsequently decreased later on (12−24 weeks). Concurrent determination of NLRP3 demonstrated that its expression was not changed significantly at the early stage (6 weeks), nor were IL-1β and IL-18 levels. At a later timeframe, NLRP3 expression was significantly increased in mouse livers, and was accompanied with the activation of IL-1β and IL-18, as well as increased serum IL-1β levels during 12−24 weeks of HFCD feeding. Hence, these results suggested that impaired mitophagy was closely associated with the activation of NLRP3 inflammasome in the progression from SFL to NASH. Therefore, we hypothesize that impaired mitophagy may trigger activation of inflammasome molecules and a pyroptotic cascade. To confirm our hypothesis, we treated primary hepatocytes with PA to investigate whether impaired mitophagy activates NLRP3 inflammasome under PA lipotoxicity in hepatocytes.
In the present study, it was confirmed by LDH release that PA exposure caused hepatocellular damage in a dose-dependent and time-dependent manner. The mitochondrial inner membrane potential of hepatocytes was collapsed within a short duration (1 h) of PA exposure, indicating that mitochondrial injury occurred. Concurrently, mRNA and protein levels of genes involved in mitophagy, such as perkin, PINK1, were elevated; so was the LC3II/Tomm20 ratio; whereas P62 expression was decreased, collectively indicating that mitophagic levels under PA exposure were enhanced during an early phase of PA treatment in comparison to the BSA controls. Of note, in mitochondrial fraction the ratios of LC3II/Tomm20 did not change significantly till 10 h; whereas P62 protein levels were increased 2 h after PA exposure. P62, the substrate of autophagy, together with LC3II/Tomm20 ratio change is considered to be a marker of autophagic flux, and increased P62 protein levels indicated that the degradation of autophagosome was reduced and the autophagic flux was defective [11]. The addition of cyclosporine A (CsA) did not significantly change LC3II/Tomm20 ratios in PA-treated cells; whereas P62 levels in mitochondrial fractions were increased significantly, confirming that increased mitophagic flux was not caused by increased mitophagy but rather by a decrease in autophagosome clearance, as reflected by accumulation of the autophagic substrate P62. In summary, PA-induced autophagy initiation and subsequent impairment were largely due to the defective autophagosomal clearance.
The NLRP3 inflammasome is a multiprotein cytoplasmic complex that serves as a pattern recognition receptor [40]. The exact mechanisms of NLRP3 inflammasome activation in hepatocytes during NASH are poorly characterized. As important sensing molecules that respond to extracellular and intracellular environmental changes, the nod-like receptors identify PAMPs/DAMPs [13, 16], and are activated to elicit a pyroptotic response [41]. In the present study mitophagy was found to be impaired to a significant extent, along with NASH progression in HFCD-fed mice. Protein and mRNA levels of NLRP3, caspase-1 and IL-1β in the liver, as well as serum IL-1β levels, were upregulated in these mice. This phenomenon was confirmed in primary hepatocytes under PA exposure for late-stage NASH. Therefore, we reason that the insufficient clearance of damaged mitochondria may trigger inflammasome activity through an undefined mechanism.
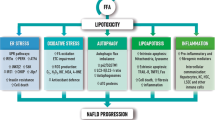
Mitochondrial renewal is part of subcellular organelle homeostasis, and is tightly regulated by mitophagy to remove damaged mitochondria and by mitogenesis through adding protein and lipid [42, 43]. The balance between mitophagy and mitogenesis contributes to the dynamical turnover of mitochondria, and normal mitochondrial turnover dynamics provides sufficient energy for entire cellular function and metabolism [43]. On the other hand, mitochondria are exposed to high levels of ROS, generating from oxidative phosphorylation for energy coupling and from other intracellular sources. Therefore, it is our speculation that activation of inflammasome molecules may occur through accelerated ROS generation when mitophagy is impaired due to fatty acid overload and toxicity. In fact, enhanced oxidant stress occurred in cytosolic and mitochondrial compartments as indicated by DCF and MitoSox detection (Supplemental Figs. 4&5, Supplemental Fig. 6C&D). Inflammasomes, such as NLRP3, are activated in response to intracellular ROS and mtROS, as well as overload of FFAs [44]. To further confirm the role of oxidant stress in the mediation of inflammasome activation, an antioxidant, N-acetyl cysteine (NAC), and a mitochondrial membrane-protective agent, acetyl-l-carnitine, were used to correct PA-induced mitophagic defects in hepatocytes. It was found that pretreatment with NAC or ALCAR mitigated PA-induced cytosolic ROS and mitochondrial ROS accumulation in hepatocytes, in turn, minimized mitochondrial DNA damage, partially restored mitophagic activity, and eventually inhibited NLRP3 inflammasome activation (Figs. 5 and 6). However, the relationship and molecular interplay between defective mitophagy and inflammasome activation remain to be better elucidated, especially under lipotoxicity. For better understanding of this complex cascade, a schematic illustration that links with lipotoxicity, oxidant stress, mitophagy, inflammasome activation, and pyroptotic consequence in hepatocytes is ditched in Fig. 7.

A schematic illustration highlighting the possible link and interaction with lipotoxicity, oxidant stress, mitophagy, inflammasome activation, and pyroptotic consequence. The hallmark of nonalcoholic steatohepatitis is steatosis with increased free fatty acid (FFAs) in hepatocytes, which results in lipotoxicity, oxidant stress and hepatocellular injury and death through apoptosis, pyroptosis or necrosis. Mitochondrion is an important subcellular organelle where fatty acid β-oxidation, the coupling of oxyphosphorylation with energy production and many other metabolic reactions take place. Mitophagy is part of mechanisms to remove damaged or degenerative mitochondrion and the impairment of mitophagy may result in accumulation of reactive oxygen species (ROS), which are generated during oxyphosphorylative process for energy production. Inflammasomes are sensing molecules that respond to changes in extracellular and intracellular microenvironment, and are activated to elicit a pyroptotic cascade, producing active caspase-1, IL-1β and IL-18, and to initiate local or systemic inflammatory responses, and hepatocellular injury or death. Palmitic acid (PA) was used to mimic fatty acid overload in hepatocytes. N-Acetyl cysteine (NAC) is a potent antioxidant, while acetyl-l-carnitine (ALCAR) is a mitochondion-protective agent, and both of them are used to minimize oxidant stress and to maintain mitochondrial hemostasis. Cyclosporine A (CsA) could stabilize mitochondrial inner membrane potential (MMP), and suppress mitophagy. MCC950 is a selective NLRP3 inhibitor, and is used to verify whether blocking NLRP3 activation may abrogate the downstream pyroptotic cascade. APGS: autophagosome, DAMP: damage-associated molecular pattern, Mito: mitochondrion
In conclusion, the findings of the present study support the hypothesis that mitophagy impairment as the result of decreased autophagosomal clearance triggers hepatic NLRP3 inflammasome activation in a murine NASH model and in primary hepatocytes. These new insights into inflammasome activation through compromised mitophagy advance our understanding of how fatty acids elicit lipotoxicity through oxidant stress and autophagy in mitochondria, and confer the molecular basis for pharmacologic intervention.
References
Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. 2017;377:2063–72.
Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328–57.
Brown GT, Kleiner DE. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism. 2016;65:1080–6.
Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016;65:1049–61.
Sfunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab. 2017;28:250–60.
Simoes ICM, Fontes A, Pinton P, Zischka H, Wieckowski MR. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol 2018;95:93–9.
Kojima H, Sakurai S, Uemura M, Fukui H, Morimoto H, Tamagawa Y. Mitochondrial abnormality and oxidative stress in nonalcoholic steatohepatitis. Alcohol Clin Exp Res 2007;31:S61–6.
Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68:280–95.
Zhan SS, Jiang JX, Wu J, Halsted C, Friedman SL, Zern MA, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006;43:435–43.
Wang L, Liu X, Nie J, Zhang J, Kimball SR, Zhang H, et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015;61:486–96.
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016;12:1–222.
Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–54.
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014;59:898–910.
Liu XJ, Duan NN, Liu C, Niu C, Liu XP, Wu J. Characterization of a murine nonalcoholic steatohepatitis model induced by high fat high calorie diet plus fructose and glucose in drinking water. Lab Invest 2018;98:1184–99.
Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol 2011;55:1086–94.
Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011;54:133–44.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011;17:179–88.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221–5.
Wu J, Li X, Zhu G, Zhang Y, He M, Zhang J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp Cell Res 2016;341:42–53.
Roberts RF, Tang MY, Fon EA, Durcan TM. Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int J Biochem Cell Biol 2016;79:427–36.
Kim MJ, Yoon JH, Ryu JH. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016;49:529–35.
Wu J, Soderbergh H, Karlsson K, Danielsson A. Protective effect of S-adenosyl-L-methionine on bromobenzene- and D-galactosamine-induced toxicity to isolated rat hepatocytes. Hepatology 1996;23:359–65.
Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012;55:222–32.
Pesce V, Nicassio L, Fracasso F, Musicco C, Cantatore P, Gadaleta MN. Acetyl-L-carnitine activates the peroxisome proliferator-activated receptor-gamma coactivators PGC-1alpha/PGC-1beta-dependent signaling cascade of mitochondrial biogenesis and decreases the oxidized peroxiredoxins content in old rat liver. Rejuvenation Res 2012;15:136–9.
Wu J, Karlsson K, Danielsson A. Effects of vitamins E, C and catalase on bromobenzene- and hydrogen peroxide-induced intracellular oxidation and DNA single-strand breakage in Hep G2 cells. J Hepatol. 1997;26:669–77.
Zhang Y, Venugopal SK, He S, Liu P, Wu J, Zern MA. Ethanol induces apoptosis in hepatocytes by a pathway involving novel protein kinase C isoforms. Cell Signal 2007;19:2339–50.
Yan HM, Ramachandran A, Bajt ML, Lemasters JJ, Jaeschke H. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci 2010;117:515–23.
Marnef A, Jady BE, Kiss T. Human polypyrimidine tract-binding protein interacts with mitochondrial tRNA(Thr) in the cytosol. Nucleic Acids Res. 2016;44:1342–53.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222–30.
Wong A, Cortopassi G. Reproducible quantitative PCR of mitochondrial and nuclear DNA copy number using the LightCycler. Methods Mol Biol. 2002;197:129–37.
Wu J, Lizarzaburu ME, Kurth MJ, Liu L, Wege H, Zern MA, et al. Cationic lipid polymerization as a novel approach for constructing new DNA delivery agents. Bioconjug Chem 2001;12:251–7.
Ye J, Li TS, Xu G, Zhao YM, Zhang NP, Fan J, et al. JCAD Promotes Progression of Nonalcoholic Steatohepatitis to Liver Cancer by Inhibiting LATS2 Kinase Activity. Cancer Res 2017;77:5287–300.
Xu G, Ye J, Liu XJ, Zhang NP, Zhao YM, Fan J, et al. Activation of pluripotent genes in hepatic progenitor cells in the transition of nonalcoholic steatohepatitis to pre-malignant lesions. Lab Invest 2017;97:1201–17.
Karasawa T, Kawashima A, Usui-Kawanishi F, Watanabe S, Kimura H, Kamata R, et al. Saturated Fatty Acids Undergo Intracellular Crystallization and Activate the NLRP3 Inflammasome in Macrophages. Arterioscler Thromb Vasc Biol 2018;38:744–56.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41.
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–5.
Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–78.
Flores-Toro JA, Go KL, Leeuwenburgh C, Kim JS. Autophagy in the liver: cell's cannibalism and beyond. Arch Pharm Res 2016;39:1050–61.
Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, et al. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta 2007;1767:1260–7.
Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol. 2009;21:194–8.
Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–32.
Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol. 2015;93:3–10.
Kiriyama Y, Nochi H. Intra- and intercellular quality control mechanisms of mitochondria. Cells 2017;7:1.
Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–85.
Acknowledgements
The authors are grateful to Mrs. Ke Qiao in the Department of Medical Microbiology and Parasitology, Fudan University School of Basic Medical Sciences for her technical support in the use of confocal microscope and Operetta High Content Imaging System. Part of this work was presented at the Annual Meeting of the American Association for the Study of Liver Disease (AASLD), Nov. 11–15, 2016, Boston, MA, USA, and published as an abstract in Hepatology 2016;64(Suppl):777A.
Funding
This work is supported by the National Natural Science Foundation of China (NSFC #81272436, 81572356, 81871997 to JW), (#81472673 and 81672720 to X-ZS), Shanghai Commission of Sciences and Technologies (#16140903700), and the Ministry of Science & Technology of China (#2016YFE0107400) to JW. Young Investigator of Zhongshan Hospital, Fudan University (#2016ZSQN03) to N-PZ.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, NP., Liu, XJ., Xie, L. et al. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest 99, 749–763 (2019). https://doi.org/10.1038/s41374-018-0177-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0177-6
This article is cited by
-
Mitochondrial control of inflammation
Nature Reviews Immunology (2023)
-
The mitophagy pathway and its implications in human diseases
Signal Transduction and Targeted Therapy (2023)
-
PINK1 ameliorates acute-on-chronic liver failure by inhibiting apoptosis through mTORC2/AKT signaling
Cell Death Discovery (2022)
-
Acetyl-L-Carnitine Exerts Neuroprotective and Anticonvulsant Effect in Kainate Murine Model of Temporal Lobe Epilepsy
Journal of Molecular Neuroscience (2022)
-
Simultaneous Modulation of NLRP3 Inflammasome and Nrf2/ARE Pathway Rescues Thioacetamide-Induced Hepatic Damage in Mice: Role of Oxidative Stress and Inflammation
Inflammation (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}