
Abstract
Bipolar disorder (BD) is a common psychiatric disorder characterized by recurrent mood swings between depression and mania, and is associated with high treatment costs. The existence of manic episodes is the defining feature of BD, during which period, patients experience extreme elevation in activity, energy, and mood, with changes in sleep patterns that together severely impair their ability to function in daily life. Despite some limitations in recapitulating the complex features of human disease, several rodent models of mania have been generated and characterized, which have provided important insights toward understanding its underlying pathogenic mechanisms. Among the mechanisms, neuronal excitatory and inhibitory (E/I) synaptic dysfunction in some brain regions, including the frontal cortex, hippocampus, and striatum, is an emerging hypothesis explaining mania. In this review, we highlight recent studies of rodent manic models having impairments in the E/I synaptic development and function. We also summarize the molecular and functional changes of E/I synapses by some mood stabilizers that may contribute to the therapeutic efficacy of drugs. Furthermore, we discuss potential future directions in the study of this emerging hypothesis to better connect the outcomes of basic research to the treatment of patients with this devastating mental illness.
Similar content being viewed by others


Introduction
Bipolar disorder (BD) is a common and devastating mental illness, characterized by recurrent mood swings between depression and mania with intervening euthymic states1. BD affects approximately 1–2.5% of the world’s population2, and the World Health Organization recognizes BD as the sixth leading cause of disability. Existence of manic episodes is the defining feature of BD, which differentiates it from unipolar major depressive disorder. The symptoms of manic episode include hyperactivity, impulsivity, elevated mood, inflated self-esteem, reduced anxiety, decreased need for sleep, and sometimes psychosis1. Both environmental and genetic risk factors contribute to the pathogenesis of mania, but the detailed molecular and cellular pathways underlying mania remain largely unknown.
So far, several rodent models of mania have been generated and characterized. Traditionally, pharmacological (e.g., psychostimulant amphetamine-induced) and environmental (e.g., sleep deprivation-induced) models were studied, but more recently various genetic models (i.e., knockout (KO), knock-in (KI), and overexpressing transgenic (TG) mice) have been developed3. Even with some limitations in satisfying all three (construct, face, and predictive) validities as a disease model3, 4, each of these rodent models has provided important insights toward understanding the pathogenic mechanisms of mania. For example, manic-like behaviors of rodents injected with amphetamine or those expressing lower levels of dopamine transporter3, together with clinical evidence of higher dopamine levels during manic episodes5, supported hyperdopaminergic activities as a major pathophysiology of mania. Nevertheless, the clinical heterogeneity of mania, such as the differential response to certain pharmacological treatments6, suggests the possibility that other pathogenic mechanisms can still exist.
Neuronal excitability is tightly controlled by excitatory and inhibitory (E/I) synaptic balance, and dysfunction of this process has been strongly associated with numerous neurodevelopmental and neuropsychiatric disorders, including autism spectrum disorders (ASDs), intellectual disability (ID), and schizophrenia (SCZ)7,8,9,10 This could involve various underlying mechanisms ranging from abnormal expression and function of pre- or postsynaptic molecules11 to impaired maturation of certain neuronal cell types, such as γ-aminobutyric acid (GABA)ergic inhibitory interneurons12. Despite some evidence suggesting abnormal GABAergic interneurons in BD13, E/I synaptic dysfunction in mania has been relatively unexplored compared to that in other brain disorders. In the current review, we highlight recent studies of rodent manic models with impairments in E/I synaptic development and function. We also summarize thus far identified molecular and functional changes of E/I synapses by some mood stabilizers. Lastly, we discuss current limitations and potential future directions of this emerging hypothesis to better connect the outcomes of basic research to the treatment of patients with BD. For more general and comprehensive coverage of animal models of mania, we refer to recent excellent reviews3,14.
Animal models of mania with E/I synaptic dysfunction
Shank3-overexpressing TG mice
The SH3 and multiple ankyrin repeat domains 3 (SHANK3, also called PROSAP2) gene encodes a core scaffold protein in postsynaptic density (PSD) of the excitatory synapse15. By interacting with hundreds of different molecules in PSD16,17, including membrane proteins, signaling molecules, and cytoskeletal components, Shank3 organizes the macromolecular protein complex and is critically involved in proper development and function of excitatory synapses. Clinically, deletions and duplications of the chromosomal region (22q13) containing SHANK3 and various point mutations of SHANK3 have been identified in patients with ASDs, ID, SCZ, BD, and attention deficit hyperactivity disorder (ADHD)18,19. Han et al.16 recently identified two patients with small 22q13 duplications that likely include only SHANK3, and found that these patients were diagnosed with hyperkinetic disorders, BD, and ADHD, respectively. Notably, the symptoms of the patient with ADHD were not improved by treatment with amphetamine, a common medication for ADHD, suggesting the possibility that the disorder could not be typical ADHD, but more likely BD. To model SHANK3 duplications, Han et al. generated Shank3 TG mice that mildly overexpress Shank3 proteins (to approximately 150%) compared to wild-type (WT) mice. Indeed, the Shank3 TG mice displayed several manic-like behaviors, including locomotor hyperactivity and hypersensitivity to amphetamine in the open-field test (OFT), reduced despair-like behavior in the tail-suspension test (TST), increased acoustic startle response, reduced prepulse inhibition (PPI), and abnormal circadian rhythms, some of which responded to valproate (VPA), a Food and Drug Administration (FDA)-approved mood stabilizer and anticonvulsant for the treatment of manic or mixed episodes in BD6,16.
The E/I synaptic morphology and function of Shank3 TG mice were characterized mainly in the hippocampus16. The number of excitatory synapses was increased, while that of inhibitory synapses was decreased in the cultured hippocampal neurons of Shank3 TG mice compared to WT neurons. Functionally, amplitude, but not frequency, of spontaneous excitatory postsynaptic currents (sEPSCs) was significantly increased, while frequency, but not amplitude, of miniature inhibitory postsynaptic currents (mIPSCs) was decreased in the CA1 pyramidal neurons of acute hippocampal slices from Shank3 TG mice. Consistent with the shifted E/I synaptic balance toward more excitation but less inhibition, the Shank3 TG mice displayed abnormal electroencephalography (EEG) patterns in the frontal cortex and hippocampus, and exhibited spontaneous seizures. The abnormal EEG was also rescued by VPA treatment. At the molecular level, Han et al. found that Shank3 interacted with various actin-regulatory proteins to promote actin polymerization in the excitatory postsynaptic sites, thereby increasing the number of dendritic spines in the CA1 pyramidal neurons of Shank3 TG mice16,20.
It was unexpected that increased expression of Shank3 proteins that exclusively localize to excitatory, but not inhibitory, postsynaptic sites caused reduced number and function of inhibitory synapses in the Shank3 TG hippocampus. One possible explanation would be impaired maturation and/or function of GABAergic inhibitory neurons. However, Lee et al. recently showed that the densities of parvalbumin (PV)- and somatostatin-positive interneurons were normal in the hippocampus, striatum, and medial prefrontal cortex (mPFC) of Shank3 TG mice21. Therefore, it is more likely that the inhibitory synaptic changes observed in Shank3 TG mice could be due to the cell-autonomous postsynaptic changes of principal neurons that may involve a shift in distribution of certain actin-regulatory proteins, such as Mena and Profilin2, from inhibitory to excitatory postsynaptic sites16.
Despite the abnormal EEG in the frontal cortex of Shank3 TG mice, it remains to be directly investigated whether the E/I synaptic morphology and function are also altered in other brain regions, such as the striatum and mPFC, of the mice. In this regard, it is notable that in the several lines of Shank3 KO and KI mice modeling ASDs and SCZ, changes in the number and function of E/I synapses in these brain regions have been observed22,23,24. Moreover, Lee et al.25 recently showed increased levels of actin filaments (F-actin) in the striatum of Shank3 TG mice, similar to the hippocampus. The detailed mechanism of how VPA treatment rescued the manic-like behaviors of Shank3 TG mice is also unknown. One possibility is that VPA enhanced the GABAergic inhibitory synaptic transmission of Shank3 TG mice by inhibiting a GABA-catabolizing enzyme, GABA transaminase, and thereby normalizing the E/I synaptic balance26.
Forebrain-specific Plcg1 KO mice
Phospholipase Cγ1 (PLCγ1) is an enzyme that, when activated by receptor tyrosine kinases, hydrolyzes membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2) to generate the second messengers diacylglycerol and inositol-1,4,5-triphosphate (IP3)27. In neurons, brain-derived neurotrophic factor (BDNF), through its receptor tropomyosin receptor kinase B (TrkB), activates PLCγ1 together with other downstream signaling components, including Ca2+/calmodulin-dependent protein kinase II (CaMKII), extracellular signal-regulated kinase, and cAMP response element-binding protein to regulate synaptic development, function, and plasticity28,29. Despite some studies suggesting the association between PLCG1 polymorphism and BD30,31, the functional roles of PLCγ1 in mature neurons in vivo has not been investigated because of the lethality of conventional Plcg1 KO mice at an early embryonic stage. To solve this problem, recently, Yang et al.32 generated forebrain-specific Plcg1 KO mice by crossing the floxed Plcg1 mice with CaMKII-Cre mice (Plcg1f/f; CaMKII mice). The Plcg1f/f; CaMKII mice exhibited several manic-like behaviors, including locomotor hyperactivity in the OFT, reduced anxiety-like behavior in the elevated plus maze (EPM) test, reduced despair-like behavior in the forced swim test (FST), hyperhedonic behavior in the sucrose preference test (SPT), and increased acoustic startle response, many of which were normalized by treatment with VPA or lithium, another FDA-approved mood stabilizer for the treatment of mania in BD33. In addition to the manic-like behaviors, the mice showed impaired learning and memory in the auditory and contextual fear conditioning tests.
The basal excitatory synaptic transmission of Plcg1f/f; CaMKII mice was normal in the hippocampal Schaffer collateral (SC)-CA1 synapses as measured by the input-output relationship and the N-methyl-d-aspartic acid (NMDA) receptor to α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor current ratio (NMDAR/AMPAR ratio)32. Moreover, amplitude and frequency of miniature EPSCs (mEPSCs) were normal in the CA1 pyramidal neurons of Plcg1f/f; CaMKII mice. Consistently, expression levels of several excitatory synaptic proteins and number of dendritic spines in the hippocampus of Plcg1f/f; CaMKII mice were comparable to those of WT mice. In contrast, the Plcg1f/f; CaMKII mice showed defects in the inhibitory synapses of the hippocampus and dorsal striatum32. Specifically, frequency, but not amplitude, of mIPSCs in the CA1 pyramidal neurons was reduced in the hippocampal slices of Plcg1f/f; CaMKII mice compared to WT mice. Notably, in the dorsal striatum, where D1 (dopamine receptor D1-expressing)- and D2 (dopamine receptor D2-expressing)-type medium spiny neurons (MSNs) account for the majority (>90%) of the neuronal population34, the amplitude of mIPSCs was reduced only in the D1 type, but not D2 type, MSN of Plcg1f/f; CaMKII mice. This is somewhat consistent with the behavioral phenotype of the mice, as activation of D1-type MSN promotes locomotor activities35.
These functional defects of inhibitory synapses were explained by a reduced number of GABAergic inhibitory presynaptic terminals on the principal neurons32. At the molecular level, both basal and BDNF-induced phosphorylation of CaMKIIα were significantly decreased at the inhibitory postsynaptic sites of Plcg1f/f; Nestin cultured hippocampal neurons32. Furthermore, the surface expression of inhibitory GABAA receptor α1 subunit, which is known to be regulated through its phosphorylation by CaMKIIα, was also decreased in the Plcg1f/f; Nestin hippocampal neurons compared to WT neurons.
In addition to the E/I synaptic dysfunction in the hippocampus and striatum, BDNF-TrkB-dependent long-term potentiation, a type of synaptic plasticity, at the SC-CA1 synapses of Plcg1f/f; CaMKII hippocampus was also defective, which could explain the impaired learning and memory of the mice. Together, these findings suggest that BDNF-TrkB-PLCγ1 signaling is required for the proper development and function of inhibitory synapses and for the normal synaptic plasticity in the striatum and hippocampus, defects of which contribute to manic-like behaviors and impaired learning and memory in the forebrain-specific Plcg1 KO mice.
Ank3-1b heterozygous mice
The ANKYRIN 3 (ANK3) gene encodes multiple isoforms of Ankyrin-G proteins that link membrane proteins to the β-spectrin/actin cytoskeleton and thereby organizing macromolecular complexes at the specialized plasma membrane compartments36. In neurons, Ankyrin-G proteins are involved in the formation and maintenance of axon initial segments (AISs) and node of Ranvier, membrane compartments critical for action potential generation and propagation, where they connect several ion channels to the cytoskeleton37. Notably, the ANK3 gene has been repeatedly associated with BD from several large-scale genome-wide association studies38,39,40.
Recently, Leussis et al.41 generated two different mouse models with reduced Ank3 expression and found that both showed some manic-like behaviors. In the first model, Ank3 expression was specifically reduced in the hippocampal dentate gyrus (DG) using the lentivirus expressing short hairpin RNAs targeting Ank3. These mice showed reduced anxiety-like behavior in multiple behavioral assays, including the EPM, light-dark transition, and novelty-suppressed feeding tasks. In addition, the mice showed increased home-cage activity during the light, but not the dark, phase. Both reduced anxiety-like behavior and increased activity were rescued by treatment with lithium41. The second model was generated by disrupting the exon 1b locus of Ank3 (Ank3-1b), which resulted in decrease of the Ankyrin-G isoforms containing exon 1b in several brain regions, including the DG, cortex, and cerebellum41,42. Because of the progressive early-onset ataxia in homozygous Ank3-1b−/− mice42, the behavioral phenotypes of heterozygous Ank3-1b+/− mice were characterized and compared to those of WT mice. Similar to the mice with DG-specific decrease of Ank3 expression, Ank3-1b+/− mice displayed reduced anxiety-like behavior in the EPM, light-dark transition, and novelty-suppressed feeding tasks. Moreover, Ank3-1b+/− mice exhibited hyperhedonic behavior in the SPT. Intriguingly, after chronic social isolation stress, the manic-like behaviors of Ank3-1b+/− mice were shifted to depression-like behaviors41. Specifically, singly housed Ank3-1b+/− mice showed increased anxiety-like behavior in the EPM test, anhedonic behavior in the SPT, and despair-like behavior in the FST, compared to singly housed WT mice. This elevated susceptibility to stress of Ank3-1b+/− mice was partly explained by the higher plasma levels of corticosterone compared to WT mice, both under basal and acute stress conditions41.
Alternative first exons (1a/1a′, 1e, and 1b) of ANK3 gene encode distinct N-terminal peptide sequences of Ankyrin-G isoforms43,44. Importantly, Lopez et al.44 revealed that PV-positive GABAergic inhibitory interneurons express only Ankyrin-G isoforms containing exon 1b, while excitatory principal neurons express the isoforms containing either exon 1e alone, or both 1e and 1b. Consistently, clustering of voltage-gated sodium channels at the AISs of PV-positive interneurons was significantly decreased in Ank3-1b+/− mice compared to WT mice. Further supporting the interneuron dysfunction, Ank3-1b+/− and Ank3-1b−/− mice exhibited abnormal EEG and seizures in an Ank3 gene dose-dependent manner (i.e., they were more severe in homozygous Ank3-1b−/− than in heterozygous Ank3-1b+/− mice). In addition, the detailed electrophysiological analysis showed reduced intrinsic excitability of the PV-positive interneurons in Ank3-1b−/− mice compared to WT mice44. Combining the abovementioned studies, it is conceivable that functional changes of PV-positive GABAergic inhibitory neurons and their synapses in some brain regions of Ank3-1b+/− mice could contribute to manic-like behaviors.
More recently, Zhu et al.45 generated and characterized a mouse model with conditional disruption of Ank3 in pyramidal neurons of adult forebrain (Ank3f/f; CaMKII). All three major isoforms of Ankyrin-G proteins (190, 270, and 480 kDa) were decreased in the forebrain regions of Ank3f/f; CaMKII mice. These mice, similar to Ank3-1b+/− mice, displayed several manic-like behaviors responsive to lithium and VPA, which could be shifted to depression-like behaviors after repeated social defeat stress. There was a loss of sodium and potassium channels at AISs of pyramidal neurons in Ank3f/f; CaMKII mice. Moreover, the number of inhibitory synapses innervating pyramidal neuron AISs was significantly decreased in the cortex of Ank3f/f; CaMKII mice. Consistent with disinhibition, c-fos expression was increased in cortical pyramidal neurons of the mice, indicating increased cortical activity45. Therefore, both Ank3-1b+/− and Ank3f/f; CaMKII mice have some defects in the inhibitory synaptic function. Nevertheless, in addition to the AISs, Ankyrin-G proteins also localize to the dendritic spines of glutamatergic excitatory postsynapse where they regulate dendritic spine morphology, and excitatory synaptic transmission and plasticity46. Therefore, morphological and functional changes of excitatory synapses in some brain regions of Ank3-1b+/− and Ank3f/f; CaMKII mice cannot be excluded, details of which remain to be investigated.
ClockΔ19 mutant mice
Abnormalities in circadian rhythms and sleep disturbance have been linked with BD and manic episodes47,48. The circadian locomotor output cycles kaput (CLOCK) gene encodes a CLOCK protein that forms a heterodimer with brain muscle ARNT-like 1 (BMAL 1) and functions as a critical transcriptional regulator in the feedback network of the molecular clock49. Notably, polymorphisms of CLOCK and BMAL 1 have been associated with BD50,51. The ClockΔ19 mutant mice have N-ethyl-N-nitrosourea-induced single base mutation at the 5′ splice donor site of intron 19, which results in loss of exon 19 during splicing and thereby produces a dominant negative CLOCK protein52. Indeed, ClockΔ19 mutant mice show significant circadian rhythm defects both in the molecular and behavioral levels53.
McClung’s group has reported a series of studies demonstrating the manic-like behaviors of ClockΔ19 mutant mice and revealing the underlying molecular and neural circuit mechanisms. ClockΔ19 mutant mice exhibited locomotor hyperactivity both in novel and familiar environments, increased response to various reward stimuli, hyperhedonic behavior in the SPT, reduced despair-like behavior in the FST, and reduced anxiety-like behavior in the EPM test, some of which were rescued by treatment with lithium54,55,56. Intriguingly, the manic-like behaviors of ClockΔ19 mutant mice were time-dependent; the behaviors were significant during the day time, but were normalized to WT levels (euthymic states) during the night time57. Hyperdopaminergic activity due to the increased firing rate of dopaminergic neurons in the ventral tegmental area (VTA) was shown as a key neuronal mechanism for the manic-like behaviors of ClockΔ19 mutant mice55. Specifically, viral expression of functional CLOCK proteins in the VTA of mutant mice was sufficient to rescue the manic-like behaviors54. Moreover, the dopaminergic neuron firing rate, tyrosine hydroxylase expression, and dopamine synthesis in the VTA of mutant mice coincided with the time-dependent behavioral changes57.
The nucleus accumbens (NAc) is a brain region of the ventral striatum critically involved in mood, reward, and addiction-related behaviors, and the MSNs of NAc get dopaminergic inputs from the VTA and excitatory glutamatergic inputs from the prefrontal cortex58. The hyperdopaminergic activity of ClockΔ19 mutant mice could lead to the circuit level changes of NAc and thereby contribute to the manic-like behaviors. Indeed, Dzirasa et al.59 revealed neurophysiological abnormalities of the NAc microcircuits in ClockΔ19 mutant mice by performing simultaneous in vivo extracellular recordings from the VTA, NAc, and prefrontal cortex. Specifically, low-gamma oscillations and single neuron phase coupling were defective in the NAc of ClockΔ19 mutant mice, which were normalized by treatment with lithium. At the molecular level, total-, phospho (S845)-, and surface expression levels of the GluA1, but not the GluA2, subunit of AMPA-type glutamate receptors were decreased in the NAc of mutant mice compared to WT mice, suggesting that increased dopamine release may indirectly lower the excitatory synaptic transmission of NAc in ClockΔ19 mutant mice3,59,60. Consistent with the biochemical changes, mEPSC amplitude, but not frequency, and AMPAR/NMDAR ratio were decreased in the MSNs of ClockΔ19 mutant NAc60. Furthermore, viral-mediated overexpression of the GluA1 subunit of the AMPA receptor in the NAc was sufficient to normalize some manic-like behaviors of the mutant mice, including reduced anxiety-like behavior in the EPM test and increased reward sensitivity in the conditioned place preference test60.
Together, reduced excitatory synaptic function of the MSNs in NAc, potentially as a consequence of increased activity of dopaminergic inputs from the VTA, could be an important mechanism for the manic-like behaviors of ClockΔ19 mutant mice. It remains to be investigated whether the inhibitory synapses of MSNs in ClockΔ19 mutant mice have also any molecular and functional changes. Moreover, whether the E/I synapses of D1- and D2-type MSNs could be differentially altered in the mutant mice is an interesting topic for future research61.
Sleep-deprived animals
Clinically, reduced sleep or sleep disturbances can trigger and worsen manic episodes62. Similarly, sleep deprivation protocols, usually obligating rodents to remain awake on a small platform surrounded by water for an extended period of time (72 h), have long been used to generate manic models63. The sleep-deprived animals indeed exhibit several manic-like behaviors, including locomotor hyperactivity, aggressive behavior, hypersexuality, and increased stereotypy, which could be normalized by treatment with lithium63,64.
A few studies have suggested that protein kinase C (PKC) could mediate important molecular mechanisms underlying the manic-like behaviors of sleep-deprived animals. The levels of PKC activity and phosphorylation of some PKC substrates were increased in the frontal cortex of sleep-deprived rats65. Moreover, treatment of quercetin, a PKC inhibitor, prevented sleep deprivation-induced locomotor hyperactivity in mice66. Among the increased PKC-dependent phosphorylation by sleep deprivation, those on the S896 of the GluN1 subunit of the NMDA receptor and the T840 of the GluA1 subunit of the AMPA receptor are notable65. S896 phosphorylation of GluN1 is involved in regulating intracellular trafficking and surface expression of NMDA receptors67. T840 phosphorylation of GluA1 could enhance channel conductance of AMPA receptors68. Therefore, it is possible that sleep deprivation could affect excitatory synaptic function and plasticity in the frontal cortex, which then contribute to manic-like behaviors.
Sleep deprivation-induced alterations in synaptic function and plasticity have been extensively investigated in different brain regions of rodents, although, in many of the studies, behavioral changes after various sleep deprivation protocols were not characterized, and therefore the synaptic changes may not be directly associated with mania. In the CA1 and DG neurons of the hippocampus, sleep deprivation resulted in impairment of long-term potentiation69,70. Consistent with the notion that NMDA receptors are critical upstream regulators of synaptic plasticity71, surface expression of NMDA receptors and NMDA receptor-mediated currents were reduced after sleep deprivation72,73. Meanwhile, AMPA receptor function, as measured by mEPSCs, was normal in the hippocampus73. Impaired hippocampal long-term potentiation, nevertheless, could be more associated with learning and memory deficits after sleep deprivation than with manic-like behaviors70. In the frontal cortex, mild sleep deprivation increased mEPSC amplitude and frequency of layer II/III pyramidal neurons74. In the deep layers (V/VI) of the mPFC, sleep deprivation decreased amplitude, but not frequency, of mEPSCs without affecting the properties of mIPSCs75. Together, sleep deprivation could result in diverse changes of E/I synaptic function, depending on the brain regions and sleep deprivation protocols. Therefore, more investigations are necessary to understand the causal relationship between E/I synaptic changes of certain brain regions and manic-like behaviors after sleep deprivation.
Molecular and functional changes of E/I synapses by mood stabilizers
Clinically, antipsychotics (such as haloperidol, risperidone, and olanzapine) and mood stabilizers (such as lithium, VPA, lamotrigine, and carbamazepine) are most commonly used for the acute and long-term management of mania, respectively1,6. Antipsychotics exhibit high-affinity antagonism toward dopamine and serotonin receptors, but their chronic treatment could also affect glutamate receptor expression76. The molecular targets and mechanisms of action for mood stabilizers are more complex than those for antipsychotics, and there are still many questions remaining in the field77,78. For example, lithium alone has multiple direct targets, including inositol monophosphatase, phosphoglucomutase, and glycogen synthase kinase-3, and additionally affects diverse downstream pathways indirectly26,79.
In Table 1, we summarize animal model and in vitro-cultured neuron studies showing E/I synaptic changes by mood stabilizers, at the molecular, cellular, and functional levels. It is notable that possibly depending on the treatment and measurement conditions, synaptic changes in a certain brain region by a single mood stabilizer can be diverse, or even opposite. Therefore, further investigations are necessary to understand whether and to what extent each of these synaptic changes contributes to the therapeutic efficacy of drugs.
Conclusions and prospects
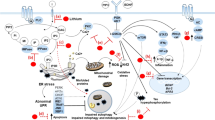
As we summarized in this review, there is an increasing number of evidence supporting the pathogenic role of E/I synaptic dysfunction in mania (Fig. 1). Nevertheless, there are many limitations and questions remaining from animal model studies. First, brain regions, neural circuits, and neuronal cell types mediating specific behavioral phenotypes of mania need to be further dissected. For example, locomotor hyperactivity and hyperhedonic behavior may be mediated by synaptic changes of different brain regions, such as the dorsal striatum and NAc, respectively35,58. Recent advances in viral-mediated gene delivery combined with optogenetic tools and brain-clearing technology have revealed the structural and functional anatomy of neural circuits critical for several depression-related behaviors80,81. Similar approaches could be applied to the abovementioned animal models of mania to test whether activation or inhibition of specific neural circuits could rescue a subset of behavioral phenotypes. Second, E/I synaptic changes preceding the onset of behavioral phenotypes may be identified and the effect of preventing such earlier synaptic changes could be investigated. In most studies so far, morphological and functional changes of the E/I synapses have been characterized at the ages when the manic-like behaviors of animal models are fully developed. Therefore, it is possible that those E/I synaptic changes may include some compensatory and homeostatic responses of neurons and/or neural circuits, and, thus, could not be causally associated with the behavioral phenotypes. Notably, in the case of schizophrenia, earlier intervention during the juvenile stage has shown to prevent adult onset of behavioral deficits in animal models82. Third, E/I synaptic mechanisms underlying the differential responses to certain mood stabilizers could be investigated using animal models. Although lithium still remains the first-line treatment for BD, only about 30% of BD patients show full responses to lithium treatment83. Despite the poorly defined neurobiological mechanism, a few genome-wide association studies have linked the variants of genes functioning in the E/I synapses, such as GRIA2 (for the glutamate receptor, ionotropic, AMPA2) and GADL1 (for the glutamate decarboxylase-like 1), to the differential responses to lithium treatment84,85. In this regard, it is intriguing that the manic-like behaviors of Shank3 TG mice were selectively rescued by VPA, but not by lithium16. More comprehensive analysis of behavioral and synaptic changes in the animal models of mania after single or mixed treatment with various drugs for BD will be an interesting direction of future study. Last but not least, the animal model studies need to be converged with clinical research on patients86. Advances in neuroimaging and induced pluripotent stem cell technologies have already narrowed the gaps between the preclinical and clinical studies of BD87,88, which, when combined, will provide important insights toward understanding the pathophysiology and potential treatment of this complex and heterogeneous mental illness.

In the hippocampus of Shank3 TG mice, excitatory synaptic function is increased, while inhibitory synaptic function is decreased. In the hippocampus and striatum of forebrain-specific Plcg1 KO mice, excitatory synaptic function is normal, but inhibitory synaptic function is decreased. In the NAc of ClockΔ19 mutant mice, excitatory synaptic function is decreased. In the brain regions of Ank3-1b+/− mice, function of the AIS of the PV-positive interneuron is impaired. EN excitatory neuron, HIP hippocampus, IN inhibitory neuron, PN principal neuron, STR striatum
References
Grande, I., Berk, M., Birmaher, B. & Vieta, E. Bipolar disorder. Lancet 387, 1561–1572 (2016).
Merikangas, K. R. et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch. Gen. Psychiatry 68, 241–251 (2011).
Logan, R. W. & McClung, C. A. Animal models of bipolar mania: the past, present and future. Neuroscience 321, 163–188 (2016).
Nestler, E. J. & Hyman, S. E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169 (2010).
Joyce, P. R. et al. Urinary catecholamines and plasma hormones predict mood state in rapid cycling bipolar affective disorder. J. Affect. Disord. 33, 233–243 (1995).
Geddes, J. R. & Miklowitz, D. J. Treatment of bipolar disorder. Lancet 381, 1672–1682 (2013).
Nelson, S. B. & Valakh, V. Excitatory/Inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698 (2015).
Lee, E., Lee, J. & Kim, E. Excitation/inhibition imbalance in animal models of autism spectrum disorders. Biol. Psychiatry 81, 838–847 (2017).
Zoghbi, H. Y. & Bear, M. F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 4, a009886 (2012).
Kehrer, C., Maziashvili, N., Dugladze, T. & Gloveli, T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 1, 6 (2008).
Sudhof, T. C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008).
Nakazawa, K. et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62, 1574–1583 (2012).
Benes, F. M. & Berretta, S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25, 1–27 (2001).
Kato, T., Kasahara, T., Kubota-Sakashita, M., Kato, T. M. & Nakajima, K. Animal models of recurrent or bipolar depression. Neuroscience 321, 189–196 (2016).
Sheng, M. & Kim, E. The Shank family of scaffold proteins. J. Cell Sci. 113, 1851–1856 (2000). (Pt 11).
Han, K. et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503, 72–77 (2013).
Lee, Y. et al. Integrative analysis of brain region-specific Shank3 interactomes for understanding the heterogeneity of neuronal pathophysiology related to SHANK3 mutations. Front. Mol. Neurosci. 10, 110 (2017).
Jiang, Y. H. & Ehlers, M. D. Modeling autism by SHANK gene mutations in mice. Neuron 78, 8–27 (2013).
Monteiro, P. & Feng, G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157 (2017).
Choi, S.-Y. & Han, K. Emerging role of synaptic actin-regulatory pathway in the pathophysiology of mood disorders. Anim. Cells Syst. (Seoul) 19, 283–288 (2015).
Lee, B. et al. Age-dependent decrease of GAD65/67 mRNAs but normal densities of GABAergic interneurons in the brain regions of Shank3-overexpressing manic mouse model. Neurosci. Lett. 649, 48–54 (2017).
Peca, J. et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442 (2011).
Lee, J. et al. Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front. Cell. Neurosci. 9, 94 (2015).
Zhou, Y. et al. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162 (2016).
Lee, Y. et al. Striatal transcriptome and interactome analysis of Shank3-overexpressing mice reveals the connectivity between Shank3 and mTORC1 Signaling. Front. Mol. Neurosci. 10, 201 (2017).
Chiu, C. T., Wang, Z., Hunsberger, J. G. & Chuang, D. M. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol. Rev. 65, 105–142 (2013).
Kim, M. J., Kim, E., Ryu, S. H. & Suh, P. G. The mechanism of phospholipase C-gamma1 regulation. Exp. Mol. Med. 32, 101–109 (2000).
Cunha, C., Brambilla, R. & Thomas, K. L. A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 3, 1 (2010).
Park, H. & Poo, M. M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23 (2013).
Turecki, G. et al. Evidence for a role of phospholipase C-gamma1 in the pathogenesis of bipolar disorder. Mol. Psychiatry 3, 534–538 (1998).
Lovlie, R., Berle, J. O., Stordal, E. & Steen, V. M. The phospholipase C-gamma1 gene (PLCG1) and lithium-responsive bipolar disorder: re-examination of an intronic dinucleotide repeat polymorphism. Psychiatr. Genet. 11, 41–43 (2001).
Yang, Y. R. et al. Forebrain-specific ablation of phospholipase Cgamma1 causes manic-like behavior. Mol. Psychiatry 22, 1473–1482 (2017).
Geddes, J. R., Burgess, S., Hawton, K., Jamison, K. & Goodwin, G. M. Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am. J. Psychiatry 161, 217–222 (2004).
Calabresi, P., Picconi, B., Tozzi, A., Ghiglieri, V. & Di Filippo, M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 17, 1022–1030 (2014).
Kravitz, A. V. et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626 (2010).
Bennett, V. & Healy, J. Membrane domains based on ankyrin and spectrin associated with cell-cell interactions. Cold Spring Harb. Perspect. Biol. 1, a003012 (2009).
Nelson, A. D. & Jenkins, P. M. Axonal membranes and their domains: assembly and function of the axon initial segment and node of Ranvier. Front. Cell. Neurosci. 11, 136 (2017).
Ferreira, M. A. et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058 (2008).
Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011).
Muhleisen, T. W. et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 5, 3339 (2014).
Leussis, M. P. et al. The ANK3 bipolar disorder gene regulates psychiatric-related behaviors that are modulated by lithium and stress. Biol. Psychiatry 73, 683–690 (2013).
Zhou, D. et al. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143, 1295–1304 (1998).
Rueckert, E. H. et al. Cis-acting regulation of brain-specific ANK3 gene expression by a genetic variant associated with bipolar disorder. Mol. Psychiatry 18, 922–929 (2013).
Lopez, A. Y. et al. Ankyrin-G isoform imbalance and interneuronopathy link epilepsy and bipolar disorder. Mol. Psychiatry 22, 1464–1472 (2017).
Zhu, S. et al. Genetic disruption of ankyrin-G in adult mouse forebrain causes cortical synapse alteration and behavior reminiscent of bipolar disorder. Proc. Natl Acad. Sci. USA 114, 10479–10484 (2017).
Smith, K. R. et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 84, 399–415 (2014).
McClung, C. A. Circadian genes, rhythms and the biology of mood disorders. Pharmacol. Ther. 114, 222–232 (2007).
McClung, C. A. How might circadian rhythms control mood? Let me count the ways. Biol. Psychiatry 74, 242–249 (2013).
Mohawk, J. A., Green, C. B. & Takahashi, J. S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 35, 445–462 (2012).
Benedetti, F. et al. Influence of CLOCK gene polymorphism on circadian mood fluctuation and illness recurrence in bipolar depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 123B, 23–26 (2003).
Nievergelt, C. M. et al. Suggestive evidence for association of the circadian genes PERIOD3 and ARNTL with bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, 234–241 (2006).
King, D. P. et al. Positional cloning of the mouse circadian clock gene. Cell 89, 641–653 (1997).
Antoch, M. P. et al. Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell 89, 655–667 (1997).
Roybal, K. et al. Mania-like behavior induced by disruption of CLOCK. Proc. Natl Acad. Sci. USA 104, 6406–6411 (2007).
McClung, C. A. et al. Regulation of dopaminergic transmission and cocaine reward by the Clock gene. Proc. Natl Acad. Sci. USA 102, 9377–9381 (2005).
Ozburn, A. R., Larson, E. B., Self, D. W. & McClung, C. A. Cocaine self-administration behaviors in ClockDelta19 mice. Psychopharmacology (Berl.). 223, 169–177 (2012).
Sidor, M. M. et al. Daytime spikes in dopaminergic activity drive rapid mood-cycling in mice. Mol. Psychiatry 20, 1406–1419 (2015).
Russo, S. J. & Nestler, E. J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 14, 609–625 (2013).
Dzirasa, K. et al. Lithium ameliorates nucleus accumbens phase-signaling dysfunction in a genetic mouse model of mania. J. Neurosci. 30, 16314–16323 (2010).
Parekh, P. K. et al. Altered GluA1 function and accumbal synaptic plasticity in the ClockΔ19 model of bipolar mania. Biol Psychiatry 2017; pii: S0006-3223(17)31721-3.
Spencer, S. et al. A mutation in CLOCK leads to altered dopamine receptor function. J. Neurochem. 123, 124–134 (2012).
Harvey, A. G. Sleep and circadian functioning: critical mechanisms in the mood disorders? Annu. Rev. Clin. Psychol. 7, 297–319 (2011).
Gessa, G. L., Pani, L., Fadda, P. & Fratta, W. Sleep deprivation in the rat: an animal model of mania. Eur. Neuropsychopharmacol. 5(Suppl), 89–93 (1995).
Valvassori, S. S. et al. Lithium ameliorates sleep deprivation-induced mania-like behavior, hypothalamic-pituitary-adrenal (HPA) axis alterations, oxidative stress and elevations of cytokine concentrations in the brain and serum of mice. Bipolar Disord. 19, 246–258 (2017).
Szabo, S. T. et al. Glutamate receptors as targets of protein kinase C in the pathophysiology and treatment of animal models of mania. Neuropharmacology 56, 47–55 (2009).
Kanazawa, L. K. et al. Quercetin reduces manic-like behavior and brain oxidative stress induced by paradoxical sleep deprivation in mice. Free Radic. Biol. Med. 99, 79–86 (2016).
Chen, B. S. & Roche, K. W. Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368 (2007).
Jenkins, M. A. et al. Regulation of GluA1 alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol. Pharmacol. 85, 618–629 (2014).
McDermott, C. M. et al. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J. Neurosci. 23, 9687–9695 (2003).
Vecsey, C. G. et al. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature 461, 1122–1125 (2009).
Malenka, R. C. & Bear, M. F. LTP and LTD: an embarrassment of riches. Neuron 44, 5–21 (2004).
Chen, C., Hardy, M., Zhang, J., LaHoste, G. J. & Bazan, N. G. Altered NMDA receptor trafficking contributes to sleep deprivation-induced hippocampal synaptic and cognitive impairments. Biochem. Biophys. Res. Commun. 340, 435–440 (2006).
McDermott, C. M., Hardy, M. N., Bazan, N. G. & Magee, J. C. Sleep deprivation-induced alterations in excitatory synaptic transmission in the CA1 region of the rat hippocampus. J. Physiol. 570, 553–565 (2006).
Liu, Z. W., Faraguna, U., Cirelli, C., Tononi, G. & Gao, X. B. Direct evidence for wake-related increases and sleep-related decreases in synaptic strength in rodent cortex. J. Neurosci. 30, 8671–8675 (2010).
Winters, B. D., Huang, Y. H., Dong, Y. & Krueger, J. M. Sleep loss alters synaptic and intrinsic neuronal properties in mouse prefrontal cortex. Brain Res. 1420, 1–7 (2011).
Miyamoto, S., Duncan, G. E., Marx, C. E. & Lieberman, J. A. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol. Psychiatry 10, 79–104 (2005).
Rapoport, S. I., Basselin, M., Kim, H. W. & Rao, J. S. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Res. Rev. 61, 185–209 (2009).
Schloesser, R. J., Martinowich, K. & Manji, H. K. Mood-stabilizing drugs: mechanisms of action. Trends Neurosci. 35, 36–46 (2012).
Quiroz, J. A., Gould, T. D. & Manji, H. K. Molecular effects of lithium. Mol. Interv. 4, 259–272 (2004).
Lammel, S. et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217 (2012).
Lammel, S., Tye, K. M. & Warden, M. R. Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav. 13, 38–51 (2014).
Marin, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 22, 1229–1238 (2016).
Alda, M. Lithium in the treatment of bipolar disorder: pharmacology and pharmacogenetics. Mol. Psychiatry 20, 661–670 (2015).
Perlis, R. H. et al. A genomewide association study of response to lithium for prevention of recurrence in bipolar disorder. Am. J. Psychiatry 166, 718–725 (2009).
Chen, C. H. et al. Variant GADL1 and response to lithium therapy in bipolar I disorder. N. Engl. J. Med. 370, 119–128 (2014).
Kaiser, T. & Feng, G. Modeling psychiatric disorders for developing effective treatments. Nat. Med. 21, 979–988 (2015).
Maletic, V. & Raison, C. Integrated neurobiology of bipolar disorder. Front. Psychiatry 5, 98 (2014).
Mertens, J. et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99 (2015).
Farooq, M. et al. Lithium increases synaptic GluA2 in hippocampal neurons by elevating the delta-catenin protein. Neuropharmacology 113, 426–433 (2017).
Tyagarajan, S. K. et al. Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc. Natl Acad. Sci. USA 108, 379–384 (2011).
Motohashi, N., Ikawa, K. & Kariya, T. GABAB receptors are up-regulated by chronic treatment with lithium or carbamazepine. GABA hypothesis of affective disorders? Eur. J. Pharmacol. 166, 95–99 (1989).
de Bartolomeis, A., Tomasetti, C., Cicale, M., Yuan, P. X. & Manji, H. K. Chronic treatment with lithium or valproate modulates the expression of Homer1b/c and its related genes Shank and Inositol 1,4,5-trisphosphate receptor. Eur. Neuropsychopharmacol. 22, 527–535 (2012).
Gideons, E. S., Lin, P. Y., Mahgoub, M., Kavalali, E. T. & Monteggia, L. M. Chronic lithium treatment elicits its antimanic effects via BDNF-TrkB dependent synaptic downscaling. Elife 6, e25480 (2017).
Ma, J. & Zhang, G. Y. Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci. Lett. 348, 185–189 (2003).
Hashimoto, R., Hough, C., Nakazawa, T., Yamamoto, T. & Chuang, D. M. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 80, 589–597 (2002).
Du, J. et al. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J. Neurosci. 24, 6578–6589 (2004).
Du, J. et al. The role of hippocampal GluR1 and GluR2 receptors in manic-like behavior. J. Neurosci. 28, 68–79 (2008).
Du, J. et al. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology 32, 793–802 (2007).
Kim, H. J. & Thayer, S. A. Lithium increases synapse formation between hippocampal neurons by depleting phosphoinositides. Mol. Pharmacol. 75, 1021–1030 (2009).
Martin, P. M. et al. DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/beta-catenin signaling. Mol. Psychiatry 23, 467–475 (2018).
Liu, Z. H., Chuang, D. M. & Smith, C. B. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int. J. Neuropsychopharmacol. 14, 618–630 (2011).
Gebhardt, C. & Cull-Candy, S. G. Lithium acts as a potentiator of AMPAR currents in hippocampal CA1 cells by selectively increasing channel open probability. J. Physiol. 588, 3933–3941 (2010).
Shim, S. S., Hammonds, M. D., Ganocy, S. J. & Calabrese, J. R. Effects of sub-chronic lithium treatment on synaptic plasticity in the dentate gyrus of rat hippocampal slices. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 343–347 (2007).
Colino, A., Garcia-Seoane, J. J. & Valentin, A. Action potential broadening induced by lithium may cause a presynaptic enhancement of excitatory synaptic transmission in neonatal rat hippocampus. Eur. J. Neurosci. 10, 2433–2443 (1998).
Valentin, A., Garcia-Seoane, J. J. & Colino, A. Lithium enhances synaptic transmission in neonatal rat hippocampus. Neuroscience 78, 385–391 (1997).
Peineau, S. et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717 (2007).
Nau, H. & Loscher, W. Valproic acid: brain and plasma levels of the drug and its metabolites, anticonvulsant effects and gamma-aminobutyric acid (GABA) metabolism in the mouse. J. Pharmacol. Exp. Ther. 220, 654–659 (1982).
Johannessen, C. U., Petersen, D., Fonnum, F. & Hassel, B. The acute effect of valproate on cerebral energy metabolism in mice. Epilepsy Res. 47, 247–256 (2001).
Hassel, B., Iversen, E. G., Gjerstad, L. & Tauboll, E. Up-regulation of hippocampal glutamate transport during chronic treatment with sodium valproate. J. Neurochem. 77, 1285–1292 (2001).
Takuma, K. et al. Chronic treatment with valproic acid or sodium butyrate attenuates novel object recognition deficits and hippocampal dendritic spine loss in a mouse model of autism. Pharmacol. Biochem. Behav. 126, 43–49 (2014).
Baldino, F. Jr. & Geller, H. M. Sodium valproate enhancement of gamma-aminobutyric acid (GABA) inhibition: electrophysiological evidence for anticonvulsant activity. J. Pharmacol. Exp. Ther. 217, 445–450 (1981).
Gean, P. W., Huang, C. C., Hung, C. R. & Tsai, J. J. Valproic acid suppresses the synaptic response mediated by the NMDA receptors in rat amygdalar slices. Brain Res. Bull. 33, 333–336 (1994).
Ko, G. Y., Brown-Croyts, L. M. & Teyler, T. J. The effects of anticonvulsant drugs on NMDA-EPSP, AMPA-EPSP, and GABA-IPSP in the rat hippocampus. Brain Res. Bull. 42, 297–302 (1997).
Martin, E. D. & Pozo, M. A. Valproate reduced excitatory postsynaptic currents in hippocampal CA1 pyramidal neurons. Neuropharmacology 46, 555–561 (2004).
Hassel, B., Tauboll, E. & Gjerstad, L. Chronic lamotrigine treatment increases rat hippocampal GABA shunt activity and elevates cerebral taurine levels. Epilepsy Res. 43, 153–163 (2001).
Sitges, M., Chiu, L. M., Guarneros, A. & Nekrassov, V. Effects of carbamazepine, phenytoin, lamotrigine, oxcarbazepine, topiramate and vinpocetine on Na+ channel-mediated release of [3H]glutamate in hippocampal nerve endings. Neuropharmacology 52, 598–605 (2007).
Sitges, M., Guarneros, A. & Nekrassov, V. Effects of carbamazepine, phenytoin, valproic acid, oxcarbazepine, lamotrigine, topiramate and vinpocetine on the presynaptic Ca2+ channel-mediated release of [3H]glutamate: comparison with the Na+ channel-mediated release. Neuropharmacology 53, 854–862 (2007).
Wang, S. J., Huang, C. C., Hsu, K. S., Tsai, J. J. & Gean, P. W. Presynaptic inhibition of excitatory neurotransmission by lamotrigine in the rat amygdalar neurons. Synapse 24, 248–255 (1996).
Lee, C. Y., Fu, W. M., Chen, C. C., Su, M. J. & Liou, H. H. Lamotrigine inhibits postsynaptic AMPA receptor and glutamate release in the dentate gyrus. Epilepsia 49, 888–897 (2008).
Cunningham, M. O. & Jones, R. S. The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous GABA release in the rat entorhinal cortex in vitro. Neuropharmacology 39, 2139–2146 (2000).
Braga, M. F., Aroniadou-Anderjaska, V., Post, R. M. & Li, H. Lamotrigine reduces spontaneous and evoked GABAA receptor-mediated synaptic transmission in the basolateral amygdala: implications for its effects in seizure and affective disorders. Neuropharmacology 42, 522–529 (2002).
Kamal, S. M. Pharmacological modulation of brain levels of glutamate and GABA in rats exposed to total sleep deprivation. J. Exp. Pharmacol. 2, 65–71 (2010).
Booker, S. A., Pires, N., Cobb, S., Soares-da-Silva, P. & Vida, I. Carbamazepine and oxcarbazepine, but not eslicarbazepine, enhance excitatory synaptic transmission onto hippocampal CA1 pyramidal cells through an antagonist action at adenosine A1 receptors. Neuropharmacology 93, 103–115 (2015).
Granger, P. et al. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 47, 1189–1196 (1995).
Ambrosio, A. F. et al. Inhibition of glutamate release by BIA 2-093 and BIA 2-024, two novel derivatives of carbamazepine, due to blockade of sodium but not calcium channels. Biochem. Pharmacol. 61, 1271–1275 (2001).
Giustizieri, M. et al. Differential effect of carbamazepine and oxcarbazepine on excitatory synaptic transmission in rat hippocampus. Synapse 62, 783–789 (2008).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government Ministry of Science, ICT & Future Planning (MISP) (NRF-2015R1C1A1A01052794), by the Brain Research Program through the NRF funded by the MISP (NRF-2015M3C7A1028790), by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (HI16C0090), and by a Korea University grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, Y., Zhang, Y., Kim, S. et al. Excitatory and inhibitory synaptic dysfunction in mania: an emerging hypothesis from animal model studies. Exp Mol Med 50, 1–11 (2018). https://doi.org/10.1038/s12276-018-0028-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-018-0028-y
This article is cited by
-
Reduction of GAS5 and FOXD3-AS1 long non-coding RNAs in patients with bipolar disorder
Scientific Reports (2023)
-
Disease-specific differences in gene expression, mitochondrial function and mitochondria-endoplasmic reticulum interactions in iPSC-derived cerebral organoids and cortical neurons in schizophrenia and bipolar disorder
Discover Mental Health (2023)
-
Functional genomic analysis delineates regulatory mechanisms of GWAS-identified bipolar disorder risk variants
Genome Medicine (2022)
-
Increased levels of TAR DNA-binding protein 43 in the hippocampus of subjects with bipolar disorder: a postmortem study
Journal of Neural Transmission (2022)
-
Adenosine A2A Receptor Blockade Ameliorates Mania Like Symptoms in Rats: Signaling to PKC-α and Akt/GSK-3β/β-Catenin
Molecular Neurobiology (2022)
