
Abstract
The mammalian Na+/H+ exchanger isoform one (NHE1), encoded by Solute Carrier Family 9, member 1 (SLC9A1), consists of 12 membrane domains and a cytosolic C-terminal domain. NHE1 plays an important role in maintaining intracellular pH homeostasis by exchanging one intracellular proton for one extracellular sodium ion. Mice with a homozygous null mutation in Slc9a1 (Nhe1) exhibited ataxia, recurrent seizures, and selective neuronal cell death. In humans, three unrelated patients have been reported: a patient with a homozygous missense mutation in SLC9A1, c.913G>A (p.Gly305Arg), which caused Lichtenstein–Knorr syndrome characterized by cerebellar ataxia and sensorineural hearing loss, a patient with compound heterozygous mutations, c.1351A>C (p.Ile451Leu) and c.1585C>T (p.His529Tyr), which caused a neuromuscular disorder, and a patient with de novo mutation, c.796A>C (p.Asn266His) which associated multiple anomalies. In this study, using whole exome sequencing, we identified a novel homozygous SLC9A1 truncating mutation, c.862del (p.Ile288Serfs*9), in two affected siblings. The patients showed cerebellar ataxia but neither of them showed sensorineural hearing loss nor a neuromuscular phenotype. The main clinical feature was similar to Lichtenstein–Knorr syndrome but deafness may not be an essential phenotypic feature of SLC9A1 mutation. Our report expands the knowledge of clinical features of SLC9A1 mutations.
Similar content being viewed by others


Introduction
The mammalian Na+/H+ exchanger isoform one (NHE1), encoded by Solute Carrier Family 9, member 1 (SLC9A1, MIM 107310), is a ubiquitously expressed membrane protein consisting of 12 membrane domains facilitating ion transportation and a cytosolic C-terminal domain regulating NHE1 activity [1]. NHE1 plays an important role in maintaining intracellular pH homeostasis by exchanging one intracellular proton for one extracellular sodium ion [2]. NHE1 activity has been implicated in several human diseases, including cardiovascular disease [2] and several forms of cancer [3, 4]. In a mouse model system, Slc9a1 (previously termed Nhe1) knockout mice exhibited ataxia, tonic-clonic seizures and selective neuronal cell death [5], suggesting a role for NHE1 in the development and/or function of the nervous system. Consistently, a homozygous missense mutation in SLC9A1, c.913G>A (p.Gly305Arg, annotated based on NM_003047.4), has been reported in patients with Lichtenstein–Knorr syndrome (MIM 616291), which is characterized by cerebellar ataxia and sensorineural hearing loss [6]. To date, only four SLC9A1 variants have been registered as disease-causing mutations (DMs) or likely DMs in the Human Gene Mutation Database (HGMD) and the clinical spectrum of patients remains undetermined. Herein, we describe a novel homozygous SLC9A1 mutation in affected siblings and detail their clinical features.
Materials and Methods
A series of patients with cerebellar atrophy were collected. Detailed clinical information was obtained by the clinicians examining the patients. A prescreening of triplet repeat disorders had not been performed. The Institutional Review Board of Yokohama City University of Medicine approved the experimental protocols. Informed consent was obtained from the patients’ guardians, in accordance with Japanese regulatory requirements.
Genomic DNA was isolated from peripheral blood leukocytes using QuickGene 610L (Wako, Osaka, Japan). A sample from patient 2 was captured using the SureSelect Human All Exon v6 (60 Mb) Kit (Agilent Technologies, Santa Clara, CA, USA), and sequenced on an Illumina HiSeq2500 (Illumina, San Diego, CA, USA) with 101 bp paired-end reads. Image analysis and base calling were performed using CASAVA software (Illumina). Sequence reads were aligned to GRCh37 and PCR duplicates were excluded using Novoalign (http://www.novocraft.com/) and Picard (http://picard.sourceforge.net/), respectively. Indel realignment and recalibration of base-quality scores were performed using the Genome Analysis ToolKit (GATK) (http://www.broadinstitute.org/gatk/). Variant calling and annotation were performed using GATK UnifiedGenotyper and ANNOVAR (http://www.openbioinformatics.org/annovar/) [7].
The average read depth of protein-coding regions had a range of 64× and at least 97.3% of target bases were sequenced by 10 or more reads. Common single-nucleotide polymorphisms (SNPs) with minor allele frequencies ≥1% in dbSNP 137 and variants that were observed in more than 5 of our 575 in-house ethnically matched control exomes were filtered out. Among the remaining rare variants, we focused on amino acid-altering or splice-affecting variants. Particular attention was paid to mutations in known causative genes associated with ataxia, cerebellar atrophy, and other neurodevelopmental disorders. SLC9A1 variants were confirmed by Sanger sequencing of PCR products with an ABI PRISM 3500xl autosequencer (Life Technologies, Carlsbad, CA, USA), using genomic DNA from the patients and their parents as a template.
Lymphoblastoid cells derived from patients were grown in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum, tylosin, and antibiotic–antimycotic solution in a 5% CO2 incubator. After incubation with dimethyl sulfoxide (DMSO) (as vehicle control) with/without 30 µM cycloheximide (Sigma) to observe the preventive effects of cyclophosphamide to nonsense-mediated mRNA decay (NMD) for 4 h, total RNA was extracted using RNeasy Plus Mini kit (Qiagen) and genomic DNA was removed using recombinant DNase I (Takara). Four milligrams of total RNA was subjected to reverse transcription using PrimeScript 1st strand synthesis kit with random hexamers (Takara). cDNA was isolated from lymphoblastoid cells from a patient with a different disease (Joubert syndrome with TMEM67 mutation) and the current patients. Relative quantity of the cDNA was confirmed by quantitative polymerase chain reaction (qPCR) with specific primers (Supplemental Table 1) The qPCR was performed using a QuantiFast SYBR Green PCR kit (Qiagen) on a Rotor-Gene Q real-time PCR cycler (Qiagen). Relative ratios of cDNA were calculated using the standard curve method with Rotor-Gene 6000 Series Software 1.7 (Qiagen) by normalizing with the autosomal internal control locus (ACTB).
Results
Clinical features
Clinical features of patients are summarized in Table 1. The two affected brothers were born to non-consanguineous Chinese parents. Patient 1 (the elder sibling) showed unstable walking and mild motor developmental delay at the age of 3 years. He did not show muscle weakness, seizures nor deafness, but showed ataxia and mild delay of talking. An auditory steady-state evoked response was normal. Initial magnetic resonance imaging (MRI) findings were unremarkable at 3 years, but a second MRI indicated mild cerebellar atrophy at the age of 5 years (Fig. 1). Patient 2 (the younger brother) showed motor developmental delay, especially in walking ability, and ataxia. MRI was not performed on him, but his clinical course was similar to his elder brother.

Brain MRI of patient I-1. Axial (a) and sagittal (b) T1-weighted brain magnetic resonance imaging (T1WI) showed normal findings at 3 years of age. At 5 years of age, axial (c) and sagittal (d) T1WI revealed mild cerebellar atrophy
Genetic analysis
Whole exome sequencing of patient 2 (I-2, Fig. 2a) revealed a homozygous truncating mutation, c.862del (p.Ile288Serfs*9) in SLC9A1. Segregation of the mutation based on an autosomal recessive model was confirmed by Sanger sequencing. The c.862del was absent from the databases of the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) and our in-house controls, and is extremely rare (MAF 0.05 %) in the Tohoku Medical Megabank Organization (ToMMo, https://ijgvd.megabank.tohoku.ac.jp/) database. The evaluation scores of the missense showed a deviation of observed variant counts from the expected number (mis-Z) of 4.37 and the probability of being loss-of-function intolerant (pLI) of 0.952.

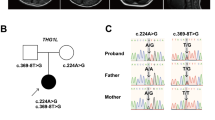
Familial pedigrees and electropherograms. Segregation of the mutation (c.862del) in patients and their parents was confirmed by Sanger sequencing. The c.862del was absent from the databases of the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) and 1000 Genome data, and extremely rare (MAF: 2/3994 = 0.05 %) in the Tohoku Medical Megabank Organization (ToMMo, https://ijgvd.megabank.tohoku.ac.jp/) database. Haplotyping indicated that the parents shared at least a 3.0 Mb haplotype, including the c.862del and three other rare homozygous variants
HomozygosityMapper (http://www.homozygositymapper.org/) showed that the younger patient (I-2) harbored 463 continuous homozygous single-nucleotide variants (SNVs) in chromosome 1 (from 24,180,806 to 27,995,533; 3.8 Mb length, Supplemental Fig. 1). SNV typing of family members indicated that the parents shared at least a 3.0 Mb haplotype, which included the c.862del and three other extremely rare variants. It implied that the c.862del was derived from a common ancestor (Fig. 2).
SLC9A1 mRNA levels was confirmed using qPCR for cDNA derived from patients lymphoblastoid cells. The samples of both patients indicated low SLC9A1 mRNA and the restoration by using cycloheximide which is one of NMD inhibitor, implied that the c.862del (p.Ile288Serfs*9) induced NMD (Fig. 3a).

SLC9A1 transcript levels in our patients and schematic presentation of NHE1 protein and current and reported mutations. (a) SLC9A1 transcript levels (averaged two partial transcripts) normalized to ACTB (beta-actin) are shown. Error bar shows standard deviation. Both of patients indicate low SLC9A1 transcript levels, and the obvious restoration by adding cycloheximide (CHX) as an NMD inhibitor. (b) Schematic presentation of NHE1 protein and current and reported variants (arrowhead and arrow, respectively). The mammalian NHE1 consists of 12 transmembrane domains (open squares with numbers 1 ~ 12) and a cytosolic C-terminal domain (gray square). Amino acid numbers of each transmembrane domain are indicated. TM transmembrane
The pathogenicity classification of mutations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines indicated that c.862del (p.Ile288Serfs*9) is pathogenic (PVS1, PM2, PM3, PP1, and PP4, Supplemental Table 2).
Discussion
To date, a total of three individuals harboring pathogenic mutations in SLC9A1 have been reported. Schematic of NHE1 and reported and current variants are shown in Fig. 3b [8, 9]. A homozygous c.913G>A (p.Gly305Arg) mutation caused Lichtenstein–Knorr syndrome (showing cerebellar ataxia and deafness) [6], and compound heterozygous mutations, c.1351A>C (p.Ile451Leu) and c.1585C>T (p.His529Tyr), caused a neuromuscular disorder [10]. One dominant mutation (de novo), c.796A>C (p.Asn266His) was reported in a patient with multiple anomalies (microcephaly, minor dysmorphisms, profound developmental delays, epilepsy, and white matter abnormalities) [11]. It remains largely unknown how different SLC9A1 mutations can cause different functional effects and thereby be associated with such distinct phenotypes. AP-1 cells expressing the p.Gly305Arg mutant (the disease-causing mutation reported in patients with Lichtenstein–Knorr syndrome) showed decreased levels of mature glycosylated NHE1 protein and its subcellular localization was also affected. [6, 12, 13]. At the cellular level, the p.Gly305Arg mutant delayed the recovery from acid treatment, indicating that defective NHE1 function affected pH homeostasis. These results suggest that the Lichtenstein–Knorr syndrome mutation (p.Gly305Arg) is loss-of-function.
In this study, we identified a homozygous truncating mutation (c.862del, p.Ile288Serfs*9) in cerebellar ataxia patients. The mRNA analysis derived lymphoblastoid cells indicated that the mutant transcript was eliminated by NMD, leading to a critical defect in NHE1 function [14]. Functional studies of truncated NHE1, similar to our case but terminated at amino acid 321, 449, 543, or 735, demonstrated a decrease of NHE1 activity and expression [15]. Therefore, we speculate that homozygous loss-of-function mutations are defective and lead to cerebellar ataxia.
Our patients showed cerebellar ataxia similar to that previously reported for Lichtenstein–Knorr syndrome (Table 1), suggesting a shared underlying pathological mechanism [6]. However, neither brother showed sensorineural hearing loss, implying that deafness might not be an essential condition for diagnosis as a cerebellar disorder resulting from SLC9A1 mutation. In addition, the patients were born to Chinese (Han) non-consanguineous parents and shared at least a 3.0 Mb loss of heterozygosity (LOH), including the pathogenic c.862del and three other extremely rare homozygous variants (Chr1:24,384,198T>A, Chr1:24,997,828T>C, and Chr1:25,775,397T>C, based on GRCh37/hg19). In a previous study of LOH in different human populations, Europeans and East Asians have very similar LOH profiles [16]. European population analysis indicated the last common ancestor of the preceding six generations resulted predominantly in LOH longer than 5 Mb, and losses of heterozygosity measuring 3 or 4 Mb are relatively common in non-consanguineous families [17]. This evidence supports the possibility of a common ancestor in a far preceding generation, because the size of the LOH is relatively small in our case.
In conclusion, our study supports a specific connection between SLC9A1 and cerebellar ataxia (but not deafness). The detailed clinical evaluation in this report provides useful information for future diagnosis of cerebellar ataxia caused by SLC9A1 mutation.
References
Lee BL, Sykes BD, Fliegel L. Structural and functional insights into the cardiac Na(+)/H(+) exchanger. J Mol Cell Cardiol. 2013;61:60–7.
Fliegel L. Regulation of the Na(+)/H(+) exchanger in the healthy and diseased myocardium. Expert Opin Ther Targets. 2009;13:55–68.
Amith SR, Fliegel L. Regulation of the Na+/H+ Exchanger (NHE1) in Breast Cancer Metastasis. Cancer Res. 2013;73:1259–64.
Parks SK, Chiche J, Pouyssegur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer. 2013;13:611–23.
Cox GA, Lutz CM, Yang CL, Biemesderfer D, Bronson RT, Fu A, et al. Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell . 1997;91:139–48.
Guissart C, Li X, Leheup B, Drouot N, Montaut-Verient B, Raffo E, et al. Mutation of SLC9A1, encoding the major Na(+)/H(+) exchanger, causes ataxia-deafness Lichtenstein-Knorr syndrome. Hum Mol Genet. 2015;24:463–70.
Miyatake S, Okamoto N, Stark Z, Nabetani M, Tsurusaki Y, Nakashima M, et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J Hum Genet. 2017;62:741–6.
Landau M, Herz K, Padan E, Ben-Tal N. Model structure of the Na+/H+ exchanger 1 (NHE1): functional and clinical implications. J Biol Chem. 2007;282:37854–63.
Koster S, Pavkov-Keller T, Kuhlbrandt W, Yildiz O. Structure of human Na+/H+ exchanger NHE1 regulatory region in complex with calmodulin and Ca2+. J Biol Chem. 2011;286:40954–61.
Farwell Hagman KD, Shinde DN, Mroske C, Smith E, Radtke K, Shahmirzadi L, et al. Candidate-gene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet Med. 2017;19:224–35.
Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–81.
Slepkov ER, Rainey JK, Li X, Liu Y, Cheng FJ, Lindhout DA, et al. Structural and functional characterization of transmembrane segment IV of the NHE1 isoform of the Na+/H+ exchanger. J Biol Chem. 2005;280:17863–72.
Alves C, Ma Y, Li X, Fliegel L. Characterization of human mutations in phosphorylatable amino acids of the cytosolic regulatory tail of SLC9A1. Biochem Cell Biol. 2014;92:524–9.
Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–77.
Li X, Augustine A, Chen S, Fliegel L. Stop codon polymorphisms in the human SLC9A1 gene disrupt or compromise Na+/H+ exchanger function. PLoS ONE. 2016;11:e0162902.
Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE. 2010;5:e13996.
McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–72.
Acknowledgements
We thank the individuals and their families for their participation in this study. We also thank Nobuko Watanabe and Mai Sato for technical assistance. We also thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported by grants from Research on Measures for Intractable Diseases; Comprehensive Research on Disability Health and Welfare; the Strategic Research Program for Brain Science (SRPBS); the Practical Research Project for Rare/Intractable Diseases; the Initiative on Rare and Undiagnosed Diseases from the Japan Agency for Medical Research and Development; a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grants-in-Aid for Scientific Research (A and B); Grant-in-Aid for Young Scientists (B); Challenging Exploratory Research from the Japan Society for the Promotion of Science; the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency; grants from the Ministry of Health, Labour and Welfare; and the Takeda Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Iwama, K., Osaka, H., Ikeda, T. et al. A novel SLC9A1 mutation causes cerebellar ataxia. J Hum Genet 63, 1049–1054 (2018). https://doi.org/10.1038/s10038-018-0488-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0488-x
This article is cited by
-
Genomic analysis of 116 autism families strengthens known risk genes and highlights promising candidates
npj Genomic Medicine (2024)
-
Structure and mechanism of the human NHE1-CHP1 complex
Nature Communications (2021)
-
The identification of two pathogenic variants in a family with mild and severe forms of developmental delay
Journal of Human Genetics (2021)
-
Role of Genetic Mutations of the Na+/H+ Exchanger Isoform 1, in Human Disease and Protein Targeting and Activity
Molecular and Cellular Biochemistry (2021)
-
A novel PAK1 variant causative of neurodevelopmental disorder with postnatal macrocephaly
Journal of Human Genetics (2020)
