
Abstract
Introduction:
Persistent pulmonary hypertension of the newborn (PPHN) is associated with substantial infant morbidity and mortality. Recently, genetic associations have been found in idiopathic pulmonary arterial hypertension.
Results:
PPHN was significantly (P < 0.05) associated with genetic variants in corticotropin-releasing hormone (CRH) receptor 1, CRHR1 and CRH-binding protein, CRHBP. Association with CRHR1 rs4458044 passed the Bonferroni threshold for significance. No mutations were found in the bone morphogenetic protein receptor type II (BMPR2) gene.
Discussion:
We describe previously unreported genetic associations between PPHN and CRHR1 and CRHBP. These findings may have implications for further understanding the pathophysiology of PPHN and treatment.
Methods:
We performed a family-based candidate gene study to examine a genetic association with PPHN and sequenced the BMPR2 gene in 72 individuals. We enrolled 110 families with infants diagnosed with PPHN based on inclusion criteria. After medical chart review, 22 subjects were excluded based on predefined criteria, and DNA samples from 88 affected infants and at least one parent per infant were collected and genotyped. Thirty-two single-nucleotide polymorphisms in 12 genes involved in vasoconstriction/vasodilation, lung development, surfactant regulation, or vascular endothelial cell function were investigated using family-based association tests.
Similar content being viewed by others


Main
Persistent pulmonary hypertension of the newborn (PPHN) is a serious and often rapidly progressive disease. If untreated, PPHN can lead to severe hypoxemia, right ventricular failure, and death. Combined with greater recognition, advances made in the treatment of neonatal pulmonary disease, such as mechanical ventilation, inhaled nitric oxide, and extracorporeal membrane oxygenation, have greatly improved the survival and outcomes of infants with PPHN. Even so, PPHN remains a disease of substantial mortality, and neonates who survive may suffer long-term consequences as a result of the associated hypoxemia and invasive therapies (1,2,3,4).
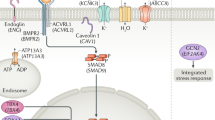
Idiopathic PPHN occurs in 1–2 per 1,000 live-born infants (1,2,5), typically presenting as hypoxemic respiratory failure within the first day of life in term or late preterm infants without associated congenital anomalies or cyanotic cardiac lesions. In normal physiology, fetal lungs have high pulmonary vascular resistance and receive only 5–10% of cardiac output. At birth, pulmonary vascular resistance normally decreases, pulmonary blood flow increases dramatically, amniotic fluid clears from the lungs, and effective gas exchange begins. In PPHN, fetal lungs fail to undergo this normal physiologic transition. Pulmonary vascular resistance remains high, restricting pulmonary blood flow, resulting in hypoxemia, which leads to vasoconstriction, decreased gas exchange, acidemia, and hypercarbia. On pathological study, neonates with PPHN are found to have marked pulmonary vascular remodeling with torturous vessels and excessive muscularization from pulmonary arteriole smooth muscle cell proliferation (6).
Pulmonary hypertension is a complex and heterogeneous disease. Although many hypotheses have been proposed, the exact etiology leading to PPHN remains unknown (6). Recent studies of pulmonary hypertension in adults (pulmonary arterial hypertension) and neonates (PPHN) have increased our understanding of the genetic basis of these distinct diseases. Genetic susceptibility to PPHN has been inferred based on studies of environmental risk factors, such as nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors taken during pregnancy and variation in the development and severity of disease (7,8,9). Genes involved in the transforming growth factor-β (TGF-β) super family (10,11), nitric oxide pathway (12,13,14), stress and inflammatory response (15,16,17), surfactant regulation (18,19), and glucocorticoids (20,21) have all been implicated in adult and/or neonatal pulmonary disease. Nearly 300 disease-causing genetic variants have been described in the gene for bone morphogenic protein receptor type II (BMPR2), associated with >70% of familial (non-neonatal) pulmonary hypertension cases (22,23). Nitric oxide synthase 3 (NOS3) polymorphisms in T-786C and Glu298Asp have been shown to contribute to altered pulmonary vascular reactivity in adults (12). We therefore evaluated 32 single-nucleotide polymorphisms (SNPs) in 12 genes with biologic plausibility of contributing to PPHN, shown in Table 3.
Results
We analyzed genotype data for 88 cases and their families. Of these, 72 cases were part of a complete triad (mother, father, and affected infant) and 16 were part of a dyad (one parent and affected infant). Our proband cohort was predominantly male (67%) and Caucasian (89%). Average gestational age was 38.3 (+/−1.8) wks. Average birth weight was 3,512 g; 31 (35%) cases were large for gestational age. Sixty (68%) cases required inhaled nitric oxide or extracorporeal membrane oxygenation. Thirty-four cases (39%) received hydrocortisone for blood pressure support in addition to vasopressors, which a total of 47 (53%) cases received. A description of the study cases and mothers is provided in Table 1 .
Associations were identified in the genes for corticotropin-releasing hormone (CRH) receptor 1, CRHR1 (rs4458044, P = 9.9 × 10−5 and rs173365, P = 0.02) and CRH-binding protein, CRHBP (rs10062367, P = 0.009 and rs10055255, P = 0.003) in infants with PPHN. The result for rs4458044 remains significant after using the Bonferroni correction (P < 0.0016). The major allele was overtransmitted in CRHR1 rs4458044 and CRHBP rs10062367 and rs10055255. The minor allele was overtransmitted in CRHR1 rs173365.
For the newborn screen analysis, there was no significant difference in gestational age, birth weight, or race between cases and controls. Mean 17-hydroxyprogesterone (17-OHP) was significantly higher in cases vs. controls (P = 1.7 × 10−4), as shown in Table 2 . Our term PPHN-affected cases had a 14% abnormal congenital adrenal hyperplasia (CAH) test result rate vs. 0% for unaffected controls (P = 0.002). In 2000, Iowa’s rate of false-positive CAH tests for all babies was 72 of 38,141 tests or 0.19%, when using the more conservative estimate of assuming all babies lost to follow-up may have had false-positive results (24).
No previously unreported consensus splice-site, missense, or nonsense mutations were found in sequenced BMPR2 exons. A previously reported missense mutation (rs2228545) was heterozygous in five cases. According to HapMap samples, this SNP has a population frequency of 5% (25). We did not find overtransmission of BMPR2 rs13010656 (P = 0.50), which is in linkage disequilibrium with the sequence variant.
Discussion
We have identified genetic variants in CRHR1 and in CRHBP associated with PPHN. Our data show a robust and highly significant association with variations in the CRHR1 gene, particularly rs4458044. CRH receptor 1 (encoded by CRHR1) and the binding protein (encoded by CRHBP) interact with CRH in the hypothalamic–pituitary–adrenal (HPA) axis and the myometrium, with cortisol as the ultimate end product. We found no evidence that mutations in BMPR2, previously associated with adult-onset pulmonary arterial hypertension, make a significant contribution to PPHN.
CRH-binding protein binds CRH, decreasing its bioavailability and hence controlling the reactivity of CRH in pregnancy. CRHBP is located on chromosome 5; SNPs significantly associated with PPHN, rs10062367 and rs10055255, are located between exons 6 and 7. In contrast, CRHR1 is a seven-transmembrane receptor with high affinity for CRH located in the anterior pituitary and myometrium, which plays an essential role in transmitting the adrenocorticotropic hormone signal for cortisol production. We investigated SNPs along CRHR1, located on chromosome 17; SNPs significantly associated with PPHN were found in intronic regions between exons 4 and 5 (rs173365) and exons 1 and 2 (rs4458044). Downstream of rs4458044, in the same intronic region, is a transcription factor binding site for peroxisome proliferator-activated receptor-γ (PPAR-γ). PPAR-γ is an essential regulator of pulmonary smooth muscle cell proliferation and vascular tone. Decreased expression of PPAR-γ has been associated with pulmonary hypertension and neonatal chronic lung disease (26). No other SNPs or genes yielded significant results.
Cortisol plays a vital role in fetal development. It is commonly accepted that maturation of the fetal HPA axis and fetal steroid production is necessary for in utero lung development and ex utero pulmonary transition (27,28). Glucocorticoid supplementation for threatened preterm birth has been an extremely effective therapy in preventing neonatal respiratory distress syndrome. Yet fetuses with conditions of deficient cortisol secretion, such as CAH, do not necessarily have lung immaturity, suggesting fetal cortisol is not requisite for fetal lung development. In mice, CRHR1 knockouts are phenotypically normal when born to a CRHR1+/− mother, but CRHR1−/− pups born to knockout females die within 48 h because of respiratory distress. On pathologic review, the pup lungs show significant dysplasia, characterized by hypercellularity, immaturity, and intra-alveolar hemorrhage. Reports do not comment on pulmonary vascular changes. This lethal neonatal phenotype is completely rescued with maternal cortisol supplementation (27,29,30). Thus, it appears that either maternal or fetal cortisol is sufficient for normal lung development, but that one source is absolutely required.
CRH, CRHR1, and CRHBP have different secretion patterns, expression, and roles in pregnancy than in other physiologic states (31,32). Given this, it is possible that the polymorphisms we describe have a significant clinical effect on the pulmonary vasculature only in utero and during the transition from fetus to neonate. In fact, although PPHN overall has a high burden of morbidity and mortality, follow-up of neonates who successfully responded to PPHN treatment found that the capacity of long-term normal development with no clinically significant pulmonary sequelae exists (33,34). If the genetic polymorphisms we describe are associated with decreased CRH binding to CRHR1 or an excessive binding by CRHBP, the resulting decreased signal by CRH would diminish the activity of the HPA axis and affect either fetal lung functional development or the capacity to adequately transition to ex utero life.
In critical illnesses, the HPA axis is activated, resulting in increased cortisol secretion, a crucial effect for homeostasis (35). Transient adrenal insufficiency, marked by low cortisol despite increased physiologic demand due to stress, has been described in both preterm (36,37) and term infants (38,39,40) and is associated with increased hemodynamic instability, poor outcomes, and longer illnesses (41,42). Studies have found that infants with severe lung disease requiring ventilation had lower rates of cortisol in the first 7 d of life and that early treatment with corticosteroids can decrease pulmonary oxidative stress and increase pulmonary arterial response to vasodilators (43,44,45). When we probed deeper into the high number of abnormal CAH newborn screen (NBS) results in our cohort (14%), we found that mean 17-OHP levels were significantly higher in term PPHN cases vs. controls (P = 1.7 × 10−4). As we noticed the abnormal NBS results only incidentally during retrospective chart review, a limitation of our study is that we were not able to measure cortisol levels to confirm whether an abnormally high 17-OHP was the result of a buildup of substrate, as the CAH screen is designed to measure, or an overall activation of the HPA axis. Clinically, many of our PPHN cases showed signs of transient adrenal insufficiency such as hypotension, hemodynamic instability, and inadequate stress response. Forty-seven (53%) cases received vasopressors, of which 34 (39%) also received postnatal glucocorticoids. Further investigation into the cortisol levels of neonates with PPHN may provide insight into the biologic consequences of genetic variation in CRHR1 and CRHBP. If adrenal insufficiency is causally linked to PPHN and/or the hemodynamic instability that often accompanies it, glucocorticoids may be an important adjuvant treatment for both PPHN and hypotension, in addition to or instead of vasopressors.
In addition to not confirming 17-OHP results with cortisol measurements, an important limitation of our study is its retrospective nature over 16 y. Although many clinical treatment advances occurred during this time, these do not affect the genetics of the affected individuals and therefore our study. Efforts were taken to minimize retrospective selection bias. All neonates diagnosed with PPHN at our center were thoroughly investigated for inclusion. Second, given that the University of Iowa Children’s Hospital is a major referral center in Iowa, our cohort was skewed toward PPHN requiring more invasive treatments, such as inhaled nitric oxide and extracorporeal membrane oxygenation. Due to power issues and our focus on genetic contributions rather than clinical outcome, we did not separate subjects for heterogeneity, such as treatment required or possible inciting factors such as meconium aspiration. Third, not all cases of PPHN were confirmed with echocardiography. Finally, our case–control study was limited to a 1:1 study. However, given statewide NBS population statistics, we do not anticipate our results would markedly differ with additional controls. Thirty-five percent of newborns in our cohort were large for gestational age, which reflects epidemiology studies that report this risk factor for PPHN (46). Strengths of the study include that it was adequately powered for an uncommon neonatal disease, allowing us to draw statistically relevant conclusions. Second, we replicated all of our significant findings with a SNP in complete linkage disequilibrium to ensure our results were not technical artifacts. Third, all tests and methods were performed at a single center, minimizing differences in understanding of standard protocols, techniques, and style.
In conclusion, whereas other studies looking at PPHN have focused primarily on vasodilators/vasoconstrictors, surfactant, vascular growth factors, and more recently serotonin receptors, we introduce the hypothesis that the HPA axis and specifically CRH signaling and bioreactivity resulting in altered cortisol levels may play a vital role in pulmonary transition from fetal to neonatal life. To our knowledge, this is the first report identifying genetic variants in CRHR1 and CRHBP associated with PPHN.
Methods
Study Design and Population
Neonates diagnosed with and treated for PPHN at the University of Iowa Children’s Hospital between 1993 and 2009 (and their families) were considered for enrollment into the study. Inclusion criteria for this family-based, candidate gene association study were a neonate with hypoxemic respiratory failure with the clinical diagnosis of pulmonary hypertension. After medical chart review, patients were excluded from genetic analysis based on predetermined exclusion criteria: gestational age <35 wks, multiple major congenital anomalies, congenital diaphragmatic hernia, cyanotic heart disease, and/or the inability to obtain a DNA sample from the neonate and at least one parent. Neonates with hypoxic respiratory failure were diagnosed with PPHN by the medical team using echocardiography, preductal/postductal oxygen saturation difference >10%, and/or a clinical response to inhaled nitric oxide. Echocardiographic findings consistent with PPHN included elevated pulmonary artery pressure as compared with systemic pressure, right-to-left or bidirectional patent ductus arteriosus shunting, and right-to-left or bidirectional shunting through the patent foramen ovale. In total, 110 families were enrolled in the study. Following medical chart review, 22 subjects were excluded from genetic analysis.
Samples were obtained retrospectively from either a DNA repository at the University of Iowa (n = 60) or recruited after medical record review (n = 28) to capture families not represented in the DNA repository. A maternal interview was conducted for families not previously enrolled in the DNA repository database. Samples were obtained from venous blood, cord blood, buccal swabs, or saliva samples. DNA was extracted using standard protocols (Qiagen Maxi Kit, Hilden, Germany).
Genotyping
Candidate genes were identified by a review of the literature, and genes associated with adult or neonatal pulmonary disease and/or with biologic plausibility were selected. Thirty-two SNPs in 12 genes were selected and genotyped utilizing Applied Biosystems (Foster City, CA) TaqMan chemistry, as noted in Table 3 . SNPs were selected according to haplotype blocks to minimize the number of SNPs selected per gene, according to high minor allele frequency to increase informativity of each SNP, and according to known functional impact. Allelic end-point fluorescence was measured and analyzed on a 7900HT PCR system (Applied Biosystems) and interpreted with Sequence Detection System software (SDS, version 2.3, Applied Biosystems). Greater than 98% of samples tested provided usable genotype information. Genotypes were entered into a laboratory database (Progeny, South Bend, IN) to generate datasets for analysis.
Statistical Analysis
Statistical analyses of the genotype data were performed with a transmission disequilibrium test, using family-based association test software (FBAT, Cambridge, MA) (47,48,49). Our primary outcome was neonatal hypoxic respiratory failure with clinical or echocardiographic findings consistent with PPHN. The transmission disequilibrium test measures overtransmission of an allele from heterozygous parents to offspring by comparing whether transmission proportions are compatible with Mendelian probabilities. This method maintains its robustness by evaluating genetic linkage only in the face of genetic association and has been shown to have sufficient power to detect even disease determinates of a relatively small effect (50). To correct for the multiple tests performed in this study, a conservative Bonferroni correction would place significance at P < 0.0016 using a standard α of 0.05. However, given the exploratory nature of this initial study, less stringent values are also of interest.
While performing subject chart reviews, we observed a high number of abnormal CAH NBS results in our cohort. This test measures the level of 17-OHP, a substrate in the steroid hormone pathway upstream of cortisol and testosterone. CAH is caused by a deficient enzyme in this pathway (usually 21-hydroxylase) leading to the buildup of 17-OHP and corresponding insufficient steroid hormone. All CAH tests were processed by the University of Iowa Hygienic Laboratory. To test the hypothesis that abnormal steroid hormone metabolism is linked to PPHN, we obtained the 17-OHP values for our PPHN-affected cases and 87 unaffected controls matched for gender, gestational age, birth weight, and year of NBS test. As very preterm infants have higher rates of false-positive CAH tests attributed to an immature HPA axis, we analyzed only term cases and controls (≥37 wks gestation; 58 cases, 60 controls) to separate gestational age effect (51). Standard clinical procedure is to repeat abnormal (borderline or presumptive positive) NBS CAH tests. We used the first-obtained 17-OHP results for our analysis. We looked at medical records and repeat 17-OHP tests to assess if any subjects were true-positive CAH. We had no true-positive CAH subjects in cases or controls and all repeat 17-OHP values normalized. Data were checked for normality and transformed using the Box-Cox statistical method (52). Analysis was performed using linear regression, adjusting for major lot changes in the assay. Categorical results were compared using Fisher’s exact test.
Sequencing
Each of the 13 exons of BMPR2 was sequenced to include at least 50 base pairs of flanking intronic sequencing using methods described previously (53). All affected cases with adequate DNA sample were sequenced (n = 72), 68 (94%) provided adequate sequencing results.
Ethics Statement
This study was approved by the University of Iowa Institutional Review Board (IRB 200307031). All participants or their guardians provided informed consent.
Statement of Financial Support
This study was funded by National Institutes of Health R01 HD-57192, R01 HD-52953, RSDP 5K12 HD-000849-23, and MSTP T32 5 GM007337.
References
Walsh-Sukys MC, Tyson JE, Wright LL, et al. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics 2000;105(1 Pt 1):14–20.
Farrow KN, Fliman P, Steinhorn RH . The diseases treated with ECMO: focus on PPHN. Semin Perinatol 2005;29:8–14.
John E, Roberts V, Burnard ED . Persistent pulmonary hypertension of the newborn treated with hyperventilation: clinical features and outcome. Aust Paediatr J 1988;24:357–61.
Clark RH, Huckaby JL, Kueser TJ,et al.; Clinical Inhaled Nitric Oxide Research Group. Low-dose nitric oxide therapy for persistent pulmonary hypertension: 1-year follow-up. J Perinatol 2003;23:300–3.
Hageman JR, Adams MA, Gardner TH . Persistent pulmonary hypertension of the newborn. Trends in incidence, diagnosis, and management. Am J Dis Child 1984;138:592–5.
Abman SH . New developments in the pathogenesis and treatment of neonatal pulmonary hypertension. Pediatr Pulmonol Suppl 1999;18:201–4.
Chambers CD, Hernandez-Diaz S, Van Marter LJ,et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med 2006;354:579–87.
Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL . Birth outcomes in pregnant women taking fluoxetine. N Engl J Med 1996;335:1010–5.
Alano MA, Ngougmna E, Ostrea EM Jr, Konduri GG . Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Pediatrics 2001;107:519–23.
Harrison RE, Berger R, Haworth SG,et al. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005;111:435–41.
Sztrymf B, Yaïci A, Girerd B, Humbert M . Genes and pulmonary arterial hypertension. Respiration 2007;74:123–32.
Casas JP, Cavalleri GL, Bautista LE, Smeeth L, Humphries SE, Hingorani AD . Endothelial nitric oxide synthase gene polymorphisms and cardiovascular disease: a HuGE review. Am J Epidemiol 2006;164:921–35.
Pearson DL, Dawling S, Walsh WF,et al. Neonatal pulmonary hypertension–urea-cycle intermediates, nitric oxide production, and carbamoyl-phosphate synthetase function. N Engl J Med 2001;344:1832–8.
Villanueva ME, Zaher FM, Svinarich DM, Konduri GG . Decreased gene expression of endothelial nitric oxide synthase in newborns with persistent pulmonary hypertension. Pediatr Res 1998;44:338–43.
Lavoie PM, Pham C, Jang KL . Heritability of bronchopulmonary dysplasia, defined according to the consensus statement of the national institutes of health. Pediatrics 2008;122:479–85.
Nickel N, Kempf T, Tapken H,et al. Growth differentiation factor-15 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2008;178:534–41.
Silverman ES, Breault DT, Vallone J,et al. Corticotropin-releasing hormone deficiency increases allergen-induced airway inflammation in a mouse model of asthma. J Allergy Clin Immunol 2004;114:747–54.
Pavlovic J, Papagaroufalis C, Xanthou M,et al. Genetic variants of surfactant proteins A, B, C, and D in bronchopulmonary dysplasia. Dis Markers 2006;22:277–91.
Kunig AM, Parker TA, Nogee LM, Abman SH, Kinsella JP . ABCA3 deficiency presenting as persistent pulmonary hypertension of the newborn. J Pediatr 2007;151:322–4.
Rogers AJ, Tantisira KG, Fuhlbrigge AL,et al. Predictors of poor response during asthma therapy differ with definition of outcome. Pharmacogenomics 2009;10:1231–42.
Melnick M, Choy HA, Jaskoll T . Glucocorticoids, tumor necrosis factor-alpha, and epidermal growth factor regulation of pulmonary morphogenesis: a multivariate in vitro analysis of their related actions. Dev Dyn 1996;205:365–78.
Sztrymf B, Coulet F, Girerd B,et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med 2008;177:1377–83.
Machado RD, Eickelberg O, Elliott CG, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2009;54: Suppl 1:S32–42.
National Newborn Screening and Genetics Resource Center (NNSGRC). Congenital adrenal hyperplasia. In: National Newborn Screening Report—2000. Austin, TX: University of Texas, 2003:121–33.
International HapMap Consortium. The International HapMap Project. Nature 2003; 426:789–96.
Simon DM, Mariani TJ . Role of PPARs and Retinoid X Receptors in the Regulation of Lung Maturation and Development. PPAR Res 2007;2007:91240.
Muglia L, Jacobson L, Dikkes P, Majzoub JA . Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature 1995;373:427–32.
Garbrecht MR, Klein JM, Schmidt TJ, Snyder JM . Glucocorticoid metabolism in the human fetal lung: implications for lung development and the pulmonary surfactant system. Biol Neonate 2006;89:109–19.
Smith GW, Aubry JM, Dellu F,et al. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 1998;20:1093–102.
Muglia LJ, Bae DS, Brown TT, et al. Proliferation and differentiation defects during lung development in corticotropin-releasing hormone-deficient mice. Am J Respir Cell Mol Biol 1999;20:181–8.
Brosnan PG . The hypothalamic pituitary axis in the fetus and newborn. Semin Perinatol 2001;25:371–84.
Grammatopoulos D, Dai Y, Chen J,et al. Human corticotropin-releasing hormone receptor: differences in subtype expression between pregnant and nonpregnant myometria. J Clin Endocrinol Metab 1998;83:2539–44.
Hoskote AU, Castle RA, Hoo AF,et al. Airway function in infants treated with inhaled nitric oxide for persistent pulmonary hypertension. Pediatr Pulmonol 2008;43:224–35.
Bernbaum JC, Russell P, Sheridan PH, Gewitz MH, Fox WW, Peckham GJ . Long-term follow-up of newborns with persistent pulmonary hypertension. Crit Care Med 1984;12:579–83.
Lamberts SW, Bruining HA, de Jong FH . Corticosteroid therapy in severe illness. N Engl J Med 1997;337:1285–92.
Martins PG, Procianoy RS . Cortisol and 17-alpha-hydroxy-progesterone levels in infants with refractory hypotension born at 30 weeks of gestation or less. Braz J Med Biol Res 2007;40:577–82.
Quintos JB, Boney CM . Transient adrenal insufficiency in the premature newborn. Curr Opin Endocrinol Diabetes Obes 2010;17:8–12.
Tantivit P, Subramanian N, Garg M, Ramanathan R, deLemos RA . Low serum cortisol in term newborns with refractory hypotension. J Perinatol 1999;19:352–7.
Fernandez EF, Watterberg KL . Relative adrenal insufficiency in the preterm and term infant. J Perinatol 2009;29: Suppl 2:S44–9.
Fernandez E, Schrader R, Watterberg K . Prevalence of low cortisol values in term and near-term infants with vasopressor-resistant hypotension. J Perinatol 2005;25:114–8.
Kamath BD, Fashaw L, Kinsella JP . Adrenal insufficiency in newborns with congenital diaphragmatic hernia. J Pediatr 2010;156:495–7.e1.
Michalaki M, Margeli T, Tsekouras A, Gogos CH, Vagenakis AG, Kyriazopoulou V . Hypothalamic-pituitary-adrenal axis response to the severity of illness in non-critically ill patients: does relative corticosteroid insufficiency exist? Eur J Endocrinol 2010;162:341–7.
Chandrasekar I, Eis A, Konduri GG . Betamethasone attenuates oxidant stress in endothelial cells from fetal lambs with persistent pulmonary hypertension. Pediatr Res 2008;63:67–72.
Ng PC, Lam CW, Lee CH,et al. Reference ranges and factors affecting the human corticotropin-releasing hormone test in preterm, very low birth weight infants. J Clin Endocrinol Metab 2002;87:4621–8.
da Costa DE, Nair AK, Pai MG, Al Khusaiby SM . Steroids in full term infants with respiratory failure and pulmonary hypertension due to meconium aspiration syndrome. Eur J Pediatr 2001;160:150–3.
Hernández-Díaz S, Van Marter LJ, Werler MM, Louik C, Mitchell AA . Risk factors for persistent pulmonary hypertension of the newborn. Pediatrics 2007;120:e272–82.
Spielman RS, McGinnis RE, Ewens WJ . Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993;52:506–16.
Horvath S, Xu X, Laird NM . The family based association test method: strategies for studying general genotype–phenotype associations. Eur J Hum Genet 2001;9:301–6.
Laird NM, Horvath S, Xu X . Implementing a unified approach to family-based tests of association. Genet Epidemiol 2000;19: Suppl 1:S36–42.
Risch N, Teng J . The relative power of family-based and case-control designs for linkage disequilibrium studies of complex human diseases I. DNA pooling. Genome Res 1998;8:1273–88.
Lee JE, Moon Y, Lee MH, Jun YH, Oh KI, Choi JW . Corrected 17-alpha-hydroxyprogesterone values adjusted by a scoring system for screening congenital adrenal hyperplasia in premature infants. Ann Clin Lab Sci 2008;38:235–40.
Box GEP, Cox DR . An analysis of transformations. J Roy Statistical Society. 1964 (Ser B) 26:211–52.
Ehn NL, Cooper ME, Orr K,et al. Evaluation of fetal and maternal genetic variation in the progesterone receptor gene for contributions to preterm birth. Pediatr Res 2007;62:630–5.
Acknowledgements
We express our sincere thanks to the families that participated in this study. Their enthusiasm and support for the project was inspiring. We also thank Susan Berends, Elise Bream, Diedre Fleener, and Laura Knosp for their invaluable assistance; Tamara Busch and Elizabeth Leslie for assistance with sequencing; Stanton Berberich, Franklin Delin, and Dari Shirazi from the State Hygienic Laboratory for their help with the Newborn Screen data; Jeffrey Segar for his comments; and the entire staff of the University of Iowa Children’s Hospital Newborn Intensive Care Unit for their tireless dedication to the patients and their support of this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Byers, H., Dagle, J., Klein, J. et al. Variations in CRHR1 are associated with persistent pulmonary hypertension of the newborn. Pediatr Res 71, 162–167 (2012). https://doi.org/10.1038/pr.2011.24
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2011.24
This article is cited by
-
Correlating SFTPC gene variants to interstitial lung disease in Egyptian children
Journal of Genetic Engineering and Biotechnology (2022)
-
The use of supplemental hydrocortisone in the management of persistent pulmonary hypertension of the newborn
Journal of Perinatology (2021)
-
Association between genetic variations in carbamoyl-phosphate synthetase gene and persistent neonatal pulmonary hypertension
European Journal of Pediatrics (2021)
-
Identification of genetic factors underlying persistent pulmonary hypertension of newborns in a cohort of Chinese neonates
Respiratory Research (2019)
-
Genetic variation in CRHR1 is associated with short-term respiratory response to corticosteroids in preterm infants at risk for bronchopulmonary dysplasia
Pediatric Research (2019)
