
Abstract
My research has focused on elucidating the allergy problem over the past two decades. The primary approach has been to uncover critical mechanisms of allergic inflammation, with particular focus on eosinophils, a hallmark cellular constituent of allergic responses. Molecular processes that bridge T helper cell type 2 (TH2) immunity with eosinophilia and key checkpoints for regulating eosinophilia have been uncovered. Notably, interleukin (IL)-5 (derived from TH2 cells) has been identified as the chief hematopoietin responsible for eosinophil expansion in the circulation. Pathways for selective eosinophil mobilization from the blood stream to the tissue have been uncovered by defining the role of the eotaxin subfamily of chemokines in eosinophil chemoattraction and activation. Finally, TH2 cell derived IL-4 and IL-13 have been defined as chief inducers of the eotaxins, and upstream orchestrators of eosinophilic inflammation. These translational studies have formulated novel therapeutic strategies (currently being tested) for a variety of eosinophilic conditions, with particular attention on hypereosinophilic syndromes and eosinophil-associated gastrointestinal disorders such as eosinophilic esophagitis.
Similar content being viewed by others

Main
During my PhD studies with Dr. K. Frank Austen under the co-mentorship of Dr. Richard Stevens, I pioneered a series of studies that led to the concept that human eosinophils were long-lived cells capable of normally surviving for several weeks after they emigrated from the bone marrow. Before this finding, it was believed that eosinophils died within hours after leaving the bone marrow. It was, thus, no surprise that Dr. Austen doubted my initial demonstration that I could culture eosinophils (derived from human blood) for over 2 wks by simple co-culture with endothelial cells (1). I subsequently identified that interleukin (IL)-3 and -5, and granulocyte-macrophage colony-stimulating factor were chief eosinophilopoeitins capable of extending their survival, inducing their activation and priming them to respond to other stimuli (2–5). Furthermore, my work was the first to identify the overproduction of IL-5 in human disease (hypereosinophilic syndromes) and to show that the pathophysiology of eosinophilic disorders involved the development of an aggressive eosinophil phenotype responding to IL-5 produced by an autocrine or paracrine mechanism (6). This identified IL-5 as a target for drug intervention in eosinophil-associated diseases, and indeed, now 20 y later, I am finally proving that a clinical agent that blocks IL-5, humanized anti-IL-5 antibody, has therapeutic utility for eosinophilic syndromes (7–9).
After 3 y of clinical training, wherein I completed two clinical research studies (10,11), I undertook a postdoctorate position in the laboratory of Professor Phil Leder, chairman of the Department of Genetics at Harvard Medical School. During this fellowship, I established the importance of eosinophil specific chemokines, characterizing the eotaxin chemokines including the original cloning of guinea pig eotaxin (12) and murine eotaxin (13), and collaborated on cloning the human homologue (14). Dr. Leder provided me with a great opportunity to pursue my own ideas by providing an open-minded, very well-funded laboratory full of other great trainees.1
In 1996, I was attracted to a recruitment offer from Cincinnati Children's Hospital Medical Center (CCHMC). A move to CCHMC represented a great opportunity to apply my extended education at Harvard in an environment that was highly supportive of pediatric-based research but was relatively devoid of formal allergy or immunology research. Indeed, I was able to make a series of seminal findings concerning the regulation and role of eosinophils in health and disease (15–19). I was the first to generate eotaxin-1 gene targeted mice and to provide definitive evidence for the importance of the eotaxin pathway in vivo (20). In addition to defining other molecules that regulate eosinophils (e.g., cloning and characterizing eotaxin-2 and generating eotaxin-2 gene targeted mice) (21–23), I established that the gastrointestinal tract was the main reservoir of eosinophils and that eosinophil homing was tightly regulated by the constitutive expression of eotaxin-1 throughout the gastrointestinal tract (24–26). Focusing on the increasing occurrence of eosinophil-associated gastrointestinal disorders (EGID), I developed several antigen- and transgene-induced models of gastrointestinal allergic diseases including eosinophilic esophagitis (EE) (27), eosinophilic gastroenteritis (26,28,29), and allergic diarrhea (30). Furthermore, I used these models to establish critical roles for eosinophils, mast cells, the eotaxin or chemokine (C-C motif) receptor 3 (CCR3) pathway, and the cytokine IL-5 in specific aspects of disease pathogenesis (22,23,27–31). In particular, I defined the spatial and temporal critical role for the eotaxin or CCR3 pathway by generating and characterizing eotaxin-1-deficient, eotaxin-2-deficient, eotaxin-1 or 2 double-deficient, and eotaxin receptor (CCR3)-deficient mice (20,22,23). I defined the intersection of the IL-13-induced allergic lung pathway with eosinophils by demonstrating that inducible IL-13 lung transgenic mice require eotaxin-mediated eosinophilia to develop lung damage (32–34). Identifying the importance of the eotaxin subfamily of chemokines (35,36), we have elucidated structural, genetic, and biochemical signaling pathways involved in this process (37–45).
Notably, my work has led to the initiation of several world-wide clinical trials testing agents that block these molecules, including our own phase I or II study examining the safety and efficacy of anti-IL-5 for hypereosinophilic syndromes (7). I was able to demonstrate that humanized anti-IL-5 is a safe and effective therapy for lowering blood and tissue eosinophilia in patients with hypereosinophilic syndromes including EE (7,8). This is a significant accomplishment, as these patients frequently require toxic steroid therapy and anti-IL-5 now appears to be a safe alternative. These encouraging findings prompted the drug manufacturer, Glaxo-SmithKline, to initiate a phase II/III trial evaluating the impact of anti-IL-5 in adult patients with hypereosinophilic syndromes and pediatric patients with EE (9); these studies are directly attributed to the foundation that I established concerning the etiology and importance of these emerging health problems. The ability to propel a project from an original patient observation (when I was an MD/PhD student and discovered elevated IL-5 contributed to hypereosinophilic phenotypes in patients) to testing in preclinical settings, and finally to a viable therapeutic intervention is a great motivation that continues to inspire my current research.
In addition to conducting basic and translational work on this disease, I have established the Cincinnati Center for Eosinophilic Disorders, the leading center in the world involved in the care of patients suffering from eosinophilic diseases (please visit the Website http://www.cchmc.org.cced). My work has been focused on unraveling the clinical features, pathogenesis, and treatment of this newly described set of diseases that we now refer to as EGID, an acronym that I coined (46). In the laboratory, we have shown that eosinophils are indeed important effector cells in eliciting cardinal features of EGID including gastromegaly, gastric dysmotility, and the epithelial hyperplasia seen in EE (27,29) Clinically, we have shown that the patients with EE report symptoms that include difficulty feeding, failure to thrive, vomiting, epigastric or chest pain, dysphagia, and food impaction (47). These symptoms appear to occur in a progressive order, as they are the presenting symptoms from infancy into adulthood, respectively (47). We have shown that EE patients are predominantly young males and have relatively high levels of eosinophils (>20–24 peak eosinophils/400× high-powered field) in the esophageal mucosa, extensive epithelial hyperplasia, a high rate of atopic disease, and normal pH monitoring of the esophagus compared with patients with gastroesophageal reflux disease (GERD) (46). Tracking the basic disease epidemiology, we have defined a disease incidence of ∼1:10,000 children and a growing prevalence that is now ∼1:1000 pediatric individuals (47). In addition, we have defined a strong familial inheritance pattern for EE and are now following over 30 multiplex families, aiming to define the dominant genetic factors involved (47).
We are accumulating substantial evidence that EE is associated with T helper cell type 2 (TH2) immune responses. In particular, elevated levels of eosinophil-active TH2 cytokines (e.g., IL-4, IL-5, and IL-13) and mast cells are present in the esophagus of EE patients (48,49). In addition, experimental models of EE can be induced in mice by allergen exposure, especially in the respiratory tract after mucosal or epicutaneous sensitization, as well as by overexpression of TH2 cytokines (IL-5 and IL-13) (27,31,50–52). Collectively, these experimental systems have established an intimate connection between the development of eosinophilic inflammation in the respiratory tract and esophagus not only in response to external allergic triggers but also to intrinsic TH2 cytokines. It is interesting to note that patients with EE sometimes report seasonal variations in their symptoms; preliminary studies have recently documented seasonal changes in esophageal eosinophil levels, especially in the proximal esophagus (53,54).
Using whole genome microarray expression profile analysis, we have described the dysregulated expression of ∼1% of the human genome in the esophageal tissue of EE patients (48). Interestingly, the EE transcript profile markedly distinguishes EE from reflux esophagitis patients; the latter mainly expresses genes comparable to normal individuals (48). Of the entire dysregulated genome, the gene with the greatest overexpression and correlation with esophageal eosinophilia is eotaxin-3, induced 50–100-fold compared with control individuals (48). Comparison of allergic and nonallergic EE patients revealed that the gene transcript signature was markedly conserved across these two major patient phenotypes. This demonstrates that the effector phase of the disease is conserved between individuals despite the driving trigger of the inflammation.
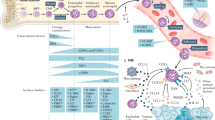
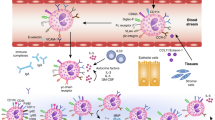
Experimental modeling in mice has established that TH2 signaling is required for induction of experimental EE. In particular, mice with the targeted deletion of STAT6 are protected from allergen and IL-13-induced experimental EE (51). Furthermore, IL-13 deficient mice have impaired allergen-induced EE (51). Of note, we have shown that IL-13 is over-expressed in the esophagus of EE patients, selectively induces eotaxin-3 in esophageal keratinocytes, and induces an EE-like transcriptome in esophageal keratinocytes (55). Notably, the esophageal tissue from EE patients markedly overexpresses eotaxin-3, but not eotaxin-1 or eotaxin-2, and IL-13-induced signaling in esophageal keratinocytes specifically induces eotaxin-3 (55). Taken together, our current working model for the disease induction (as shown in Fig. 1) is that food-antigen-driven TH2 cells produce IL-13 that subsequently induces local eotaxin-3, which is then responsible for the recruitment and activation of blood derived eosinophils.

A proposed model to explain molecular and cellular mechanisms involved in EE pathogenesis, eotaxin-3-associated eosinophil recruitment and treatments. Aeroallergen, food allergen and skin sensitization have been implicated in EE pathogenesis. Elemental diet, glucocorticoids, and anti-IL-5 treatments improve the microscopic features of EE acting at different levels on the disease pathogenesis. Proton pomp inhibitors (PPI) inhibit H+ secretion by parietal cells of the stomach and can only partially improve EE features. Hyperplasic epithelial cells of the esophagus overexpress eotaxin-3 likely in response to IL-13. Eotaxin-3 overexpression allows chemoattraction of CCR3+ cells. Inheritance of EE disease suggests a genetic predisposition. A SNP in the eotaxin-3 gene has been associated with EE. The Linkage disequilibrium pattern (ID'I) of the SNPs in Eotaxin-3 gene, based on the genotype data in a European population from the HapMap database, is presented. ID'I is the measure of the correlation between two SNPs, it ranges from 0 (in linkage equilibrium) to 1 (in strong linkage disequilibrium). Except for the SNP pair +76 and −5419 with the ID'I = 0.71, the other SNP pairs (in black) are in strong linkage disequilibrium (ID'I = 1). As such, the genotype of one SNP speaks for the others. Figure adapted from Blanchard C et al., J Allergy Clin Immunol 118:1054–1059 Copyright © 2006 American Academy of Allergy, Asthma and Immunology Published by Mosby, Inc., with permission.
Our genetic study of a single nucleotide polymorphism (SNP) (+2496T → G, rs2302009) in the eotaxin-3 gene has shown association with EE by both population-based case-control comparison and family-based transmission disequilibrium test (48,49). Of note, this SNP located in the 3′ UTR of the eotaxin-3 mRNA might affect mRNA stability. Notably, the induction of inflammatory cytokines is often controlled at the level of mRNA stability. Moreover, this SNP may disrupt a putative AU-rich element sequence and thus modify the mRNA levels and/or responsiveness to glucocorticoids. Interestingly, eotaxin-3 (+2496T → G) is in strong linkage disequilibrium with upstream genetic variants, making a ∼7 kb haplotype block in the white population (http://www.hapmap.org) (Fig. 1). Taken together, this genetic variant (+2496T → G), or the one(s) in strong linkage disequilibrium with it, is likely to contribute to EE susceptibility.
Evidence is emerging that lymphocytes and mast cells are also involved in EE pathogenesis (48). Indeed, esophageal lymphocytes and mast cell levels are increased in EE. This increase in cell numbers is corroborated by the increased expression of lymphocytes and mast cell-specific genes. TH2 cell-derived IL-5 appears to have a fundamental role in EE, as demonstrated by the ablation of allergen and IL-13-induced esophageal eosinophilia in response to neutralizing anti-IL-5 antibody treatment and/or IL-5 gene targeting (27,50). Consistent with these observations, we have shown that allergen-induced experimental EE in mice is dependent upon lymphocytes (56).
We are now developing agreed upon therapy for EE (57). At present, therapy for EE is based on antigen elimination trials, anti-inflammatory approaches, and physical dilatation when strictures are present. From the onset, anti-GERD therapy is indicated for the initial treatment of EE because acid can trigger esophageal eosinophilia, albeit generally of lower magnitude than that associated with EE. If anti-GERD therapy is unsuccessful, specific food allergen elimination (a restricted diet) or an exclusive elemental (amino acid based) formula is recommended. Although very efficient, these are often unsatisfactory or practically difficult. Although a diet consisting of an exclusive elemental (amino acid based) formula is often effective, this may not be well tolerated (especially in older individuals) because it frequently requires a surgically placed feeding tube, can be financially costly (e.g., thousands of dollars per month), and is unpalatable. Glucocorticoids (systemic or topical) have been used with satisfactory results in some patients. Systemic steroids are used for acute exacerbations, whereas topical steroids are used to provide long-term control. However, we recently have shown that a significant fraction of patients are steroid resistant (58,59). We have recently completed the first controlled clinical trial for EE testing the efficacy of swallowed fluticaonse (880 μg per day) compared with placebo in a double blind study. Results from our 3 m clinical study revealed that 1) the drug is effective compared with the control; 2) the drug only works in ∼50% of patients identifying bona fide steroid resistance in EE; and 3) the placebo effect occurs in ∼10% of study patients (59). We have also taken other approaches to treat EE based on mechanism-based targets identified through our research. In particular, we have shown that anti-IL-5 therapy is very effective at ablating the development of experimental EE in murine models, and appears to also improve eosinophil infiltration in the esophagus in an early clinical trial (7,8); a large-scale anti-IL-5 trial for EE is currently underway. In a preclinical trial, anti-human IL-13 has been shown to be useful, and it will be of interest to eventually examine the impact of IL-13 blockade in EE patients (52). In addition, we have shown that CCR3 gene target mice are protected from the development of experimental EE (48). Other anti-inflammatory approaches, such as leukotriene receptor antagonists, have been advocated, but they have not been shown to reverse esophageal pathology. Because IgE effector cells, such as mast cells and basophils, are a source of proinflammatory chemokines, cytokines, and proteases, it is also possible that anti-IgE therapy would have anti-inflammatory effects in EE. It is important to note that EE is a chronic disorder that requires ongoing therapy; the disease almost uniformly returns when therapy is discontinued (e.g., glucocorticoids are stopped or diet is liberated).
Taken together, these findings have now provided molecular diagnostic criteria and pathways for disease intervention. Indeed, eotaxin-3 and CCR3 blockers are now being considered for EE.
My research also encompasses several other areas of investigation, such as elucidating the pathogenesis of experimental asthma in mice; we were one of the first investigators to define the “asthma genome” based on global expression profile analysis in mice (60). I have led studies analyzing the transcriptional regulation of the “asthma genome” (61). My work has been focused on defining the importance of several new pathways including the provocative finding that arginine is primarily metabolized by arginase in the asthmatic lung. Before our work, arginine was thought to be mainly metabolized by nitric oxide synthase, but my colleagues and I have proposed a new paradigm in which arginase and the arginine transport protein CAT2 coordinately regulate several processes that appear to be germane in asthma pathogenesis (62). Recently, we have extended these findings by defining the critical role of the arginine transporter protein in regulating homeostatic lung immunity (63).
Future.
As a physician scientist, my work is guided by the burning desire to truly improve human life through medical research. Fortunately, my work has already influenced how we view the allergy problem, specifically focused on eosinophils and the involvement of this enigmatic cell in a variety of debilitating inflammatory diseases, especially gastrointestinal allergy. I have moved molecular findings into animal models and from there have even translated these findings into a variety of therapeutic approaches for the treatment of eosinophilic disorders. Gratifyingly, one of these approaches has already demonstrated benefit in humans, while others are on the near horizon. Although much work has been done, the greatest is yet to come. I am hopeful that the current amazing burst of scientific knowledge, combined with the fortunate colleagues, scientific, and translational environment that I have immersed myself among at CCHMC, will facilitate my mission to understand and treat eosinophilic disorders.
Abbreviations
- CCR3:
-
chemokine (C-C motif) receptor 3
- EE:
-
eosinophilic esophagitis
- EGID:
-
eosinophil associated gastrointestinal disorders
- GERD:
-
gastroesophageal reflux disease
- SNP:
-
single nucleiotide polymorphism
References
Rothenberg ME, Owen WF Jr Silberstein DS, Soberman RJ, Austen KF, Stevens RL 1987 Eosinophils cocultured with endothelial cells have increased survival and functional properties. Science 237: 645–647
Owen WF, Rothenberg ME, Silberstein DS, Gasson JC, Stevens RL, Austen KF, Soberman RJ 1987 Regulation of human eosinophil viability, density, and function by granulocyte/macrophage colony-stimulating factor in the presence of 3T3 fibroblasts. J Exp Med 166: 129–141
Rothenberg ME, Owen WF Jr Silberstein DS, Woods J, Soberman RJ, Austen KF, Stevens RL 1988 Human eosinophils have prolonged survival, enhanced functional properties, and become hypodense when exposed to human interleukin 3. J Clin Invest 81: 1986–1992
Rothenberg ME, Petersen J, Stevens RL, Silberstein DS, McKenzie DT, Austen KF, Owen WF 1989 IL-5-dependent conversion of normodense human eosinophils to the hypodense phenotype uses 3T3 fibroblasts for enhanced viability, accelerated hypodensity, and sustained antibody-dependent cytotoxicity. J Immunol 143: 2311–2316
Rothenberg ME, Pomerantz JL, Owen WF, Avraham S, Soberman RJ, Austen KF, Stevens RL 1988 Characterization of a human eosinophil proteoglycan, and augmentation of its biosynthesis and size by interleukin 3, interleukin 5, and granulocyte/macrophage colony stimulating factor. J Biol Chem 263: 13901–13908
Owen WF, Rothenberg ME, Petersen J, Weller PF, Silberstein D, Sheffer AL, Stevens RL, Soberman RJ, Austen KF 1989 Interleukin 5 and phenotypically altered eosinophils in the blood of patients with the idiopathic hypereosinophilic syndrome. J Exp Med 170: 343–348
Garrett JK, Jameson SC, Thomson B, Collins MH, Wagoner LE, Freese DK, Beck LA, Boyce JA, Filipovich AH, Villanueva JM, Sutton SA, Assa'ad AH, Rothenberg ME 2004 Anti-interleukin-5 (mepolizumab) therapy for hypereosinophilic syndromes. J Allergy Clin Immunol 113: 115–119
Stein ML, Collins MH, Villanueva JM, Kushner JP, Putnam PE, Buckmeier BK, Filipovich AH, Assa'ad AH, Rothenberg ME 2006 Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol 118: 1312–1319
Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, Simon H-U, Schwartz LB, Rossenwasser LJ, Ring J, Griffin EF, Haig AE, Frewer PIH, Parkin JM, Gleich GJ 2008 Treatment of hypereosinophilic syndrome with mepolizumab. N Eng J Med 358: 1215–1228
Rothenberg ME, White FV, Chilmonczyk B, Chatila T 1995 A syndrome involving immunodeficiency and multiple intestinal atresias. Immunodeficiency 5: 171–178
Rothenberg ME, Weber WE, Longtine JA, Hafler DA 1996 Cytotoxic gamma delta T lymphocytes associated with an Epstein-Barr virus-induced posttransplantation lymphoproliferative disorder. Clin Immunol Immunopathol 80: 266–272
Rothenberg ME, Luster AD, Lilly CM, Drazen JM, Leder P 1995 Constitutive and allergen-induced expression of eotaxin mRNA in the guinea pig lung. J Exp Med 181: 1211–1216
Rothenberg ME, Luster AD, Leder P 1995 Murine eotaxin: an eosinophil chemoattractant inducible in endothelial cells and in interleukin 4-induced tumor suppression. Proc Natl Acad Sci USA 92: 8960–8964
Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD 1996 Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med 2: 449–456
Rothenberg ME 1998 Eosinophilia. N Engl J Med 338: 1592–1600
Rothenberg ME 1999 Eotaxin, an essential mediator of eosinophil trafficking into mucosal tissues. Am J Respir Cell Mol Biol 21: 291–295
Rothenberg ME, Mishra A, Brandt EB, Hogan SP 2001 Gastrointestinal eosinophils in health and disease. Adv Immunol 78: 291–328
Rothenberg ME, Mishra A, Brandt EB, Hogan SP 2001 Gastrointestinal eosinophils. Immunol Rev 179: 139–155
Rothenberg ME, Hogan SP 2006 The eosinophil. Annu Rev Immunol 24: 147–174
Rothenberg ME, MacLean JA, Pearlman E, Luster AD, Leder P 1997 Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J Exp Med 185: 785–790
Zimmermann N, Hogan SP, Mishra A, Brandt EB, Bodette TR, Pope SM, Finkelman FD, Rothenberg ME 2000 Murine eotaxin-2: a constitutive eosinophil chemokine induced by allergen challenge and IL-4 overexpression. J Immunol 165: 5839–5846
Pope SM, Fulkerson PC, Blanchard C, Akei HS, Nikolaidis NM, Zimmermann N, Molkentin JD, Rothenberg ME 2005 Identification of a cooperative mechanism involving interleukin-13 and eotaxin-2 in experimental allergic lung inflammation. J Biol Chem 280: 13952–13961
Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME 2005 The eotaxin chemokines and CCR3 are fundamental regulators of allergen-induced pulmonary eosinophilia. J Immunol 175: 5341–5350
Matthews AN, Friend DS, Zimmermann N, Sarafi MN, Luster AD, Pearlman E, Wert SE, Rothenberg ME 1998 Eotaxin is required for the baseline level of tissue eosinophils. Proc Natl Acad Sci USA 95: 6273–6278
Mishra A, Hogan SP, Lee JJ, Foster PS, Rothenberg ME 1999 Fundamental signals that regulate eosinophil homing to the gastrointestinal tract. J Clin Invest 103: 1719–1727
Mishra A, Hogan SP, Brandt EB, Wagner N, Crossman MW, Foster PS, Rothenberg ME 2002 Enterocyte expression of the eotaxin and interleukin-5 transgenes induces compartmentalized dysregulation of eosinophil trafficking. J Biol Chem 277: 4406–4412
Mishra A, Hogan SP, Brandt EB, Rothenberg ME 2001 An etiological role for aeroallergens and eosinophils in experimental esophagitis. J Clin Invest 107: 83–90
Hogan SP, Mishra EB, Brandt E, Foster PS, Rothenberg ME 2000 A critical role for eotaxin in experimental oral antigen-induced eosinophilic gastrointestinal allergy. Proc Natl Acad Sci USA 97: 6681–6686
Hogan SP, Mishra A, Brandt EB, Royalty MP, Pope SM, Zimmermann N, Foster PS, Rothenberg ME 2001 A pathological function for eotaxin and eosinophils in eosinophilic gastrointestinal inflammation. Nat Immunol 2: 353–360
Brandt EB, Strait RT, Hershko D, Wang Q, Muntel EE, Scribner TA, Zimmermann N, Finkelman FD, Rothenberg ME 2003 Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest 112: 1666–1677
Mishra A, Hogan SP, Brandt EB, Rothenberg ME 2002 Interleukin-5 promotes eosinophil trafficking to the esophagus. J Immunol 168: 2464–2469
Fulkerson PC, Fischetti CA, Hassman LM, Nikolaidis NM, Rothenberg ME 2006 Persistent effects induced by IL-13 in the lung. Am J Respir Cell Mol Biol 35: 337–346
Fulkerson PC, Fischetti CA, Mcbride ML, Hassman LM, Hogan SP, Rothenberg ME 2006 A central regulatory role for eosinophils and the eotaxin/CCR3 axis in chronic experimental allergic airway inflammation. Proc Natl Acad Sci USA 103: 16418–16423
Fulkerson PC, Fischetti CA, Rothenberg ME 2006 Eosinophils and CCR3 regulate IL-13-transgene-induced pulmonary remodeling. Am J Pathol 169: 2117–2126
Rothenberg ME, Zimmermann N, Mishra A, Brandt E, Birkenberger LA, Hogan SP, Foster PS 1999 Chemokines and chemokine receptors: their role in allergic airway disease. J Clin Immunol 19: 250–265
Zimmermann N, Hershey GK, Foster PS, Rothenberg ME 2003 Chemokines in asthma: cooperative interaction between chemokines and IL-13. J Allergy Clin Immunol 111: 227–242
Zimmermann N, Daugherty BL, Stark JM, Rothenberg ME 2000 Molecular analysis of CCR-3 events in eosinophilic cells. J Immunol 164: 1055–1064
Zimmermann N, Daugherty BL, Kavanaugh JL, El-Awar FY, Moulton EA, Rothenberg ME 2000 Analysis of the CC chemokine receptor 3 gene reveals a complex 5′ exon organization, a functional role for untranslated exon 1, and a broadly active promoter with eosinophil-selective elements. Blood 96: 2346–2354
Zimmermann N, Conkright JJ, Rothenberg ME 1999 CC chemokine receptor-3 undergoes prolonged ligand-induced internalization. J Biol Chem 274: 12611–12618
Zimmermann N, Colyer JL, Koch LE, Rothenberg ME 2005 Analysis of the CCR3 promoter reveals a regulatory region in exon 1 that binds GATA-1. BMC Immunol 6: 7
Zimmermann N, Bernstein JA, Rothenberg ME 1998 Polymorphisms in the human CC chemokine receptor-3 gene. Biochim Biophys Acta 1442: 170–176
Zimmermann N, Rothenberg ME 2003 Receptor internalization is required for eotaxin-induced responses in human eosinophils. J Allergy Clin Immunol 111: 97–105
Fulkerson PC, Zimmermann N, Hassman LM, Finkelman FD, Rothenberg ME 2004 Pulmonary chemokine expression is coordinately regulated by STAT1, STAT6, and IFN-gamma. J Immunol 173: 7565–7574
Fulkerson PC, Zimmermann N, Brandt EB, Muntel EE, Doepker MP, Kavanaugh JL, Mishra A, Witte DP, Zhang H, Farber JM, Yang M, Foster PS, Rothenberg ME 2004 Negative regulation of eosinophil recruitment to the lung by the chemokine monokine induced by IFN-gamma (Mig, CXCL9). Proc Natl Acad Sci USA 101: 1987–1992
Fulkerson PC, Zhu H, Williams DA, Zimmermann N, Rothenberg ME 2005 CXCL9 inhibits eosinophil responses by a CCR3- and Rac2-dependent mechanism. Blood 106: 436–443
Rothenberg ME 2004 Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 113: 11–28
Noel RJ, Putnam PE, Rothenberg ME 2004 Eosinophilic esophagitis. N Engl J Med 351: 940–941
Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, Jameson SC, Kirby C, Konikoff MR, Collins MH, Cohen MB, Akers R, Hogan SP, Assa'ad AH, Putnam PE, Aronow BJ, Rothenberg ME 2006 Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest 116: 536–547
Blanchard C, Wang N, Rothenberg ME 2006 Eosinophilic esophagitis: pathogenesis, genetics, and therapy. J Allergy Clin Immunol 118: 1054–1059
Mishra A, Rothenberg ME 2003 Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology 125: 1419–1427
Akei HS, Mishra A, Blanchard C, Rothenberg ME 2005 Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology 129: 985–994
Blanchard C, Mishra A, Saito-Akei H, Monk P, Anderson I, Rothenberg ME 2005 Inhibition of human interleukin-13-induced respiratory and oesophageal inflammation by anti-human-interleukin-13 antibody (CAT-354). Clin Exp Allergy 35: 1096–1103
Onbasi K, Sin AZ, Doganavsargil B, Onder GF, Bor S, Sebik F 2005 Eosinophil infiltration of the oesophageal mucosa in patients with pollen allergy during the season. Clin Exp Allergy 35: 1423–1431
Fogg MI, Ruchelli E, Spergel JM 2003 Pollen and eosinophilic esophagitis. J Allergy Clin Immunol 112: 796–797
Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, Collins MH, Putnam PE, Wells SI, Rothenberg ME 2007 IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibilty with glucocorticoids. J Allergy Clin Immunol 120: 1292–1300
Mishra A, Schlotman J, Wang M, Rothenberg ME 2007 Critical role for adaptive T cell immunity in experimental eosinophilic esophagitis in mice. J Leukoc Biol 81: 916–924
Furuta GT, Liacouras CA, Collins MH, Gupta SK, Justinich C, Putnam PE, Bonis P, Hassall E, Straumann A, Rothenberg ME 2007 Eosinophilic esophagitis in children and adults: a systematic review and consensus recommendations for diagnosis and treatment. Gastroenterology 133: 1342–1363
Noel RJ, Putnam PE, Collins MH, Assa'ad AH, Guajardo JR, Jameson SC, Rothenberg ME 2004 Clinical and immunopathologic effects of swallowed fluticasone for eosinophilic esophagitis. Clin Gastroenterol Hepatol 2: 568–575
Konikoff MR, Noel RJ, Blanchard C, Kirby C, Jameson SC, Buckmeier B, Akers R, Cohen MB, Collins MH, Assa'ad AH, Aceves SS, Putnam PE, Rothenberg ME 2006 A randomized double-blind-placebo controlled trial of fluticasone proprionate for pediatric eosinophilic esophagitis. Gastroenterology 131: 1381–1391
Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, Muntel EE, Witte DP, Pegg AA, Foster PS, Hamid Q, Rothenberg ME 2003 Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest 111: 1863–1874
Zimmermann N, Mishra A, King NE, Fulkerson PC, Doepker MP, Nikolaidis NM, Kindinger LE, Moulton EA, Aronow BJ, Rothenberg ME 2004 Transcript signatures in experimental asthma: identification of STAT6-dependent and -independent pathways. J Immunol 172: 1815–1824
Vercelli D 2003 Arginase: marker, effector, or candidate gene for asthma?. J Clin Invest 111: 1815–1817
Rothenberg ME, Doepker MP, Lewkowich IP, Chiaramonte MG, Stringer KF, Finkelman FD, MacLeod CL, Ellies LG, Zimmermann N 2006 Cationic amino acid transporter 2 regulates inflammatory homeostasis in the lung. Proc Natl Acad Sci USA 103: 14895–14900
Richard JP, Rothenberg ME, Jencks WP 1984 Formation and stability of ring-substituted 1-phenylethyl carbocations. J Am Chem Soc 106: 1361–1372
Rothenberg ME, Richard JP, Jencks WP 1985 Equilibrium constants for the interconversion of substituted 1-phenylethyl alcohols and esters. A measurement of intramolecular electrostatic interactions. J Am Chem Soc 107: 1340–1346
Drazen JM 2004 Presentation of the Kober Medal to K. Frank Austen. J Clin Invest 114: 1174–1176
Acknowledgements
My research career nearly started from the cradle, as my childhood was spent pursuing curiosities. Although my father and mother did not graduate high school and college, respectively, they seemed to always have the answers, or they encouraged me to find them. I am thus grateful for their love and guidance and the opportunities that they provided. My first formal research experience was a summer project with Dr. Alan Abramson (Long Island Jewish Hospital) where I aimed to optimize tissue culture conditions for growing human epithelial cells. As a former lifeguard and swimmer, Dr. Abramson made the condition of my summer employment require that I continue to lifeguard when I was not in the laboratory. Indeed, it was on the beach that I often came up with scientific ideas, and until this day, my most creative moments are in unconventional places (like the shower).
I was fortunate to attend a small liberal arts strong research institution, Brandeis University. This provided a fantastic environment with talented and dedicated professors and most importantly, an environment that encouraged undergraduate research pursuits. My college experience was marked by a 2-year apprenticeship in the laboratory of the National Academy of Science member late Professor William P. Jencks. Although formally trained as a medical doctor (at Harvard), Dr. Jencks' research centered on understanding mechanisms of enzyme catalysis; he was considered the foremost authority in this area for two decades. I published my first two articles with Dr. Jencks regarding the mechanism of nucleophilic substitution reactions (64,65). I attribute Dr. Jencks with instilling within me the quest to break complex scientific questions into simple (but fundamental) research inquiries. My encounter with him instilled inner humility within me (because of the humbleness he displayed in light of his vast knowledge). Subsequently, Harvard Medical School was a fantastic think tank composed of brilliant and fascinating students and an unlimited supply of world-renown Professors. After dabbling in a couple of laboratories, I was fortunate to become engaged by a lively professor's lecture on the complement cascade. Directly after the lecture, the Professor invited me to his laboratory and gave me a personal tour of his two floors of research laboratories. With his arm around my back, I was struck by both his grandfatherly-like love and vast knowledge; indeed, I signed up for a research rotation in his laboratory. This began my 25-y relationship with Professor K. Frank Austen (also a member of the National Academy of Sciences), one of the most influential immunologists and inflammation researchers over the past half-century [see description of Austen's Kober Medal award (66)]. I would like to thank Professor K. Frank Austen who continually provides me with the highest possible standard to emulate and who has been a motivational driving force in my career. Figure 2 shows a photograph of Drs. Austen and Boat and myself. This photograph was taken in 2005 when I proudly presented our division to Professor Austen as part of our external Scientific Advisory Committee review.

Photograph of Drs. Tom Boat (left), K. Frank Austen (middle), and Marc Rothenberg (right). The photograph was taken in 2006 when Dr. Austen visited the Cincinnati Children's Hospital Medical Center as part of the scientific advisory council annual review process.
There is not enough space in this article to thank all of my amazing mentors, friends, family, colleagues, patients (and their families), foundations, grant agencies, and trainees to whom I am grateful. First, I thank my wife, Joy, who has been my undivided supporter and has given me the encouragement and support to pursue my research passions. Second, I thank Dr. Tom Boat, the department chairperson who originally recruited me to CCHMC and helped develop my professional career in a caring manner. I thank Dr. Boat for successfully nominating me for this prestigious award and for having confidence in me. Next, I also thank my other primary research mentors Drs. Phil Leder and the late William P. Jencks, as well as my role models Drs. Raif Geha, Sam Lux, James Ferrara, Alan Ezekowitz, and David Nathan. I am grateful to my local colleagues including Drs. Fred Finkelman, Simon Hogan, Nives Zimmermann, Anil Mishra, Jeffrey Whitsett, David Williams, Mitchell Cohen, Philip Putnam, Amal Assa'ad, Arnold Strauss, my current and past trainees, the CCED staff, Andrea Lippelman, my outside colleagues including Drs. Andrew Luster, Paul Foster, Robert Wilmott, and Glenn Furuta, and Hashem. I am grateful to the grant agencies that have supported my research, including most importantly, the National Institutes of Health (NIAID and NIDDK), the Burroughs Welcome Fund, the Food Allergy and Anaphylaxis Network (FAAN), and the FDA Orphan Drug Grant program. I am very grateful to Ellyn Kodroff for setting up the CURED (Campaign Urging Research For Eosinophilic Diseases) Foundation, Dave and Denise Bunning for establishing the Food Allergy Project, and the Buckeye Foundation that support our research in part; I am motivated on a daily basis to find the best treatment and cure for eosinophilic diseases because of their personal significance to so many wonderful people that I have been privileged to meet and help through these foundations and other contacts.
Finally, I am grateful to the Society of Pediatric Research for selecting me for this prestigious award, especially in view of the distinguished past recipients.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rothenberg, M. 2007 E. Mead Johnson Award: Scientific Pursuit of the Allergy Problem. Pediatr Res 64, 110–115 (2008). https://doi.org/10.1203/PDR.0b013e3181794507
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181794507
