
Abstract
The mitochondrial DNA (mtDNA) point mutation T8993G has been associated with maternally inherited Leigh syndrome (MILS) when very abundant (>95%). MILS patients are usually severely affected and die in early infancy. In 1993, a novel T8993C point mutation was described in a juvenile form of Leigh syndrome (LS) characterized by a less aggressive clinical course. We describe four unrelated T8993C patients who had diverse, relatively mild, clinical manifestations. Polymerase chain reaction-restriction fragment length polymphorphism analysis showed that the heteroplasmic T8993C point mutation was very abundant in several tissues from all four patients (94.2 ± 1.5%) but was less copious in blood from 20 maternal relatives. ATP production in mitochondria isolated from skin fibroblasts in three patients was normal, whereas in one patient it was decreased to 20-35% of controls. These findings suggest that the T8993C mutation is less severe than the more common T8993G mutation.
Similar content being viewed by others


Main
LS is a genetically determined neurodegenerative disorder of infancy or childhood characterized by developmental delay with psychomotor regression, signs of brainstem dysfunction such as abnormal respiration and nystagmus, and lactic acidosis(1). The diagnosis of LS rests on neuroradiologic or autopsy evidence of bilateral and symmetrical necrotizing lesions in basal ganglia, thalamus, brainstem, and spinal cord. The pathophysiology of LS is related to altered energy metabolism and deficiencies of COX, an autosomal recessive condition, and of PDHC E1α subunit, inherited as X-linked trait, have been associated with LS(2–4). More recently, a mtDNA point mutation at nucleotide 8993 (T8993G) has been shown to be a common cause of MILS(5, 6). This mutation may impair ATP production by altering the electric charge in the proton channel of the F1F0-ATP synthase complex(7). In 1993, a novel mutation in the same nucleotide (T8993C) was described in four siblings with late-onset LS(8). We proposed that the distinct base change could act differently(9).
We now describe clinical, biochemical, and molecular genetic studies in four unrelated patients with the T8993C point mutation. Comparison of clinical and biochemical features in patients with the T8993C or the T8993G mutation confirms that there are significant differences between the two syndromes.
METHODS
Family pedigrees and percentage of mutant mtDNA are outlined in Figure 1.

Family pedigree and percentage of mutant mtDNA in four T8993C patients. Percentages are in blood samples from maternal relatives.B, blood; M, muscle; SF, skin fibroblasts.
Case 1. This 5-y-old boy was reported previously(9).
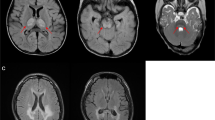
Case 2. A 13-y-old boy presented with peripheral neuropathy, absent ankle tendon reflexes, ataxia, progressive visual loss due to retinitis pigmentosa, and mild developmental delay. These findings fit the clinical criteria for the NARP syndrome(10). His milestones were delayed. He sat at 9 mo, walked at 18 mo, and spoke in sentences at 5 y. Since age 3 y, he had an unsteady gait, which worsened during febrile episodes. Brain MRI showed a small cerebellar vermis. Retinitis pigmentosa was diagnosed at 7 y of age, after which his vision gradually deteriorated. The maternal grandmother had visual problems of uncertain cause and onset, and a maternal cousin had mental retardation.
Case 3. A 23-y-old woman presented with weakness, ataxia, generalized seizures, and chronic anxiety. Although early motor development had been normal, she appeared clumsy, and did not walk independently until 18 mo of age. At 12 y, she had a generalized tonic seizure, developed progressive gait ataxia, and a spinocerebellar syndrome was diagnosed. In the following years she deteriorated gradually. At age 21, neurologic examination showed short stature, nystagmus, diffuse weakness and hypotonia, and impaired coordination with generalized clumsiness. Tendon reflexes were absent in the legs but brisk in the arms. Hammertoe deformities and Babinski signs were present bilaterally. Blood lactate was mildly elevated (2.7 mM/L, normal<1.8 mM/L). A MRI scan showed a prominent cisterna magna extending rostrally between the cerebellar hemispheres and bilateral abnormalities in the anterior aspect of the putamen. Muscle biopsy showed fiber size variation, with scattered “pale” fibers by COX histochemistry. Family history was unremarkable.
Case 4. This 4-y-old boy developed normally until age 18 mo, when he manifested hypotonia and gait ataxia. Symptoms worsened during intercurrent febrile illnesses. At age 3 y, a T2-weighted MRI scan showed bilateral basal ganglia lucencies. Occasional subsarcolemmal accumulations of mitochondria were revealed by ultrastructural examination of biopsied muscle. Neurologic examination revealed decreased tone, poor coordination, hyperactive knee jerks, and bilateral Babinski signs. Parents believed that L-carnitine (750 mg b.i.d.) improved his coordination, but his gait remained broad-based, and he developed choreoathetosis of arms. The patient's 16-mo-old brother had psychomotor delay.
Biochemical analyses. In two patients, electron transport chain enzymes and citrate synthase activities were assayed in muscle extracts as previously reported(11). Mitochondria were prepared by scraping monolayer cultures of skin fibroblasts in 30 mL of isolation buffer(0.27 M mannitol, 0.1 mM EDTA, 0.05% BSA, 10 mM Tris-HCl, pH 7.3) and ATP synthesis during oxidative phosphorylation was measured in terms of ADP phosphorylation by inorganic phosphate (32Pi), as described previously(12). Mitochondria (100-200 μg of protein) were incubated 1 h at 30°C in 0.3 mL of 30 mM Tris-acetate, pH 7.4, 10 mM MgCl2, 10 mM phosphate-Tris (105 cpm of 32Pi), 6 mM substrate, 20 mM glucose, 20 U of hexokinase, 1 mM ADP, and 50 μM adenosine pentaphosphate. Substrates included succinate, malate,α-keto-glutarate, and pyruvate plus malate. ATP hydrolysis was determined spectrophotometrically at 25°C using an ATP-regenerating system containing pyruvate kinase and an excess of phosphoenol-pyruvate in 50 μM Tris-acetate, pH 7.4, 1 μM MgCl2 buffer in a final volume of 1 mL, as described elsewhere(13).
Molecular genetic analyses. Total DNA extraction from blood, skin fibroblasts, or muscle, detection of the nucleotide 8993 point mutation, quantitative analysis of the mutant genome, and direct sequencing of the two mtDNA encoded ATPase genes (ATPase 6 and 8) were performed as reported elsewhere(9). We also performed quantitative analysis of the T8993C point mutation in mitochondria isolated from skin fibroblasts, as described previously(13).
RESULTS
The T8993C mutation resulted in heterogeneous clinical presentation. Cases 1 and 4 had courses reminiscent of late-onset MILS. Reversible exacerbations associated with inter-current illnesses occurred in the context of a rather stable developmental delay. In contrast, case 2 had the clinical features of NARP, although the proportion of mutant genomes was higher than in patients with the T8993G-NARP cases(10). Case 3 appeared to have an early onset form of spinocerebellar ataxia, although the presence of hyperintense symmetrical lesions in the putamen were suggestive of LS.
The activities of respiratory chain complexes in muscle homogenates from patients 1 and 3 were normal. ATP synthesis in isolated fibroblast mitochondria was normal in patients 1 and 2, and mildly reduced in patient 3. However, values were markedly decreased with all substrates in patient 4, with residual activities ranging from 25 to 35% of normal (Table 1). ATP hydrolysis was normal in all patients.
Direct sequencing and RFLP analysis showed a T8993C point mutation in all tissues tested from our four patients. The mutation was very abundant (94.2± 1.5) in blood, muscle, and skin fibroblasts as well as in isolated fibroblast mitochondria (>98%) from the patients but was less abundant in blood from 20 maternal relatives, with a mean percentage of 43.5 ± 28.3% (see Fig. 1).
DISCUSSION
Point mutations in mtDNA are associated with a growing list of maternally inherited neurodegenerative disorders. Two distinct pathogenic mutations at the very same mtDNA nucleotide (T8993G and T8993C) are a common cause of MILS. The genetic interrelationship and the pathogenic mechanism(s) of these mutations remain to be clarified.
To investigate whether the different base changes cause distinct clinical or biochemical phenotypes, we examined four unrelated patients in which the T8993C mutation was present in similarly high proportions. We found that the T→ C mutation was associated with heterogeneous clinical presentations ranging from a typical NARP syndrome (usually associated with lower abundance of the T8993G mutation), to spinocerebellar dysfunction, to a moderately severe and slowly progressive neurodegenerative disorder. When we compared these clinical features with those reported in patients with the T8993G mutation, or with deficiencies of PDHC or COX and onset in infancy, we found some overlaps among the four groups (Table 2). However, patients with the T8993G mutation or PDHC deficiency have earlier onset and a more rapidly progressive course. Seizures are commonly associated with both mutations at nucleotide 8993 and with PDHC-LS, whereas they are rarely seen in COX-LS. Retinitis pigmentosa, especially in combination with seizures, appears to be virtually diagnostic of the 8993 mutations. Respiratory distress and a rapid clinical evolution occur often in the T8993G patients. It is worth reiterating that the T8993C mutation, even when present in similar abundance as the T → G base change, causes less severe clinical manifestations and a more protracted course.
Disorders associated with mtDNA mutations are typically heterogeneous among family members. The presence of the mutation in several apparently healthy individuals with increased percentage in subsequent generations distinguishes the T8993C mutation from the more common T8993G, which usually shows a more rapid segregation. This suggests that very high levels of T8993C mutation are required for clinical penetrance.
Clinical syndromes due to mtDNA defects also show different patterns of tissue involvement in one individual. This phenomenon is related to heteroplasmy, stemming from different proportions of mutated genomes in the oocyte and their random segregation during embryogenesis. The preferential involvement of tissues such as brain in case 1 or retina in case 2, which may harbor similar high proportions of mutated genomes, is probably due to these organs' high dependence on oxidative metabolism.
Although the pathophysiology of LS is unclear, the presence of lactic acidosis in most patients had long suggested that cerebral energy failure may be a common underlying mechanism. The discovery of several mitochondrial enzyme defects, such PDHC and COX deficiencies, has supported this concept. The biochemical and functional consequences of the T8993G and T8993C mutations remain to be clarified. As both mtDNA mutations alter a highly conserved amino acid in the C terminus region of the ATPase 6 gene, ATP synthesis would be expected to be similarly impaired in the two conditions. The T8993G mutation converts Leu to Arg at position 156 in the F1F0 portion of the ATP synthase complex. Tatuch and Robinson(7) found that ATP production was markedly decreased in mitochondria isolated from lymphoblastoid cell lines from three T8993G patients, and their results have been confirmed in mitochondria isolated from skin fibroblasts(13). As those authors suggested, the T8993G mutation may interfere with the energy-driven mechanism of ATP synthesis by altering the electric charge of the proton channel. Although the T8993C mutation(converting Leu-156 to Pro) does not alter the charge of the ATPase proton channel, it could impair ATP synthesis by altering the channel structure, possibly by inserting a kink in the α-helical domain. However, ATP formation tested with different respiratory chain substrates was normal in fibroblast mitochondria from three of the four patients in this study. Although ATP production was markedly decreased only in the most severely affected patient, residual activity was still considerably higher than observed in patients with the T8993G mutation(13). These results agree with data obtained in experiments of site-directed mutagenesis of the α subunit of Escherichia coli F1F0-ATP synthase(14). When Leu-207, which is homologous to human Leu-156, was substituted by Arg (or Tyr or Cys), ATP synthesis through oxidative phosphorylation was abolished due to altered charge in the F0 portion of the proton channel. On the other hand, changing Leu-207 to Pro caused only partial reduction of ATP synthesis(40-50%) (Brian D. Cain, personal communication to Eric A. Schon). The proportion of mutated mtDNA needed to cause biochemical abnormality could vary from case to case, possibly because of other inherited or acquired factors. At any rate, in our experience, the severity of the biochemical defect seems to correlate with clinical manifestations, as shown by patient 4, who was the most severely affected and whose fibroblast mitochondria had the lowest level of ATP production (though not as low as in patients with the more severe T8993G mutation[13]). Additional studies in mutant cell lines or in rho0 cybrids containing variable percentages of the T8993C mutation will be necessary to provide further insights into the deleterious effects of this mutation.
In conclusion, our findings provide strong evidence for clinical heterogeneity in patients with the T8993C mutation. Despite the abundance of mutated genomes in affected tissues, the more protracted clinical course in these patients suggests that the T8993C mutation is better tolerated by tissues. These observations lead us to speculate that other cellular factors modulate the pathogenicity of this mtDNA mutation.
Abbreviations
- mtDNA:
-
mitochondrial DNA
- MILS:
-
maternally inherited Leigh syndrome
- T8993G or T8993C:
-
T-to-G or T-to-C mutations at nucleotide 8993 in the mtDNA
- LS:
-
Leigh syndrome
- COX:
-
cytochrome c oxidase
- PDHC:
-
pyruvate dehydrogenase complex
- NARP:
-
neurogenicm uscle weakness, ataxia, retinitisp igmentosa
- MRI:
-
magnetic resonance imaging
References
Leigh D 1951 Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 14: 216–221.
Willems Jl, Monnens LA, Trijbels LMF, Veerkamp JH, Meyer AE, Van Dam K, van Haelst U 1977 Leigh's encephalomyelopathy in a patient with cytochrome c oxidase deficiency in muscle tissue. Pediatrics 60: 850–857.
Brown RM, Dahl HHM, Brown GK 1989 X-chromosome localization of the functional gene for the E1α subunit of human pyruvate dehydrogenase complex. Genomics 4: 174–181.
Van Erven PM, Gabreels FJ, Ruitenbeek W, Den Hartog MR, Fischer JC, Renier WO, Trijbels JM, Sloof JL, Janssen AJ 1985 Subacute necrotizing encephalomyelopathy (Leigh syndrome) associated with disturbed oxidation of pyruvate, malate and 2-oxoglutarate in muscle and liver. Acta Neurol Scand 72: 36–42.
Tatuch Y, Christodoulou J, Feigenbaum A, Clarke JT, Wherret J, Smith C, Rudd N, Petrova-Benedict R, Robinson BH 1992 Heteroplasmic mtDNA mutation (T g) at 8993 can cause Leigh's disease when the percentage of abnormal mtDNA is high. Am J Hum Genet 50: 852–858.
Santorelli F, Shanske S, Macaya A, De Vivo DC, DiMauro S 1993 The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh's syndrome. Ann Neurol 34: 827–834.
Tatuch Y, Robinson BH 1993 The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem Biophys Res Commun 192: 124–128.
de Vries DD, van Engelen BG, Gabreels FJ, Ruitenbeek W, van Oost BA 1993 A second missense mutation in the mitochondrial ATPase6 gene in Leigh's syndrome. Ann Neurol 34: 410–412.
Santorelli F M, Shanske S, Jain KD, Tick D, Schon EA, DiMauro S A T>C mutation at nt 8993 of mitochondrial DNA in a child with Leigh syndrome. Neurology 44: 972–974.
Holt IJ, Harding AE, Petty RKH, Morgan-Hughes JA 1990 A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 46: 428–433.
Di Mauro S, Servidei S, Zeviani M, DiRocco M, De Vivo DC, DiDonato S, Uziel G, Berry K, Hoganson G, Johnsen SD, Johnson PC 1987 Cytochrome c oxidase in Leigh syndrome. Ann Neurol 22: 498–506.
Tuena de Gomez-Puyou M, Ayala G, Darszon A, Gomez-Puyou A 1984 Oxidative phosphorylation and the Pi-ATP exchange reaction of submitochondrial particles under the influence of organic solvents. J Biol Chem 259: 9472–9478.
Vazquez-Memije ME, Shanske S, Santorelli FM, Kranz-Eble P, Davidson E, De Vivo DC, DiMauro S 1996 Comparative biochemical studies in fibroblasts from patients with different forms of Leigh syndrome. J Inher Metab Dis ( in press)
Hartzog PE, Cain BD 1993 The alpha Leu207Arg mutation in F1F0-ATP synthase from Escherichia coli. J Biol Chem 268: 12250–12252.
Author information
Authors and Affiliations
Additional information
Supported in part by program project PO1 HD32062 from the National Institute of Child Health and Human Development (NICHD), by grants from the Muscular Dystrophy Association, and the Colleen Giblin Charitable Foundation. F.M.S. is supported by a scholarship from Telethon-Italia. M.E.V.-M. is supported by a grant from Unidad de Investigación en Genetica Humana, Centro Médico Nacional IMSS, Mexico, DF.
Rights and permissions
About this article
Cite this article
Santorelli, F., Mak, SC., Vazquez-Memije, M. et al. Clinical Heterogeneity Associated with the Mitochondrial DNA T8993C Point Mutation. Pediatr Res 39, 914–917 (1996). https://doi.org/10.1203/00006450-199605000-00028
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199605000-00028
This article is cited by
-
Mitochondrial disorders
Japanese Journal of Human Genetics (1997)
-
Mitochondrial encephalomyopathies: what next?
Journal of Inherited Metabolic Disease (1996)
