
Abstract
MRPS23 is a nuclear gene encoding a mitochondrial ribosomal protein. A patient with a mitochondrial disorder was found to carry a variant in MRPS23. More cases are necessary to establish MRPS23 as a mitochondrial disease gene. Of 5134 exomes performed in our center, we identified five independent patients who had similar clinical manifestations and were homozygous for the same germline variant c.119C>T; p.P40L in MRPS23. Detailed clinical findings, mitochondrial enzyme activity assays from cultured skin fibroblasts, PCR-Sanger-sequencing, and variant age estimation were performed. Their available family members were also studied. Eight members homozygous for the MRPS23 p.P40L were identified. All were from Hmong hilltribe. Seven presented with alteration of consciousness and recurrent vomiting, while the eighth who was a younger brother of a proband was found pre-symptomatically. Patients showed delayed growth and development, hearing impairment, hypoglycemia, lactic acidosis, and liver dysfunction. In vitro assays of cultured fibroblasts showed combined respiratory chain complex deficiency with low activities of complexes I and IV. PCR-Sanger-sequencing confirmed the variant, which was estimated to have occurred 1550 years ago. These results establish the MRPS23-associated mitochondrial disorder inherited in an autosomal recessive pattern and provide insight into its clinical and metabolic features.
Similar content being viewed by others
Introduction
Mitochondrial disorders are caused by pathogenic variants in nuclear or mitochondrial genes leading to impaired ATP generation and inadequate energy production. They have a worldwide prevalence of 5 to 20 in 100,0001 and present with a variety of symptoms including growth failure, lactic acidosis, neuropathy, cardiomyopathy, hepatopathy, and myopathy2. Although variants in at least 343 genes have been reported to cause mitochondrial disorders (http://genomit.eu/), evidence for some genes as disease-causing is limited, and their clinical and metabolic features are not well-described.
In a previous study by Kodha et al. in 2016, a missense variant in the MRPS23 gene was identified in a Japanese patient with hypoglycemia, liver dysfunction, and combined respiratory chain complex deficiency with low levels of complexes I and IV activities3. To date, this is the only report of an MRPS23 germline variant, highlighting the need for more patients and detailed clinical and metabolic features to establish MRPS23 as a disease gene and understand its phenotypic spectrum. In this study, we identified eight patients from five Hmong families with the same MRPS23 c.119C>T; p.P40L germline variant and characterized their clinical manifestations and metabolic profiles from cultured fibroblasts.
Materials and methods
Patients and the obtainment of blood samples
Of 5134 exomes performed in our center, we identified five independent patients who were homozygous for the same germline variant c.119C>T; p.P40L in MRPS23. They were from five different medical centers in Thailand including Maharaj Nakhon Chiang Mai Hospital, Siriraj Hospital, Queen Sirikit National Institute of Child Health, Nakornping Hospital and King Chulalongkorn Memorial Hospital. Their medical records were reviewed. Their blood samples were obtained and sent to our center at Chulalongkorn University. All participants have been processed in accordance with the Declaration of Helsinki, and informed consents were obtained from all participants and their legal guardians. The study was approved by the Institutional Review Board of the Faculty of Medicine of Chulalongkorn University (IRB#264/62).
Variant analysis
After performing whole exome sequencing on the DNA samples extracted from leukocytes, carried out by Macrogen Inc. (Seoul, Korea), and analyzing the data by filtering variants located in exons or flanking introns, as well as rare nonsynonymous variants with a minor allele frequency of less than 1% in the Genome Aggregation Database (gnomAD), Sanger sequencing was utilized to validate and analyze the identified variant, c.119C>T; p.P40L in MRPS23 for all patients and their available family members.
Variant dating
The SNP array using HumanOmniZhongHua-8 BeadChip was performed in 13 individuals (All individuals with a horizontal line above each individual symbol in Fig. 1A, except II-3 and III-2 in Family 1, II-2 in Family 4 and Family 5). The variant dating was estimated using a web tool https://shiny.wehi.edu.au/rafehi.h/mutation-dating/4.

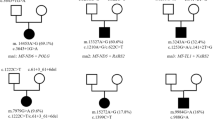
Genetic and metabolic features of the MRPS23-associated mitochondrial disorder. (A) Pedigrees of the five independent families. Dark boxes represented affected patients. (B) Chromatogram of the wild type, heterozygous and homozygous MRPS23 c.119C>T; p.P40L variant. (C) Size of homozygous regions around the MRPS23 c.119C>T; p.P40L variant in five patients (F1 II-9 and F1 III-1 in Family 1, F2 II-1 in Family 2, and F3 II-3 and F3 II-4 in Family 3). (D) Functional studies of fibroblasts from patients and their parents in Family 3. Mitochondrial respiratory chain enzyme activity. (E) The Blue Native-PAGE of protein lysate from mitochondrial’s fibroblasts. Twenty microgram of mitochondria from two controls and four patients were loaded in each lane; after electrophoresis by BlueNative-PAGE, they were transferred to PVDF membranes for western blotting. The membranes were cut into separate pieces prior to hybridization with antibodies for complex I (Invitrogen, MA, USA, 459100; NDUFA9 antibody), III (Invitrogen MA, USA, 459140; UQCRC1 antibody), IV (Invitrogen MA, USA, 459600; MTCO1 antibody), II (Invitrogen MA, USA, 459600; MTCO1 antibody) and were incubated with the respective antibodies for detection (see Supplementary Information 1). (F) Oxygen consumption rate (OCR) by Seahorse.
Skin fibroblast culture
The skin punch biopsy of the two affected siblings and their parents of Family 3 was performed. Skin fibroblasts were cultured at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM with 4.5 g/L glucose; Nacalai Tesque; Kyoto, Japan) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. Cells were harvested after they reached 80% confluence and used for enzyme activity assay.
Enzyme activity assay
All these enzyme activity assay data were compared with citrate synthase, which is an intrinsic indicator of mitochondria and used to assess enzyme activity. Mitochondrial respiratory chain enzyme activity was analyzed using skin fibroblasts derived from patients and their parents of Family 3.
The OCR (oxygen consumption rate) by Seahorse and BlueNative-PAGE from skin fibroblasts
The protocol from Ogawa et al. and Invernizzi et al. was used for cell preparation and OCR experiment5,6. The mitochondrial oxygen consumption of skin fibroblasts derived from MRPS23 patients and fHDF (fetal human dermal fibroblast) as a control was measured, and the maximum respiratory rate (MRR) was compared. Blue Native-PAGE was performed to fractionate protein complexes in their native state. To enable probing for proteins of a specific molecular weight range, Western blot membranes were cut into separate pieces prior to antibody hybridization.
Declaration
During the preparation of this work the authors used https://chat.openai.com in order to improve language and readability. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Results
Clinical characterization of patients with homozygous c.119C>T; p.P40L variant in MRPS23
The clinical characteristics of all eight individuals from five independent families were presented in Table 1. They all were of Hmong hill tribe. Since some lived in a very remote area with limited access to medical care, their clinical information was incomplete. One patient (F4 II-1) died at age 2 years and 4 months due to fever from an unknown cause, which might be also resulted from limited medical access. Seven patients homozygous for the variant manifested with alteration of consciousness and recurrent vomiting, except individual F3 II-4, who was diagnosed pre-symptomatically after his older brother (F3 II-3) was given a molecular diagnosis. Other clinical features included delayed growth (50%), delayed development (60%), hearing impairment (33%), hypoglycemia (67%), lactic acidosis (83%) and transaminitis (40%). Ebstein anomaly, a congenital heart defect, was found in one patient, F3 II-4. In addition, one proband, F4 II-1, underwent an MRI scan of the brain, which showed no appreciable abnormalities.
Variant studies
Whole exome sequencing revealed that all five probands were homozygous for the c.119C>T; p.P40L variant in MRPS23. Among the 10,268 alleles analyzed from our dataset of 5134 exomes, only a single heterozygous MRPS23 c.119C>T allele was identified in the unaffected father of a patient with a chromosome abnormality hailing from Chiang Mai province. Based on the family name, there is a possibility that he belongs to the Hmong Clans. PCR-Sanger sequencing confirmed its presence and identified more patients. Genotype of each individual is shown in Fig. 1A and exemplified in Fig. 1B.
Variant dating
Haplotype sharing of five patients was shown in Fig. 1C. The homozygous regions spanned around 500–2000 kb. The age of this haplotype following Gandolfo LC’s method was estimated to be 62.1 generations ago, with a confidence interval of 22–187.2. Assuming 25 years per generation, this variant occurred 1550 years ago.
Enzyme activity assay, the OCR (oxygen consumption rate) by Seahorse and BlueNative-PAGE from skin fibroblasts
The enzyme activity assay of OXPHOS enzyme complexes I, II, II + III, III and IV was presented in Fig. 1D. Mitochondrial respiratory chain enzyme activity was analyzed using skin fibroblasts derived from two patients and their parents in Family 3. The enzyme activity measurements were confirmed based on the diagnostic criteria described in the literature7,8. The range of 40–100% (black line with upper and lower range in Fig. 1D) was considered as the reference range, and the activity was considered to be decreased if it was below 40%. As shown in Fig. 1D, the enzymatic activity of complex I in fibroblasts of the two patients and their parents and that of complex IV in II-3 were found to be decreased compared to the reference ranges.
For BlueNative-PAGE, mitochondria were isolated from fibroblasts of patients and their parents in Family 3. As the result in Fig. 1E, complexes I, III, and IV band concentrations of both patients were decreased compared to the normal control. In their mother, only the complex IV band concentration was decreased. In their father, there was a slight decrease in the concentration of complex I band, but a decrease in the concentration of complexes III and IV bands. However, no decrease in the concentration of complex II band was observed.
The mitochondrial oxygen consumption of skin fibroblasts derived from the two patients and their parents in Family 3 and fHDF as a control was measured. The bar graph (Fig. 1F) was a comparison of the maximum respiratory rate (MRR) of fibroblasts, and it was found that the OCR of MRPS23-derived fibroblasts was considerably lower than that of control cells in both glucose and galactose medium.
Discussion
In this article, we provide a comprehensive description of the clinical, metabolic, and genetic features of the MRPS23-related mitochondrial disorder. While a previous study identified a patient with the c.119C>G; p.P40R variant in the MRPS23 gene, there was limited information on phenotypic features. We present eight additional individuals homozygous for a pathogenic variant, confirming MRPS23 as a gene causing mitochondrial disorder, and provide a more detailed account of the phenotypic spectrum and metabolic disturbances.
The MRPS23 gene, located on chromosome 17q22, encodes a protein component of the mitoribosome small subunit, which plays a role in mitochondrial translation9. Deficiencies in complexes I, II, III, and IV are the most common causes of mitochondrial OXPHOS deficiency. Moreover, mitoribosome deficiency can also cause mitochondrial disease10. Variants in other members of the MRPS family, such as bi-allelic MRPS34 and MRPS2 variants, can cause Leigh-like syndrome and OXPHOS deficiency3,11.
Our study identified eight individuals from five independent families who were homozygous for the c.119C>T; p.P40L variant in MRPS23. Their available parents were found to carry one mutant allele, indicating an autosomal recessive mode of inheritance. All five families had Hmong ancestry, and the fact that they share the same variant suggests a founder effect. Despite clan exogamy being the primary kinship principle, ethnic endogamy in the Hmong population resulted from cultural preference could lead to the accumulation of variants of autosomal recessive disorders12,13. Using long homozygous regions, we estimated that the variant occurred 62.1 generations or 1550 years ago.
The clinical features of these eight patients are typical of disorders caused by variants in genes encoding mitoribosomal subunits10. These include delayed growth, delayed development, hearing impairment, hypoglycemia, lactic acidosis, and transaminitis. Notably, not all patients had delayed growth or development, indicating that with proper management, patients with MRPS23-related disorder could have normal growth and development. Although presymptomatic diagnosis in individual F3 II-4 was expected to yield an even more favorable outcome, growth and developmental delay were observed due to the co-occurrence of congenital heart disease.
There are several lines of evidence to demonstrate that the variant in the nuclear MRPS23 gene is responsible for the disease. First, all probands with similar clinical findings were homozygous for the same MRPS23 c.119C>T variant, while there were no other individuals in our in-house exome database who were homozygous of the variants. Secondly, the MRPS23 variant segregates with the disease in all identified five families. All parents were heterozygous and none of the unaffected members were homozygous for the variants. Thirdly, the amino acid residue 40 is likely to impact protein functions, as supported by a previously reported patient with a pathogenic p.P40R variant at the same codon. Fourthly, this variant was predicted to be pathogenic by six different prediction software tools. Specifically, it received a “deleterious” designation with a score of 0.00 from SIFT, a “probably damaging” classification with a score of 1.000 from PolyPhen-2, a “possibly pathogenic” assessment with a score of 0.038 from MCAP, and was considered “most likely to be deleterious and potentially pathogenic” with a score of 26.8 (exceeding the threshold of 20) according to CADD. Additionally, it was labeled as “pathogenic strong” with a score of 0.875 by MutPred and deemed “disease causing” by MutationTaster2.
This variant is also classified as likely pathogenic (PP3 PM5 PM2 PP5) according to the American College of Medical Genetics and Genomics (ACMG) classification system. Fifthly, the proline at amino acid residue 40 is highly conserved from c. elegans to humans, as shown by evolutionary conservation analysis performed by the MutationTaster2 software14 (see Supplementary Information 2).
To determine metabolic disturbances, we used skin fibroblasts from patients (homozygous TT) and parents (heterozygous CT) in Family 3 for enzyme activity assays, oxygen consumption rate by Seahorse, and BlueNative-PAGE experiments. No induced-mutation cells were used for functional studies in this report. Western blotting experiments showed decreased band intensities of complex I in both patients and those of complex IV in both patients and their father compared to the control. Complex II did not show any difference among patients, their parents and controls. This is probably because complex II, unlike complexes I, III, IV, and V, does not contain any subunits which are encoded by mitochondrial genome3,10. Both patients and their parents exhibited similar aberrations in the enzyme activity assay, BlueNative-PAGE, OCR by glucose medium, and MRR of glucose medium; therefore all these assays cannot differentiate patients carrying biallelic loss-of-function variants from carriers who carry monoallelic variants. Only the Western blot determining the complex I level can distinguish patients from carriers. This situation resembles the acid beta-glucosidase enzyme activity test used in Gaucher disease15. Since the complex I deficiency was detected in the parents, determination of the complex I cannot be used to diagnosed MRPS23-associated mitochondrial disorder. This emphasizes the importance of molecular diagnosis.
This study highlights the specific MRPS23 variant associated with lactic acidosis and hypoglycemia, which has significant implications for individuals of Hmong ancestry carrying the homozygous c.119C>T; p.P40L variant. Symptomatic considerations and pre-symptomatic warnings are both vital in clinical management. Given the high prevalence of this variant, it is crucial to prioritize MRPS23-related disorder as the primary diagnosis when Hmong patients present symptoms of lactic acidosis and hypoglycemia.
In conclusion, we establish MRPS23 as a nuclear gene causing a mitochondrial disorder inherited in an autosomal recessive manner and characterize its clinical manifestations and metabolic profiles.
Data availability
This variant was submitted into the ClinVar database (Accession SCV004015155). The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
References
Gorman, G. S. et al. Mitochondrial diseases. Nat. Rev. Dis. Primers 2, 16080. https://doi.org/10.1038/nrdp.2016.80 (2016).
Ylikallio, E. & Suomalainen, A. Mechanisms of mitochondrial diseases. Ann. Med. 44, 41–59. https://doi.org/10.3109/07853890.2011.598547 (2012).
Kohda, M. et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 12, e1005679. https://doi.org/10.1371/journal.pgen.1005679 (2016).
Gandolfo, L. C., Bahlo, M. & Speed, T. P. Dating rare mutations from small samples with dense marker data. Genetics 197, 1315–1327. https://doi.org/10.1534/genetics.114.164616 (2014).
Invernizzi, F. et al. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion 12, 328–335. https://doi.org/10.1016/j.mito.2012.01.001 (2012).
Ogawa, E. et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 40, 685–693. https://doi.org/10.1007/s10545-017-0042-6 (2017).
Bernier, F. P. et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 59, 1406–1411. https://doi.org/10.1212/01.wnl.0000033795.17156.00 (2002).
Murayama, K. et al. Intractable secretory diarrhea in a Japanese boy with mitochondrial respiratory chain complex I deficiency. Eur. J. Pediatr. 168, 297–302. https://doi.org/10.1007/s00431-008-0753-7 (2009).
Mai, N., Chrzanowska-Lightowlers, Z. M. & Lightowlers, R. N. The process of mammalian mitochondrial protein synthesis. Cell Tissue Res. 367, 5–20. https://doi.org/10.1007/s00441-016-2456-0 (2017).
Gardeitchik, T. et al. Bi-allelic mutations in the mitochondrial ribosomal protein MRPS2 cause sensorineural hearing loss, hypoglycemia, and multiple OXPHOS complex deficiencies. Am. J. Hum. Genet. 102, 685–695. https://doi.org/10.1016/j.ajhg.2018.02.012 (2018).
Liu, C., Zhou, W., Liu, Q. & Peng, Z. Hypoglycemia with lactic acidosis caused by a new MRPS2 gene mutation in a Chinese girl: A case report. BMC Endocr. Disord. 22, 15. https://doi.org/10.1186/s12902-021-00924-1 (2022).
Lee, S. Diasporic kinship hegemonies and transnational continuities in the Hmong diaspora. Identities 27, 229–247. https://doi.org/10.1080/1070289X.2018.1457347 (2020).
Sun, B., Wen, Y. F., Culhane-Pera, K. A., Lo, M. & Straka, R. J. Pharmacogenomic variabilities in geo-ancestral subpopulations and their clinical implications: Results of collaborations with Hmong in the United States. Front. Genet. 13, 1070236. https://doi.org/10.3389/fgene.2022.1070236 (2022).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. https://doi.org/10.1038/nmeth.2890 (2014).
Dardis, A. et al. Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1. Orphanet J. Rare Dis. 17, 442. https://doi.org/10.1186/s13023-022-02573-6 (2022).
Acknowledgements
The authors would like to thank all patients and their families to participate in this study. This project was funded by the Health Systems Research Institute (66-122).
Author information
Authors and Affiliations
Contributions
Conceptualization: V.S., K.S., R.I., C.I.; Funding Acquisition: V.S.; Investigation (clinical): C.K., N.V., W.D., N.T., P.K., W.K.; Investigation (genetics and expression study): R.I., C.I., T.M., M.S., Y.S., T.O., A.O., K.M.; Writing-original draft: R.I. and C.I.; Writing-review and editing: V.S. and K.S. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ittiwut, C., Ittiwut, R., Kuptanon, C. et al. Genetic, metabolic and clinical delineation of an MRPS23-associated mitochondrial disorder. Sci Rep 13, 22005 (2023). https://doi.org/10.1038/s41598-023-49161-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-49161-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
