
Abstract
Whole exome sequencing (WES) is an effective tool for the genetic diagnosis of mitochondrial disorders due to various nuclear genetic defects. In this study, three patients affected by extremely rare mitochondrial disorders caused by nuclear genetic defects are described. The medical records of each patient were reviewed to obtain clinical symptoms, results of biochemical and imaging studies, and muscle biopsies. WES and massive parallel sequencing of whole mtDNA were performed for each patient. The oxygen consumption rate (OCR) and complex activity I and IV was measured. Patients 1 and 2 had exhibited global developmental delay and seizure since early infancy. Blood lactate, the lactate-to-pyruvate ratio, and urinary excretion of Krebs cycle intermediates were markedly elevated. Patient 1 also was noted for ophthalmoplegia. Patient 2 had left ventricular hypertrophy and ataxia. Patient 3 developed dysarthria, gait disturbance, and right-side weakness at age 29. Brain magnetic resonance imaging demonstrated abnormal signal intensity involving the bilateral thalami, midbrain, or pons. Based on WES, patient 1 had p.Glu415Gly and p.Arg484Trp variants in MTO1. In patient 2, p.Gln111ThrfsTer5 and RNA mis-splicing were identified in TSFM. Patient 3 carried p.Met151Thr and p.Met246Lys variants in AARS2. Skin fibroblasts of three patients exhibited decreased OCRs and complex 1 activity, and mitochondrial DNA was normal. These results demonstrate the utility of WES for identifying the genetic cause of extremely rare mitochondrial disorders, which has implications for genetic counseling.
Similar content being viewed by others


Introduction
Mitochondrial disorders can result from genetic defects in nuclear or mitochondrial DNA (mtDNA). mtDNA variants are responsible for 10–30% of these disorders. More than 1000 nuclear genes also encode mitochondrial proteins [1, 2], and genetic defects in more than the 250 nuclear genes have been reported in mitochondrial disorders, featuring multiple respiratory chain defects [3].
Mitochondrial dysfunction can affect single or multiple organs and systems, primarily those with the highest energy demands. The clinical features are diverse, including lactic acidosis, seizures, encephalopathy, intellectual disability/developmental delay, ataxia, sensorineural deafness, optic atrophy, diabetes mellitus, cardiomyopathy, hepatopathy, and myopathy. A scoring system, such as the mitochondrial disease criteria (MDC), is useful for the diagnostic process [4]. However, clinical severity is quite variable among patients and clinical suspicion is not plausible in certain patients.
In addition, the genetic diagnosis of mitochondrial disorders has been challenging because mtDNA analyses cannot identify nuclear genetic defects and nuclear genetic loci are widely heterogeneous. Recently, advanced methodology, including next-generation sequencing (NGS) and whole exome sequencing (WES), has provided effective tools for the diagnosis of mitochondrial disorders [5,6,7,8,9]. The diagnostic rate of mitochondrial disorders by WES is 16–60%, depending on the patient population [5,6,7,8].
In this study, three patients with extremely rare mitochondrial disorders caused by nuclear genetic defects were evaluated; the results provide additional clinical and genetic information related to these extremely rare disorders.
Materials and methods
Patients
The medical records for each patient were reviewed to obtain their clinical symptoms, MDC score [4], results of biochemical and imaging studies, and muscle biopsy. The study was approved by the Institutional Review Board for Human Research of Asan Medical Center (IRB number: 2017-0260).
Molecular analysis
Informed consent was obtained, and blood samples were collected from the patients and parents. Total genomic DNA was extracted from peripheral blood lymphocytes using the Gentra Puregene Kit (Qiagen Inc., Germantown, MD, USA).
To detect mitochondrial genome rearrangements, m.5835-15978, m.14837-6642, m.591-9397, and m.8602-1411 were amplified using primers targeting each region. Primers covering m.7908-14268, m.14081-2669, and m.2480-8095 were used to detect a common deletion, m.8470_13446del4977 (4977 bp).
Whole mtDNA parallel sequencing was performed as previously described [10]. Briefly, entire mtDNA was amplified by single- or two-fragment PCRs (TAKARA LA Taq, Cat. No. RR02AG). The quantified PCR products were used for library preparation by the Nextera XT DNA Kit (Illumina, San Diego, CA, USA). Sequencing was performed using the Illumina MiSeq platform (Stem Cell Center, ASAN Medical Center), and the data were analyzed using NextGENe software with MitoMAP (https://www.mitomap.org//MITOMAP). The reference alignment was NC_012920.1, which was used for variant calling.
For patients 1 and 2, WES was performed using SureSelect Human All Exon V4 + UTR 71 Mb (Agilent, Santa Clara, CA, USA), following the manufacturer’s protocol. The exome library was sequenced with the 100 bp paired-end mode of the HiSeq SBS Kit. Read quality was checked using fastQC (v.0.10.1), and low-quality bases (below Q20) were removed using Cutadapt (v.1.8.1). High-quality reads were then aligned to the human reference genome hg19 using Burrows–Wheeler Aligner (BWA) (v.0.7.12) with a minimum seed of 45, and duplicated reads were further removed using PicardTools (v.1.98). For unique reads, SNP and indel variants for 33 genes were identified using GenomeAnalysisTK (v.2.3.9) and the detailed annotation of the variants was performed using SnpEff (v.4.1).
For patient 3, targeted exome sequencing (4813 OMIM genes) was performed. Exomes were captured using the TruSight One Panel (Illumina Inc.), which enriches a 12 Mb region spanning 4813 genes. Sequencing was performed using the Next Seq platform (Illumina Inc.). Sequence reads were aligned to the reference genome, hg19, using BWA (v.0.7.12, MEM algorithm). Mean depth of coverage was 90× (>10× = 98%).
Sanger sequencing of the genomic variants identified by exome sequencing was performed for all patients and their families. For the clinical interpretation of variants, allele frequency data were obtained from various databases, including gnomAD (http://gnomad.broadinstitute.org/) and ExAc (http://exac.broadinstitute.org/). The pathogenicity of variants was assessed using in silico prediction tools, such as PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), and MutationTaster (http://www.mutationtaster.org) and Human Splicing Foundation (http://www.umd.be/HSF/). Variants were classified according to The American College of Medical Genetics and Genomics (ACMG) [11].
Assessment of mitochondrial respiratory function and complex activity
Mitochondrial respiratory function was measured using the XF Cell Mito Stress Test Kit and the Seahorse Extracellular Flux (XF) 24 Analyzer (Seahorse Bioscience, Inc., North Billerica, MA, USA) as previously described [10]. The oxygen consumption rate (OCR) was continuously quantified in real time in skin or muscle fibroblast cell lines. Mitochondrial respiration included basal respiration, ATP-linked respiration, maximal respiratory capacity, and spare respiratory capacity. OCR was measured on 4 × 104 cells in each well under basal conditions and then with sequential additions of oligomycin (1.5−2 μM), FCCP (0.5−1.0 μM), rotenone (0.5−1.0 μM), and antimycin A (0.5−1.0 μM) at the indicated times to determine mitochondrial parameters.
The activities of cellular mitochondrial complexes I and IV were analyzed spectrophotometrically using Abcam’s microplate assay kits (Abcam Inc., Cambridge, UK, ab109721, ab109909) according to the manufacturer’s instructions [12]. Briefly, extracted proteins from 1 × 106 were added to wells of premium Nunc MaxiSorp™ modular microplates. After 3 h, target enzyme was captured in the well. Specific substrates were added in each well and mitochondrial complex I and IV activity were measured at 450 and 550 nm, respectively.
Results
Clinical description of patients
Patient 1
The male patient was the first child of nonconsanguineous Korean parents (Table 1). He was born after 40 gestational weeks with a birth weight of 2840 g (standard deviation [SD], −1.2) and a height of 47 cm (SD, −1.1). Immediately after birth, he was admitted to the neonatal intensive care unit due to spontaneous pneumothorax and generalized tonic–clonic seizure. The blood lactate level was 6.4 mmol/L (reference [ref.], 0.56–1.39), the lactate-to-pyruvate (LP) ratio was 44.3 (pyruvate, 1.3 mg/dL; ref., 0.3–0.7), and urinary excretion of 4-hydroxyphenyl lactic acid (387.6 mmol/mol Cr.; ref., 0–28.7) and 4-hydroxyphenyl pyruvic acid (90.8 mmol/mol Cr.; ref., 0–33.8) was elevated. Electroencephalogram (EEG) showed intermittent suppression-burst background and several spike discharges from the central head area. Echocardiography showed a 6-mm-sized atrial septal defect. Under the suspicion of mitochondrial disease, he had been given coenzyme Q10 (5 mg/kg/day), l-carnitine (50 mg/kg/day), and dichloroacetate (DCA, 30 mg/kg/day). At age 11 months, severe metabolic acidosis (pH 7.1) with high blood lactate levels (9−15 mmol/L) developed. At this point, ophthalmoplegia, bilateral ptosis, and strabismus were noted. In brain magnetic resonance imaging (MRI), T2-prolonged and nonenhancing lesions involving the bilateral thalami, midbrain, and posterior pons were observed (Fig. 1a). He was barely able to sit alone at 1 year 4 months old and step with support at 2 years old. He could speak only a few words at 2.7 years old. At the most recent evaluation (6 years and 4 months old), his height was 106 cm (SD, −2.5) and body weight was 15.7 kg (SD, −3.2). He could walk about 20 steps and speak a maximum of five words. His younger sibling died at 7 days after birth due to severe lactic acidosis.

Brain MRI of the three patients. a Patient 1 at 11 months old; brain MRI shows T2-prolonged and nonenhancing lesions (arrow) involving the bilateral thalami, midbrain, and posterior pons in T2 FLAIR. b Patient 2 at 6 months old; symmetric high signal intensity foci in T2-weighted image (arrow) were observed in the dorsal portion of the pons. c Patient 3 at 29 years old; bilateral symmetric T2 high signal intensity is observed in the periventricular and deep white matter of the cerebral hemispheres, corpus callosum, dorsolateral thalamus, cerebral peduncles, and pons along the corticospinal tracts
Patient 2
The male patient was the first child of nonconsanguineous Korean parents (Table 1). He was born after 40 gestational weeks with a birth weight of 2730 g (SD, −1.9) and a height of 47 cm (SD, −1.1). Pregnancy, labor, and vaginal delivery were uneventful. Recurrent hypoglycemia, hypertriglyceridemia, and hepatomegaly had been observed since 3 months of age. At age 5 months, he experienced severe metabolic acidosis, high blood lactate (12.8 mmol/L), and an L/P ratio of 36. In urine organic acid analysis, lactic acid was 1386.4 mmol/mol Cr. (ref., 3.1–119.3), 3-hydroxybutyric acid was 757.0 mmol/mol Cr. (ref., 0–211.4), fumaric acid was 147.3 mmol/mol Cr. (ref., 0–28.3), and malic acid was 103.6 mmol/mol Cr. (ref., 0–28.3). In the T2-weighted image in brain MRI at age 6 months, symmetric high signal intensity foci were detected in a dorsal portion of the pons (Fig. 1b). Echocardiography revealed left ventricular (LV) hypertrophy (LV mass, 129.75 g/m2). Coenzyme Q10 (10 mg/kg/day), l-carnitine (50 mg/kg/day), and DCA (15 mg/kg/day) had been prescribed. He developed infantile spasm at 1 year old. Gastrostomy was performed owing to poor oral intake, failure to thrive, and recurrent hypoglycemia at 1 year 5 months old. He had truncal ataxia. In addition, optic atrophy was observed at the age of 5 years. At 7 years old, his LV mass decreased to 69.9 g/m2. At the most recent evaluation (9 years and 1 month old), his height was 117.6 cm (SD, −2.6) and body weight was 20.5 kg (SD, −2.3). He could barely walk a few steps alone and had severe mental retardation.
Patient 3
Male patient 3 was born at full term to healthy Korean parents (Table 1). His development and growth were normal until 27 years old when he was accidentally hit by a brick and lost consciousness for less than a minute. No specific findings were obtained in brain MRI. He developed dysarthria at 28 years old. His neurological symptoms progressed, and gait disturbance and right-side weakness were noticed at 29 years old. Muscle spasticity and ankle clonus were also noticed. T2-weighted brain MRI demonstrated bilateral symmetric high signal intensity in the periventricular and deep white matter of the cerebral hemispheres, corpus callosum, dorsolateral thalamus, cerebral peduncles, and pons along the corticospinal tracts (Fig. 1c). Muscle biopsy, echocardiography, audiometry, ophthalmologic examination, and biochemical findings were normal.
MDC scores for patient 1 at 5 years old, patient 2 at 5 years old, and patient 3 at 29 years old were 8 (definite), 7 (probable), and 2 (possible), respectively [4].
Genetic analysis
Whole mtDNA sequencing
No pathogenic variants or genomic deletion were observed in mtDNA in any patient.
Exome sequencing
Whole or targeted (4813 genes) exome sequencing was performed at the ages of 5 years (patient 1), 5 years (patient 2), and 29 years (patient 3).
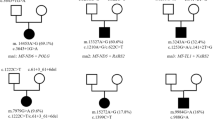
Patient 1 had compound heterozygous variants in MTO1 (NM_012123.3), c.1244A > G (p.Glu415Gly) inherited from the father and c.1450C > T (p.Arg484Trp) inherited from the mother (Fig. 2a). The MTO1 gene encodes mitochondrial-tRNA modifier 1. c.1244A > G and c.1450C > T are expected to significantly alter the protein function based on pathogenicity predictions (PolyPhen-2, 1.0; SIFT, 0.0; and MutationTaster, disease-causing). c.1450C > T is a previously reported [13], and c.1244A > G has not been reported. Both variants were categorized as “likely pathogenic” according to the ACMG guidelines [11].

Sanger pherogram of variants in the three patients. a MTO1 variants in patient 1 and his parents. b TSFM variants in patient 2: one frameshift variant and one variant resulting in exon 5 skipping at the mRNA level. c AARS2 variants in patient 3 and his parents. Arrow: location of variant
Patient 2 had two variants in the TSFM gene (NM_005726.5), which encodes mitochondrial translation elongation factor Ts (EFTs); c.331_340delCAGGAAGGAA (p.Gln111ThrfsTer5) and c.57G > A. c.331_340delCAGGAAGGAA were inherited from his mother, but c.57G > A was not found in his mother or father. c.57G > A variant is located on the last codon of exon 1 and was predicted to alter the splicing process, according to the Human Splicing Foundation. Indeed, c.57G > A causes aberrant splicing during the RNA processing of TSFM, resulting in exon 5 skipping (Fig. 2b). These two variants have not been reported previously. Both variants were categorized as “pathogenic” according to the ACMG guidelines [11].
In patient 3, compound heterozygous variants were found in the AARS2 gene (NM_020745.3) encoding mitochondrial alanyl-tRNA synthetase protein; c.452T > C (p.Met151Thr) was inherited from the mother, and c.737T > A (p.Met246Lys) was inherited from the father (Fig. 2c). Both variants were pathogenic based on in silico analyses (PolyPhen-2, 1.0 and 0.971; SIFT, 0.0; and MutationTaster, disease-causing). The allele frequency of c.452T > C was 0.006% in gnomAD, and c.737T > A has not been reported previously. Both variants were categorized as “likely pathogenic” according to the ACMG guidelines [11].
Mitochondrial function in skin fibroblasts of the three patients
Mitochondrial function in skin fibroblasts and muscle of the three patients was lower than that of the control (Fig. 3a, b, c-1). Patient 1 exhibited significantly lower basal respiration (19.6 ± 5.3 vs. 33.4 ± 11.7 pmol/min/µg DNA, P = 0.034), ATP production (14.7 ± 4.7 vs. 26.3 ± 7.8 pmol/min/µg DNA, P = 0.013), maximal respiration (34.0 ± 7.7 vs. 62.4 ± 22.1 pmol/min/µg DNA, P = 0.015), and spare respiratory capacity (14.4 ± 3.5 vs. 28.9 ± 11.2 pmol/min/µg DNA, P = 0.013) than the patient’s mother (Fig. 3a-2). The OCR for patient 2 indicated significantly lower basal respiration (48.7 ± 10.4 vs. 69.2 ± 11.2 pmol/min/µg DNA, P = 0.025), ATP production (36.5 ± 7.9 vs. 59.4 ± 8.8 pmol/min/µg DNA, P = 0.008), maximal respiration (72.6 ± 12.1 vs. 111.3 ± 72.6 pmol/min/µg DNA, P = 0.006), and spare respiratory capacity (23.9 ± 6.9 vs. 42.0 ± 9.6 pmol/min/µg DNA, P = 0.007) than a healthy control (Fig. 3b-2). Muscle and skin fibroblasts of patient 3 showed lower basal respiration (muscle: 24.8 ± 6.7 vs. 54.0 ± 12.9 pmol/min/µg DNA, P = 0.02; skin: 22.8 ± 3.5 vs. 54.0 ± 12.9 pmol/min/µg DNA, P = 0.03), ATP production (muscle: 20.5 ± 5.9 vs. 37.5 ± 10.6 pmol/min/µg DNA, P = 0.01; skin: 16.9 ± 3.0 vs. 37.5 ± 10.6 pmol/min/µg DNA, P = 0.027), maximal respiration (muscle: 38.3 ± 9.4 vs. 66.5 ± 15.3 pmol/min/µg DNA, P = 0.016; skin: 38.0 ± 5.9 vs. 66.5 ± 15.3 pmol/min/µg DNA, P = 0.017) than skin fibroblasts of a healthy control, but there was no significant difference in the spare respiratory capacity (Fig. 3c-2).

Comparison of the overall oxygen consumption rate (OCR) patterns and key parameters of mitochondrial respiration in patients’ fibroblast and muscle and controls’ fibroblast. a-1 Comparison of OCR in fibroblasts of patient 1 and the patient’s healthy mother. a-2 Comparison of parameters of mitochondrial respiration in fibroblasts of patient 1 and the patient’s healthy mother. b-1 Comparison of OCR in fibroblasts of patient 2 and a healthy control. b-2 Comparison of parameters of mitochondrial respiration in fibroblasts of patient 2 and a healthy control. c-1 Comparison of OCR in fibroblasts of patient 3, muscle cells of patient 3, and a healthy control’s fibroblasts. c-2 Comparison of parameters of mitochondrial respiration in fibroblasts of patient 3, muscle cells of patient 3, and a healthy control’s fibroblast. *P < 0.05
Complex I activity (0.8 ± 0.001 vs. 2.9 ± 0.001 mOD/min, P < 0.001) and complex I/complex II of patient 1’s fibroblasts (0.5 ± 0.01 vs. 1.6 ± 0.01 mOD/min, P < 0.001) were lower than those of healthy control’s fibroblasts (Fig. 4a). Complex I activity (0.9 ± 0.01 vs. 2.9 ± 0.01 mOD/min, P < 0.001) and complex I/complex II of patient 2’s fibroblasts (0.582 ± 0.07 vs. 1.586 ± 0.07 mOD/min, P < 0.001) were lower than those of healthy control’s fibroblasts (Fig. 4b). Skin fibroblasts of patient 3 showed lower complex I (1.7 ± 0.01 vs. 2.7 ± 0.01 vs. 2.9 ± 0.01 mOD/min, P < 0.001) and complex I/complex II than muscle of patient 3 and healthy control’s fibroblasts (0.9 ± 0.01 vs. 1.8 ± 0.01 vs. 1.6 ± 0.01 mOD/min, P < 0.001) (Fig. 4c). There was no significant difference of complex activity between control’s muscle cell and patient 3’s muscle cell (Fig. 4d). No significant difference was identified in complex IV activity and complex IV/complex II in all patients (Fig. 4).

Comparison of complex 1 and IV activity in fibroblast of patients and controls. a Comparison of complex I and IV activity in fibroblast of patient 1 and the patient’s healthy mother. b Comparison of complex I and IV activity in fibroblast of patient 2 and a healthy control. c Comparison of complex I and IV activity of patient 3, muscle cells of patient 3, and a healthy control’s fibroblast. (d) Comparison of complex I and IV activity of muscule cells of patient 3 and a healthy control’s muscle cell. ***P < 0.001
Discussion
The major clinical manifestations of mitochondrial disorders include ophthalmological, otologic, cardiac, endocrinological, myopathic, and neurological features. In general, affected patients present with dysfunction in multiple organs but usually exhibit myopathic and neurological manifestations. The MDC scoring system is a useful tool for the clinical diagnosis of mitochondrial diseases. The major clinical features in the three patients in our report were ophthalmological (two patients), cardiac (one patient), endocrinological (one patient), myopathic (two patients), and neurological (three patients). However, based on the overall clinical features as well as the MDC scores, clinical suspicion for mitochondrial disease was possible in patients 1 and 2 but not in patient 3.
Extensive investigations of mtDNA were performed by Sanger sequencing, massively parallel sequencing, and long-range PCR analyses. However, these analyses did not reveal any pathogenic point variants or large genomic alterations. The genetic diagnosis of mitochondrial disease was made by WES in all three patients. Genetic confirmation improves patient care and enables adequate genetic counseling for family members. Optimal nutritional support as well as pharmacological supplementation, such as coenzyme Q10, l-carnitine, l-arginine, vitamin B complex, vitamins C and E, alpha-lipoic acid, and dichloroacetate, can increase mitochondrial energy production and reduce the production of reactive oxygen species. These supplements might help to stabilize disease progression when curative therapeutic options are lacking [14]. Considering the disease severity for the three patients described in the current report, genetic diagnosis was very important for future reproductive genetic counseling. For families with mtDNA variants, prenatal genetic testing or preimplantation genetic diagnosis is difficult owing to the poor correlation or predictability of the heteroplasmy of mtDNA variants. Preimplantation genetic diagnosis or prenatal testing is possible in families with known nuclear genetic defects.
The nuclear genetic defects identified in the present study can be classified according to their role in the translational process [15]. MTO1 is involved in the chemical modification of mt-tRNA, and AARS2 plays an important role in the initiation of translation by mt-tRNA synthetase [15, 16]. TSFM is associated with the initiation, elongation, and termination of aminoacyl mt-tRNA [15, 17]. The measurement of OCR in fibroblasts revealed the mitochondrial defect in each patient and supported the predicted pathological consequences of the variants in the three patients.
Mitochondrial diseases associated with these three genes are extremely rare; 35 patients with MTO1 variants [13], 6 patients with TSFM variants [17,18,19,20,21], and about 20 patients with AARS2 variants have been reported [22,23,24,25]. c.1244A > G in MTO1, c.331_340delCAGGAAGGAA and c.57G > A in TSFM, and c.737T > A in AARS2 have not been reported previously. The most common features of patients with MTO1 variants are hypertrophic cardiomyopathy (HCMP), lactic acidosis, and a variable degree of developmental delay; optic atrophy, feeding difficulties, and seizure may also occur [13]. Patient 1 also presented with the most severe clinical symptoms in the neonatal period; after the infantile period, his general condition stabilized without cardiomyopathy with supportive care until 6 years of age. The c.1244A > G and c.1450C > T variants were located in evolutionarily conserved amino acid residues [26] and appeared to alter the protein function according to in silico analyses. Patients with TSFM variants also exhibit fatal encephalomyopathy, HCMP, and infantile hepatic failure [18, 20]. Their long-term complications include developmental delay, ataxia, dysarthria, tremor, and optic atrophy [17,18,19,20]. The clinical manifestations in patient 2 were compatible with those in previous cases; with supportive care, he has remained alive until 7 years of age. The frameshift TSFM variant c.331_340delCAGGAAGGAA was inherited from his mother. The other variant, c.57G > A, was de novo or a case of paternal germline mosaicism. The c.57G > A variant is synonymous, but located at the end of exon 1. It has been recurrently observed, in a number of genes, that the synonymous variants at the end of an exon caused the aberrant splicing. The c.57G > A variant also led to the skipping of exon 5. The variants in AARS2 were found in severe infantile cardiomyopathy in two families in 2012 [23]. Patients with AARS2 variants experience progressive leukoencephalopathy and cognitive impairment in their second or third decade of life, similar to patient 3 in the present study [22, 24]. The c.452T > C variant in our patient is found at an extremely low frequency in the general population (gnomAD), and c.737T > A has not been reported. The two variants are located in a highly conserved amino acid residue and are predicted to alter protein function significantly based on an in silico analysis.
In MTO1 mutants, isolated complex IV deficiency and a combination of complex I and complex IV deficiencies are most commonly observed [13]. All patients with TSFM deficiencies show defects in complex IV activity, but each patient has difficulty performing a different set of complex activities [17,18,19,20]. In AARS2 mutants, patients with cardiomyopathy show combined complex I and complex III deficiencies, while patients with progressive leukodystrophy show an isolated complex IV deficiency [23, 24]. Although the activity of all complex was not determined in the current study, decreased overall OCR and complex I activity indicated the contributions of various mitochondrial complexes and key parameters of mitochondrial function.
In conclusion, WES successfully identified the genetic causes of extremely rare mitochondrial disorders, and the results of OCR measurement strongly supported a causal association between clinical symptoms and genetic defects. The long-term clinical courses of these extremely rare disorders need to be improved by genetic diagnosis at right time and optimal supportive care.
References
Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. 2004;27:349–62.
Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–7.
Mayr JA, Haack TB, Freisinger P, Karall D, Makowski C, Koch J, et al. Spectrum of combined respiratory chain defects. J Inherit Metab Dis. 2015;38:629–40.
Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, Rodenburg RJ, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–6.
Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, Mensenkamp AR, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–6.
Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. J Am Med Assoc. 2014;312:68–77.
Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis. 2015;38:437–43.
Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, Karkucinska-Wieckowska A, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174.
Murayama K, Shimura M, Liu Z, Okazaki Y, Ohtake A. Recent topics: the diagnosis, molecular genesis, and treatment of mitochondrial diseases. J Hum Genet. 2019;64:113–25.
Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, et al. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell. 2016;18:625–36.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants. Genet Med. 2015;17:405–24.
Merz TM, Pereira AJ, Schurch R, Schefold JC, Jakob SM, Takala J, et al. Mitochondrial function of immune cells in septic shock: a prospective observational cohort study. PLoS ONE. 2017;12:e0178946.
O’Byrne JJ, Tarailo-Graovac M, Ghani A, Champion M, Deshpande C, Dursun A, et al. The genotypic and phenotypic spectrum of MTO1 deficiency. Mol Genet Metab. 2018;123:28–42.
Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol. 2009;11:414–30.
Boczonadi V, Horvath R. Mitochondria: impaired mitochondrial translation in human disease. Int J Biochem Cell Biol. 2014;48:77–84.
Powell CA, Nicholls TJ, Minczuk M. Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front Genet. 2015;6:79.
Ahola S, Isohanni P, Euro L, Brilhante V, Palotie A, Pihko H, et al. Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy. Neurology. 2014;83:743–51.
Smeitink JA, Elpeleg O, Antonicka H, Diepstra H, Saada A, Smits P, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869–77.
Emperador S, Bayona-Bafaluy MP, Fernandez-Marmiesse A, Pineda M, Felgueroso B, Lopez-Gallardo E, et al. Molecular-genetic characterization and rescue of a TSFM mutation causing childhood-onset ataxia and nonobstructive cardiomyopathy. Eur J Hum Genet. 2016;25:153–56.
Vedrenne V, Galmiche L, Chretien D, de Lonlay P, Munnich A, Rotig A. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol. 2012;56:294–7.
Traschutz A, Hayer SN, Bender B, Schols L, Biskup S, Synofzik M. TSFM mutations cause a complex hyperkinetic movement disorder with strong relief by cannabinoids. Parkinsonism Relat Disord. 2018. https://doi.org/10.1016/j.parkreldis.2018.09.031.
Lynch DS, Zhang WJ, Lakshmanan R, Kinsella JA, Uzun GA, Karbay M, et al. Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol. 2016;73:1433–39.
Gotz A, Tyynismaa H, Euro L, Ellonen P, Hyotylainen T, Ojala T, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:635–42.
Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ, Salomons GS, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology. 2014;82:2063–71.
Lee JM, Yang HJ, Kwon JH, Kim WJ, Kim SY, Lee EM, et al. Two Korean siblings with recently described ovarioleukodystrophy related to AARS2 mutations. Eur J Neurol. 2017;24:e21–e22.
Meyer S, Scrima A, Versees W, Wittinghofer A. Crystal structures of the conserved tRNA-modifying enzyme GidA: implications for its interaction with MnmE and substrate. J Mol Biol. 2008;380:532–47.
Funding
This study was supported by a grant from the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (NRF-2018M3A9H1078335), and the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea (Nos. 2017-756 and 2018-756).
Author information
Authors and Affiliations
Contributions
BHL and EK designed the study. GHS, EK, and BHL drafted the manuscript and analyzed data. GHS, AO, H-WY, and BHL were the treating pediatricians and conducted all clinical analyses. The genetic and functional analyses were performed by GHK, EK, YL, JP, ENK and TK. BHL and EK revised the manuscript. All authors were involved in analyzing and interpreting data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Seo, G.H., Oh, A., Kim, E.N. et al. Identification of extremely rare mitochondrial disorders by whole exome sequencing. J Hum Genet 64, 1117–1125 (2019). https://doi.org/10.1038/s10038-019-0660-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0660-y
