
Abstract
Single-chain crosslinked star polymers with multiple hydrophilic short arms and a hydrophobic core were created as novel microgel star polymers of single polymer chains. The synthetic process involves the intramolecular crosslinking of self-folding amphiphilic random copolymers in water. For this process, amphiphilic random copolymers bearing hydrophilic poly(ethylene glycol) (PEG) and hydrophobic olefin pendants were synthesized by ruthenium-catalyzed living radical copolymerization of PEG methyl ether methacrylate, dodecyl methacrylate and hydroxyl-functionalized methacrylates, and the in situ or postesterification of the hydroxyl pendants of the resulting copolymers with methacryloyl chloride. The olefin-bearing copolymers with 20–40 mol% hydrophobic units efficiently self-folded because of hydrophobic interactions in water. These folded structures were then crosslinked intramolecularly using a free radical initiator or a ruthenium catalyst to selectively yield single-chain crosslinked star polymers, whereas a counterpart containing 50 mol% hydrophobic units induced bimolecular aggregation in water to give double-chain crosslinked star polymers. The primary structure of the star polymers can be precisely controlled with random copolymer precursors. Owing to the PEG arm units, the star polymers further showed thermosensitive solubility in water.
Similar content being viewed by others

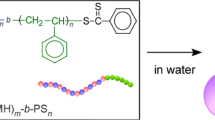

Introduction
Local crosslinking of polymer chains and their aggregates is a key technology to build soluble functional macromolecules of stable three-dimensional architectures in various solvents and environments.1, 2, 3, 4, 5 Microgel-core star polymers1, 2, 3, 4, 6, 7, 8, 9, 10, 11, 12 are representative core–shell macromolecules carrying a microgel core that is covered by multiple linear arm polymers. The crosslinked core not only serves to maintain the star-branched structure in various environments but also provides unique nano-compartments for catalysis,8, 9, 10, 11 and molecular encapsulation and release.12 Such functionalized star polymers are efficiently obtained via the local crosslinking of living linear polymers (or macroinitiators) with functional crosslinking agents (for example, divinyl compounds) and monomers in living radical polymerization.13 However, the preparation of microgel-core star polymers with control over the number of arm chains and in-core functionality remains challenging because the core-forming process (microgelation) competitively undergoes both ‘intermolecular’ and ‘intramolecular’ crosslinking of multiple polymer chains; the crosslinking efficiency thus depends on the total concentration of arm polymers ([arm]0) and monomers ([linker]0, [monomer]0) in addition to the molar ratio of monomers to arm chains ([linker]0/[monomer]0/[arm]0).
In contrast, single-chain folding polymers, single-chain polymeric nanoparticles and unimer micelles literally comprise single polymer chains that self-fold through ‘intramolecular’ physical association and/or covalent linking in solution.14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 They can therefore provide functional nanospaces based on single polymer chains.17, 18, 25, 27 Recently, we synthesized various amphiphilic and functional random copolymers via living radical polymerization to create single-chain folding (self-folding) polymers with hydrophobic and hydrogen-bonding interactions in water.23, 24, 25, 26, 27 Compared with conventional microgel-core star polymers, single-chain folding polymers have several inherent features: (1) the primary structure (for example, molecular weight, monomer composition and sequence and terminal structure and number) of single-chain folding polymers is identical to that of random copolymer precursors; (2) folding properties are tunable by monomer composition, monomer species and degree of polymerization; and (3) the folded structure can be dynamically and reversibly varied by external stimuli.
For instance, amphiphilic random copolymers of hydrophilic poly(ethylene glycol) methyl ether methacrylate (PEGMA: CH2=CMeCO2(CH2CH2O)8.5CH3, Mn=475) and hydrophobic dodecyl methacrylate (DMA) showed unique self-folding and aggregation properties in water through DMA content control. The copolymers with 20–40 mol% DMA selectively form self-folding unimer micelles because of hydrophobic interactions in water, whereas copolymers with 50 mol% DMA form a double-chain aggregate in water.23 Such self-folding polymers have dynamic and hydrophobic cores of polymethacrylate backbones and dodecyl pendants that are stabilized by multiple short PEG chains. They are therefore structurally regarded as single-chain dynamic star polymers. With these intriguing features, PEGMA/DMA-based copolymers would be one of the most promising scaffolds for precisely functionalized nanospaces, where the folded structure is formed only in water and is easily unfolded (random coil) in organic solvents.
Herein, we produced single-chain crosslinked star polymers via the intramolecular crosslinking (patching) of self-folding amphiphilic random copolymers in water (Scheme 1). This is a new class of microgel star polymers, whereby the number of arms, functionality and the crosslinking density are efficiently and directly controlled using random copolymer precursors that are prepared by living radical polymerization. The single-chain crosslinked star polymers can further maintain their compact folded structure even in organic solvents.
Single-chain crosslinked star polymers were prepared via the following four steps: (1) polymerization, (2) pendant olefin introduction, (3) single-chain folding and (4) intramolecular crosslinking. Hydroxyl-functionalized amphiphilic random copolymers (P1–P7) were first synthesized by living radical copolymerization of PEGMA, DMA and hydroxyl-functionalized monomers (12-hydroxydodecyl methacrylate (HDMA) or 2-hydroxylethyl methacrylate (HEMA)). The composition of hydrophobic monomers (DMA and HDMA) was precisely controlled: 20, 40 and 50 mol% per chain. The hydroxyl-functionalized copolymers were then quantitatively esterified with methacryloyl chloride (MAC) to give olefin-bearing amphiphilic random copolymers (P1-O–P7-O). P1-O–P7-O underwent self-folding or double-chain aggregation because of hydrophobic interactions in water before being homogeneously crosslinked with a free radical initiator or a ruthenium catalyst to give star polymers (S1–S7). The selective intramolecular crosslinking without macroscopic gelation was attributed to the local accumulation of hydrophobic olefin pendants into the central cores and the efficient isolation of their polymer chains by multiple hydrophilic PEG chains.
Experimental procedure
Materials
For polymerization, PEGMA (Aldrich, St Louis, MO, USA) and DMA (Wako, Osaka, Japan; purity >95%) were purified by column chromatography charged with inhibitor remover (Aldrich) and degassed by triple vacuum–argon purge cycles before use. HEMA (Aldrich; purity >99%), ethyl 2-bromoisobutyrate (1; TCI, Tokyo, Japan; purity >98%) and ethyl 2-chloro-2-phenylacetate (4; Aldrich; purity >97%) were distilled under reduced pressure before use. HDMA, a bifunctional initiator (2) and a trifunctional initiator (3) were synthesized as shown below. MAC (TCI; purity >80.0%) and triethylamine (TCI; purity >99.0%) were purified by distillation before use. 1,12-Dodecanediol (Aldrich; purity >99%), 2-bromo-2-methylpropanoyl bromide (Aldrich; purity >98%), 1,4-bis(2-hydroxyethoxy)benzene (TCI; purity>95%) and 1,1,1-tris(hydroxymethyl)ethane (Aldrich; purity >99%) were degassed by triple vacuum–argon purge cycles before use. Ru(Ind)Cl(PPh3)2 (Aldrich), RuCp*Cl(PPh3)2 (Cp*: pentamethylcyclopentadienyl; Aldrich; purity >97%) and (4-hydroxyphenyl)diphenylphosphine (PPh2(C6H4OH); Aldrich; purity >98%)36 were used as received, and [RuCp*(μ3-Cl)]4 was synthesized according to a published procedure.37 These ruthenium complexes and a ligand were handled in a glove box under moisture- and oxygen-free argon (H2O<1 p.p.m.; O2<1 p.p.m.). 1,2,3,4-Tetrahydronaphthalene (tetralin; Kisida Chemical, Osaka, Japan; purity >98%), an internal standard for monomer conversion determined by proton nuclear magnetic resonance (1H NMR), was dried over calcium chloride overnight and distilled twice over calcium hydride. n-Bu3N (TCI; purity >99%), 4-dimethylamino-1-butanol (4-DMAB; TCI; purity >98%),38 2,2’-azobis(2-methylpropionamidine) dihydrochloride (V-50; Wako; purity >95%), dimethyl 2,2’-azobis(isobutyrate) (MAIB; Wako; purity >97%), ethanol (Wako, dehydrated) and pure water (Wako) were degassed before use. Toluene was purified before use by passing it through a purification column (Glass Contour Solvent Systems: SG Water USA, Nashua, NH, USA). Dry tetrahydrofuran (THF; Wako, dehydrated) and dry diethyl ether (Wako; purity >99.5%) were used without further purification.
Characterization
The molecular weight distribution curves, number-average molecular weight (Mn), peak molecular weight (Mp) and dispersity (Mw/Mn) of the polymers were measured by size-exclusion chromatography (SEC) in dimethylformamide (DMF) containing 10 mM LiBr at 40 °C (flow rate: 1 ml min−1) equipped with three linear-type polystyrene gel columns (KF-805L, Shodex, Tokyo, Japan; exclusion limit=4 × 106 mol g−1, particle size=10 μm, pore size=5000 Å, 0.8 cm i.d. × 30 cm) connected to a Jasco PU-2080 precision pump, a Jasco RI-2031 refractive index detector and a Jasco UV-2075 Ultraviolet–visible (UV–vis) detector set at 270 nm (Jasco, Tokyo, Japan). The columns were calibrated against 10 standard poly(MMA) samples (Polymer Laboratories, Church Stretton, UK; Mn=1000–1200 000 g mol−1, Mw/Mn=1.06–1.22). 1H NMR spectra were recorded in CDCl3, acetone-d6, DMF-d7 and D2O on a JEOL JNM-ECA500 spectrometer operating at 500.16 MHz (JEOL, Tokyo, Japan). Electrospray ionization mass spectrometry was performed on a Waters Quattro micro API (Waters, Milford, MA, USA). The absolute weight-average molecular weight (Mw) was determined using multiangle laser light scattering (MALLS) with SEC in DMF on a Dawn E instrument (Wyatt Technology, Santa Barbara, CA, USA; Ga-As laser; λ=690 nm). SEC was performed in DMF containing 10 mM LiBr at 40 °C (flow rate: 1 ml min−1) equipped with three linear-type polystyrene gel columns (Shodex KF-805L) connected to a Jasco PU-2080 precision pump, a Jasco RI-1530 refractive index detector and a Jasco UV-1570 UV–vis detector set at 270 nm. The refractive index increment (dn/dc) was measured in DMF at 40 °C on an Optilab DSP refractometer (Wyatt Technology; λ=690 nm, c<2.5 mg ml−1). Dynamic light scattering was performed on an Otsuka Photal ELSZ-0 equipped with a semiconductor laser (wavelength: 658 nm) at 25 °C (Otsuka, Osaka, Japan). The measuring angle was 165° and the data were analyzed by the CONTIN method. UV–vis spectra were obtained using a Shimadzu MultiSpec-1500 or UV-1800 in H2O/acetone (19:1) and acetone at 25 °C (optical path length=1.0 cm) (Shimadzu, Kyoto, Japan). Transmission electron microscopy images were taken on a JEOL JEM-2000EXII at an acceleration voltage of 100 kV. The aqueous polymer solutions were applied to a carbon-coated Cu grid, negatively stained with 2% uranyl acetate and the excess fluid was removed with filter paper.
Synthesis of HDMA
In a 100 ml round-bottomed flask filled with argon, MAC (18.6 mmol, 1.8 ml) was slowly added to a solution of 1,12-dodecanediol (27.9 mmol, 5.6 g) and triethylamine (20.8 mmol, 2.9 ml) in dry THF (25 ml) at room temperature. The reaction mixture was then stirred at 25 °C for 18 h. After evaporation, diethyl ether (50 ml) and distilled water (50 ml) were poured into the crude mixture. The aqueous phase was separated and extracted by diethyl ether (50 ml), and the ether extracts were combined with the organic layer. The combined organic phase was washed with water three times, ammonia water and brine and was dried over anhydrous Na2SO4 overnight. After the ether was removed in vacuo, the crude product was purified by silica gel column chromatography with hexane/ethyl acetate (80:20, v/v) to give HDMA as a colorless liquid (0.98 g, 20% yield). 1H NMR [500 MHz, CDCl3, 25 °C, δ=0 TMS] δ=6.08 (s, 1H), 5.45 (m, 1H), 4.09 (t, 2H, J=6.7 Hz), 3.50 (t, 2H, J=6.7 Hz), 1.90 (s, 3H), 1.61 (quin, 2H, J=6.7 Hz), 1.47 (quin, 2H, J=6.7 Hz), 1.35–1.22 (16H). 13C NMR [125 MHz, CDCl3, 25 °C, δ=77.0 CDCl3] δ=167.4, 136.4, 125.2, 64.7, 62.7, 32.6, 29.5-29.3, 29.1, 28.5, 25.8, 25.6, 18.2. Electrospray ionization mass spectrometry m/z ([M+Na]+) calcd. for C12H22O2Na 221.2, found 221.4.
Synthesis of a bifunctional initiator (2)
In a 100 ml round-bottomed flask filled with argon, 2-bromo-2-methylpropanoyl bromide (6.5 mmol, 0.80 ml) was added to a solution of 1,4-bis(2-hydroxyethoxy)benzene (1.8 mmol, 0.35 g) and triethylamine (6.5 mmol, 0.91 ml) in dry THF (20 ml) at 0 °C. The reaction mixture was stirred at 25 °C for 18 h. After the evaporation of the reaction solution, diethyl ether (50 ml) and distilled water (50 ml) were poured into the flask. The aqueous phase was separated and extracted by diethyl ether (50 ml), and the ether extracts were combined with the organic layer. The combined organic phase was washed with water three times, ammonia water and brine and was dried over anhydrous Na2SO4 overnight. After the ether was removed in vacuo, a pure solid product (bifunctional initiator: 2) was obtained (0.72 g, 80% yield). 1H NMR [500 MHz, CDCl3, 25 °C, δ=7.26 (CHCl3)] δ=6.86 (s, 4H), 4.49 (t, 4H, J=5.3 Hz), 4.17 (t, 4H, J=4.8 Hz), 1.93 (s, 12H). 13C NMR [125 MHz, CDCl3, 25 °C, δ=77.0 (CDCl3)] δ=171.6, 153.1, 115.9, 66.4, 64.3, 55.5, 30.7.
Synthesis of a trifunctional initiator (3)
In a 100 ml round-bottomed flask filled with argon, 2-bromo-2-methylpropanoyl bromide (16 mmol, 1.98 ml) was added to a solution of 1,1,1-tris(hydroxymethyl)ethane (3.5 mmol, 0.42 g) and triethylamine (16 mmol, 2.2 ml) in dry THF (60 ml) at 0 °C. The reaction mixture was stirred at 25 °C for 18 h. After the evaporation of the reaction solution, diethyl ether (50 ml) and distilled water (50 ml) were poured into the flask. The aqueous phase was separated and extracted by diethyl ether (50 ml), and the ether extracts were combined with the organic layer. The combined organic phase was washed with water three times, ammonia water and brine and was dried over anhydrous Na2SO4 overnight. After the ether was removed in vacuo, a pure solid product [1,1,1-tris(2-bromoisobutyryloxymethyl)ethane: 3] was obtained (0.99 g, 50% yield). 1H NMR [500 MHz, CDCl3, 25 °C, δ=7.26 (CHCl3)] δ=4.12 (s, 6H), 1.94 (s, 18H), 1.17 (s, 3H). 13C NMR [125 MHz, CDCl3, 25 °C, δ=77.0 (CDCl3)] δ=171.2, 66.6, 55.4, 39.7, 30.7, 16.9.
Synthesis of olefin-bearing amphiphilic copolymers
The synthesis of the olefin-bearing amphiphilic copolymers (P1-O–P7-O) was carried out by the syringe technique under argon in baked glass flasks or tubes equipped with a three-way stopcock via ruthenium-catalyzed living radical polymerization and in situ or postesterification.
P2-O (via in situ esterification): in a 30 ml glass tube, Ru(Ind)Cl(PPh3)2 (0.008 mmol, 6.4 mg) was placed. Then, THF (4.6 ml), tetralin (0.1 ml), a 400 mM THF solution of n-Bu3N (0.4 ml, n-Bu3N=0.16 mmol), PEGMA (2.4 mmol, 1.1 ml), DMA (1.2 mmol, 0.35 ml), a 308 mM THF solution of HDMA (1.3 ml, HDMA=0.4 mmol) and a 143 mM THF solution of 2 (0.11 ml, 2=0.016 mmol) were added sequentially into the tube at 25 °C under argon. The total volume of the reaction mixture was 8.0 ml. The tube was placed in an oil bath maintained at 60 °C. At predetermined intervals, the mixture was sampled with a syringe under dry argon, and the reaction was terminated by cooling the solution to –78 °C. After 25 h, the conversion of PEGMA/DMA+HDMA reached 80%/80%, respectively, determined by 1H NMR using tetralin as an internal standard. Into this solution, MAC (0.19 ml, 2 mmol) was directly added at 0 °C. The mixture was then stirred for 16 h at 25 °C. Then, the reaction was quenched with dry ethanol (5 ml). After the solvent was removed in vacuo, the crude polymer was purified by silica gel column chromatography with toluene as an eluent and precipitated into hexane to give P2-O. SEC (DMF, poly(methyl methacrylate) (PMMA) std.): Mn=58 500 g mol−1; Mw/Mn=1.27. SEC-MALLS (DMF, 0.01 M LiBr): Mw=126 000 g mol−1. 1H NMR [500 MHz, acetone-d6, 25 °C, δ=2.04 (CD2HCOCD3)]: δ 6.95–6.85 (aromatic), 6.09–6.04 (olefin), 5.65–5.60 (olefin), 4.25–4.06 (–COOCH2CH2O–, CH2=C(CH3)COOCH2CH2CH2–), 4.06–3.90 (–COOCH2CH2CH2–), 3.81–3.43 (–OCH2CH2O–), 3.36–3.28 (–OCH3), 2.16–1.78 (–CH2C(CH3)–), 1.93 (CH2=C(CH3)COO–), 1.74–1.63 (–COOCH2CH2(CH2)9CH3), 1.53–1.27 (–COOCH2CH2(CH2)9CH3), 1.17–0.85 (–COO(CH2)11CH3, –CH2C(CH3)–); PEGMA/DMA/HEMA-olefin=157/76/23; Mn (NMR)=102 000. P3-O, P4-O and P5-O were similarly synthesized.
P7-O (via postesterification): RuCp*Cl(PPh3)2 (0.0075 mmol, 6.0 mg) was placed in a 100 ml round-bottom flask. Then, ethanol (29 ml), tetralin (0.5 ml), a 400 mM toluene solution of 4-DMAB (2 ml, 4-DMAB=0.8 mmol), PEGMA (16 mmol, 7.0 ml), DMA (4.0 mmol, 1.2 ml), HEMA (1.0 mmol, 0.12 ml) and a 486 mM toluene solution of ECPA (0.16 ml, ECPA=0.08 mmol) were added sequentially into the tube at 25 °C under argon. The total volume of the reaction mixture was 40 ml. The flask was then placed in an oil bath maintained at 40 °C. At predetermined intervals, the mixture was sampled with a syringe under dry argon, and the reaction was terminated by cooling the solution to –78 °C. After 47 h, the conversion of PEGMA/DMA/HEMA reached 74%/78%/80%, respectively, determined by 1H NMR using tetralin as an internal standard. The quenched solution was evaporated to dryness. The crude polymer was purified by silica gel column chromatography with toluene as an eluent and precipitated into hexane to give P7. SEC (DMF, PMMA std.): Mn=57 000 g mol−1; Mw/Mn=1.20.
In a 100 ml round-bottom flask filled with argon, MAC (5.0 mmol, 0.48 ml) was added to the solution of P7 (5.6 g, [OH]=~1 mmol) and triethylamine (6.0 mmol, 0.83 ml) in dry THF (20 ml) at 0 °C. The reaction mixture was stirred at 25 °C for 18 h. Then, the reaction was quenched with dry ethanol (5 ml). After the solvent was removed in vacuo, the crude product was purified by silica gel column chromatography with toluene as an eluent and precipitated into hexane to give P7-O. SEC (DMF, PMMA std.): Mn=58 800 g mol−1; Mw/Mn=1.18. SEC-MALLS (DMF, 0.01 M LiBr): Mw=109 000 g mol−1. 1H NMR [500 MHz, CD3OD, 25 °C, δ=3.30 (CD2HOD)]: δ 7.35–7.20 (aromatic), 6.21–6.16 (olefin), 5.77–5.70 (olefin), 4.46–4.20 (–COOCH2CH2OCO–), 4.18–4.05 (–COOCH2CH2O–), 4.05–3.90 (–COOCH2CH2CH2–), 3.82–3.47 (–OCH2CH2O–), 3.39–3.35 (–OCH3), 2.00 (CH2=C(CH3)COO–), 2.20–1.75 (–CH2C(CH3)–), 1.75–1.60 (–COOCH2CH2(CH2)9CH3), 1.58–1.20 (–COOCH2CH2(CH2)9CH3), 1.20–0.70 (–COO(CH2)11CH3, –CH2C(CH3)–); PEGMA/DMA/HEMA-olefin=141/35/8.3; Mn (NMR)=77 000. P1-O and P6-O were similarly synthesized.
Intramolecular crosslinking of olefin-bearing amphiphilic copolymers in water
The synthesis of star polymers (S1–S7) was carried out by the syringe technique under argon in baked glass tubes or flasks equipped with a three-way stopcock.
S2 (with a ruthenium catalyst): in a 30 ml glass tube, [RuCp*Cl]4 (0.43 mg, 0.0004 mmol) and PPh2(C6H4OH) (0.89 mg, 0.0032 mmol) were mixed in toluene (0.72 ml) at 80 °C for 12 h under argon. The solution was then evaporated in vacuo at 25 °C to give a solid ruthenium complex. Into the tube, the ethanol solution of P2-O (90 mg ml−1, 0.4 ml, P2-O=36 mg, olefin=0.008 mmol), ethanol (0.4 ml) and H2O (7.2 ml) were added at 25 °C under argon. The mixture was kept at 25 °C and sampled with a syringe at predetermined periods to determine the conversion of the olefin by 1H NMR (63% conversion after 120 h). After the solution was evaporated in vacuo in the presence of toluene, the crude product was purified by silica gel column chromatography with toluene as an eluent and precipitated into hexane to give S2. SEC (DMF, PMMA std.): Mn=48 900 g mol−1; Mw/Mn=1.22. SEC-MALLS (DMF, 0.01 M LiBr): Mw=120 000 g mol−1. 1H NMR [500 MHz, CDCl3, 25 °C, δ=7.26 (CHCl3)]: δ 6.10–6.09 (olefin), 5.55–5.53 (olefin), 4.17–4.0 (–COOCH2CH2O–), 4.0–3.82 (–COOCH2CH2CH2–), 3.72–3.46 (–OCH2CH2O–), 3.39–3.34 (–OCH3), 2.13–1.70 (–CH2C(CH3)–), 1.70–1.55 (–COOCH2CH2(CH2)9CH3), 1.45–1.20 (–COOCH2CH2(CH2)9CH3), 1.18–0.70 (–COO(CH2)11CH3, –CH2C(CH3)–). S1 and S3–S5 were similarly synthesized with their corresponding copolymers.
S7 (with a free radical initiator): in a 100 ml round-bottom flask, P7-O (250 mg, olefin=0.027 mmol), V-50 (25 mg, 0.092 mmol) and H2O (25 ml) were added at 25 °C under argon (total volume: 25 ml). The mixture was kept at 25 °C under UV irradiation (375 nm) for 72 h (77% conversion: determined by 1H NMR). After the water was removed in vacuo in the presence of toluene, the crude product was purified by silica gel column chromatography with toluene as an eluent and precipitated into hexane to give S7. SEC (DMF, PMMA std.): Mn=50 200 g mol−1; Mw/Mn=1.23. SEC-MALLS (DMF, 0.01 M LiBr): Mw=118 000 g mol−1. 1H NMR [500 MHz, DMF-d7, 25 °C, δ=8.01 (DMF)]: δ 7.41–7.25 (aromatic), 6.22–6.15 (olefin), 5.86–5.75 (olefin), 4.55–4.26 (–COOCH2CH2OCO–), 4.26–4.07 (–COOCH2CH2O–), 4.07–3.93 (–COOCH2CH2CH2–), 3.85–3.40 (–OCH2CH2O–), 3.39–3.26 (–OCH3), 2.30-1.77 (–CH2C(CH3)–), 1.77–1.63 (–COOCH2CH2(CH2)9CH3), 1.60–1.27 (–COOCH2CH2(CH2)9CH3), 1.21–0.70 (–COO(CH2)11CH3, –CH2C(CH3)–). S6 was similarly obtained with S6-O and V-50.
Results and discussion
Design of olefin-bearing amphiphilic random copolymers
Olefin-bearing amphiphilic random copolymers (P1-O–P7-O) were synthesized as precursors for star polymers (S1–S7) via the following two steps: (1) preparation of hydroxyl-functionalized amphiphilic random copolymers (P1–P7) via ruthenium-catalyzed living radical copolymerization and (2) introduction of methacrylate units into P1–P7 via in situ or postesterification of the hydroxyl pendants with methacryloyl chloride (MAC) (Scheme 1). Hydrophilic PEG (-(CH2CH2O)8.5Me) and hydrophobic dodecyl groups (–C12H24- or -C12H25) were introduced into the side chains of P1–P7 to induce efficient self-folding and/or association of corresponding P1-O–P7-O in water.
Polymerization
P1–P7 were synthesized by the ruthenium-catalyzed copolymerization of PEGMA (Mn=475), DMA and HDMA or HEMA with alkyl halide initiators (1–4) (Table 1). Here, three types of catalytic systems were employed: Ru(Ind)Cl(PPh3)2/n-Bu3N in THF at 60 °C (P2–P5), Ru(Ind)Cl(PPh3)2/n-Bu3N in toluene at 80 °C (P6) and RuCp*Cl(PPh3)2/4-DMAB in ethanol at 40 °C (P1 and P7). The Ru(Ind)Cl(PPh3)2/n-Bu3N system with THF allowed us to directly prepare olefin-bearing copolymers (P2-O–P5-O) by the in situ esterification of the generated copolymers (P2–P5) via the sequential addition of MAC without isolation. HDMA in P1–P6 served as a hydrophobic monomer that placed the olefin far from the methacrylate backbones via a long dodecyl linkage (–C12H24–), whereas HEMA in P7 resulted in the olefin being in close proximity to the polymer backbone via a short ethylene spacer (–C2H4–). In addition to monofunctional bromide or chloride initiators (1, 4), dibromide (2) or tribromide (3) initiators24 were employed to investigate the effects of active terminal numbers and branched structures on the single-chain crosslinking properties (discussed later). The following parameters were systematically varied: the feed ratio of monomers to an initiator (l=[PEGMA]0/[initiator]0, m=[DMA]0/[initiator]0, n=[HDMA or HEMA]0/[initiator]0); the total degree of polymerization (DP=l+m+n)=200 (P1), 250 (P2–P6), 263 (P7); the molar ratio of their hydrophobic monomers [100 × (m+n)/DP]=20 (P6, P7), 40 (P1–P4), 50 (P5) mol%; the molar ratio of hydroxyl-functionalized monomers for crosslinking units [100 × n/DP]=5 (P7), 10 (P1–P3, P5), 20 (P6), 40 (P4) mol%.
As shown in Figure 1, all copolymerizations proceeded smoothly via the simultaneous consumption of monomers up to 74–93% conversion, independent of the ruthenium catalysts, initiators, monomers and solvents, to provide hydroxyl-functionalized copolymers with controlled molecular weight and narrow molecular weight distribution (P1–P7, Mn=47 400–73 600 g mol−1, Mw/Mn=1.2–1.4, determined by SEC in DMF with PMMA std. calibration, Table 1).

Synthesis of hydroxyl-functionalized amphiphilic copolymers (P2, P3, P6, P7) via ruthenium-catalyzed living radical polymerization of poly(ethylene glycol) methyl ether methacrylate (PEGMA), dodecyl methacrylate (DMA) and 12-hydroxydodecyl methacrylate (HDMA) or 2-hydroxylethyl methacrylate (HEMA) with 2 (P2: a, e), 3 (P3: b, f) and 4 (P6: c, g; P7: d, h). (a–d) Time-conversion curves and (e–h) size-exclusion chromatography (SEC) curves. (e–h) Esterification of the hydroxyl groups of P2, P3, P6 and P7 with methacryloyl chloride into olefin-bearing amphiphilic copolymers ((e) P2-O, (f) P3-O, (g) P6-O, (h) P7-O). MW, molecular weight; PMMA, poly(methyl methacrylate).
Pendant olefin introduction
Esterification of the hydroxyl pendants of P1–P7 was examined with MAC in the presence of triethylamine in THF at 25 °C ([OH]0/[MAC]0=1/5). P1, P6 and P7 were isolated by preparative SEC (the removal of ruthenium catalysts and monomers) and then esterified with MAC, whereas P2–P5 were sequentially esterified via the direct addition of MAC into the polymerization solutions. After mixing the solutions for 16–18 h, the products were analyzed by 1H NMR in acetone-d6 or methanol-d4.
As typically shown in Figure 2, P1-O–P7-O exhibited proton signals from the olefin (n: 6.2–6.1, 5.7–5.6 p.p.m.) and methyl groups (o: 2.0–1.9 p.p.m.) of the pendant methacrylates, in addition to those from the PEG units (c: 4.2–4.1 p.p.m., d: 3.8–3.4 p.p.m., e: 3.4–3.3 p.p.m.), dodecyl pendants (–C12H25 or -C12H24-, f, j: 4.1–3.9 p.p.m., g, k: 1.7 p.p.m., h, l: 1.5–1.3 p.p.m., i: 1.0–0.7 p.p.m.), ethyl pendants (–C2H4-, j’: 4.5–4.1 p.p.m.) and methacrylate backbones (a: 1.2–0.7 p.p.m., b: 2.2–1.8 p.p.m.). The peak area ratio of the pendant olefin (n) to respective monomer units (PEGMA (c, e), DMA (f), HDMA or HEMA (j)) confirmed that P1–P7 were almost quantitatively esterified with MAC into P1-O–P7-O. It should be noted that the in situ esterification was as effective as the postesterification counterpart. P2-O, P4-O–P7-O further exhibited aromatic proton signals (p) derived from the initiating sites of 2 (~6.9 p.p.m.) or 4 (7.3–7.2 p.p.m.) (Figures 2c and d). Estimated from the peak area ratio of the monomer units to the initiators (p), the DP of the respective monomers (l/m/n,DP) was close to the DP calculated from their feed ratios (l/m/n,calcd) (Table 2). The number-average molecular weight for P2-O, P4-O–P7-O (Mn (NMR)) was thus determined as 77 700–107 000 g mol−1 by 1H NMR.

Proton nuclear magnetic resonance (1H NMR) spectra of (a) P1, (b) P1-O, (c) P2-O and (d) P7-O in acetone-d6 (a–c) or methanol-d4 (d) at 25 °C.
After esterification, P1-O–P7-O still maintain narrow molecular weight distribution (Mw/Mn=1.2–1.4) and the number-average molecular weight was almost identical to the precursors (P1–P7) (Figure 1 and Table 2). The absolute weight-average molecular weight (Mw,O) was determined by SEC-MALLS: Mw,O=109 000–160 000 g mol−1. Mw,O of P2-O, P4-O–P7-O were in good agreement with the values calculated from Mn (NMR) and Mw/Mn [Mw,O (calcd)=Mn (NMR) × Mw/Mn].
Intramolecular crosslinking of self-folding copolymers in water
PEGMA/DMA random copolymers with 20–40 mol% hydrophobic DMA units efficiently self-fold in water because of hydrophobic interactions.23 Before intramolecular crosslinking, the self-folding properties of P7-O (19 mol% DMA) in water were investigated by dynamic light scattering. As expected, the hydrodynamic radius of P7-O in water (Rh=6.4 nm) was smaller than that in CH2Cl2 (8.5 nm), indicating that P7-O self-folds in water to form unimer micelle that locally accumulate the hydrophobic pendant groups within the interior. Given these features, we examined the synthesis of single-chain crosslinked star polymers in water via these two strategies: (1) free radical crosslinking with an azo initiator and (2) living radical crosslinking from a polymer terminal with a ruthenium catalyst (Scheme 1 and Table 3).
Free radical crosslinking
The intramolecular crosslinking of P6-O or P7-O was investigated with a water-soluble azo initiator (2,2’-azobis(2-methylpropionamidine) dihydrochloride: V-50) in water under UV irradiation (375 nm) at 25 °C ([polymer]0=10 mg ml−1, Figure 3 and Table 3). P6-O and P7-O possess comparable hydrophobicity (~20 mol% HDMA or DMA), whereas the olefin content and positions are different: P6-O contains 20 mol% olefin (per total DP) dangling via a dodecyl (HDMA) spacer, whereas P7-O has 5 mol% olefin via an ethylene (HEMA) spacer. Both crosslinking reactions homogeneously proceeded without any macroscopic gelation up to 89% olefin conversion in 16 h (P6-O) and 77% in 72 h (P7-O) (confirmed by 1H NMR) to provide S6 and S7 with narrow molecular weight distributions (Mw/Mn=1.2–1.3, by SEC in DMF, Figures 3a and c). Importantly, the Mn of S6 and S7 was smaller than that of the corresponding precursors (P6-O and P7-O) obtained by SEC, whereas the absolute Mw of the products by SEC-MALLS is in good agreement with that of the precursors (Mw,O) (Table 3, Mw=132 000 (S6), 118 000 (S7) g mol−1); the ratio of Mw to Mw,O (Mw/Mw,O) was thus close to 1. These results demonstrate that P6-O and P7-O are intramolecularly fixed to give single-chain crosslinked star polymers (S6 and S7) with many short PEG arms (140–160). It should be noted that this synthetic strategy allows us to precisely and directly control the primary structure (for example, molecular weight, monomer composition) of the single-chain crosslinked star polymers using olefin-bearing copolymers.

Synthesis of single-chain crosslinked star polymers (solid lines) via intramolecular crosslinking of (a, b) P6-O or (c, d) P7-O with (a, c) V-50 in water or (b, d) dimethyl 2,2’-azobis(isobutyrate) (MAIB) in toluene at 25 °C under ultraviolet (UV) irradiation (375 nm): [olefin in precursor]0=4.2 (P6-O), 1.1 (P7-O) mM, [V-50]0=3.7 mM, [MAIB]0=4.3 mM ([polymer]0/[initiator]0=10/1.0 mg ml−1). MW, molecular weight; PMMA, poly(methyl methacrylate).
The compactness was further evaluated from the ratio of the peak molecular weight of S6 and S7 (Mp) to their corresponding precursors (Mp,O: P6-O, P7-O) (Table 3 and Figures 3a and c). This ratio (Mp/Mp,O) indicates a shrinking index of the products against their precursors. Mp/Mp,O for S6 (0.72) was smaller than that for S7 (0.87), meaning that S6 was more compact than S7 in comparison with their corresponding precursors. This is because P6-O has fully mobile pendant olefins in high concentration that are efficiently crosslinked into S6. Selective single-chain crosslinking of P7-O in water was achieved even in high concentrations (up to 80 mg ml−1). Analyzed by dynamic light scattering, the Rh of S7 in CH2Cl2 (6.7 nm) was close to that of P7-O in D2O (6.4 nm). This demonstrates that S7 is effectively crosslinked to maintain the compact self-folding structure in CH2Cl2.
Beyond our expectations, even in toluene (good solvent), P6-O and P7-O did not induce gelation with MAIB under UV irradiation (375 nm) at 25 °C ([polymer]0=10 mg ml−1). The pendant olefins of P6-O and P7-O were homogeneously consumed up to a conversion of 86% and 69% in 12 h, respectively. However, the SEC peaks of the products were identical to those of P6-O or P7-O (Figures 3b and d), indicating that the products still maintain the random-coil structure of the precursors. This unique phenomenon suggests that in a good solvent, the pendant olefins should be predominantly consumed via the addition of a MAIB fragment radical and the subsequent coupling between the adduct radical and a MAIB fragment radical. Such a homogeneous reaction in toluene further supports that the selective crosslinking of single chains in water is attributed to not only intramolecular hydrophobic interactions in water but also the steric hindrance of PEG chains that effectively isolate polymer chains.
S6 was further analyzed by transmission electron microscopy (Figure 4). The negatively stained micrograph of S6, cast on a carbon-coated Cu grid from an aqueous solution, exhibited small white dots below 10 nm without any large aggregates. Thus, selective intramolecular crosslinking of P6-O was also visibly confirmed.

Transmission electron micrograph of S6 cast from an aqueous solution ([S6]=1.0 × 10−4 g l−1). The sample is negatively stained with 2% uranyl acetate (white dots: S6; black: background).
Living radical crosslinking
As an alternative to a free radical initiator, a hydrophilic ruthenium catalyst [RuCp*Cl(PPh2(C6H4OH))2] was combined with olefin-bearing precursors (P1-O–P5-O) in water. In this system, the terminal halogens (~C-Br) of the self-folding precursors could be reversibly activated by the ruthenium catalyst (Ru) to generate carbon radicals (~C•+Br-Ru) that induce intramolecular crosslinking through the pendant olefins. The ruthenium complex was prepared by mixing [RuCp*Cl]4 and PPh2(C6H4OH) in toluene at 80 °C for 12 h. After evaporation, the crude ruthenium was directly utilized for P1-O–P5-O in water/ethanol (9:1, v/v) at 25 °C ([polymer]0=5 or 20 mg ml−1).
P1-O–P5-O were homogeneously crosslinked without any gelation to give S1–S5 with narrow molecular weight distributions (Mw/Mn=1.2–1.3) and molecular weights (Mn, Mp) smaller than the corresponding precursors (Figure 5 and Table 3). The crosslinking efficiency (olefin conversion), the association number of precursor chains in the products (intra- or inter-molecular linking) and the compactness of products against the precursors were dependent on the following factors: the number of bromine terminals (initiator: 1, 2, 3), olefin content (n) and hydrophobic monomer content (m+n).

Synthesis of star polymers via the intramolecular crosslinking of P1-O, P2-O, P3-O, P4-O and P5-O with a ruthenium catalyst in water: [olefin in precursor]0/[RuCp*Cl(PPh2(C6H4OH))2]0=1.0/0.2 (S1-S3), 4.0/0.8 (S4, S5) mM in H2O/ethanol (9:1, v/v) at 25 °C. (a, b) Mp/Mp,O as a function of conversion and (c) size-exclusion chromatography (SEC) curves. MW, molecular weight; PMMA, poly(methyl methacrylate).
Owing to the low radical concentration produced via the reversible activation of the carbon–bromine terminus, a ruthenium catalyst induced the crosslinking of P1-O–P5-O (120–360 h) much slower than a free radical initiator for P6-O and P7-O (16–72 h, Table 3). However, the pendant olefins of P2-O–P5-O with bifunctional and trifunctional initiators (2, 3) were efficiently consumed up to a conversion of 63–70%, whereas those of P1-O with a monofunctional 1 were consumed up to a conversion of ~30%.
To estimate the association numbers of the polymer chains, the final products (S1–S3, S5) were analyzed by SEC-MALLS (Table 3). P1-O–P3-O, the precursors for S1–S3, consist of ~40 mol% hydrophobic monomers (100 × (m+n)/DP) with ~10 mol% olefin units (100 × n/DP), whereas P5-O, the precursor for S5, carries ~50 mol% hydrophobic monomers with ~10 mol% olefin units. The absolute Mw of S1 and S2 were determined to be 128 000 and 120 000 g mol−1, respectively, and these are in good agreement with those for the corresponding precursors (Mw,O). Thus, S1 and S2 are single chain-crosslinked star polymers. This result is consistent with the efficient self-folding of PEGMA/DMA random copolymers with 40 mol% DMA in water.23 S3 also consists predominantly of unimolecularly crosslinked star polymers of the branched precursor owing to the Mw that is relatively close to that of P3-O. In contrast, the Mw of S5 (296 000 g mol−1) was approximately twice that of P5-O (128 000 g mol−1), demonstrating that S5 consists of two polymer chains of P5-O. A PEGMA/DMA (100/100) random copolymer with 50 mol% DMA is known to form bimolecular associations in water.23 Thus, P5-O would also form a bimolecular aggregate in water during the crosslinking process to yield a unique double-chain crosslinked star polymer with narrow molecular weight distribution (S5, Mw/Mn=1.15).
The shrinking process and compactness of the products was further assessed from the SEC peak molecular weight ratio between the products (Mp) and the corresponding precursors (Mp,O) [Mp/Mp,O]. Figures 5a and b plot Mp/Mp,O for the products (intermediates) obtained from S1-O–P5-O against conversion of the pendant olefin. In all cases, Mp/Mp,O decreased with increasing conversion, indicating that the products gradually shrink with crosslinking. Mp/Mp,O for S1–S3 obtained from P1-O–P3-O (n: ~10 mol%) was close to ∼0.7, whereas Mp/Mp,O for S4 from P4-O (n: 33 mol%) reached a final value of 0.49, much smaller than that for S2 (Table 3). This is because P4-O can be crosslinked more tightly than P2-O because of a large olefin content. Thus, the compactness of single-chain and unimolecularly crosslinked star polymers (S1–S4) is dependent on the olefin content (n) of the precursors with comparative hydrophobicity (m+n: 33–40%).
Properties and functions
Mobility
To evaluate the mobility of the crosslinked cores, single-chain star polymers (S6, S7) were analyzed by 1H NMR spectroscopy in DMF-d7 and D2O at 25 °C in comparison with a PEGMA/DMA (160/40) random copolymer (Random)23 (Figure 6). All samples exhibited broader proton signals of the hydrophobic methacrylate backbones (a, b) and dodecyl pendants (c, d, e) in D2O than in DMF-d7. This indicates that these segments aggregate within their inner compartments via hydrophobic interactions in water resulting in restricted mobility. In detail, the dodecyl proton signal (d) broadened in this order: Random<S7<S6; the mobility of the hydrophobic compartments thus decreased in that order. S6, in particular, showed quite broad signals not only in D2O but also in DMF-d7 (Figures 6e and f). This is because the HDMA linkage efficiently fixed and stabilized the self-folding structure even in a good solvent such as DMF-d7. In contrast, S7 exhibited dodecyl proton signals (c, d, e; Figure 6b) that are as sharp as the Random structure (Figure 6a), consistent with the free mobility of the non-crosslinked dodecyl pendants.

Proton nuclear magnetic resonance (1H NMR) spectra of (a, b) a PEGMA/DMA (160/40) random copolymer (Random), (c, d) S7 and (e, f) S6 in DMF-d7 (a, c, e) and D2O (b, d, f) at 25 °C. DMA, dodecyl methacrylate; PEGMA, poly(ethylene glycol) methyl ether methacrylate.
Hydrophobicity
Solvatochromism is effective in evaluating the polarity of the microenvironments of polymeric materials and aggregates. A pyridinium N-phenolate betaine dye, the so-called Reichardt’s dye (RD), is well known to show a negative solvatochromic shift (blue shift) of UV–vis absorption originating from the intramolecular charge transfer with increasing solvent polarity.23, 24, 39 Actually, the maximum wavelength (λmax) of RD shifted to 455 nm in H2O/acetone (19:1, v/v) from 675 nm in acetone (Figure 7a).

(a) Ultraviolet–visible (UV–vis) spectra of Reichardt’s dye (RD) (long dash), RD with S7 (black) and RD with P7-O (gray) in H2O/acetone (19:1, v/v) and RD in acetone (dash) at 25 °C, [polymer]0/[RD]0=0.045/0.45 mM. (b) Effect of polymer concentration (S6: filled square; S7: open square; P7-O: filled circle; poly(poly(ethylene glycol) methyl ether methacrylate) (PPEGMA): open circle) on λmax of RD: [polymer]0/[RD]0=0.0045/0.45–0.090/0.45 mM in H2O/acetone (19:1, v/v) at 25 °C.
Thus, the hydrophobicity of single-chain crosslinked star polymers was evaluated by UV–vis measurements of the aqueous solution of RD with S6 or S7 in H2O/acetone (19:1, v/v). To investigate the effect of crosslinking on the hydrophobicity, S6 and S7 were compared with a non-crosslinked P7-O (precursor for S7); all of the samples contain ~20 mol% of a hydrophobic monomer (HDMA or DMA).
S6, S7 and P7-O induced a negative solvatochromic shift of RD more effectively than PPEGMA (homopolymer), and had an almost identical λmax of 545 nm ([polymer]0/[RD]0=0.045/0.45 mM) (Figure 7). This means that RD was encapsulated into the crosslinked or self-folding compartments to give UV–vis absorptions reflecting the hydrophobic microdomains. Importantly, the hydrophobicity of the polymer compartments is only dependent on the hydrophobic monomer content and independent of the mobility. However, in contrast to a non-crosslinked P7-O, S6 and S7 effectively induced a blue shift in RD, even using a small feed of polymers against RD ([λmax=536 (S6), 524 (S7), and 485 (P7-O) nm, [polymer]0/[RD]0=0.0045/0.45 mM). This importantly demonstrates that single-chain crosslinked star polymers (S6 and S7) enclose the hydrophobic RD within the fixed compartments more efficiently and stably than a non-crosslinked counterpart.
Thermosensitive solubility
PEG and related materials often show thermosensitive solubility in water to induce lower critical solution temperature-type phase separation upon heating.7, 11, 23, 24, 40 Thus, the cloud point (Cp) of an olefin-bearing precursor (P1-O) and the crosslinked star polymer (S1) was determined by UV–vis measurements of their aqueous solutions scanning from 70 to 95 °C at a heating rate of 1 °C per min ([polymer]=4 mg ml−1). As shown in Figure 8, both of the solutions turned turbid upon heating. However, the Cp of S1 (~81 °C; ~90% transmittance) was clearly higher than the Cp of P1-O (~76 °C; ~90% transmittance) despite perfectly identical composition of their polymers. This unique phenomenon likely occurs because the hydrophobic dodecyl pendants in S1 are confined stably within the crosslinked inner core, to hardly promote the dehydration of PEG chains.

Transmittance of P1-O and S1 aqueous solutions as a function of temperature (heating rate: 1 °C per min, heating range: 70–95 °C): [polymer]=4 mg ml−1.
Conclusion
In conclusion, we successfully produced single-chain crosslinked star polymers as a new class of star polymers via the intramolecular crosslinking of self-folding amphiphilic copolymers in water. For this process, amphiphilic random copolymers carrying hydrophilic PEG chains and hydrophobic dodecyl and olefin pendants were synthesized via ruthenium-catalyzed living radical polymerization of PEGMA, DMA, and HDMA or HEMA, and in situ or postesterification with methacryloyl chloride. Olefin-bearing precursors with 20–40 mol% hydrophobic units efficiently self-folded in water via hydrophobic interactions to form unimer micelles placing olefin pendants within the inner cores. The self-folding precursors were thus intramolecularly crosslinked with a free radical initiator or a ruthenium catalyst in water to give single-chain crosslinked star polymers. In contrast, an olefin-bearing precursor with 50 mol% hydrophobic units gave double-chain crosslinked star polymers via the bimolecular association of the precursor in water. The primary structure of these star polymers can be precisely controlled with amphiphilic random copolymer precursors. Thus, the star polymers developed herein would be promising as scaffolds for novel functional macromolecules and precision nanospaces.

(a) Synthesis of olefin-bearing amphiphilic random copolymers (P1-O–P7-O) via ruthenium-catalyzed living radical polymerization of poly(ethylene glycol) methyl ether methacrylate (PEGMA), dodecyl methacrylate (DMA) and 12-hydroxydodecyl methacrylate (HDMA) or 2-hydroxylethyl methacrylate (HEMA) with 1–4, followed by the esterification of hydroxyl-functionalized amphiphilic random copolymers (P1–P7) with methacryloyl chloride. (b) Synthesis of single-chain crosslinked star polymers (S1–S7) via the intramolecular crosslinking of self-folding precursors (P1-O–P7-O) with an azo initiator or a ruthenium catalyst in water.
References
Terashima, T. Functional spaces in star and single-chain polymers via living radical polymerization. Polym. J. 46, 664–673 (2014).
Terashima, T. & Sawamoto, M. Microgel-core star polymers as functional compartments for catalysis and molecular recognition. ACS Symp. Ser. 1101, 65–80 (2012).
Blencowe, A., Tan, J. F., Goh, T. K. & Qiao, G. G. Core cross-linked star polymers via controlled radical polymerisation. Polymer 50, 5–32 (2009).
Gao, H. Development of star polymers as unimolecular containers for nanomaterials. Macromol. Rapid Commun. 33, 722–734 (2012).
Elsabahy, M. & Wooley, K. L. Strategies toward well-defined polymer nanoparticles inspired by nature: chemistry versus versatility. J. Polym. Sci. A Polym. Chem. 50, 1869–1880 (2012).
Terashima, T., Motokawa, R., Koizumi, S., Sawamoto, M., Kamigaito, M., Ando, T. & Hashimoto, T. In situ and time-resolved small-angle neutron scattering observation of star polymer formation via arm-linking reaction in ruthenium-catalyzed living radical polymerization. Macromolecules 43, 8218–8232 (2010).
Terashima, T., Nishioka, S., Koda, Y., Takenaka, M. & Sawamoto, M. Arm-cleavable microgel star polymers: a versatile strategy for direct core analysis and functionalization. J. Am. Chem. Soc. 136, 10254–10257 (2014).
Terashima, T., Ouchi, M., Ando, T. & Sawamoto, M. Transfer hydrogenation of ketones catalyzed by PEG-armed ruthenium-microgel star polymers: microgel-core reaction space for active, versatile and recyclable catalysis. Polym. J. 43, 770–777 (2011).
Terashima, T., Nomura, A., Ito, M., Ouchi, M. & Sawamoto, M. Star-polymer-catalyzed living radical polymerization: microgel-core reaction vessel by tandem catalyst interchange. Angew. Chem. Int. Ed. 50, 7892–7895 (2011).
Chi, Y., Scroggins, S. T. & Fréchet, J. M. J. One-pot multi-component asymmetric cascade reactions catalyzed by soluble star polymers with highly branched non-interpenetrating catalytic cores. J. Am. Chem. Soc. 130, 6322–6323 (2008).
Kanaoka, S., Yagi, N., Fukuyama, Y., Aoshima, S., Tsunoyama, H., Tsukuda, T. & Sakurai, H. Thermosensitive gold nanoclusters stabilized by well-defined vinyl ether star polymers: reusable and durable catalysts for aerobic alcohol oxidation. J. Am. Chem. Soc. 129, 12060–12061 (2007).
Koda, Y., Terashima, T. & Sawamoto, M. Fluorous microgel star polymers: selective recognition and separation of polyfluorinated surfactants and compounds in water. J. Am. Chem. Soc. 136, 15742–15748 (2014).
Ouchi, M., Terashima, T. & Sawamoto, M. Transition metal-catalyzed living radical polymerization: toward perfection in catalysis and precision polymer synthesis. Chem. Rev. 109, 4963–5050 (2009).
Ouchi, M., Badi, N., Lutz, J.-F. & Sawamoto, M. Single-chain technology using discrete synthetic macromolecules. Nat. Chem. 3, 917–924 (2011).
Altintas, O. & Barner-Kowollik, C. Single-chain folding of synthetic polymers by covalent and non-covalent interactions: current status and future perspectives. Macromol. Rapid Commun. 33, 958–971 (2012).
Sanchez-sanchez, A., Pérez-Baena, I. & Pomposo, J. A. Advances in click chemistry for single-chain nanoparticle construction. Molecules 18, 3339–3355 (2013).
Terahsima, T. & Sawamoto, M. Sequence-regulated polymers via living radical polymerization: from design to properties and functions. ACS Symp. Ser. 1170, 255–267 (2014).
Artar, M., Huerta, E., Meijer, E. W. & Palmans, A. R. A. Dynamic single chain polymeric nanoparticles: from structure to functions. ACS Symp. Ser. 1170, 313–325 (2014).
Li, L., Raghupathi, K., Song, C., Prasad, P. & Thayumanavan, S. Self-assembly of random copolymers. Chem. Commun. 50, 13417–13432 (2014).
Lyon, C. K., Prasher, A., Hanlon, A. M., Tuten, B. T., Tooley, C. A., Frank, P. G. & Berda, E. B. A brief user’s guide to single-chain nanoparticles. Polym. Chem. 6, 181–197 (2015).
Morishima, Y., Nomura, S., Ikeda, T., Seki, M. & Kamachi, M. Characterization of unimolecular micelles of random copolymers of sodium 2-(acrylamide)-2-methylpropanesulfonate and methacrylamides bearing bulky hydrophobic substituents. Macromolecules 28, 2874–2881 (1995).
Yusa, S., Sakakibara, S., Yamamoto, T. & Morishima, Y. Fluorescence studies of pH-responsive unimolecular micelles formed from amphiphilic polysulfonates possessing long-chain alkyl carboxyl pendants. Macromolecules 35, 10182–10188 (2002).
Terashima, T., Sugita, T., Fukae, K. & Sawamoto, M. Synthesis and single-chain folding of amphiphilic random copolymers in water. Macromolecules 47, 589–600 (2014).
Sugita, T., Matsumoto, K., Terashima, T. & Sawamoto, M. Synthesis of amphiphilic three-armed star random copolymers via living radical polymerization and their unimolecular folding properties in water. Macromol. Symp. 350, 76–85 (2015).
Terashima, T., Mes, T., de Greef, T. F. A., Gillissen, M. A. J., Besenius, P., Palmans, A. R. A. & Meijer, E. W. Single-chain folding of polymers for catalytic systems in water. J. Am. Chem. Soc. 133, 4742–4745 (2011).
Gillissen, M. A. J., Terashima, T., Meijer, E. W., Palmans, A. R. A. & Voets, I. K. Sticky supramolecular grafts stretch single polymer chains. Macromolecules 46, 4120–4125 (2013).
Arter, M., Terashima, T., Sawamoto, M., Meijer, E. W. & Palmans, A. R. A. Understanding the catalytic activity of single-chain polymeric nanoparticles in water. J. Polym. Sci. A Polym. Chem. 52, 12–20 (2014).
Foster, E. J., Berda, E. B. & Meijer, E. W. Metastable supramolecular polymer nanoparticles via intramolecular collapse of single polymer chains. J. Am. Chem. Soc. 131, 6964–6966 (2009).
Mes, T., van der Weegen, R., Palmans, A. R. A. & Meijer, E. W. Single-chain polymeric nanoparticles by stepwise folding. Angew. Chem. Int. Ed. 50, 5085–5089 (2011).
Hosono, N., Gillissen, M. A. J., Li, Y., Sheiko, S. S., Palmans, A. R. A. & Meijer, E. W. orthogonal self-assembly in folding block copolymers. J. Am. Chem. Soc. 135, 501–510 (2013).
Altintas, O., Lejeune, E., Gerstel, P. & Barner-Kowollik, C. Bioinspired dual self-folding of single polymer chains via reversible hydrogen bonding. Polym. Chem. 3, 640–651 (2012).
Croce, T. A., Hamilton, S. K., Chen, M. L., Muchalski, H. & Harth, E. Macromolecules 40, 6028–6031 (2007).
Cherian, A. E., Sun, F. C., Sheiko, S. S. & Coates, G. W. Formation of nanoparticles by intramolecular cross-linking: following the reaction process of single polymer chains by atomic force microscopy. J. Am. Chem. Soc. 129, 11350–11351 (2007).
Murray, B. S. & Fulton, D. A. Dynamic covalent single-chain polymer nanoparticles. Macromolecules 44, 7242–7252 (2011).
Chao, D., Jia, X., Tuten, B., Wang, C. & Berda, E. B. Controlled folding of a novel electroactive polyolefin via multiple sequential orthogonal intra-chain interactions. Chem. Commun. 49, 4178–4180 (2013).
Ouchi, M., Yoda, H., Terashima, T. & Sawamoto, M. Aqueous metal-catalyzed living radical polymerization: highly active water-assisted catalysis. Polym. J. 44, 51–58 (2012).
Ouchi, M., Ito, M., Kamemoto, S. & Sawamoto, M. Highly active and removable ruthenium catalysts for transition-metal-catalyzed living radical polymerization: design of ligands and cocatalysts. Chem. Asian J. 3, 1358–1364 (2008).
Yoda, H., Nakatani, K., Terashima, T., Ouchi, M. & Sawamoto, M. Ethanol-mediated living radical homo- and copolymerizations with Cp*-ruthenium catalysts: active, robust, and universal for functionalized methacrylates. Macromolecules 43, 5595–5601 (2010).
Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 94, 2319–2358 (1994).
Lutz, J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: toward new generations of smart biocompatible materials. J. Polym. Sci. A Polym. Chem. 46, 3459–3470 (2008).
Acknowledgements
This research was supported by the Ministry of Education, Science, Sports and Culture through Grant-in-Aids for Scientific Research (A: 24245026, C: 26410134) and Young Scientist (B: 24750104). We also thank Mr Akihiro Uesaka and Professor Syunsaku Kimura (Department of Material Chemistry, Kyoto University) for transmission electron microscopy measurements.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Terashima, T., Sugita, T. & Sawamoto, M. Single-chain crosslinked star polymers via intramolecular crosslinking of self-folding amphiphilic copolymers in water. Polym J 47, 667–677 (2015). https://doi.org/10.1038/pj.2015.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2015.54
This article is cited by
-
Amphiphilic thermoresponsive copolymer bottlebrushes: synthesis, characterization, and study of their self-assembly into flower-like micelles
Polymer Journal (2021)
-
Structurally nanoengineered antimicrobial peptide polymers: design, synthesis and biomedical applications
World Journal of Microbiology and Biotechnology (2021)
-
PJ ZEON Award for outstanding papers in Polymer Journal 2015
Polymer Journal (2016)
