
Abstract
The self-assembly of block copolymers into 1D, 2D and 3D nano- and microstructures is of great interest for a wide range of applications. A key challenge in this field is obtaining independent control over molecular structure and hierarchical structure in all dimensions using scalable one-pot chemistry. Here we report on the ring opening polymerization-induced crystallization-driven self-assembly (ROPI-CDSA) of poly-L-lactide-block-polyethylene glycol block copolymers into 1D, 2D and 3D nanostructures. A key feature of ROPI-CDSA is that the polymerization time is much shorter than the self-assembly relaxation time, resulting in a non-equilibrium self-assembly process. The self-assembly mechanism is analyzed by cryo-transmission electron microscopy, wide-angle x-ray scattering, Fourier transform infrared spectroscopy, and turbidity studies. The analysis revealed that the self-assembly mechanism is dependent on both the polymer molecular structure and concentration. Knowledge of the self-assembly mechanism enabled the kinetic trapping of multiple hierarchical structures from a single block copolymer.
Similar content being viewed by others

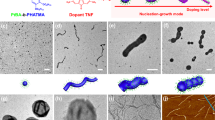
Introduction
Self-assembly of block copolymers (BCP) in solution has received significant interest in the areas of drug delivery, medical imaging, catalysis, and templated synthesis1,2. The properties and performance of these materials are intrinsically linked to both their molecular and hierarchical structure3. Consequently, research in this area has been focused on providing a fundamental understanding of how the molecular structure and assembly environment can be tailored to control the hierarchical assembly process4. Self-assembly processes can be either thermodynamically or kinetically controlled5,6. Thermodynamically controlled processes will adopt the hierarchical structure with the lowest free energy regardless of the starting conformation of the BCPs. Thermodynamically controlled processes tend to result in predictable and well-defined structures. However, their application is limited by their sensitivity to changes in the environment, which can significantly alter the formed structures7,8. Kinetically controlled processes tend to result in stable structures, but the final structures can be highly sensitive to the assembly method5,7,9. While the final structural dependence on assembly methodology offers the flexibility to form multiple hierarchical structures from a single BCP, it necessitates strict control of the synthetic conditions3,7,10.
Typically, BCP self-assembly is achieved via direct dissolution, solvent-switch, and thin-film hydration methods11,12,13. However, with these methods, it is challenging to control the formation of anisotropic structures, they involve multi-step processes that are difficult to scale up reproducibly, and result in low concentration BCP solutions (typically ≤1% solids w/w)14,15,16. To address these challenges, crystallization-driven self-assembly (CDSA) and polymerization-induced self-assembly (PISA) have emerged as promising alternatives. CDSA utilizes BCPs with a semi-crystalline core-forming block to form anisotropic 1D and 2D structures with high precision, where crystallization of the BCP core is the dominant driving force of self-assembly17,18,19,20,21,22,23. Whereas amorphous BCPs typically assemble into spheres, worms, and vesicles14,24, crystalline BCPs will typically result in morphologies with low curvature such as 2D platelet lamellae or 1D nanorods15, which introduces anisotropy to the system. In CDSA, the insoluble block is typically crystallized by dissolving the polymer in a selective solvent to facilitate /induce crystallization18,24, often utilizing heat-cool cycles to control the crystallization process15,23,24. However, as with traditional self-assembly methods, CDSA typically occurs in dilute solutions (~1% solids w/w)14,20,24. The PISA method utilizes controlled polymer chemistry to generate the BCP directly in the selective solvent system11,13,14,25,26,27. Conceptually, a homopolymer is chain-extended with a co-monomer and as the core-forming block grows it becomes increasingly insoluble, triggering self-assembly of the BCP. PISA is a scalable method that affords control over the BCP structure at high concentrations (10–50% solids w/w)14,25,28,29,30. To date, most PISA processes have been reversible-deactivation radical polymerizations (RDRP)1,2,11,14,31, primarily reversible addition-fragmentation chain-transfer polymerization (RAFT-PISA)11,14,31,32,33,34. However, PISA can theoretically be extended to all types of living polymerizations and has been demonstrated with living anionic polymerization35, ring-opening metathesis (ROMP) of norbornenes13,29, radical ring-opening copolymerization (rROP) of cyclic ketenes36, and ring-opening polymerization (ROP) of N-carboxyanhydrides37. PISA has also been combined with CDSA, termed polymerization-induced crystallization-driven self-assembly (PI-CDSA), to generate crystalline self-assemblies in high concentrations (10–25% solids w/w)15,16,38. Currently, PI-CDSA has been demonstrated for sequential living anionic polymerization15,16, and the ring-opening metathesis of organometallic polymers38. Polylactones, being semi-crystalline in nature, are excellent candidates for the development of a fully organic PI-CDSA to produce biocompatible, biodegradable BCP materials at high concentration in a scalable process. Polylactones, including poly(L-lactide) (PLLA) are the most widely known class of sustainable polymers39, and are incorporated into multiple FDA-approved formulas40,41,42. To date, ROP is the only controlled method to synthesize polylactone-based BCPs2,28. Thus, the development of a ring-opening PI-CDSA (ROPI-CDSA), would address the production limitations of polylactone-based crystalline-nanoparticles facilitating their use commercially.
The absence of ring-opening PI-CDSA of lactones is likely due to the stringent requirements for ROP reactions and the limited monomer/polymer/solvent combinations available39,43,44. Foremost, ROP of lactones cannot occur in protic solvents, as these solvents would compete with initiation species. However, some aprotic organic solvents, including aromatics like toluene, have induced crystallization in PLLA at room temperature45.
Here, we present the ring opening PI-CDSA of poly-(L-lactide)-block-polyethylene glycol (PLLA-b-PEG) BCPs in toluene. Kinetics studies of the polymerization and self-assembly show that ROPI-CDSA results in a non-equilibrium assembly process. Structural and morphological evolution of self-assembly is tracked with cryogenic-transmission electron microscopy (cryo-TEM), wide-angle X-ray scattering (WAXS), UV/Vis spectroscopy, and Fourier transform infrared (FTIR) spectroscopy, revealing a hierarchical (1D→2D→3D) growth mechanism. The data reveal that the assembly process and the final meta-stable structures can be controlled by alterations in both BCP molecular structure and concentration. Knowledge of the mechanism enables the trapping of materials with different structures and dimensionalities.
Results
ROPI-CDSA design and synthetic parameters
ROPI-CDSA experiments were performed using L-lactide as the monomer, mono-functional polyethylene glycol (mPEG45) as the initiator, toluene as the solvent and triazabicyclodecene (TBD) the catalyst (Fig. 1a)44,46,47,48. While other organocatalysts can achieve better dispersity (Ð) than TBD, they require high catalyst loading (1–10% mol vs 0.1% mol) and longer reaction times48. TBD is a highly active ROP catalyst that can achieve high conversion in seconds39,47. If reaction mixtures are left too long the Ɖ will broaden from transesterification47. Therefore, to limit transesterification reactions were quenched after 90 s for concentrations of 10% solids w/w and 60 s for concentrations greater than 10% solids w/w. As shown in Table 1, a library of 19 polymers was synthesized with a variation in the degree of polymerization (DP) of the PLLA block and total solids concentration (Supplementary Tables 1 and 2 for additional information on synthesis). For PLLA10-b-PEG45, 1–4 (5–20% solids w/w), no self-assembly was observed visually (i.e., solutions remain clear/no observed turbidity) or by cryo-TEM. At 20% solids w/w (PLLA45-b-PEG45 10, PLLA90-b-PEG45 16, and PLLA135-b-PEG45 19), control over Ð worsens, most likely due to transesterification from higher TBD solution concentrations (see also Supplementary Table 1). The 20% solids w/w samples macroscopically phase separate forming distinct solution and gel-like phases. Above 20% solids w/w, mPEG45 is not fully soluble in toluene. At 20% solids, limited PEG solubility may have also contributed to transesterification. For samples 5–19, the polymerizations reached >92% (Supplementary Fig. 1) conversion within 60–90 s and resulted in well-defined (Ð < 1.2) BCP (Supplementary Fig. 2). The resulting solutions became turbid at various time points, ranging from during the polymerization for 16 (PLLA90-b-PEG45 20% solids w/w) and 19 (PLLA135-b-PEG45) to more than 24 h post-polymerization for 5 (PLLA25-b-PEG45 10% solids w/w) and 7 (PLLA45-b-PEG45 5% solids w/w). This indicates that in most instances the relaxation time is significantly longer than the polymerization time and is dictated by the molecular structure of the BCP. Here, relaxation time refers to the time taken for the assembled structures to reach a low energy configuration and stop reorganizing. Control experiments were performed by synthesizing PLLA homopolymers in dichloromethane (where PLLA is fully soluble) and toluene (where PLLA is partially soluble), using ethanol as an initiator. In dichloromethane, >95% conversion was achieved in 90 s, whereas in toluene, only 25 and 28% conversion were achieved after 90 s for PLLA45 and PLLA90, respectively (Supplementary Table 3). This demonstrates that the PEG chains promote the polymerization by increasing the solubility of the growing PLLA chain in toluene. This is similar to previous PISA reports which have shown that self-assembly during polymerization can enhance the rate of polymerization of the selective block14. A second control was performed to determine if the polymerization is required for initiation of the self-assembly process, PLLA-b-PEG BCPs synthesized in dichloromethane were purified and re-dispersed directly in toluene. The polymers did not fully dissolve/disperse even when left over a period of months, indicating that direct polymerization in toluene is required to form stable assemblies at room temperature and that the structures formed in ROPI-CDSA are under kinetic control. Heat-cool cycles or a solvent-switch would likely also lead to the formation of stable assemblies, however, the purpose of the second control was to determine if the samples are pathway dependent. Pathway dependence indicates that it should be possible to control the ROPI-CDSA structures by modification of both the thermodynamics and kinetics of the process (see “Discussion” section on non-equilibrium PISA).

a ROPI-CDSA scheme. b Turbidity measurements (UV/Vis 600 nm) for PLLA DP 45, 60, 75, and 90 at 10% solids w/w, with PEG DP 45 standard error calculated from 3 runs. The data show that the self-assembly kinetics vary as a function of PLLA DP, where increasing DP results in a faster rate of self-assembly. c Photographs showing the difference in turbidity for samples 9 (PLLA45-b-PEG45 10% solids w/w left) and 15 (PLLA90-b-PEG45 10% solids w/w right).
Self-assembly and crystallization kinetics
To track how PLLA DP influences the self-assembly kinetics, UV/Vis measurements were performed to track the turbidity changes post polymerization for polymers PLLA45-b-PEG45 9, PLLA45-b-PEG45 11, PLLA45-b-PEG45 12, and PLLA45-b-PEG45 15, all at 10% solids w/w. (Fig. 1b). Measurements were performed at 600 nm, a wavelength in which no molecular species present in ROPI-CDSA absorb. The data showed that with increasing PLLA DP, the initial rate of self-assembly increases and results in more turbid final solutions.
To determine if crystallization of the PLLA block is a driving force in the self-assembly, the solutions for PLLA45-b-PEG45(10% solids w/w 9) and PLLA90-b-PEG45 (10% solids w/w 15) were freeze-dried at various time points between 5 min and 24 h post polymerization. The dried powders were analyzed by WAXS and FTIR spectroscopy to monitor the crystallization behavior (Fig. 2). Crystallinity is tracked over time by comparing the area of the crystalline peaks to the total area in WAXS patterns (Supplementary Figs. 3–5 and Supplementary Discussion on % crystallinity calculations). In both cases, metastable crystalline intermediates signified by broad peaks were observed at early time points giving way to sharper crystalline peaks at later time points (Fig. 2). For PLLA45-b-PEG45 (10% solids w/w 9), the PLLA crystallinity increased rapidly during the first three hours to 51% crystalline and then increased slowly, to a maximum of approximately 81% crystalline around 24 h. For PLLA90-b-PEG45 (10% solids w/w 15), the crystallinity increased at a faster rate, reaching 48% crystallinity within the first h and then increased slowly to 63% at 24 h. The WAXS data is consistent with the development of the α crystalline form of PLLA, which is considered the more thermodynamically stable polymorph (Fig. 2a, b)49,50,51. To compare BCP crystallization to homopolymer crystallization, two PLLA samples with DP = 45 and DP = 90 were crystallized in toluene, which achieved 87 and 86% crystallinity, respectively. This indicates that the presence of the PEG block could inhibit crystallization for longer PLLA blocks. The FTIR spectra show the carbonyl-stretch at 1749 cm−1 broadening to form two peaks (1749 cm−1 and 1754 cm−1), which is also consistent with the crystallization of PLLA (Fig. 2c, d)49,50,52. Peak ratios of 1754 cm−1 to 1749 cm−1 indicate significant changes in the carbonyl environment occurring in the early time points in PLLA45-b-PEG45 (10% solids w/w 9) and PLLA90-b-PEG45 (10% solids w/w 15), supporting the WAXS data.

a, b WAXS pattern for PLLA45-b-PEG45 (10% solids w/w 9) (a) and PLLA90-b-PEG45 (10% solids w/w 15) (b) over time and c, d FTIR spectra of the carbonyl shift for 9 (c) and 15 (d). Note for (a), from 24 h to 168 h, no significant difference is observed. For (b), from 3 h to 24 h, no significant difference is observed. Note that for (a), 5 min and 1 h, and 2b, 5 min show metastable crystalline intermediates. The data show a development in the crystallinity over time indicating that the self-assembly process measured in Fig. 1 is related to the crystallization of the PLLA core-forming block.
Self-assembly mechanism
To monitor the morphological evolution, the freeze-dried samples were re-dispersed in water (a non-solvent for PLLA) at 0.5% solids w/w and analyzed by cryo-TEM. Previous examples of CDSA of PLLA-based BCPs in water demonstrate that for self-assembly to occur, solutions must be heated in water (>65 °C) and cooled20,21. Thus, at room temperature, we expect that PLLA-b-PEG nanoparticles are stable in water. Furthermore, it is well known that crystallinity and nanostructure of PLLA-based BCP are stable to dehydration and resuspension53.To confirm that the BCP structures formed in toluene are stable to freeze drying and re-dispersion, several control experiments were performed (Supplementary Figs. 6–11 and Supplementary Discussion on controls). All of these control experiments showed the structures to be stable. Additionally, cryo-TEM analysis of the ROPI-CDSA structures which had been in water several weeks after resuspension, and water resuspensions from dehydrated BCPs aged several months showed stable structures. This indicates that the BCPs form stable kinetically-trapped structures, consistent with previous literature of PLLA-based BCPs in water (Supplementary Fig. 12)19,20,22. This also demonstrates that ROPI-CDSA can be used to produce a range of hierarchal PLLA-b-PEG structures by kinetically trapping in water at various time points during the self-assembly process.
The cryo-TEM data for PLLA45-b-PEG45 (10% solids w/w 9, Fig. 3a–c) shows that the assembly process proceeds via the formation of spheres (t = 1 h diameter = 14.4 ± 2.6 nm Supplementary Fig. 13), that evolve into rods (t = 3 h diameter = 15.8 ± 2.3 nm and length = 56.1 ± 42.4 nm Supplementary Figs. 14 and 15), which appear to aggregate and form lamellae (t = 24 h thickness = 24.7 ± 2.2 nm length = 3212 ± 927 nm width = 859 ± 308 nm Supplementary Fig. 16). For most time-points, the samples showed a mixture of structures (e.g., spheres and rods or rods and lamellae), which is consistent with a kinetically controlled process54,55. On several occasions the rods appear to be aligned (Fig. 3b inset) and in some cases they appear to be connected by long fibers (diameter = 10 nm, length > 1 μm, Supplementary Fig. 17). The distinction between nanorods and nanofibers is typically related to their aspect ratios, where nanorods typically have ratios of 3–15 and fibers typically are >> 1556. Here, the fibers appear to act as nucleation sites for the spheres and rods, directing their formation. The cryo-TEM data for sample PLLA90-b-PEG45 (10% solids w/w 15, Fig. 3d–f) shows a different formation pathway. The initial structures appear to be poorly-defined rod-like precursors (diameter = 29 ± 12 nm length = 90 ± 27 nm Supplementary Fig. 18) which evolve into lamellae (width = 360 ± 250 nm length = 1270 ± 910 nm thickness = 22 ± 2 nm Supplementary Fig. 19) with a second population of rod-like structures (diameter = 22 ± 6 nm length = 101 ± 35 nm Supplementary Fig. 20). After 6 h, the rod-like structures are no longer present, with only lamellae (width = 360 ± 230 nm length = 1300 ± 700 nm thickness = 23 ± 2 nm Supplementary Fig. 21) and 3D lamellae stacks present, confirmed through a tilt series (Supplementary Fig. 22).

a–c PLLA45-b-PEG45 (10% solids w/w 9), a = 1 h, b = 6 h, c = 24 h. d–f PLLA90-b-PEG45 (10% solids w/w 15), d = 5 min, e = 3 h, f = 6 h. The data show two distinct assembly mechanisms that appear to be a result of unimer addition (sample 9) and particle aggregation (sample 15), resulting in lamellae with different morphologies.
Phase diagram
The above data show that the structure of the PLLA-b-PEG assemblies varies as a function of both molecular structure and time post polymerization. To construct a phase diagram and verify the reproducibility of ROPI-CDSA11,14,26, samples were left for several days post polymerization. However, it is important to note that each sample will likely have its own self-assembly pathway to reach these relaxed, meta-stable structures. Representative cryo-TEM and SEM images of various morphologies are shown in Fig. 4. The samples from Table 1 are plotted as a function of PLLA DP and % solids w/w and constitute a phase diagram shown in Fig. 5. Samples 7 and 8 (PLLA45-b-PEG45 5% and 7.5% solids w/w, respectively, Fig. 4a, b) formed short rods (width 20–30 nm, length 20–70 nm), sample 5 (PLLA25-b-PEG45 10% solids w/w) (Fig. 4c) formed fibers (diameter ≈ 10 nm, length > 1 micron) similar to the structure found in Fig. 3b and Supplementary Fig. 17, samples 6 (PLLA25-b-PEG45 20% solids w/w), 9–10 (PLLA45-b-PEG45 10 and 20% solids w/w, respectively), 11 (PLLA60-b-PEG45 10% solids w/w) and (Fig. 4d–f) formed a mixture of rods (width 20–30 nm length 100–1000 nm) and lamellae 12 (PLLA75-b-PEG45 10% solids w/w) (width 200–1000 nm length 500 nm to 4 μm), and samples 13 (PLLA90-b-PEG45 5% solids w/w) and 14 (PLLA90-b-PEG45 7.5% solids w/w) (Fig. 4g, h) formed lamellae (width 100–1000 nm length 200 nm to 3.5 μm). Samples 15–16 (PLLA90-b-PEG45 10 and 20% solids w/w respectively), 17 (PLLA120-b-PEG45 7.5% solids w/w), and 18–19 (PLLA135-b-PEG45 10 and 20% solids w/w, respectively) (Fig. 4i–l) formed organogels in the toluene solution as confirmed by oscillatory rheology (Supplementary Fig. 23) which indicated that a 3D network formed in solution57. Stacks or aggregates of lamellae were observed by cryo-TEM (Fig. 4i, j). Freeze drying of the samples and analyzing the bulk powders by SEM revealed the formation of a 3D porous material. Here individual lamellae are not clearly discernable, although it appears that the materials are likely composed of individual lamellae measuring ~5 μm in length The SEM images are in good agreement with cryo-TEM images collected from the gels, both showing the presence of lamellae and an abundance of 3D stacked lamellae with lengths of 500 nm to 5 μm.

Selected cryo-TEM (a–j) and SEM (k–l) images for representative morphologies of the phase diagram, a–c is for 1D systems, d–h are for 2D systems, i–l are for 3D systems. Images are from the following samples: a, b: 7, c: 5, d, e: 9, f: 6, g, h: 13, i: 16, j: 19, k, l: 17. (See Table 1 for a sample guide). The data show that ROPI-CDSA can be used to form block copolymer materials with a wide range of morphologies, dimensions, and length scales.

The phase diagram shows hierarchical ordering for samples 1–19 (Table 1). No SA refers to no self-assembly observed. The phase diagram shows a clear trend where higher dimensional structures are favored at higher solids content and higher PLLA DPs.
Discussion
This work tracks the evolution of PLLA-b-PEG morphologies immediately following polymerization, revealing a hierarchical evolution from 0D spheres to 1D rods and fibers to 2D lamellae and, in select cases, to 3D porous networks consisting of stacked or aggregated lamellae. Lamellae are likely favored as polymer crystallinity tends to promote morphologies with low curvature17,18, while stacking can minimize surface energies. Comparison of the kinetic data obtained from UV-Vis, WAXS and FTIR shows that crystallization is coincident with self-assembly for both PLLA45-b-PEG45 (10% solids w/w 9) and PLLA90-b-PEG45 (10% solids w/w 15), despite the kinetics for both samples being significantly different (Fig. 6). This suggests that the self-assembly is driven by the crystallization of the PLLA block; although, our control experiments show that polymerization is promoted by the presence of the PEG initiator. Consequently, we propose that the assembly process is driven by both the amphiphilicity of the BCP, and the crystallization of the PLLA block24. The data showed that the length of the PLLA block strongly influences the self-assembly kinetics and morphology, with longer blocks favoring faster self-assembly (Figs. 1 and 6), and the formation of higher dimensional structures (Fig. 5). This kinetic observation is consistent with previous reports on the CDSA of poly(L-lactide)-block-poly(N,N-dimethylacrylamide) (PLLA-b-PDMA) diblock copolymers in alcohols, where the kinetic differences were attributed to the difference in solubility of the BCP chain (i.e., more soluble polymers crystallize more slowly)19,23. However, the morphological results revealed the more solvophobic PLLA-b-PDMA polymers formed lower dimensional structures (1D vs 2D)19,23, in contrast to what we report here. For ROPI-CDSA of PLLA-b-PEG we see two fundamentally different assembly mechanisms for PLLA45-b-PEG45 (10% solids w/w 9) and PLLA90-b-PEG45 (10% solids w/w 15). PLLA45-b-PEG45 (10% solids w/w 9) (more soluble polymer) appears to initially form high aspect ratio fibers that act as nucleation sights for the crystalline rods, directing the growth to occur along the fiber in one dimension. As rod growth occurs during the time period where most of the crystallinity develops, we propose that these rods form by unimer addition from either the solution or by Ostwald ripening of the fibers58. At later time points, these rods aggregate, resulting in 2D growth and the formation of the lamellae (Fig. 7a). PLLA90-b-PEG45 (10% solids w/w 15) (less soluble polymer) initially appears to form irregularly shaped particles that grow through a particle aggregation mechanism. This results in an initial 2D growth, forming lamellae, and later in 3D growth, forming stacked lamellae and the hierarchical porous network (Fig. 7b). The morphological difference between the lamellae formed in PLLA45-b-PEG45 (10% solids w/w 9) and PLLA90-b-PEG45 (10% solids w/w 15) are also consistent with proposed mechanistic differences (Fig. 3). Cryo-TEM images of PLLA60-b-PEG45 (10% solids w/w 11) and PLLA75-b-PEG45 (10% solids w/w 12) show morphologies that are consistent with both mechanisms acting simultaneously. The lamellae structures formed by 11 and 12 have characteristics of both 9, as they are composed of aligned rods, and 15, in that the lamellae are irregularly shaped (Supplementary Fig. 24).

Composite graphs showing the change in turbidity (measured at 600 nm), % crystallinity of PLLA block, and carbonyl peak ratio for a PLLA45-b-PEG45 (10% solids w/w 9) and b PLLA90-b-PEG45 (10% solids w/w 15). The standard error of 3 runs is plotted for turbidity measurements and a calculated error is plotted for crystallinity and carbonyl peak ratio values, see Supplementary information for more details. For the PLLA crystallinity (%) data, early time point metastable crystalline precursors are highlighted with a yellow circle. The data show that the self-assembly kinetics are coincident with the crystallinity kinetics.

a Proposed mechanism for PLLA45-b-PEG45 (10% solids w/w 9), favoring unimer growth. First, fibers form as a template for 1D growth, giving spheres and rods. Later, these rods aggregate to form 2D lamellae. b Proposed mechanism for PLLA90-b-PEG45 (10% solids w/w 15), favoring aggregation. First, ill-defined 2D rod-like structures form and grow via aggregation. Later, 2D lamellae aggregate, giving 3D lamella stacks.
These two different initial growth mechanisms (templated unimer addition and particle aggregation) are consistent with growth models previously reported for small molecules and inorganic crystals58,59, where the observed differences in formation pathways are explained by considering the relative energy barriers for either pathway. In our work, the longer PLLA block reduces the energy barrier for crystallization and raises the energy barrier for unimer exchange. However, one important consideration for macromolecular self-assembly that is not present for small molecules or inorganic crystals is the influence of chain stretching.
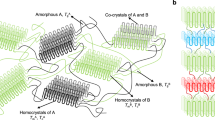
The dimensions of self-assembled BCP structures are determined by the aggregation number and the stretching or coiling of the polymer chains17,60. For spheres, rods, and lamellae, the degree of polymer chain stretching (ω) can be determined by comparing the measured radius (r) (or half the lamella thickness) to the maximum length of the polymer chain (Lmax) (Fig. 8) as shown in Eq. (1).

a When ω = 1 giving a dense corona and b when ω < 0.5 giving significant coiling. In ROPI-CDSA, PLLA45-b-PEG45 (10% solids w/w 9) has a denser corona, in-between ω = 1 and ω < 0.5. In contrast, PLLA90-b-PEG45 (10% solids w/w 15) has a ω < 0.5 exhibiting more coiling.
From distance calculations using CrystalMaker® software, the molecular length of a PLLA monomer is 3.69 Å Supplementary Fig. 25 and Supplementary Methods). The total fully extended PLLA backbone lengths (Lmax) are 16.6 nm and 33.2 nm for PLLA45-b-PEG45 (10% solids w/w 9) and PLLA90-b-PEG45 (10% solids w/w 15), respectively. This would result in a theoretical maximum fiber/rod diameter and/or lamella thickness of 33.2 nm for PLLA45-b-PEG45 and 66.4 nm for PLLA90-b-PEG45. For 9, the fibers have an ω of 0.33 (0.29–0.37); for the rods and lamella, ω changes over time from ω1hr = 0.42 (0.34–0.50) to ω24 hr = 0.74 (0.68–0.81), becoming more stretched out during the self-assembly process. (N.B. lamella interdigitation is ruled out as we observed ω > 0.5). For 15, ω stays relatively constant, at about 0.35 (0.32–0.37). In comparison to PLLA45-b-PEG45 (10% solids w/w 9), PLLA90-b-PEG45 (10% solids w/w 15) has a longer PLLA block and a smaller ω. This results in particles of 15 having a substantially lower corona density than 9. We propose that this promotes particle aggregation events by lowering the barrier to inter-lamellar core-core interactions61. We propose that this also facilitates stacking and the formation of structures in 3D.
An interesting feature of PISA experiments is that the molecular structure of the species assembling changes during the assembly process. As it is well known that the thermodynamics and kinetics of self-assembly change with molecular structure8,62,63, PISA processes occur as a result of the evolving energy landscape. As the initial homopolymer and monomer are soluble, the system is initially ‘in-equilibrium’ (and dissolved). Every addition of monomer to the end of the homopolymer chain creates a building block (the growing block copolymer) that is higher in free energy (due to its increasingly amphiphilic nature). The majority of PISA experiments can be categorized as either thermodynamically-controlled14,26,64 or kinetically-trapped2,26,27,34,64,65,66, which differ based on the type of evolving energy landscape. In the thermodynamically controlled systems, the energy landscape is relatively smooth compared to kBT, resulting in very short relaxation times. With each monomer addition, the landscape changes and the system relaxes to its thermodynamic minimum. Consequently, the system evolves down a thermodynamic pathway and the design of these systems is based on thermodynamic considerations28. In the kinetically-trapped systems, the landscape evolves such that it becomes rough compared to kBT, at which point the system gets trapped in its configuration29,34. In this system, the polymer evolves thermodynamically until the morphology gets locked in place by the changing energy landscape. The key feature of both these types of PISA processes (thermodynamically and kinetically controlled) is that the relaxation time is much faster than the polymerization time67. Consequently, after each monomer addition, the system relaxes to a lower energy state and the overall reaction coordinate proceeds downhill (Fig. 9) and the assembly is finished once the polymerization finishes. In the system described here, the roughness of the energy landscape is on the order of kBT. The consequence of this is that the relaxation times (driven by the crystallization kinetics) are long (hours to days). As the polymerization times are short (seconds to minutes), this creates a situation where with each monomer addition, the system becomes increasingly further from its minimum free energy organization as the system does not have time to relax to a lower energy state (Fig. 9). Therefore, we propose that during ring-opening polymerization, it is the release of the lactide ring strain, which drives a non-equilibrium self-assembly evolution (See Supplementary Discussion on non-equilibrium assembly)55,68. Although not all of the chemical energy stored in the monomer will be converted into free energy to drive self-assembly (some being lost as heat energy), as the growing polymer chain is unstable, it has a higher free energy than the soluble homopolymer. Interestingly, the ring strain release during lactide polymerization (−23 kJ/mol)69, is similar to the energy stored in GDP-rich microtubules, a classic out-of-equilibrium biological system (−22 kJ/mol)70,71. Microtubule assembly is a classic dissipative assembly process. The dissipative nature of the assembly is related to the reversibility of the chemistry, which is common in out-of-equilibrium systems71,72,73. In the presented PLLA-b-PEG systems, while the self-assembly is initiated through a modification of the molecular structure, the chemistry is non-reversible, which is advantageous for using the process to trap the meta-stable structures. Here, this is achieved by re-dispersing the structures in water, where the energy landscape (at room temperature) is rough compared to kBT. Consequently, we can divide the process into two stages. Stage 1 is the non-equilibrium assembly process that occurs during polymerization (Fig. 9, stage 1). Stage 2 is the relaxation process that occurs as the high energy structures relax to a lower energy configuration (Fig. 9, stage 2). In this paper, we have only investigated the relaxation process; however, our control experiments show that the presence of the PEG block is essential for the polymerization to occur efficiently, indicating that some assembly is taking place during stage 1. This is justified from the evidence that kinetically-trapped BCPs structures, are highly dependent on the assembly mechanisms61,74,75,76. Furthermore, comparing kinetically-trapped structures in water using the same BCP but different methods (solvent switch and CDSA, see also Supplementary Figs. 7–10) shows that ROPI-CDSA forms by a different pathway. Our hypothesis is that this pathway is dependent on the non-equilibrium stage of the assembly process (Fig. 9, stage 1), and can provide access to meta-stable precursors that do not occur during the other assembly methods. Our data show two mechanisms by which meta-stable precursors can direct self-assembly. First, the formation of the highly coiled amorphous fibers which directs the location and dimensionality of the crystallization process. Second, the formation of precursor particles with low corona densities which facilitates 2D and 3D growth through particle aggregation. Although more work is needed to investigate the non-equilibrium assembly process, the prospect of controlling PISA assembly mechanisms by changing the relative rates of polymerization and self-assembly provides an exciting opportunity to create polymeric materials based on kinetic considerations (rather than thermodynamic considerations). For example, Khor et al.77. demonstrated that changing the rate of polymerization in emulsion RAFT PISA experiments resulted different morphological outcomes. Specifically, that a faster rate of polymerization provides access to polymer vesicles instead of spheres (achieved at slower rates of polymerization). Although not discussed in the text, a plausible explanation for this is that the faster rate of polymerization provides access to a non-equilibrium state that enables relaxation to vesicles. This study is an important example which, in addition to the work presented here, highlights that further study of the non-equilibrium behavior in PISA experiment can provide access to unique assembly processes and potentially to unique structures.

The dashed lines represent the situation where the relaxation time is short compared to the polymerization time. Consequently, with each monomer addition, the growing block copolymer relaxes to a lower energy configuration. The result is that the reaction proceeds downhill and finishes when the polymerization terminates. The solid line represents the situation where the polymerization time is much shorter than the relaxation time. Consequently, with each monomer addition, the growing block copolymer cannot fully relax to a lower energy configuration. The result is that the reaction initially proceeds uphill (Stage 1) and then relaxes post polymerization (Stage 2). In Stage 2, the energy barriers represent morphological transitions (e.g., spheres to rods or rods to lamellae).
In conclusion, ROP of L-lactide in toluene, using a monofunctionalized PEG initiator, and TBD as a catalyst, results in the polymerization-induced crystallization-driven self-assembly of PLLA-b-PEG block copolymers. This method, termed ROPI-CDSA, can produce 1D, 2D, and 3D structures with concentrations ranging from 5 to 20% solids w/w. As the rates of the polymerization are faster than the rates of self-assembly and crystallization, the initial structures generated post polymerization are very far from equilibrium. Over time, driven by crystallization and solvent compatibility, the system relaxes to a lower energy configuration. The hierarchical growth is controlled by three processes operating on different time-scales, polymerization, amphiphilic self-assembly, and crystallization. Polymerization creates an amphiphile which can self-assemble by microphase separation, and a polymer capable of undergoing crystallization-driven self-assembly. The interplay between these three processes is complex but provides a rich design space to create block hierarchical copolymer materials. The growth mechanism can occur via a predominantly unimer addition or particle aggregation mechanism. Understanding of the mechanism and kinetics of the relaxation process allows a range of meta-stable structures to be trapped by freeze drying, from a single BCP. The freeze-dried structures can then be stored and redispersed in water as required. This work provides a basis for the use of ROPI-CDSA to generate hierarchical 1D, 2D, and 3D materials using biocompatible and biodegradable polymers with a scalable one pot strategy. It additionally outlines an approach for utilizing PISA as a method to create non-equilibrium self-assembly processes and trap unique meta-stable structures.
Methods
Materials
mPEG45 (MW = 2000) (Sigma Aldrich) was azeotropically distilled ×2 in toluene and high-vacuumed overnight. L-lactide (TCI) was recrystallized in toluene ×3. Anhydrous toluene (99.8%), and triazabicyclodecene (TBD) were purchased from Sigma Aldrich and were used without further purification. Benzoic acid (Fisher Chemical) was used without further purification. Milli-Q water (ρ > 18 MΩ cm) was used as the solvent for all aqueous solutions. Chemicals were stored in a dry-N2 atmosphere glove box. Reactions were performed in a N2 glove box.
PLLA-b-PEG synthesis and self-assembly
Procedure adapted from Waymouth et al.43 L-lactide (64.9 mg, 0.45 mmol, PLLA target DP = 45) was added to a solution of mPEG45 (40 mg, 20 μmol) in 1.08 mL of toluene (10% solids w/w). 15 μL (0.1% mol) TBD from a toluene stock solution (4.3 mg/mL) was then added. The solution was stirred for 90 s and subsequently quenched with 0.05 mL benzoic acid stock solution (100 mg/mL). We noticed that the rate of stirring influenced the self-assembly kinetics and therefore stirring was kept at 400 rpm for reproducibility (For full results see also Supplementary Table 1, for additional synthetic information see Supplementary Table 2). 1H NMR (500 MHz, CDCl3) δ 5.16 (q, J = 7.0 Hz, CH, PLLA backbone), 5.03 (q, J = 6.7 Hz, CH L-lactide), 3.72–3.59 (m, CH2 PEG backbone), 3.54 (dd, J = 5.6, 3.6 Hz, CH2, PEG), 3.37 (s, 3H, terminal CH3 PEG), 1.67 (dd, J = 6.7, 1.5 Hz, CH3 L-lactide), 1.58 (d, J = 6.7 Hz, CH3 PLLA backbone), 1.50 (dd, J = 14.7, 7.0 Hz, terminal CH3 PLLA) (see also Supplementary Fig. 1).
Preparation methods of aqueous solutions
Lyophilized powders were obtained by freezing the toluene solutions in a round bottom flask with liquid nitrogen followed by sublimation using a vacuum pump. Resuspension of the powders was aided by sonication for 30 min in a Branson 3800 Ultrasonic Cleaner. Solvent extractions were performed by dropping a few (5–10) μL of toluene solution into excess water and vortexing for 10 s. In both cases aqueous solutions with a concentration of 0.5 mg/mL (0.5% solids w/w) were obtained and cryo-TEM analysis showed similar results for both preparations.
Structural characterization
Proton nuclear magnetic resonance (1H NMR) spectra were collected on a 500 MHz Bruker Avance spectrometer in CDCl3. Chemical shifts are given in ppm, calibrated from residual CHCl3. Gel permeation chromatography (GPC) was performed in DMF using an Agilent 1100 chromatograph equipped with RID detector and a PL gel 5 μm 300 × 7.5 mm mixed column. Samples were calibrated against polystyrene standards (see also Supplementary Fig. 2 for GPC traces).
Turbidity measurements
Self-assembly kinetics were measured with UV/Vis spectroscopy on a Thermo Scientific NanoDrop 2000c. Changes in turbidity were measured at 600 nm every 15 s for 720 min with a moderate stirring rate. Triplicate runs were taken of each sample (Supplementary Fig. 26). Plots shown in paper are binned by a factor of 20.
Crystallization measurements
Wide-angle X-ray scattering (WAXS) patterns were measured on a Rigaku Smart lab X-ray diffractometer in Bragg-Brentano diffraction mode utilizing X-rays generated at 40 kV and 44 mA with Cu Kα irradiation (step size 0.2 deg, speed 1.0, IS 2/3 deg, RS1 2/3 deg, RS2 0.3 mm). Approximately 20 mg of a lyophilized sample was used in measurements. PLLA crystallinity was originally calculated using Smart Lab (Rigaku) software (see also Supplementary Fig. 4 and Supplementary Tables 4 and 5), but due to the possibility of overlapping PEG peaks, a deconvolution routine was employed to separate PEG and PLLA peaks (see also Supplementary Fig. 5 and Supplementary Tables 6 and 7). Using Smart Lab software, following background correction, amorphous peak and crystalline peak areas were defined using the previous peak assignment of PLLA-b-PEG51,78. Additional crystallinity analysis was conducted using a custom peak fitting python script. The area of interest was restricted between 10 and 30° and a constant background was subtracted. After which a model function was created as the sum of a mixture of Voigt and Gaussian components. The model was subsequently fitted to the experimental data by optimizing the individual component parameters (peak center, amplitude, sigma, and gamma). The quality of the fit was assessed by measuring the mean square error between the model and the experimental data. The parameters and script used to fit the data is discussed further in the SI. In both cases, crystallinity was calculated from the area of crystalline peaks as a percentage of the total peak area (For more details on peak assignment see SI). The percentage of crystallinity was normalized by the mass ratio of the PLLA segment. Fourier transform infrared (FTIR) absorbance spectra were collected on a Jasco 4700 FTIR from lyophilized samples (Supplementary Figs. 27 and 28).
Cryogenic-transmission electron microscopy (cryo-TEM)
Cryo-TEM samples were prepared from resuspended or extracted solutions onto Quantifoil R2/2 (Electron Microscopy Sciences) grids. Grids were glow discharged for 70 s to increase hydrophilicity prior to sample loading. Vitrification was carried out by an Automatic Plunge Freezer ME GP2 (Leica Microsystems) with 3 μL of sample. Grid preparation was performed at 95% humidity and the grids were blotted for 3 s prior to plunging into liquid propane. Cryo-TEM samples were then placed on a Gatan Cryo-TEM holder and imaged on a JEOL 2100F TEM using a Schottky type field emission gun operating at 200 keV. Images were recorded using DigitalMicrograph (Gatan) software with a Gatan OneView CMOS camera at 4k × 4k resolution.
Scanning electron microscopy (SEM)
Samples were prepared from lyophilized samples which were freeze-cracked in liquid N2 and coated ex-situ with 3 nm of iridium (Quorum, Q150T Plus). Secondary electron images were collected on a FEI, Quanta 3D FEG with Everhart-Thornley detector, using a 5 kV acceleration potential, and a probe current of 200 pA.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
The code for deconvoluting the WAXS data is available from the corresponding author on reasonable request.
References
Khor, S. Y., Quinn, J. F., Whittaker, M. R., Truong, N. P. & Davis, T. P. Controlling nanomaterial size and shape for biomedical applications via polymerization-induced self-assembly. Macromol. Rapid Commun. 40, e1800438 (2019).
Le, D., Keller, D. & Delaittre, G. Reactive and functional nanoobjects by polymerization-induced self-assembly. Macromol. Rapid Commun. 40, 1800551 (2019).
Gerbelli, B. B. et al. Hierarchical self-assembly of peptides and its applications in bionanotechnology. Macromol. Chem. Phys. 220, 1900085 (2019).
Qiu, H., Hudson, Z. M., Winnik, M. A. & Manners, I. Multidimensional hierarchical self-assembly of amphiphilic cylindrical block comicelles. Science 347, 1329 (2015).
Lohr, A., Lysetska, M. & Würthner, F. Supramolecular stereomutation in kinetic and thermodynamic self-assembly of helical merocyanine dye nanorods. Angew. Chem. Int. Ed. 44, 5071–5074 (2005).
Wang, J., Liu, K., Xing, R. & Yan, X. Peptide self-assembly: thermodynamics and kinetics. Chem. Soc. Rev. 45, 5589–5604 (2016).
van Rijt, M. M. J. et al. Designing stable, hierarchical peptide fibers from block co-polypeptide sequences. Chem. Sci. 10, 9001–9008 (2019).
Nicolai, T., Colombani, O. & Chassenieux, C. Dynamic polymeric micelles versus frozen nanoparticles formed by block copolymers. Soft Matter 6, 3111–3118 (2010).
Tanoue, R. et al. Thermodynamically controlled self-assembly of covalent nanoarchitectures in aqueous solution. Acs Nano 5, 3923–3929 (2011).
He, X. et al. Complex and hierarchical 2D assemblies via crystallization-driven self-assembly of poly(l-lactide) homopolymers with charged termini. J. Am. Chem. Soc. 139, 9221–9228 (2017).
Blanazs, A., Ryan, A. J. & Armes, S. P. Predictive phase diagrams for RAFT aqueous dispersion polymerization: effect of block copolymer composition, molecular weight, and copolymer concentration. Macromolecules 45, 5099–5107 (2012).
Yeow, J. & Boyer, C. Photoinitiated polymerization-induced self-assembly (photo-PISA): new insights and opportunities. Adv. Sci. 4, 1700137 (2017).
Wright, D. B., Touve, M. A., Adamiak, L. & Gianneschi, N. C. ROMPISA: ring-opening metathesis polymerization-induced self-assembly. ACS Macro Lett. 6, 925–929 (2017).
Canning, S. L., Smith, G. N. & Armes, S. P. A critical appraisal of RAFT-mediated polymerization-induced self-assembly. Macromolecules 49, 1985–2001 (2016).
Boott, C. E., Gwyther, J., Harniman, R. L., Hayward, D. W. & Manners, I. Scalable and uniform 1D nanoparticles by synchronous polymerization, crystallization and self-assembly. Nat. Chem. 9, 785–792 (2017).
Oliver, A. M. et al. Scalable fiber-like micelles and block co-micelles by polymerization-induced crystallization-driven self-assembly. J. Am. Chem. Soc. 140, 18104–18114 (2018).
Vilgis, T. & Halperin, A. Aggregation of coil-crystalline block copolymers: equilibrium crystallization. Macromolecules 24, 2090–2095 (1991).
He, W.-N. & Xu, J.-T. Crystallization assisted self-assembly of semicrystalline block copolymers. Prog. Polym. Sci. 37, 1350–1400 (2012).
Inam, M. et al. 1D vs. 2D shape selectivity in the crystallization-driven self-assembly of polylactide block copolymers. Chem. Sci. 8, 4223–4230 (2017).
Petzetakis, N., Dove, A. P. & O’Reilly, R. K. Cylindrical micelles from the living crystallization-driven self-assembly of poly(lactide)-containing block copolymers. Chem. Sci. 2, 955–960 (2011).
Petzetakis, N., Walker, D., Dove, A. P. & O’Reilly, R. K. Crystallization-driven sphere-to-rod transition of poly(lactide)-b-poly(acrylic acid) diblock copolymers: mechanism and kinetics. Soft Matter 8, 7408–7414 (2012).
Pitto-Barry, A., Kirby, N., Dove, A. P. & O’Reilly, R. K. Expanding the scope of the crystallization-driven self-assembly of polylactide-containing polymers. Polym. Chem. 5, 1427–1436 (2014).
Yu, W., Inam, M., Jones, J. R., Dove, A. P. & O’Reilly, R. K. Understanding the CDSA of poly(lactide) containing triblock copolymers. Polym. Chem. 8, 5504–5512 (2017).
Ganda, S. & Stenzel, M. H. Concepts, fabrication methods and applications of living crystallization-driven self-assembly of block copolymers. Prog. Polym. Sci. 101, 101195 (2020).
Charleux, B., Delaittre, G., Rieger, J. & D’Agosto, F. Polymerization-induced self-assembly: from soluble macromolecules to block copolymer nano-objects in one step. Macromolecules 45, 6753–6765 (2012).
Warren, N. J. & Armes, S. P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 136, 10174–10185 (2014).
Sun, J.-T., Hong, C.-Y. & Pan, C.-Y. Formation of the block copolymer aggregates via polymerization-induced self-assembly and reorganization. Soft Matter 8, 7753–7767 (2012).
Penfold, N. J. W., Yeow, J., Boyer, C. & Armes, S. P. Emerging trends in polymerization-induced self-assembly. Acs Macro Lett. 8, 1029–1054 (2019).
Varlas, S., Foster, J. C. & O’Reilly, R. K. Ring-opening metathesis polymerization-induced self-assembly (ROMPISA). Chem. Commun. 55, 9066–9071 (2019).
D’Agosto, F., Rieger, J. & Lansalot, M. RAFT-mediated polymerization-induced self-assembly. Angewandte Chemie International Edition, https://doi.org/10.1002/anie.201911758 (2020).
Parkatzidis, K., Wang, H. S., Truong, N. P. & Anastasaki, A. Recent developments and future challenges in controlled radical polymerization: A 2020. Update Chem. 6, 1575–1588 (2020).
Figg, C. A. et al. Polymerization-induced thermal self-assembly (PITSA). Chem. Sci. 6, 1230–1236 (2015).
Fu, C., Xu, J. & Boyer, C. Photoacid-mediated ring opening polymerization driven by visible light. Chem. Commun. 52, 7126–7129 (2016).
Cunningham, V. J. et al. Poly(glycerol monomethacrylate)–poly(benzyl methacrylate) diblock copolymer nanoparticles via RAFT emulsion polymerization: synthesis, characterization, and interfacial activity. Macromolecules 47, 5613–5623 (2014).
Tanaka, H. et al. In situ and real-time small-angle neutron scattering studies of living anionic polymerization process and polymerization-induced self-assembly of block copolymers. Phys. B: Condens. Matter 385-386, 742–744 (2006).
Guégain, E., Zhu, C., Giovanardi, E. & Nicolas, J. Radical ring-opening copolymerization-induced self-assembly (rROPISA). Macromolecules 52, 3612–3624 (2019).
Grazon, C. et al. Aqueous ring-opening polymerization-induced self-assembly (ROPISA) of N-carboxyanhydrides. Angew. Chem. Int. Ed. 59, 622–626 (2020).
Sha, Y. et al. ROMPI-CDSA: ring-opening metathesis polymerization-induced crystallization-driven self-assembly of metallo-block copolymers. Chem. Sci. 10, 9782–9787 (2019).
Dove, A. P. Organic catalysis for ring-opening polymerization. ACS Macro Lett. 1, 1409–1412 (2012).
Bobo, D., Robinson, K. J., Islam, J., Thurecht, K. J. & Corrie, S. R. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res. 33, 2373–2387 (2016).
He, X. et al. Poly(ethylene glycol)-block-poly(ε-caprolactone)-and phospholipid-based stealth nanoparticles with enhanced therapeutic efficacy on murine breast cancer by improved intracellular drug delivery. Int J. Nanomed. 10, 1791–1804 (2015).
Wang, J. et al. Poly(ethylene glycol)-polylactide micelles for cancer therapy. Front Pharm. 9, 202–202 (2018).
Lohmeijer, B. G. G. et al. Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters. Macromolecules 39, 8574–8583 (2006).
Kamber, N. E. et al. Organocatalytic ring-opening polymerization. Chem. Rev. 107, 5813–5840 (2007).
Sato, S., Gondo, D., Wada, T., Kanehashi, S. & Nagai, K. Effects of various liquid organic solvents on solvent-induced crystallization of amorphous poly(lactic acid) film. J. Appl Polym. Sci. 129, 1607–1617 (2013).
Chuma, A. et al. The reaction mechanism for the organocatalytic ring-opening polymerization of L-lactide using a guanidine-based catalyst: Hydrogen-bonded or covalently bound? J. Am. Chem. Soc. 130, 6749–6754 (2008).
Pratt, R. C., Lohmeijer, B. G., Long, D. A., Waymouth, R. M. & Hedrick, J. L. Triazabicyclodecene: a simple bifunctional organocatalyst for acyl transfer and ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 128, 4556–4557 (2006).
Pratt, R. C. et al. Exploration, optimization, and application of supramolecular thiourea−amine catalysts for the synthesis of lactide (co)polymers. Macromolecules 39, 7863–7871 (2006).
Pan, P. & Inoue, Y. Polymorphism and isomorphism in biodegradable polyesters. Prog. Polym. Sci. 34, 605–640 (2009).
Saeidlou, S., Huneault, M. A., Li, H. & Park, C. B. Poly(lactic acid) crystallization. Prog. Polym. Sci. 37, 1657–1677 (2012).
Kricheldorf, H. R. & Meier-Haack, J. Polylactones, 22 ABA triblock copolymers of L-lactide and poly(ethylene glycol). Die Makromol. Chem. 194, 715–725 (1993).
Lan, Q. & Li, Y. Mesophase-mediated crystallization of poly(l-lactide): deterministic pathways to nanostructured morphology and superstructure control. Macromolecules 49, 7387–7399 (2016).
Petzetakis, N. et al. Hollow block copolymer nanoparticles through a spontaneous one-step structural reorganization. Acs Nano 7, 1120–1128 (2013).
Exploring the energy landscape. Nat. Nanotechnol. 10, 101, https://doi.org/10.1038/nnano.2015.20 (2015).
Mattia, E. & Otto, S. Supramolecular systems chemistry. Nat. Nanotechnol. 10, 111–119 (2015).
Yu, H.-Y. et al. From cellulose nanospheres, nanorods to nanofibers: various aspect ratio induced nucleation/reinforcing effects on polylactic acid for robust-barrier food packaging. ACS Appl. Mater. Interfaces 9, 43920–43938 (2017).
Suzuki, M. & Hanabusa, K. Polymer organogelators that make supramolecular organogels through physical cross-linking and self-assembly. Chem. Soc. Rev. 39, 455–463 (2010).
De Yoreo, J. J. et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 349, aaa6760 (2015).
De Yoreo, J. J. & Vekilov, P. G. Principles of crystal nucleation and growth. Rev. Mineral. Geochem. 54, 57–93 (2003).
Patterson, J. P., Robin, M. P., Chassenieux, C., Colombani, O. & O’Reilly, R. K. The analysis of solution self-assembled polymeric nanomaterials. Chem. Soc. Rev. 43, 2412–2425 (2014).
Parent, L. R. et al. Directly observing micelle fusion and growth in solution by liquid-cell transmission electron microscopy. J. Am. Chem. Soc. 139, 17140–17151 (2017).
Wright, D. B. et al. Blending block copolymer micelles in solution; obstacles of blending. Polym. Chem. 7, 1577–1583 (2016).
Choi, S.-H., Lodge, T. P. & Bates, F. S. Mechanism of molecular exchange in diblock copolymer micelles: hypersensitivity to core chain length. Phys. Rev. Lett. 104, 047802 (2010).
Zhou, W., Qu, Q., Xu, Y. & An, Z. Aqueous polymerization-induced self-assembly for the synthesis of ketone-functionalized nano-objects with low polydispersity. ACS Macro Lett. 4, 495–499 (2015).
Lowe, A. B. RAFT alcoholic dispersion polymerization with polymerization-induced self-assembly. Polymer 106, 161–181 (2016).
Truong, N. P., Dussert, M. V., Whittaker, M. R., Quinn, J. F. & Davis, T. P. Rapid synthesis of ultrahigh molecular weight and low polydispersity polystyrene diblock copolymers by RAFT-mediated emulsion polymerization. Polym. Chem. 6, 3865–3874 (2015).
Takahashi, R. et al. Unraveling the kinetics of the structural development during polymerization-induced self-assembly: decoupling the polymerization and the micelle structure. Polym. Chem. 11, 1514–1524 (2020).
Marolf, D. M. & Jones, M. R. Measurement challenges in dynamic and nonequilibrium nanoscale systems. Anal. Chem. https://doi.org/10.1021/acs.analchem.9b02702 (2019).
Duda, A. & Penczek, S. Thermodynamics of L-lactide polymerization. Equilib. monomer concentration. Macromolecules 23, 1636–1639 (1990).
Desai, A. & Mitchison, T. J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 13, 83–117 (1997).
Ragazzon, G. & Prins, L. J. Energy consumption in chemical fuel-driven self-assembly. Nat. Nanotechnol. 13, 882–889 (2018).
Tena‐Solsona, M. & Boekhoven, J. Dissipative self‐assembly of peptides. Isr. J. Chem. https://doi.org/10.1002/ijch.201900018 (2019).
van Rossum, S. A. P., Tena-Solsona, M., van Esch, J. H., Eelkema, R. & Boekhoven, J. Dissipative out-of-equilibrium assembly of man-made supramolecular materials. Chem. Soc. Rev. 46, 5519–5535 (2017).
Ianiro, A. et al. Liquid–liquid phase separation during amphiphilic self-assembly. Nat. Chem. 11, 320–328 (2019).
Cui, H., Chen, Z., Zhong, S., Wooley, K. L. & Pochan, D. J. Block copolymer assembly via kinetic control. Science 317, 647 (2007).
Yan, Y., Huang, J. & Tang, B. Z. Kinetic trapping—a strategy for directing the self-assembly of unique functional nanostructures. Chem. Commun. 52, 11870–11884 (2016).
Khor, S. Y., Truong, N. P., Quinn, J. F., Whittaker, M. R. & Davis, T. P. Polymerization-induced self-assembly: the effect of end group and initiator concentration on morphology of nanoparticles prepared via RAFT aqueous emulsion polymerization. ACS Macro Lett. 6, 1013–1019 (2017).
Aldén, M., Lydén, M. & Tegenfeldt, J. Effect of counterions on the interactions in solid dispersions between polyethylene glycol, griseofulvin and alkali dodecyl sulphates. Int. J. Pharmaceutics 110, 267–276 (1994).
Acknowledgements
Cryo-TEM, SEM, and WAXS data were acquired using the instrumentation at the IMRI (ps.uci.edu/imri/) facilities at UCI. FTIR data were acquired using the UCI laser spectroscopy lab (chem.uci.edu/~dmitryf/index.html). Valuable assistance with some data processing from Jovany Merham is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
P.J.H. designed and performed the synthetic procedures, collected the cryoTEM, FTIR, WAXS, UV-Vis data, and wrote the paper. A.M.R. collected the SEM data and processed the WAXS data. J.P.P conceived the project and supervised the study. All authors discussed the results and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hurst, P.J., Rakowski, A.M. & Patterson, J.P. Ring-opening polymerization-induced crystallization-driven self-assembly of poly-L-lactide-block-polyethylene glycol block copolymers (ROPI-CDSA). Nat Commun 11, 4690 (2020). https://doi.org/10.1038/s41467-020-18460-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-18460-2
This article is cited by
-
Non-classical crystallization in soft and organic materials
Nature Reviews Materials (2024)
-
Beyond nothingness in the formation and functional relevance of voids in polymer films
Nature Communications (2024)
-
Sequence structure controllable polymerization-induced self-assembly
Science China Chemistry (2024)
-
Lateral growth of cylinders
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
