
Abstract
Grade IV astrocytoma or glioblastoma multiforme (GBM) is one of the most aggressive and lethal tumors affecting humans. ADAR2-mediated A-to-I RNA editing, an essential post-transcriptional modification event in brain, is impaired in GBMs and astrocytoma cell lines. However, the role of ADAR2 editing in astrocytomas remains to be defined. Here, we show that ADAR2 editing rescue in astrocytomas prevents tumor growth in vivo and modulates an important cell cycle pathway involving the Skp2/p21/p27 proteins, often altered in glioblastoma. We demonstrate that ADAR2 deaminase activity is essential to inhibit tumor growth. Indeed, we identify the phosphatase CDC14B, which acts upstream of the Skp2/p21/p27 pathway, as a novel and critical ADAR2 target gene involved in glioblastoma growth. Specifically, ADAR2-mediated editing on CDC14B pre-mRNA increases its expression with a consequent reduction of the Skp2 target protein, as shown both in vitro and in vivo. We found that, compared to normal brain, both CDC14B editing and expression are progressively impaired in astrocytomas from grade I to IV, being very low in GBMs. These findings (1) demonstrate that post-transcriptional A-to-I RNA editing might be crucial for glioblastoma pathogenesis, (2) identify ADAR2-editing enzyme as a novel candidate tumor suppressor gene and (3) provide proof of principle that ADAR2 or its substrates may represent a suitable target(s) for possible novel, more effective and less toxic approaches to the treatment of GBMs.
Similar content being viewed by others


Introduction
Astrocytomas are tumors affecting the central nervous system and are classified by the World Health Organization (WHO) in increasing grades of malignancy from grade I to IV.1 Grade IV astrocytoma or glioblastoma multiforme (GBM) is a highly aggressive brain tumor, with a median survival that rarely exceeds 18 months in both children and adults.2, 3 GBM is resistant to conventional radiotherapy and chemotherapy and, in contrast to other cancers, patient outcomes have only marginally improved in the past decades.4 This is mainly due to the lack of comprehensive knowledge of the molecular pathways involved in GBM.5, 6 Several studies have focused on the genomic characterization of mutations in genes important in human astrocytomas, and different lines of evidence have been accumulated regarding the molecular pathways altered in these tumors.7, 8 The Skp2 protein, a critical component of the Skp2-SCF complex, is frequently impaired in GBMs.9, 10 This protein acts as an E3-ligase on specific target genes such as p27Kip1 and p21Cip1/Waf1/Sdi1 by driving their ubiquitylation and degradation.11 Notably, Skp2 overexpression is frequently observed in human cancers including glioblastomas.12, 13 Conversely, its target genes p21 and p27, known to have a pivotal role at the G1/S checkpoint, are found down-expressed.14, 15 Interestingly, all these genes were rarely found mutated in cancers,15, 16 highlighting the importance of post-transcriptional mechanisms in GBM physiopathology.
In mammals, A-to-I RNA editing is mediated by three different ADAR (adenosine deaminase acting on RNA) enzymes. ADAR1 and ADAR2 are ubiquitously expressed and catalytically active enzymes, whereas ADAR3 is only expressed in the brain and catalytically inactive.17, 18, 19 ADARs bind dsRNA through their N-terminal RNA-binding domains and convert adenosine into inosine via their C-terminal catalytic domain (deaminase domain).17, 18, 19 Inosine is recognized as guanosine by both splicing and translational machineries.20, 21 Consequently, ADARs have the potential to ‘fine-tune’/modulate genomic information with important consequences on several RNAs and proteins.
A-to-I RNA editing is highly frequent in mammalian brain compared with other tissues.18, 22 In particular, ADAR2-editing activity is crucial for the function of many proteins expressed in central nervous system23, 24, 25, 26 and is essential for normal brain development.27 Indeed, Adar2−/− mice die shortly after birth, displaying a severe neurological phenotype.28
The importance of A-to-I RNA editing on coding regions has been largely documented.23, 24, 25, 26 The vast majority of editing events, however, lies within non-coding sequences of pre-mRNAs, such as introns and untranslated regions, and are usually clustered within dsRNA structures formed by the pairing of inverted-repeated Alu elements.29, 30, 31, 32, 33 The biological meaning of such events is yet to be understood, though the emerging picture is that editing in non-coding regions might modulate gene expression by altering splicing, localization and degradation of target RNAs.34, 35
A variety of human cancers exhibits global hypo-editing in both coding and non-coding regions of several transcripts.36, 37, 38 Moreover, ADAR2 editing is impaired in GBM and astrocytoma cell lines.37, 38, 39 However, we still do not understand whether the loss of ADAR2 editing is relevant to astrocytoma tumor growth.
In this study, we show that ADAR2 inhibits astrocytoma tumor growth in vivo; furthermore, we have identified a new ADAR2 target gene and an ADAR2-responsive molecular pathway that are involved in cell proliferation through cell cycle modulation.
Results
ADAR2-editing activity inhibits astrocytoma growth in vitro and in vivo
We and others have previously shown that high-grade astrocytomas and astrocytoma cell lines lack ADAR2-editing activity as tested on numerous target genes.36, 37, 38 We thus investigated whether ADAR2 in astrocytoma cells would affect in vivo tumor growth.
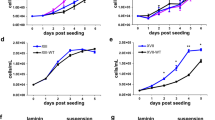
To this end, we generated a novel polyclonal astrocytoma derivative U118 cell line (EGFP-ADAR2, hereafter referred to as ADAR2), expressing the exogenous ADAR2 protein at almost 3-fold over the endogenous protein levels (Figures 1a and b), differently from the U118 cell lines previously used in which ADAR2 was expressed >10-fold over the endogenous enzyme.37 This level of ADAR2 protein was adequate to rescue normal editing levels at specific sites, such as those found in normal white matter (Supplementary Figures S1a and b) and primary astrocytes (data not shown). As controls, we generated polyclonal cell lines expressing identical levels of the inactive version of ADAR2 (EGFP-ADAR2 E/A, hereafter referred to as ADAR2 E/A; Figures 1a and b) and the empty vector (EGFP, hereafter referred to as vector). ADAR2 E/A was generated by a single point mutation in the catalytic domain that changes the key glutamate residue (E396) into an alanine, while the RNA-binding capability of the protein remains unaffected.40, 41, 42 The endogenous expression of ADAR1 was not modified by ADAR2 upregulation (Supplementary Figure S2). The modified U118 cell lines (ADAR2, ADAR2 E/A and vector) and untreated U118 cells were tested in parallel for cell proliferation, cell cycle progression and apoptosis (from day 1 to 4) (Figures 1c–e). The in vitro assays demonstrated that a threefold expression of the active ADAR2 induces a statistically significant decrease in cell proliferation (∼40% at 96 h compared to the controls) (Figure 1c) with a reduction of cell percentage in S-phase and a correlated increase of the G1-phase cell population (30% at 48 h) (Figure 1d). There was no evidence of either apoptosis (Figure 1e) or toxic effects (MTT test, data not shown).

ADAR2-editing activity regulates astrocytoma cell proliferation through cell cycle modulation. (a) ADAR2 immunoblotting of total protein extracts from U118, ADAR2 and ADAR2 E/A cells. (b) Relative protein expression is normalized using β-actin and expressed as arbitrary unit compared with the endogenous protein arbitrary set to 1. (c) Proliferation curve of 5 × 104 U118 cells untreated (light gray) or stably transfected with vector (dark gray), ADAR2 (red) and ADAR2 E/A (black) from day 1 to 4. Error bars indicate s.d. of five independent experiments. Mean±s.d. (n=5), *P<0.05, **P<0.01 when ADAR2 and ADAR2 E/A cells are compared. (d) 5 × 104 cells were tested for cell cycle distribution (BrdU plus PI) and analyzed by FACSCanto II flow cytometer from day 1 to 4. Experiment of ADAR2 cells (red) and the inactive ADAR2 E/A (black) cells at 24–48 h after seeding are shown. Mean±s.d. (n=3), *P<0.05, **P<0.01. Identical results were obtained using the Click-iT EDU Flow Cytometry Assay Kit (Invitrogen). (e) 5 × 104 cells were tested for apoptosis from day 1 to 4. ADAR2 cells (red) and the inactive ADAR2 E/A (black) cells are shown. Mean±s.d. (n=3). Of note, 5 × 104 cells were seeded simultaneously and proliferation (c), cell cycle (d) and apoptosis (e) were performed in parallel.
Next, we tested whether the ADAR2-editing activity may restrict astrocytoma growth in vivo as well. To address this issue, we subcutaneously injected 2.5 × 106 cells from each cell line (with EGFP cells showing >99% positivity with identical mean intensity of fluorescence) into the flank of nude mice (20 mice per cell line, in two independent experiments). Tumor growth was monitored every 2 days for over 1 month (Figure 2a).

ADAR2-editing activity inhibits astrocytoma tumor growth. (a) Tumor growth curve of 2.5 × 106 U118, vector, ADAR2 and ADAR2E/A cells. Tumor volume was measured and plotted as the fold increase relative to the first tumor measurement set as 1 (y-axis). Days post injections are shown on the x-axis. Mean±s.d. (n=20), *P<0.01 when ADAR2 and ADAR2 E/A tumors are compared. (b) Kaplan–Meier plot analysis of cumulative survival of indicated mice (2.5 × 106 U118 cell lines). (c) Immunohistochemistry analysis of ADAR2 and ADAR2 E/A tumors dissected at the exponential growth phase (15 days post-injection). Representative fields of tumor sections stained with anti-GFP, anti-Ki67 and TUNEL (arrows show apoptotic cells) antibodies are shown in the left panel. Percentage of positive cells for GFP, Ki67 and TUNEL in untreated U118 (light gray), vector (dark gray), ADAR2 (red) and ADAR2 E/A (black) tumors are shown in the right panel. Mean±s.d. (n=10), *P<0.01.
The mice injected with ADAR2 E/A cells developed an actively growing solid tumor, with an average size ranging from 0.092 cm3±0.008 (at day 15 post-injection) to 0.161 cm3±0.014 (at day 26 p.i., tumor growth peak), with a 1.75-fold increase (Figure 2a). In contrast, all 20 mice injected with ADAR2 cells developed an extremely small tumor mass, with an average size ranging from 0.062 cm3±0.009 (at day 15 p.i.) to 0.070 cm3±0.005 (at day 26 p.i.), with a 0.13-fold increase (Figure 2a). The difference in tumor growth between mice injected with ADAR2-U118 cells and those injected with all control cell lines (ADAR2 E/A, vector and untreated) was statistically significant (P<0.01; Figure 2a). Moreover, mice injected with cells expressing the active ADAR2 survived longer than controls; in particular, all control mice died within 48 days p.i., while mice injected with ADAR2-U118 cells survived in healthy conditions for 60 days (ADAR2 versus ADAR2 E/A, P<1 × 10−8; Figure 2b). To rule out the possibility that a higher number of cells overexpressing active ADAR2 might develop a growing tumor, 8 × 106 ADAR2 cells were also injected in parallel with the control cell lines (10 mice per cell line). Again, we observed that the cells overexpressing active ADAR2 did not give rise to growing tumors, while the three control cell lines already produced a visible solid tumor 6 days p.i. (data not shown). As ADAR2 modulates the cell cycle at the G1/S transition in vitro (Figure 1d), we also analyzed the cell cycle profile of cells (U118 untreated and ADAR2) isolated directly from xenograft tumors at day 15 p.i. Our results demonstrated that ADAR2 slows-down cell entry at the G1/S transition also in vivo (Supplementary Figure S3).
Biopsies from tumors in the exponential growth phase (day 15 p.i., 2.5 × 106 cells) were dissected from mice and immunohistochemistry was performed. We confirmed the presence of EGFP-tagged proteins in the tumor masses by GFP staining (Figure 2c, GFP and Supplementary Figure S4). We also examined the proliferative activity of tumor cells (Ki67 index), finding that tumors overexpressing ADAR2 showed a 50% decrease in cell proliferation as compared with controls (ADAR2 versus ADAR2 E/A, P<0.01) (Figure 2c, Ki67 and Supplementary Figure S4). Next, we estimated the apoptotic index using the TUNEL method, finding no statistically significant differences among tumors (Figure 2c, TUNEL and Supplementary Figure S4). Similar results were also obtained from IHC analysis of the tumors at day 26 p.i. (data not shown).
In conclusion, active ADAR2 decreases cell proliferation in vitro and in vivo by the modulation of the cell cycle at G1/S transition. Moreover, cells overexpressing ADAR2 considerably prolonged mice survival and significantly inhibited astrocytoma growth in vivo.
ADAR2 modulates the key cell cycle molecules Skp2 and p21Cip/p27Kip
As ADAR2 modulates the cell cycle at the G1/S transition in vitro and in vivo (Figure 1d; Supplementary Figure S3), we tested whether ADAR2 activity may affect specific proteins involved in this checkpoint, such as p27, p21 and Skp2, which are often altered in GBMs. In particular, Skp2 is frequently upregulated in cancers, while its target proteins p27 and p21 are strongly repressed.10, 43
We found that the functionally active ADAR2, but not the inactive ADAR2 E/A, significantly increased p27 and p21 protein levels, both in vitro (Figure 3a) and in vivo (Figures 3b and c; ADAR2 versus ADAR2 E/A, P<0.01). Conversely, active ADAR2 strongly decreased Skp2 protein level both in vitro (Figure 3a) and in vivo (Figures 3b and c; ADAR2 versus ADAR2 E/A, P<0.01). The modulation of p27, p21 and Skp2 protein levels was not due to variations in their mRNA levels (Figure 3d).

Analysis of p27/p21/Skp2 in vitro and in vivo upon ADAR2 expression (a) p27, p21 and Skp2 immunoblotting analysis of total protein extracts from vector, ADAR2 and ADAR2 E/A cell lines. (b) Immunohistochemistry of p27, p21 and Skp2 on ADAR2 and ADAR2 E/A tumor xenografts dissected at 15 days p.i.: representative fields are shown. (c) Percentage of p27, p21 and Skp2 positive cells detected by IHC analysis in tumor xenografts (15 days p.i.) originated from untreated U118 (light gray), vector (dark gray), ADAR2 (red) and ADAR2 E/A (black) cells. Mean±s.d., (n=10), *P<0.01. (d) qRT–PCR analysis of p27, p21 and Skp2 mRNAs from vector, ADAR2 and ADAR2 E/A cell lines. Mean±s.d. (n=4). (e) Immunoblotting of CDK4, CDK2, Cyclin D1 and Cyclin E proteins of total protein extracts from vector, ADAR2 and ADAR2 E/A cell lines.
Of note, ADAR2 deaminase activity specifically affected the Skp2/p21/p27 pathway, as other proteins, also involved in the G1/S checkpoint, such as CDK4, CDK2, Cyclin D1 and Cyclin E, were unaffected by ADAR2 overexpression (Figure 3e and data not shown).
Taken together, our data demonstrate that ADAR2 deaminase activity inhibits cell proliferation through the modulation of specific proteins (Skp2, p21 and p27), controlling the G1/S transition of cell cycle.
ADAR2 edits CDC14B and modulates its expression in astrocytomas
As the editing activity of ADAR2 is required to restrict astrocytoma tumor growth (Figures 1, 2, 3), we searched for ADAR target genes potentially involved in astrocytoma. We took advantage of a newly available database that includes validated or predicted editable substrates (DARNED; http://darned.ucc.ie)44 and of the ExpEdit web server (http://www.caspur.it/ExpEdit).45 No editing events were predicted in p21, p27 and Skp2 transcripts (data not shown). Among different possible ADAR2 target genes identified (data not shown), we focused on CDC14B phosphatase, as recent studies have demonstrated that it can act upstream of Skp2/p21/p27 pathway. In detail, CDC14B promotes Skp2 degradation through Ser64 dephosphorylation, preventing p21/p27 protein degradation.46
We therefore tested whether ADAR2 edits CDC14B, as predicted in silico. Among several intronic Alu elements potentially targeted by ADARs within CDC14B pre-mRNA, for example, within intron 7–8 and intron 13–14, we focused on a portion of CDC14B intron 7–8, encompassing AluJr and AluSx. These inverted Alu elements can be folded back in a stable dsRNA secondary structure, as calculated by the Zuker algorithm (free energy ΔG=−391)47 (Supplementary Figure S5). Next, we evaluated the presence of editing events in the AluJr region by direct sequencing of cDNA pools obtained from ADAR2-U118 cells and normal brain white matter. A total of 13 adenosines were found to be edited (1–13I) to a similar extent in the two samples (Figure 4a; Supplementary Figure S5d, respectively, and data not shown), while undetectable editing levels were found in U118 and modified U118 cell lines (vector and ADAR2 E/A; Figure 4a and data not shown). Editing on CDC14B pre-mRNA occurred upon ADAR2 expression in at least three astrocytoma cell lines (U118, A172, U87; Figure 4a and data not shown), thus increasing CDC14B mRNA and protein levels (Figures 4b–d; Supplementary Figure S6 and data not shown), while decreasing CDC14B pre-mRNA at least in U118 cells (Figure 4b). A recovered expression of CDC14B, due to ADAR2-editing activity, was also demonstrated in astrocytoma xenografts compared with the controls (ADAR2 E/A) (Figure 4e).

RNA editing mediated by ADAR2 enzyme modulates CDC14B expression in vitro and in vivo. (a) Sequence chromatograms of a portion of the endogenous CDC14B intronic sequence (intron 7–8) isolated from U118 gDNA/cDNA and U118 ADAR2 cDNA. Editing (herein identified as 1–4I and 13I) appears as a double peak of adenosine (in green) and guanosine (in black) in cDNA pools. Editing positions and percentages are indicated. (b) Representative experiment of semi-quantitative RT–PCR of CDC14B pre-mRNA and mRNA in ADAR2 and ADAR2 E/A cells. Levels of expression are expressed in arbitrary units calculated as a relative-fold increase compared to ADAR2 E/A sample arbitrarily set to 1. Samples are normalized to β-actin. Primer sequences are shown in Supplementary Table S1. (c) qRT–PCR of CDC14B mRNA in vector, ADAR2 and ADAR2 E/A cells. Mean±s.d., (n=3), *P<0.01. (d) CDC14B immunoblotting analysis of total protein extracts from vector, ADAR2 and ADAR2 E/A cell lines (left panel) and relative densitometric analysis (right panel). (e) Immunohistochemistry on ADAR2 and ADAR2 E/A tumor xenografts with anti-CDC14B antibody. Representative fields are shown.
This intriguing connection between ADAR2-mediated editing in a non-coding sequence of CDC14B and CDC14B expression was further confirmed by a reverse experiment in which we reduced the level of exogenous ADAR2 in the ADAR2-U118 cells by RNAi (Figure 5a). Stable silencing of ADAR2 decreased both editing at all the identified sites within the CDC14B transcript (Figure 5b and data not shown) and CDC14B mRNA expression levels (Figure 5c; scramble versus siADAR2, P<0.01). Moreover, siADAR2 cells showed a significant re-acceleration of cell proliferation rate when compared with scramble cells and ADAR2 cells (Figure 5d). Of note, siADAR2 cells behaved similarly to the untreated U118 cells (Figure 5d).

ADAR2 editing increases CDC14B expression and modulates cell proliferation. (a) ADAR2 immunoblotting analysis of total protein extracts from ADAR2-U118 cells and the same cell lines stably transfected with either scramble (scr) or siADAR2 plasmids. (b) Sequence chromatograms of the endogenous CDC14B transcript and editing levels of randomly selected sites are shown. Arrows indicate editing positions and percentages. (c) qRT–PCR of CDC14B in ADAR2, scrADAR2 and siADAR2 cells. Mean±s.d. (n=2), **P<0.01. (d) Proliferation curve of untreated U118, ADAR2, scrADAR2 and siADAR2 cells. Mean±s.d. (n=3), *P<0.05, **P<0.01 when scrADAR2 and siADAR2 are compared. (e) qRT–PCR of both ADAR2 and CDC14B at 24, 48 and 72 h after silencing of the endogenous ADAR2 in the A172 astrocytoma cell line. Mean±s.d. (n=2), *P<0.05, **P<0.01 when scrADAR2 and siADAR2 are compared. (f) Proliferation curves of A172 cells silenced for endogenous ADAR2 and A172 scramble. Mean±s.d. (n=2), **P<0.01.
Finally, transient silencing of endogenous ADAR2 was performed in the A172 astrocytoma cell line. These cells are malignant but not tumorigenic and, in contrast to U118, display a measurable editing level (∼80%) at the GluR-B Q/R site (data not shown). Editing at this ADAR2-specific site is a useful indicator of the effect/extent of ADAR2 activity. A172 cells silenced for ADAR2 (70% reduction of ADAR2 at 72 h post transfection; Figure 5e) showed a decrease of CDC14B expression level (scramble versus siADAR2, P<0.01 at 72 h p.t.; Figure 5e) and displayed a significant increase of cell proliferation (Figure 5f).
The above data suggest that ADAR2-mediated editing increases CDC14B expression in astrocytoma cells with a consequent inhibition of cell proliferation.
Importance of ADAR2-mediated CDC14B expression in astrocytoma cell proliferation
To investigate the role of CDC14B expression in cell proliferation, we directly transfected an equal amount of either the active CDC14B-flag or the inactive C314S-CDC14B-flag, which is unable to dephosphorylate Skp2,46 in two astrocytoma cell lines (U118 and U87) (Supplementary Figure S7a). We monitored cell proliferation and Skp2 levels. We found that the overexpression of the active CDC14B mimicks ADAR2 action with a significant reduction of cell proliferation and a decrease of Skp2 protein level when compared to its inactive form, in both astrocytoma cell lines (Figures 6a–d; P<0.05 in U118 and P<0.01 in U87 at 72 h p.t.).

Importance of ADAR2-mediated CDC14B expression in astrocytoma cell proliferation (a) Western blotting analysis of Skp2 protein at 48 and 72 h after transfection of either CDC14B-flag or C314S-CDC14B-flag in U118; in (b) proliferation curve of the U118 cell lines transfected as in (a). Mean±s.d., (n=2), *P<0.05. (c) Western blotting analysis of Skp2 protein at 48 and 72 h after transfection of either CDC14B-flag or C314S-CDC14B-flag in U87; in (d) proliferation curve of the U87 cell lines transfected as in (c). Mean±s.d., (n=2), *P<0.05, **P<0.01. (e) Schematic design for silencing and overexpression of CDC14B. Stably transfected ADAR2-U118 cells were silenced for the endogenous CDC14B at time 0 and, 24 h later (when we observed already a ∼90% reduction of the endogenous CDC14B, see Supplementary Figure S7b), were transfected with equal amounts (see Supplementary Figure S7c) of either CDC14B-flag (in red) or C314S-CDC14B-flag (in black) plasmids. (f) Proliferation curve of ADAR2-U118 cells silenced for endogenous CDC14B (in gray) and transfected 24 h later with either CDC14B-flag (in red) or C314S-CDC14B-flag (in black) plasmids. As a control of proliferation, we used the ADAR2-U118 cell line (in blue). Mean±s.d., (n=2), **P<0.01 when ADAR2 C314S-CDC14B-flag was compared with ADAR2 CDC14B-flag at 72 and 96 h; *P<0.05 when we compared ADAR2-U118 to ADAR2 C314S-CDC14B-flag or to ADAR2 CDC14B-flag at 72 and 96 h. (g) Western blotting analysis of Skp2 protein at 72 and 96 h after transient tranfection of CDC14B-flag and C314S-CDC14B-flag in ADAR2-U118 cells silenced for endogenous CDC14B.
To elucidate the contribution of CDC14B in ADAR2-mediated astrocytoma proliferation arrest, we first silenced the endogenous CDC14B in an ADAR2 background (ADAR2-U118 cells). Twenty-four hours later, we ectopically reintroduced an identical amount of either the active CDC14B-flag or the inactive C314S-CDC14B-flag (Figure 6e). Experimental controls for silencing of the endogenous CDC14B and overexpression of the exogenous CDC14B-flag are shown in Supplementary Figures S7b and c, respectively. A statistically significant difference in cell proliferation was found between the stably transfected ADAR2-U118 cells (which promote an endogenous upregulation of CDC14B; blue line in Figure 6f) and the ADAR2-C314S-CDC14B-flag (in which only the inactive CDC14B was present, as the endogenous CDC14B was silenced; black line in Figure 6f; P<0.05 at 72 and 96 h). These data imply that ADAR2-mediated CDC14B upregulation is sufficient to significantly decrease cell proliferation (difference in cell proliferation between blue and black lines in Figure 6f). An additional upregulation of active CDC14B-flag in ADAR2-U118 cells (red line in Figure 6f) further inhibited cell proliferation compared with the ADAR2-U118 control cells (P<0.05 at 72–96 h). Importantly, Skp2 protein was analyzed in the same cells and was found to be downregulated only upon the expression of CDC14B-flag (Figure 6g).
Overall, these data demonstrate that the over expression of the active CDC14B has an important role in astrocytoma proliferation, and that the level of CDC14B mediated by ADAR2 in these cells is sufficient to significantly inhibit cell proliferation through Skp2 modulation.
Direct correlation between ADAR2-editing activity and CDC14B expression in astrocytomas
The existence of a direct correlation between editing and expression of CDC14B was investigated in 19 astrocytomas at different grades of malignancy and in 3 samples of control normal brain tissue (Table 1). Overall, the highest editing levels of the CDC14B transcript were found in normal tissues, while a progressive decrease of editing was found in astrocytoma tumor samples, which correlates with tumor malignancy (from grade I to IV), being very low in GBMs (Figures 7a and b and Table 1). Together with a progressive loss of editing, we found a concomitant decrease of CDC14B mRNA expression in the same tumor samples, with the lowest levels in GBMs (Figure 7c and Table 1).

Editing and expression levels of CDC14B transcript in astrocytomas and control brain tissues. (a) The editing percentage (y axis) at each site (1–13I) (x axis) within the intron 7–8 AluJr of CDC14B is shown in control tissues (mean of 3 samples, in black), grade I (mean of 7 samples, in dark gray) and grade IV astrocytomas (mean of 12 samples, in light gray). (b) Representative sequence chromatograms of endogenous CDC14B (intron 7-8) from a white matter (Ctrl) and two astrocytoma tissues (grade I and IV) cDNAs. Top trace is gDNA from the same white matter sample. Arrows indicate editing positions and percentages. (c) qRT–PCR of CDC14B in control tissues (mean of 3 samples, in black), astrocytomas grade I (mean of 7 samples, in dark gray) and grade IV (mean of 12 samples, in light gray). Mean±s.d. (n=3), *P<0.05, **P<0.01. (d) Immunohistochemistry for CDC14B in representative samples of astrocytomas grade IV, astrocytomas grade I and normal brain tissues (Ctrl) (see Table 1). Endothelium is the internal positive control for CDC14B as shown in the bottom right panel. (e) Correlation of RNA-editing levels (y axis) and mRNA expression of CDC14B transcript (x axis) was analyzed in controls (black dots), astrocytoma grade I (dark gray dots) and grade IV (light gray dots) tissues. Examples of statistically significant correlation are shown. Non parametric Sperman’s Rho correlation was used. A P value less than 0.05 was set as the limit for statistical significance for each editing site.
CDC14B protein expression was also investigated in the same astrocytoma samples (Figure 7d and Table 1). Specifically, CDC14B protein was expressed in normal astrocytes and localized in both nuclei and cytoplasm (almost 100% of positive cells; Figure 7d, top panel). Low-grade astrocytomas showed a similar distribution of CDC14B, with ⩽90% of positive cells (Figure 7d, bottom left panel). GBM tissues appeared to be virtually negative for CDC14B staining, with just 5% of positive cells (Figure 7d, bottom right panel).
Of note, we found a statistically significant correlation between editing and mRNA expression of CDC14B in human astrocytoma tumors as found in 5/13 editing sites (site 1, 3, 6, 7 and 12) using a non-parametric Spearman’s correlation (Figure 7e).
In summary, we found that ADAR2 is active and CDC14B is both edited and expressed in normal white matter; on the contrary, a progressive reduction of ADAR2 activity and of CDC14B editing and expression was detected with increased astrocytoma tumor progression (from grade I to IV) and aggressiveness (Ki67, KPS and outcome) (Table 1).
Discussion
A-to-I RNA editing displays low variability among humans,48 indicating that the editing machinery is tightly regulated in healthy individuals. However, deranged RNA editing levels were found in many different human cancers including glioblastomas.49, 50
It has been shown that ADAR2-editing activity decreases in GBMs37, 38 and editing at the GluR-B Q/R site is essential in inhibiting astrocytoma invasiveness.51 Adar2 knockout mice die soon after birth due to severe neurological defects 28 and hence do not develop tumors. However, it is possible that ADAR2 ablation alone may not be sufficient to start a tumorigenic process (gliomagenesis), but multiple tumorigenic events/inputs are necessary, as occurs for several well-known tumor suppressor genes.52, 53, 54 Despite the fact that the reduction of ADAR2 editing seems to be important in glioblastoma cells, the role of this enzyme remains to be fully elucidated.
Herein, we explored whether ADAR2 has an onco-suppressive role in astrocytoma growth and which substrates/molecular pathways are eventually involved. As ADARs may have editing-independent functions,55 we investigated the role of both the wild-type and the deaminase-inactive version of ADAR2 in vivo.
We first found that fully active ADAR2 significantly decreases the ability of astrocytoma cells to generate growing tumors in a mouse model, while the catalytically inactive ADAR2 E/A does not (Figure 2a). Moreover, mice injected with active ADAR2 astrocytoma cells survived statistically longer than controls (Figure 2b). We investigated the possible mechanisms underlining these observations, finding that the deaminase activity of ADAR2 is essential to cell proliferation control.
Indeed, ADAR2-overexpressing cells accumulate in the G1-phase of cell cycle (Figure 1d; Supplementary Figure S3). The Skp2/p21/p27 molecular pathway, often impaired in GBMs, was found to be specifically modulated by ADAR2 activity, while other proteins (CDK4, CDK2, Cyclin D1 and Cyclin E) also important in this cell cycle checkpoint were found unaltered (Figure 3). In particular, active ADAR2 decreased Skp2 protein (usually upregulated in cancers) and conversely increased p21/p27 proteins (usually downregulated in a variety of tumors) in three astrocytoma cell lines and tumor xenografts (Figure 3). The finding that p21 and p27, two well-known tumor suppressor genes, respond significantly to ADAR2 is intriguing and opens new questions on whether/how A-to-I RNA editing might be involved in other tumors.11, 16
The observation that the ADAR2 deaminase activity is essential to specifically modulate the Skp2/p21/p27 pathway suggests that ADAR2 target gene(s) exist, which act upon this pathway in astrocytomas. As ADAR2 may edit several substrates in astroglia, we sought to identify new potential target genes involved in cell proliferation and cell cycle modulation in silico. The CDC14B transcript emerged as a new candidate, as it can regulate the S-phase entry in a Skp2-dependent manner.46 Indeed, we found that CDC14B pre-mRNA undergoes multiple editing events in non-coding regions in normal human brain (Supplementary Figure S5d and data not shown). Moreover, editing on CDC14B decreases in GBM tissues (Figures 7a and b, and data not shown) and in astrocytoma cell lines (Figure 4a and data not shown), while it is rescued upon ADAR2 overexpression (Figure 4a and data not shown).
ADAR-mediated RNA editing in coding regions of mRNA targets is a well-known mechanism able to recode the genetic information by amino-acid substitutions.33 On the contrary, the biological significance of RNA editing in non-coding regions of pre-mRNAs, such as Alu repeats, is still unclear. The idea is emerging that this type of editing may have an important role in RNA processing and in the modulation of gene-expression.35, 48, 56
Here, we report that ADAR2-mediated editing increases both CDC14B mRNA and protein levels in vitro and in vivo (Figures 4, 5, 6). This intriguing connection between CDC14B editing and expression was established in vitro by overexpressing either the active or the inactive ADAR2 in three astrocytoma cell lines (Figure 4; Supplementary Figure S6) and by silencing the overexpressed ADAR2 in ADAR2-U118 cells (Figures 5a–d) and the endogenous ADAR2 in the A172 astrocytoma cell line (Figures 5e–f). Noteworthy, mice xenografts and human astrocytoma tissues further supported our in vitro results (Figures 4e and 7d and Table 1).
How ADAR2 enhances CDC14B expression is not completely clear and further studies are necessary to address this question. We observed an increase of CDC14B mRNA and a concomitant decrease of its pre-mRNA in U118. Thus, we propose that editing events on CDC14B, as the ones identified herein, may accelerate its maturation process (Figure 4b and data not shown). Indeed, A-to-I editing has the potential to alter directly pre-mRNA sequences important for splicing events,30 as the ones identified in the CDC14B pre-mRNA sequences that undergo editing (Supplementary Figure S8). Despite these observations, we cannot rule-out the possibility that an indirect effect of ADAR2 editing might exist (such as editing-mediated pre-mRNA stability and/or editing on specific miR and/or editing on RNA/protein involved in splicing events).
Another novel piece of information emerging from our studies is the fact that CDC14B is both edited and modulated by ADAR2, as previously shown for GluR-B and GluR-A transcripts28 and as suggested for Munc18-1 and synaptotagmin-7 transcripts.56 How does CDC14B regulate astrocytoma cell proliferation? Our findings show that ADAR2-mediated-CDC14B upregulation significantly inhibits astrocytoma proliferation both in vitro and in vivo (Figures 4, 5, 6, 7, Table 1) and that this occurs through the regulation of Skp2 protein (Figure 6). The contribution of this novel ADAR2 substrate to astrocytoma tumor growth inhibition was demonstrated by the significant difference in cell proliferation between cells without a functional CDC14B in an ADAR2 background and ADAR2 cells (in which an active CDC14B was present). Indeed, ADAR2-mediated expression of CDC14B is sufficient to decrease consistently cell proliferation (Figure 6f). Interestingly, a stronger upregulation of the active CDC14B in ADAR2-cells further decreased U118 cell proliferation (Figure 6f), probably due to the simultaneous action of this protein on the different cell cycle checkpoints G1/S46 and G2/M.57 This observation may imply that a higher expression of ADAR2 can further decrease cell proliferation as previously observed,37 may be due to the action of CDC14B over multiple/different cell cycle check points.
Indeed, CDC14B phosphatase seems to be implicated in several cell pathways, albeit sometimes with contradictory functions.46, 58, 59 How this protein may act at multiple levels is at present unclear; most likely, CDC14B protein levels and/or different isoforms might be involved. An intriguing possibility is that RNA editing may be part of the equation, because we demonstrate that A-to-I RNA editing modulates CDC14B expression at least in brain. In line with these considerations, we observed that CDC14B editing and expression are connected in tissue samples, both being impaired in human astrocytomas. It is noteworthy that the decrease of CDC14B editing and expression correlates with the increase of astrocytoma aggressiveness and with poor patient outcome (Table 1).
Based on our findings, we propose that ADAR2-editing enzyme has the potential to inhibit astrocytoma growth through the upregulation of CDC14B, which in turn affects the Skp2/p21-p27 pathway (Figure 8). However, the existence of additional ADAR2 substrates, beyond CDC14B, implicated in the tumorigenic process remains highly likely.

Schematic representation of a model for the role of ADAR2 enzyme in astrocytoma cell proliferation. ADAR2 promotes CDC14B editing and overexpression (directly and/or indirectly) in astrocytoma cells. CDC14B leads to Skp2 degradation and consequent upregulation of p21 and p27 proteins, with cells accumulating in the G1 phase of the cell cycle. See also Supplementary Figure S5a.
Malignant gliomas are the most common primary brain tumor in adults and the most frequent solid neoplasia in children. These tumors are highly resistant to conventional treatments, highly aggressive and often located in critical areas of the brain and consequently difficult to treat by neurosurgery. Therefore, approaches aimed at identifying new molecular pathways and potential tumor suppressor genes are needed to design alternative gene-therapy strategies and more effective and less toxic treatments. Our study reveals the importance of the ADAR2 RNA-editing enzyme in brain cancer and represents an in vivo demonstration of the gene-therapy potential of ADAR2 for the treatment of gliomas. To this end, we can hypothesize that gene therapy-based approaches, aimed at introducing a wild-type copy of the ADAR2 gene or its downstream target molecules such as CDC14B into tumor cells, may represent a novel and effective therapy for these highly aggressive brain tumors.
Materials and methods
For additional materials and methods see Supplementary Information.
Tumor and control tissues
Astrocytoma tissue samples (Table 1) were dissected from tumor core and malignancy grade (from grade I to IV) was assessed according to the WHO classification. Control samples of normal white matter tissue were dissected close to the tumor during surgery. The study was approved by the local committee on the use of human samples for experimental studies and informed consent was obtained from each subject or subject’s guardian.
Immunohistochemistry
Immunohistochemistry was performed on 3 μm sections using the streptavidin–biotin method. The percentage of stained cells was measured by scoring at least 6–10 fields per section (at least 5 sections were considered) by two blind observers and positive cells were divided by the total number of the hematoxylin–eosin-stained cells. Images were acquired through the LUCIA software, version 4.81 (Nikon, Sesto Fiorentino, Florence, Italy) with a Nikon Digital Camera DXM1200F.
Tumorigenicity assay in nude mice
2.5 × 106 or 8 × 106 cells (U118 and polyclonal cell lines stably transfected with EGFP vector, EGFP-ADAR2 and EGFP-ADAR2 E/A) were subcutaneously injected in the flank of 6-week-old female nude mice (nu/nu, Charles River, Wilmington, MA, USA) after less than six serial passages in vitro. A total of 10 mice/cell line were injected on both flanks with untreated, EGFP, ADAR2 E/A and ADAR2 U118 cells, in two independent experiments (with a total of 20 mice/cell line). Tumor size was assessed every 2 days by caliper measurement. Tumor volume was calculated as follows: volume (mm3)=D × d2 × π/6, where D and d are the longest and the shortest diameters, respectively. For survival analysis, the mice injected with 2.5 × 106 cells were monitored for 60 days. Mice injected with the three tumorigenic cell lines died or were killed for ethical reasons (500 mm3 tumor size) within 48 days p.i. In particular, 62.5% of mice injected with the untreated cells, 50% of mice injected with the vector cells and 37.5% of mice injected with the ADAR2 E/A cells reached the 500 mm3 tumor size. Differently, all the mice injected with ADAR2 cells developed extremely small tumors (with a tumor mass never reaching 500 mm3) and survived in healthy conditions.
siADAR2 cell line and siCDC14B cell line
To obtain an ADAR2-U118 cell line stably silenced for ADAR2, we used the BLOCK-iT inducible Pol II miR RNAi Expression Vector Kit by EmGFP (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
ADAR2-U118 cells (3 × 105) were seeded and then transiently silenced for endogenous CDC14B (ON-TARGETplus SMART pool, Thermo Scientific Dharmacon, Lafayette, CO, USA). After 24 h the silenced cells were transfected with 4 μg of CDC14B-flag and C314S-CDC14B-flag inactive constructs (kindly provided by G Rodier) using Lipofectamine 2000 (Invitrogen) and cell proliferation was monitored for 3 days.
References
Kleihues P, Burger PC, Scheithauer BW . The new WHO classification of brain tumours. Brain Pathol 1993; 3: 255–268.
Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev 2001; 15: 1311–1333.
Zhu Y, Parada LF . The molecular and genetic basis of neurological tumours. Nat Rev Cancer 2002; 2: 616–626.
Stupp R, van den Bent MJ, Hegi ME . Optimal role of temozolomide in the treatment of malignant gliomas. Curr Neurol Neurosci Rep 2005; 5: 198–206.
Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res 2008; 68: 3286–3294.
Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell 2005; 8: 119–130.
Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol 2010; 12: 49–57.
Lee SH, McCormick F . Downregulation of Skp2 and p27/Kip1 synergistically induces apoptosis in T98G glioblastoma cells. J Mol Med 2005; 83: 296–307.
Saigusa K, Hashimoto N, Tsuda H, Yokoi S, Maruno M, Yoshimine T et al. Overexpressed Skp2 within 5p amplification detected by array-based comparative genomic hybridization is associated with poor prognosis of glioblastomas. Cancer Sci 2005; 96: 676–683.
Schiffer D, Cavalla P, Fiano V, Ghimenti C, Piva R . Inverse relationship between p27/Kip.1 and the F-box protein Skp2 in human astrocytic gliomas by immunohistochemistry and Western blot. Neurosci Lett 2002; 328: 125–128.
Kitagawa K, Kotake Y, Kitagawa M . Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci 2009; 100: 1374–1381.
Nakayama KI, Nakayama K . Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol 2005; 16: 323–333.
Chiarle R, Fan Y, Piva R, Boggino H, Skolnik J, Novero D et al. S-phase kinase-associated protein 2 expression in non-Hodgkin's lymphoma inversely correlates with p27 expression and defines cells in S phase. Am J Pathol 2002; 160: 1457–1466.
Coqueret O . New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 2003; 13: 65–70.
Abukhdeir AM, Park BH . p21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med 2008; 10: 1–18.
Hershko DD . Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008; 112: 1415–1424.
Nishikura K . Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol 2006; 7: 919–931.
Bass BL . RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 2002; 71: 817–846.
Keegan LP, Gallo A, O'Connell MA . The many roles of an RNA editor. Nat Rev Genet 2001; 2: 869–878.
Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S . Synthetic polynucleotides and the amino acid code. V. Proc Natl Acad Sci USA 1962; 48: 613–616.
Rueter SM, Dawson TR, Emeson RB . Regulation of alternative splicing by RNA editing. Nature 1999; 399: 75–80.
Paul MS, Bass BL . Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J 1998; 17: 1120–1127.
Greger IH, Khatri L, Ziff EB . RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 2002; 34: 759–772.
Ohlson J, Pedersen JS, Haussler D, Ohman M . Editing modifies the GABA(A) receptor subunit alpha3. RNA 2007; 13: 698–703.
Seeburg PH, Higuchi M, Sprengel R . RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev 1998; 26: 217–229.
Seeburg PH . A-to-I editing: new and old sites, functions and speculations. Neuron 2002; 35: 17–20.
Wahlstedt H, Daniel C, Enstero M, Ohman M . Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 2009; 19: 978–986.
Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000; 406: 78–81.
Chen LL, Carmichael GG . Gene regulation by SINES and inosines: biological consequences of A-to-I editing of Alu element inverted repeats. Cell Cycle 2008; 7: 3294–3301.
Athanasiadis A, Rich A, Maas S . Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2004; 2: 2144–2158.
Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 2004; 22: 1001–1005.
Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009; 324: 1210–1213.
Nishikura K . Editing the message from A to I. Nat Biotechnol 2004; 22: 962–963.
Nishikura K . Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 2010; 79: 321–349.
Farajollahi S, Maas S . Molecular diversity through RNA editing: a balancing act. Trends Genet 2010; 26: 221–230.
Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res 2007; 17: 1586–1595.
Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L et al. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem 2008; 283: 7251–7260.
Maas S, Patt S, Schrey M, Rich A . Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci USA 2001; 98: 14687–14692.
Galeano F, Leroy A, Rossetti C, Gromova I, Gautier P, Keegan LP et al. Human BLCAP transcript: new editing events in normal and cancerous tissues. Int J Cancer 2010; 127: 127–137.
Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL . Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 2005; 309: 1534–1539.
Gallo A, Keegan LP, Ring GM, O'Connell MA . An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. Embo J 2003; 22: 3421–3430.
Lai F, Drakas R, Nishikura K . Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J Biol Chem 1995; 270: 17098–17105.
Halfter H, Friedrich M, Resch A, Kullmann M, Stögbauer F, Ringelstein EB et al. Oncostatin M induces growth arrest by inhibition of Skp2, Cks1, and cyclin A expression and induced p21 expression. Cancer Res 2006; 66: 6530–6539.
Kiran A, Baranov PV . DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 2010; 26: 1772–1776.
Picardi E, D'Antonio M, Carrabino D, Castrignano T, Pesole G . ExpEdit: a webserver to explore human RNA editing in RNA-Seq experiments. Bioinformatics 2011; 27: 1311–1312.
Rodier G, Coulombe P, Tanguay PL, Boutonnet C, Meloche S . Phosphorylation of Skp2 regulated by CDK2 and Cdc14B protects it from degradation by APC(Cdh1) in G1 phase. Embo J 2008; 27: 679–691.
Zuker M . Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31: 3406–3415.
Greenberger S, Levanon EY, Paz-Yaacov N, Barzilai A, Safran M, Osenberg S et al. Consistent levels of A-to-I RNA editing across individuals in coding sequences and non-conserved Alu repeats. BMC Genomics 2010; 11: 608.
Gallo A, Locatelli F . ADARs: allies or enemies? The importance of A-to-I RNA editing in human disease: from cancer to HIV-1. Biol Rev Camb Philos Soc 2011; 87: 95–110.
Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G . Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis 2011; 32: 1569–1577.
Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 2002; 8: 971–978.
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P . Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82: 675–684.
Jackson RJ, Engelman RW, Coppola D, Cantor AB, Wharton W, Pledger WJ . p21Cip1 nullizygosity increases tumor metastasis in irradiated mice. Cancer Res 2003; 63: 3021–3025.
Martin-Caballero J, Flores JM, Garcia-Palencia P, Collado M, Serrano M . Different cooperating effect of p21 or p27 deficiency in combination with INK4a/ARF deletion in mice. Oncogene 2004; 23: 8231–8237.
Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM et al. Editing independent effects of ADARs on the miRNA/siRNA pathways. Embo J 2009; 28: 3145–3156.
Yang L, Zhao L, Gan Z, He Z, Xu J, Gao X et al. Deficiency in RNA editing enzyme ADAR2 impairs regulated exocytosis. Faseb J 2010; 24: 3720–3732.
Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M . The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008; 134: 256–267.
Tumurbaatar I, Cizmecioglu O, Hoffmann I, Grummt I, Voit R . Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS One 2011; 6: e14711.
Chiesa M, Guillamot M, Bueno MJ, Malumbres M . The Cdc14B phosphatase displays oncogenic activity mediated by the Ras-Mek signaling pathway. Cell Cycle 2011; 10: 1607–1617.
Acknowledgements
This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC, Milan, Italy) grant to A Gallo and a special project 5 × 1000 AIRC grant to F Locatelli. The authors are grateful to Geneviève Rodier for the CDC14B clones, to Alekos Athanasiadis and to Ernesto Picardi for bioinformatics support and to Mattia Locatelli for statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Galeano, F., Rossetti, C., Tomaselli, S. et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 32, 998–1009 (2013). https://doi.org/10.1038/onc.2012.125
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.125
Keywords
This article is cited by
-
RNA modification: mechanisms and therapeutic targets
Molecular Biomedicine (2023)
-
The epitranscriptome of high-grade gliomas: a promising therapeutic target with implications from the tumor microenvironment to endogenous retroviruses
Journal of Translational Medicine (2023)
-
Deciphering glioma epitranscriptome: focus on RNA modifications
Oncogene (2023)
-
RNA modifications in cancer
British Journal of Cancer (2023)
-
Genome-wide perturbations of Alu expression and Alu-associated post-transcriptional regulations distinguish oligodendroglioma from other gliomas
Communications Biology (2022)
